

Abstract
Rampant phospholipid peroxidation initiated by iron causes ferroptosis unless this is restrained by cellular defences. Ferroptosis is increasingly implicated in a host of diseases, and unlike other cell death programs the physiological initiation of ferroptosis is conceived to occur not by an endogenous executioner, but by the withdrawal of cellular guardians that otherwise constantly oppose ferroptosis induction. Here, we profile key ferroptotic defence strategies including iron regulation, phospholipid modulation and enzymes and metabolite systems: glutathione reductase (GR), Ferroptosis suppressor protein 1 (FSP1), NAD(P)H Quinone Dehydrogenase 1 (NQO1), Dihydrofolate reductase (DHFR), retinal reductases and retinal dehydrogenases (RDH) and thioredoxin reductases (TR). A common thread uniting all key enzymes and metabolites that combat lipid peroxidation during ferroptosis is a dependence on a key cellular reductant, nicotinamide adenine dinucleotide phosphate (NADPH). We will outline how cells control central carbon metabolism to produce NADPH and necessary precursors to defend against ferroptosis. Subsequently we will discuss evidence for ferroptosis and NADPH dysregulation in different disease contexts including glucose-6-phosphate dehydrogenase deficiency, cancer and neurodegeneration. Finally, we discuss several anti-ferroptosis therapeutic strategies spanning the use of radical trapping agents, iron modulation and glutathione dependent redox support and highlight the current landscape of clinical trials focusing on ferroptosis.
Subject terms: Cell death in the nervous system, Cell biology, Neurological disorders
Introduction
Ferroptosis is regarded as a cell death modality of metabolism. The biochemical mechanisms of ferroptosis involve a complex interaction between oxidative stress, lipid metabolism, and iron homeostasis that results in the peroxidation of polyunsaturated fatty acid (PUFA)-containing phospholipids to produce phospholipid peroxide radicals.1–3 These lipid peroxide radicals can react with other PUFAs that, in turn, generate additional peroxide radicals, which propagate as a chain-reaction throughout the phospholipid bilayer, leading to cell rupture. Briefly, ferroptosis-prone PUFAs contain bis-allylic hydrogen atoms that are liable to removal from the PUFA scaffold, exposing carbon moieties that can react with ferric iron or oxygen radicals either directly, or subsequent to attachment of molecular oxygen, form lipid (hydro)peroxides.4 The oxidation of PUFAs in the cell membrane can be initiated by reactive oxygen species (ROS) such as hydroxyl radicals, which can be generated by labile iron in the Fenton reaction.5 Hence, iron levels are strictly regulated by a variety of storage, transport and export proteins, including ferritin, transferrin, hepcidin, ferroportin and transferrin receptor 1 (TFR1) to avoid excess iron-derived ROS generation.6 Yet, the peroxidation of PUFAs is unavoidable, and this would result in ferroptosis if not continually interdicted by cellular defences. Thus, ferroptosis is distinct from other cell death modalities, where the initiation of cell death is an active event (e.g., apoptosis); ferroptosis, rather, usually is initiated by the withdrawal of cellular antioxidant defences.
Several antioxidant systems target upstream and downstream pathways of lipid peroxidation, with glutathione peroxidase 4 (GPX4) being the principal enzyme responsible for inhibiting ferroptosis.7 GPX4 is the only known enzyme that can detoxify phospholipid hydroperoxides directly in membranes.8 GPX4 consumes glutathione (GSH) when it performs this function, and it is supported by certain metabolites (e.g. ubiquinone) and vitamins (vitamins E, K, A) that can reduce phospholipid peroxides. These metabolites are recycled by enzymes that all consume nicotinamide adenine dinucleotide phosphate (NADPH): thioredoxin reductases (TR), glutathione reductase (GR), Ferroptosis suppressor protein 1 (FSP1), Dihydrofolate reductase (DHFR), NAD(P)H Quinone Dehydrogenase 1 (NQO1) and retinal reductases. Thus, NADPH is the foundational metabolite fuelling ant-ferroptotic defence through the above-mentioned metabolic intermediators. NADPH is depleted during ferroptosis and must be regenerated to avoid cell death.1,9–12 The reductive potential of NADPH is obtained by carbon metabolism, which underscores the inherent coupling of metabolism with ferroptosis.
After discussing the fundamentals of ferroptosis, including how it is modelled, where it occurs in the cell, and the role of lipids (the fuel of ferroptosis) and iron (the fire of ferroptosis), this review will focus on defence strategies to directly modulate and regulate lipids and iron in conjunction with key ferroptosis defence enzymes and metabolites that depend on NADPH. Subsequently, we will discuss evidence for ferroptosis in parallel with evidence for NADPH dysregulation in degenerative diseases.
Modelling ferroptosis
In vitro models of ferroptosis have been developed to help understand the complex mechanisms underpinning this cell death modality. Key strategies to induce ferroptosis include 1.) system xc- inhibition by erastin or glutamate; which reduces cystine importation into cells leading to a lowering of intracellular glutathione. 2) Inhibition of glutathione synthesis by buthionine sulphoximine (BSO); which inhibits gamma-glutamylcysteine synthetase.1,13,14 3.) GPX4 direct inhibition via RSL3, depletion by FIN56 or indirect inhibition by FINO2.1,15–17 4.) iron-dependent lipid peroxidation; the addition of excess iron can induce ROS, lipid peroxidation and subsequent cell death, although toxicity is not always specific to ferroptosis18,19 and 5.) inhibition of ferroptosis suppressor protein 1 (FSP1) which prevents the production of reduced CoQ10 and other vitamins that possess lipophilic radical-trapping antioxidant properties.20
While existing models are useful tools to explore the biochemical mechanisms and risk factors leading to ferroptosis, there is a lack of translation into a clinical context, with no specific ferroptosis inhibitors or activators approved yet for clinical use. The measurement of ferroptosis in vivo is also limited by the lack a specific biomarker. Cell culture studies are often conducted in hyperoxic conditions with an abundance of metabolites exceeding physiological levels leading to a physiological translation gap. Cysteine deficiency is a foundational paradigm to initiate ferroptosis in vitro, yet we currently lack examples of chronic disease where cysteine deficiency is established. Apart from the brain, under physiological conditions system Xc-, composed of the transporter subunit, xCT (SLC7A11) and the regulatory subunit, SLC3A2, is modestly expressed in most tissues and Slc7a11 knockout mice appear healthy with a normal lifespan and have no clear adverse phenotype.21–24 However, this changes when metabolism is hijacked in cancer with SCL7A11 levels rising to meet a new demand of cysteine for glutathione synthesis.25 Hence, new models of metabolic disturbances relevant to diseases are required to understand ferroptosis vulnerability and/or resistance which is likely underpinning manifestations of ferroptosis in vivo.
Organelles involved in ferroptosis
Recent research has revealed that ferroptosis can be invoked and propagated by excessive lipid peroxidation at phospholipid-membrane-bound organelles, namely the mitochondria,26 endoplasmic reticulum,27 and Golgi apparatus.28 Since the peroxisome can synthesise and incorporate PUFA into the phospholipid membrane, peroxisomes may serve as not only a site of lipid peroxidation, but also a vehicle for propagating lipid peroxidation essential for ferroptosis progression.29 Conversely, increasing the number and size of lipid droplets (LD) is a strategy to sequester and shield PUFA from peroxidation, thus protecting the cells from ferroptosis.30,31 The activities of the nucleus, including (post)-transcriptional and cell cycle regulation, additionally serve to modulate ferroptosis in proliferative cancer cells. For instance, oxidative stress induces the translocation nuclear factor erythroid 2-related factor 2 (NRF2) from the cytosol to the nucleus where NRF2 transcriptionally activates the expression of enzymes involved in antioxidant defence system to minimise lipid peroxidation.32,33 It is also interesting to note that cell cycle arrest can either enhance ferroptosis sensitivity via stabilising p53 and CDK4/6 inhibition34 or promote resistance via inducing lipid droplet formation.35 As such, the role of nucleus in mediating either ferroptosis sensitivity or resistance appears context specific.
Like the nucleus, the lysosome plays a pleiotropic role in mediating ferroptosis. One of the first links between the lysosome and ferroptosis was drawn from the lysosomal-dependent autophagic process, which is regarded as an accelerator of ferroptosis via NCOA4-depedent ferritinophagy36,37 or RAB7A-dependent lipophagy, leading to the accumulation of reactive Fe2+ and lipid peroxides (see38 for a comprehensive review on autophagy-driven ferroptosis). Nonetheless, recent studies uncover that under cyst(e)ine deprivation, cells not only activate ATF4 stress response pathway to mobilise lysosomal cysteine storage,39 but also increase the uptake and breakdown of cysteine-rich albumin in the lysosome by cathepsin B (CTSB) to export of cysteine.40 Cysteine is then used as a substrate for the synthesis of GSH, which is essential for most antioxidant enzymatic activity. Thus, the lysosome plays an important role in maintaining the intracellular cysteine pool and so also serves as a checkpoint for ferroptosis. Together, these studies demonstrate that whether lysosome represents an accelerator or a brake for ferroptosis is context-specific, dependent on the types of stress.
Regardless of internal sites of lipid peroxidation, the termination of ferroptosis converges on the plasma membrane where cell rupture is facilitated by plasma membrane pores.41,42 Studies have also shown intercellular propagation of death following treatment with ferroptosis inducing agents.1,11,43 The concept of ferroptotic cell death propagation was recently explored in a muscle remodelling limb development system.44 Co et al. demonstrated that a ferroptotic death signal primed cells to become redox bistable enabling ROS amplification-diffusion events causing ferroptosis to spread via trigger waves (self-regenerating chemical fronts that spread rapidly over extended distances45,46) subsequently causing mass cell death. This study highlighted the critical need for ferroptosis defence systems to prevent tissue damage. Collectively, ferroptosis occurs because of a collapse of cellular antioxidant defence system leading to excessive lipid peroxidation at the phospholipid membrane of various internal organelles and plasma membrane.
Lipid peroxidation and ferroptosis susceptibility
Ferroptosis occurs because of lipid peroxidation. The composition of phospholipids in the plasma membrane dictates ferroptosis vulnerability and the resources required to defend against it. These lipid classes are defined by double long-chain hydrocarbon attached to a glycerol backbone. The glycerol molecule contains a phosphate (3-position) that can be conjugated to different head groups. The four most common headgroups (choline, ethanolamine, serine and inositol) have different biophysical and chemical properties to provide diverse building blocks for a flexible asymmetric curved lipid bilayer: since ethanolamine is smaller than choline, ethanolamine head groups dominate in the inner leaflet of lipid bilayers, whereas the larger choline has a greater abundance in the outer leaflet.47 These favourable biophysical properties of phosphatidylethanolamines are complicated by an increased propensity toward lipid peroxidation. In addition to being located medially where they are exposed to intracellular free radicals and Fe2+, phosphatidylethanolamines often have a higher abundance of PUFA tail groups that are more susceptible to oxidation.47 Indeed, when ferroptosis is induced these are prominently oxidised.48
The biochemistry of lipid peroxidation was characterised over 25 years ago and consists of three key events 1.) Initiation 2.) Propagation cycles and 3.) Termination.49–51 Initiation occurs when an electron oxidant/free radical (i.e., hydroxyl, alkoxyl or hydroperoxyl radicals) abstracts a hydrogen atom from a lipid fatty acid to produce a carbon centred radical.52 Different phospholipid species have varying vulnerabilities to initiation due to variable degrees of difficulty in abstracting a hydrogen atom: polyunsaturated fatty acids (PL-PUFAs) are highly susceptible due to the presence of bis-allylic hydrogens that are more easily abstracted than hydrogens in monounsaturated fatty acids (MUFAs) or fully saturated fatty acids (SFAs). This carbon centred radical reacts with dioxygen to produce a lipid peroxyl radical L-OO⋅, or PL-PUFA-OO⋅ which is responsible for a series of propagation cycles since PL-PUFA-OO⋅ can produce new radicals by abstracting hydrogen from adjacent phospholipids forming PL-PUFA-OOH.52 PL-PUFA-OOHs are positioned at a ferroptosis-intersection whereby they can either 1.) react with labile Fe2+ (Fenton-like reaction) to produce a PL-PUFA-O⋅ which can propagate a peroxidation chain reaction on adjacent phospholipid, ultimately leading to ferroptosis or 2.) be converted by GPX4 to a lipid alcohol, a chemically inert species that is not susceptible to Fe2+ and radical propagation. Like the initiation step, the rate of propagation lipid peroxidation is influenced by the strength of carbon-hydrogen bond dissociation energies favouring weaker bonds. The weakest bonds are those at the bis-allylic methylene positions, followed by monoallylic hydrogen and alkyl C-H bonds.49 Substitution of hydrogen with deuterium atoms at the bis-allylic position reduces peroxidation susceptibility and consequently decreases ferroptosis vulnerability.4
Lipid peroxidation of membrane phospholipids is the executioner of ferroptosis. Using an oxidative lipidomics approach with florescent probes (BODIPY 581/591 and dihydrorhodamine 123), Vanden Berghe et al. (2020) distinguished ferroptosis from other regulated cell death modalities (apoptosis, necroptosis and pyroptosis) by demonstrating greater levels of lipid peroxidation and predominance of oxidised phosphatidylethanolamine species (oxPE) followed by oxidized phosphatidylserine (oxPS) and phosphatidylinositol (oxPI).48 Preferential oxidation of specific phospholipids in ferroptosis reveals that ferroptosis vulnerability depends on the presence or absence of oxidative-sensitive lipids. Hence, ferroptosis sensitivity is inextricably linked to phospholipid species composition and distribution.
Modulation of phospholipid sensitivity to ferroptosis
Phospholipids can be synthesised de novo, but an efficient strategy to reduce (or increase) the ferroptosis risk of a phospholipid bilayer is via phospholipid remodelling. Through the Lands cycle, phospholipids can selectively substitute out and replace an acyl chain (sn-2 position). This occurs in two simplified stages 1.) phospholipases in the A2 family (PLA2) cleave the fatty acid at the sn-2 position to liberate a free fatty acid and lysophospholipid and 2.) a lysophospholipid acyl-transferase (LPLAT) esterifies the sn-2 position of the LPL with a new fatty acid.53,54 Acyl-CoA synthase long-chain (ACSL) family proteins are also required as they esterify CoA groups onto free fatty acids which enables their incorporation into phospholipids.
In 2015, Stockwell’s group conducted a genetic screen in haploid cells and discovered acyl-CoA synthetase long-chain family member 4 (ACSL4) and LPCAT3 as key proteins modulating lipid metabolism in ferroptosis.55 Similarly, MBOAT2 (also known as LPCAT4) was highlighted by a whole genome CRISPR activation screen as a ferroptosis supressing gene.56 ACSL4, LPCAT3 and MBOAT1/2 are enzymes that modulate polyunsaturated fatty acids (PUFAs) in membrane phospholipids, with varying phospholipid preferences, to modulate phospholipid sensitivity to ferroptosis.12,55,57 Specifically, MBOAT2 suppresses ferroptosis by selectively transferring MUFAs into Lyso-PE, thus decreasing availability of PE-PUFA, a preferred substrate for phospholipid peroxidation.56 LPCAT3 (also known as MBOAT5), preferentially introduces polyunsaturated acyl groups onto lyso-PC (sn-2 position) and ACSL4 (in conjunction with an LPCAT) sensitizes to ferroptosis by specifically esterifying arachidonic acid and adrenic acid into PE thus increasing the risk of oxidation and ferroptosis.12,57,58 Knockout of ACSL4 provided greater protection than knock out of LPCAT3 in inducible Gpx4-/- murine embryonic fibroblasts (Pfa1 cells), which the authors suggest implies a more dominant role of ACSL4 in ferroptosis induction, however also demonstrates the redundancy of other LPCATs.12
Ferroptosis sensitive phospholipids can also be modulated to decrease ferroptosis risk. Phospholipase PLA2G6 (PNPLA9, iPLA2beta) metabolises hydroperoxide phosphatidylethanolamines to lyso-phosphatidylethanolamines and oxidized fatty acid, thus mitigating ferroptosis vulnerability.59 iPLA2beta genetic or pharmacological inactivation removes this layer of lipid peroxide defence and sensitises cells to ferroptosis.60 If not modulated to a ferroptosis resistant species, phospholipid hydroperoxides can also fracture into secondary products with short fatty acyl residues esterified in parental phospholipid (sn-2 position). These truncated oxidised phospholipids are structurally and functionally similar to platelet-activating factor (thus also known as PAF-like phospholipids), which has recently been shown to initiate and propagate ferroptosis.61 Ferroptosis could be suppressed by PAF-acetylhydrolase (II) (PAFAH2), another enzyme capable of modulating phospholipids by converting the short acyl chain into lyso-phospholipids, thus acting similar to iPLA2beta to remove oxidised lipids and protect against oxidative stress and ferroptosis risk.61,62
Evidence of the impact of phospholipid modulation on ferroptosis vulnerability has also been shown in cells treated with exogenous MUFAs.63 MUFA-treated cells displayed a ferroptosis resistant phenotype which was dependent on MUFA activation by acyl-coenzyme A synthetase long-chain family member 3 (ACSL3) and displacement of PUFAs from the plasma membrane. The protection was associated with a reduction in lipid reactive oxygen species and levels of phospholipids containing oxidizable PUFAs. Recently, the protection of MUFA treatment via phospholipid modulation has been confirmed in vitro.64 Mice fed a diet enriched in oleic acid (a MUFA) had reduced iron-overload induced liver lipid peroxidation and damage. Protection was associated with decreased levels of polyunsaturated fatty acyl phospholipids and ether-linked phospholipids.
Phospholipids can also be protected from autoxidation via 7-dehydrocholesterol, a cholesterol precursor synthesized by sterol C5-desaturase (SC5D) showing potent anti-ferroptotic activity.65,66 In the oncogenic environment, 7-dehydrocholesterol supports ferroptosis prevention by using the conjugated diene to prevent phospholipid autoxidation consequently protecting mitochondria and plasma membranes from phospholipid autoxidation and ferroptosis.65,66
The role of iron in initiating ferroptosis
Iron is an abundant metal on earth that almost all lifeforms depend upon.67 This transition element is important for a plethora of biological functions due to its capacity to redox cycle in two oxidation states within physiological parameters, Fe2+ and Fe3+, which enables the delivery and storage of oxygen, acid-base reactions and the conduction of electrons in the electron transfer chain.68 This same iron chemistry that biology exploits for a host of cellular functions also inadvertently causes oxidative stress and lipid peroxidation (Fig. 1).
Fig. 1.

Central role of iron in reactive oxygen species generation and lipid peroxidation. Iron is involved directly and indirectly at several points to produce reactive oxygen species and lipid peroxidation. Indirectly, iron containing proteins in the electron transport chain (ETC) generate O2-• which is reduced to H2O2 by superoxide dismutase (SOD). H2O2 can either be quenched by catalase (CAT) or react with iron via the Fenton reaction, to generate hydroxyl radicals (•OH). The Fenton reaction can also catalyse the production of lipid peroxyl radicals (LOO• /LO•) from lipid hydroperoxide (LOOH). Radical trapping agents (RTAs) can quench lipid peroxyl radicals. Indirectly, iron contained in lipoxygenases (LOX) catalyse oxygenation of polyunsaturated fatty acids (PUFAs) and lipids to produce lipid hydroperoxide (LOOH). Glutathione peroxidase 4 (GPX4) can siphon lipid hydroperoxides away from fuelling lipid peroxidation and propagation by reducing PLOOH (high ferroptosis risk) to benign lipid alcohols (LOH). The reducing power of GPX4 is fuelled by reduced glutathione (GSH) which is dependent on NADPH to be recycled from its reduced form glutathione disulfide (GSSG). The breakdown of iron storage protein ferritin (FTH) can result in increased labile iron to facilitate these reactions. Figure created using Biorender.coms
If iron homeostasis is not in balance, the unique chemical properties of free iron can hamper cellular functions, primarily through the generation of oxidative stress and lipid peroxidation.2 The term “ferroptosis” which incorporates reference to iron (“ferrum”, the latin word for iron) was named in 2012 by Stockwell’s group based on the characterised “iron dependent” modality of cell death.1 This was based on the concept of labile iron being a catalyst for lipid peroxidation via Fenton- and Haber-Weiss–like reactions, in which H2O2 is reductively cleaved by ferrous iron to produce hydroxyl radicals that are then able to abstract a labile hydrogen from PUFAs.69 Iron containing enzymes can also facilitate the formation of PLOOH. Non-heme iron-containing lipoxygenases (LOXs) can also generate PUFA lipid hydroperoxides.57,70 The pro-ferroptotic role of LOXs is further evidenced by studies showing that knockdown (via siRNAs) or pharmacological inhibition of LOXs renders cells resistant to ferroptosis.4,71 However, LOX inhibitors have been proven to be potent radical-trapping antioxidants that protect lipids from autoxidation thus questioning the extent of which LOXs induce ferroptosis. While LOXs may contribute to the load of LOOH within the cell and potentiate ferroptosis vulnerability,72–74 the involvement of LOXs in initiating ferroptosis is still unclear.
While NADPH fuels several anti-ferroptotic proteins, there are two iron containing NADPH-dependent enzymes that can contribute to lipid peroxidation: (i) Heme containing NADPH oxidases (NOXs) which transfer electrons from cytosolic NADPH during the production of ROS which promotes lipid peroxidation1,75 and (ii) NADPH-dependent cytochrome P450 oxidoreductase (POR) which enables membrane polyunsaturated phospholipid peroxidation.76
Iron regulation for ferroptosis defence
To combat excess intracellular labile iron and consequently reduce ferroptosis vulnerability, the cell has two key defence strategies, 1. Sequester iron in ferritin: a “safe” non-toxic storage protein or 2. Control iron flux: Increase iron export and reduce import (Fig. 2).
Fig. 2.

Cellular labile iron pool regulation. The labile iron pool is regulated by several proteins including i.) transferrin receptor 1 (TFR1) that facilitates iron influx in the form of transferrin, ii.) ferroportin (FPN) an iron channel facilitating iron export, iii) Ferritin which can store labile iron or release iron after lysosomal degradation which is mediated by nuclear receptor activator 4 (NCOA4) and/or iv.) Heme degradation by heme oxygenase 1 (HMOX-1) which releases iron and produces byproducts Biliverdin (BVD) and carbon monoxide (CO). The iron response protein/Iron response element (IRP/IRE) system responds to labile iron concentrations and subsequently regulates the expression of several proteins involved in iron regulation. Figure created using Biorender.com
Ferritin and iron storage
The upregulation of ferritin to store labile iron mitigates free radical-mediated damage via labile iron and the Fenton reaction.77–79 There are two ferritin subunits, H and L, that facilitate iron detoxification and long-term storage in a redox-silent oxidised species.80 FTH1 has ferroxidase activity and converts reactive Fe2+ to a more stable Fe3+, which enables iron entry into the ferritin mineral core which is mediated by FTL.81 A single ferritin cage which consists of 24 H- or L- subunits can hold up to 4500 iron atoms.82
The iron response element/iron responsive protein (IRE/IRP) system is responsible for regulating the translation and subsequent expression of ferritin.83,84 The IRE/IRP system consists of a 5’ untranslated region of ferritin mRNA (IRE) and two RNA binding proteins (IRP1 and IRP2).83,84 IRP1 and IRP2 both respond to cellular iron levels but in different ways; IRP1 assembles an Fe-S cluster, turning it into an aconitase that cannot bind IREs, and IRP2 is targeted for proteasomal degradation by iron-dependent stabilisation of FBXL5, an E3 ligase that facilitates ubiquitination of IRP2.85 In both cases, increased iron leads to decreased IRE-binding capacity by IRP thus inhibiting the IRP from binding the IRE and repressing translation, resulting in increased ferritin synthesis along with other iron responsive proteins (i.e. ALAS-1).86 In contrast, under iron deprivation ferritin breakdown is activated by active nuclear receptor activator 4 (NCOA4) that binds and flags ferritin for lysosomal degradation and ferritin translation is repressed.87 Consequentially, degenerative models presenting with high tissue iron are often attempting to compensate with increased levels of ferritin.88,89
Autophagic degradation of iron storage processes also occur in response to ferroptosis induction.90,91 There are up to 35 autophagy related genes that contribute to the core autophagy machinery.36,92 Gao et al. used RNAi screening with genetic analysis to identify 11 autophagy related genes among other genes engaged in the pentose phosphate pathway and iron homeostasis as positive regulators of ferroptosis. Blockage of autophagy prevented the accumulation of labile iron and reactive oxygen species, thus preventing progression of ferroptotic cell death.93 Similarly, knockdown or knockout of autophagy related 5 and 7 (Atg5, Atg7) in fibroblasts and cancer cells decreased intracellular ferrous iron and lipid peroxidation in response to erastin, subsequently blunting ferroptotic cell death.92
The importance of iron sequestration and reduction is evidenced by attenuation of disease progression with either genetic (ferritin overexpression) or pharmacological modes (iron chelation).90,91 This is also true in the context of ferroptosis inducers where inhibition of NCOA4 prevented ferritin degradation and suppressed ferroptosis while overexpression of NCOA4 increased ferritin degradation and promoted ferroptosis.36,92 Other indirect ways of inhibiting ferritinophagy, such as via increased ApoE, have also shown protection against cysteine deficiency-induced ferroptosis.19 In addition to cytosolic ferritin, mitochondrial ferritin also exerts protection against erastin-induced ferroptosis.94
Iron efflux and influx: transferrin receptor 1 and ferroportin
Iron influx is largely controlled by the expression of the Transferrin Receptor (TFRC/TFR1), a dimeric glycoprotein receptor for iron-loaded transferrin in the plasma. Transferrin binds to iron in the blood in a tight but reversible configuration and transports it systemically.95 TFRC extracellular domains have high affinity for iron-loaded transferrin to form a TF-TFRC complex which is internalized via receptor-mediated endocytosis.95 Iron is reduced in the endosome from Fe3+ to Fe2+ by the NAD(P)H-dependent transmembrane ferrireductase STEAP3 or by intraluminal ascorbate and released into cytosol via solute carrier family 11 member 2 (SLC11A2/DMT1).95–98 Subsequently, TFRC and TF are recycled back to the cell membrane and extracellular fluid, respectively. The dependence of iron delivery by transferrin and transferrin receptor was confirmed in a study that investigated serum factors that induced ferroptosis: both transferrin and transferrin receptor were required for serum dependent ferroptosis.99
Stockwell’s group conducted an antibody screen to detect ferroptosis in mice immunised with erastin treated membranes from lymphoma cells.100 Interestingly, they identified an antibody (3F3 ferroptotic membrane antibody) with a human transferrin receptor 1 protein antigen that was effective as a ferroptosis staining reagent, leading to the proposal that transferrin receptor is a selective ferroptosis indicator. Indeed, under some pathological conditions, cells that are susceptible to ferroptosis have an upregulation of TFR1 and down regulation of ferritin.2,101 In contrast, downregulation of TFRC has been shown to attenuate the ferroptosis by reducing iron import.101
There is only one known transmembrane exporter of non-heme iron, Ferroportin (SLC40A1/ferroportin/FPN1).84 Erastin induces the downregulation of ferroportin, which is prevented by ferroptosis inhibitors (iron chelation, ferrostatin-1 and N-acetyl cysteine).102 Ferroportin knockdown exacerbates erastin-induced ferroptosis, whereas genetic or pharmacological overexpression renders protection.102,103 In vivo, ferroportin surface expression is dictated by hepcidin, a protein secreted primarily by hepatocytes into the circulation where it binds to its receptor ferroportin causing its internalisation and degradation.104 Hepcidin is regulated by several factors including i.) HFE, a MHC class I-like protein that binds beta-2 microglobulin and TFRC in its extracellular α1-α2 domain,105–107 ii.) hemojuvelin (HJV), a membrane protein that acts as a co-receptor for bone morphogenetic protein (BMP) to signal via the SMAD pathway to regulate hepcidin expression108,109 and iii.) transferrin receptor 2 (TfR2), which acts as an iron sensor that can bind iron-loaded transferrin in the bloodstream, and hepatocytes leading to hepcidin upregulation.110,111 Mutations in these key hepcidin regulating genes that leads to a reduced production of hepcidin, or mutations in hepcidin and ferroportin, can lead to an iron overload disorder called hereditary hemochromatosis (HH). If left untreated, hemochromatosis leads to iron accumulation in the skeletal muscle, liver, heart, pancreas, and joints leading to fatigue, cirrhosis, arrhythmias, diabetes, and arthritis.112–116 Due to pathological iron overload, ferroptosis has been implicated as a mechanism of HH complications. In two hemochromatosis mouse models that develop severe iron overload (Hjv–/– and Smad4Alb/Alb mice), elevated liver iron was associated with increased lipid peroxidation (MDA), decreased NADPH and liver damage that was attenuated with ferrostatin-1 treatment.24 This study also conducted microarray analyses of iron-treated bone marrow–derived macrophages and identified Slc7a11 as a candidate gene of ferroptosis in hemochromatosis, however future studies are required to characterise ferroptosis vulnerability in the human hemochromatosis population.
Exosomal transport of ferritin is a non-canonical cellular mechanism for exporting iron.117,118 Mammary epithelial and breast carcinoma cells survive in response to pharmacological and physiological ferroptotic stress due to an upregulation of a pathway involving multivesicular body/exosome expulsion of ferritin and iron out of the cell.118 This was shown to be mediated by the pentaspanin protein prominin2, which facilitated ferroptosis resistance via the formation of ferritin containing exosomes.118 Importantly, this mechanism introduced the concept of rapid modulation of intracellular iron levels ( < 2 h). Thus, controlling the labile iron pool via iron flux and storage is central to influencing ferroptosis susceptibility. However, some cancer cells that are reprogrammed to rapidly import iron for rapid proliferation are paradoxically resistant to ferroptosis.119 This is due to an additional layer of ferroptosis defence dictated by several systems utilising enzyme and metabolite coupling.
Iron mediated ferroptosis defence in infection, inflammation and immunity
Iron dysregulation has recently been implicated as an initiating factor of ferroptosis in a range of different infectious diseases.120–122 Fundamentally, iron is a necessary element for successful infection.123–125 As a response, host defence mechanisms activated during infection attempt to restrict iron from pathogens. Mucosal surfaces that act as an entry point to many pathogens are coated with a fine layer of fluid that contains a high concentration of lactoferrin and lipocalin 2 that sequester iron to restrict the abundance of iron to microbes.126,127 Lactoferrin is structurally and functionally similar to transferrin as an iron transport molecule, however unlike transferrin that releases iron in acidified endosomes ( < pH 5.5), lactoferrin does not release iron even at a low pH (i.e., pH of 3.5), ensuring that iron restriction occurs in infected tissues that are often characterised by a highly acidic environment.128,129 Lipocalin-2 (pseudonyms; siderocalin or NGAL for neutrophil gelatinase-associated lipocalin) is secreted in humans and mice by epithelia, activated neutrophils and macrophages, to confiscate bacterial siderophores including enterobactin (secreted by a subset of E.coli and other Gram-negative bacteria) that bind ferric iron, thus sequestering ferric iron from the invading bacteria.130,131 A lack of lipocalin-2, conceivably a first line iron chelation defence against ferroptosis, increases the mortality in mice during E.coli sepsis or pneumonia.132,133
In response to infection or inflammatory stimuli, a cytokine-driven increase in hepcidin results in a drop of plasma iron, a response known as ‘hypoferremia of inflammation’.129,134 A drop in serum iron has been reported in several diseases associated with ferroptosis including Alzheimer’s disease,135 Parkinson’s disease136 and multiple sclerosis137 (discussed in depth later). Hepcidin downregulates ferroportin, thus decreasing iron export from cells. This is particularly beneficial for preventing the release from macrophages that actively collect and recycle iron. In hepcidin KO mice, hypoferremia of inflammation is absent or significantly reduced.138,139 Iron overload disorders (i.e., hereditary hemochromatosis or β-thalassemia), compromise host induced iron restriction due to impaired hepcidin action, and subsequently cause increased susceptibility to infections with microbes that can exploit this weakness.129 Hepcidin mutation is one cause of familial hemochromatosis.140 Beta-thalassemia suppresses hepcidin production due to an over population of erythroid precursors that release erythroferrone,141 a hormone that inhibits hepcidin transcription by inhibiting bone morphogenetic protein signalling in hepatocytes.142,143
Amaral et al. found that M. tuberculosis increased both labile iron and lipid peroxidation in infected macrophages.120,144 Initially described as necroptosis, the dying macrophages displayed a clear ferroptosis signature of high oxidised lipids and low GPX4 expression. Since cell death was also prevented by ferrostatin-1 or iron chelation, Amaral et al. redefined the cell death as ferroptosis. M. tuberculosis has also been shown to promote dissemination of ferroptosis by the secretion of protein tyrosine phosphatase A, which enters a host cell nucleus to promote asymmetric dimethylation of histone H3 arginine 2 via targeting protein arginine methyltransferase 6 leading to the inhibition of GPX4 expression.145,146 While key findings have been replicated in a mouse model of tuberculosis,144 the translation to human tuberculosis remains to be investigated. Due to the iron-scavenging properties of macrophages, they are also inherently vulnerable to ferroptosis (reviewed elsewhere147). The ferroptosis inhibitor ferrostatin-1 is reported to reduce cell death in ferric citrate challenged bone marrow-derived macrophages24 and to mitigate erythrophagocytosis in red pulp macrophages from a rodent model of transfusion.148
GPX4 expression is essential for the function of a range of different immune cells including CD8+ and CD4+ T cells, which fail to expand and protect against acute lymphocytic choriomeningitis virus and Leishmania major parasite infections when lacking Gpx4.149 Dendritic cells fail to secrete pro-inflammatory cytokines (TNF and IL6) and express MHC class I in response to the maturation signal of lipopolysaccharide upon GPX4 inhibition by RSL3.150
While not a focus of this review about ferroptosis defence, the immune response can also act to induce ferroptosis in pathological cells. CD8+ T cells or natural killer cells, key regulators of antitumor host immunity, release IFNγ, which has been shown to exaggerate glutathione depletion, lower mRNA and protein levels of two subunits of system xc− (SLC3A2 and SLC7A11), and increase lipid peroxidation, so increasing sensitivity to ferroptosis activators.151 Ferroptosis in cancer cells is accompanied with elevated expression of PTGS2 and the release of prostaglandin E(2),152 which when certain levels are reached, play an immunosuppressive response.153 In addition, cancer cells dying from ferroptosis, in contrast to necroptosis, have also been shown to impede subsequent dendritic anti-tumour mechanisms.154 Thus, cancer cells may counteract ferroptosis with immunomodulation to progress tumour growth.155
Enzyme metabolite coupling in ferroptosis defence
Several antioxidant systems target upstream and downstream pathways of lipid peroxidation. Antioxidant systems involve a complex interaction between reducing agents that can be proteins, metabolites, or vitamins. Several studies have investigated the implication of vitamin supplementation on ferroptosis; however, we will discuss the role of key vitamins (vitamin A, E, K and C) in their respective enzyme-metabolite coupling. A common thread uniting all key enzymes and metabolites that combat lipid peroxidation during ferroptosis is a dependence on a key cellular reductant, nicotinamide adenine dinucleotide phosphate (NADPH). Here, we will review the key ferroptotic defence enzymes and metabolites that depend on NADPH; 1.) glutathione reductase (GR), 2.) Ferroptosis suppressor protein 1 (FSP1), 3.) NAD(P)H Quinone Dehydrogenase 1 (NQO1), 4.) Dihydrofolate reductase (DHFR), 5.) retinal reductases and 6.) thioredoxin reductases (TR) (Fig. 3).
Fig. 3.

The reducing power of NADPH fuels ferroptosis defence. Each nicotinamide adenine dinucleotide phosphate (NADPH) molecule can donate two electrons. Electrons donated by NADPH reduce key anti-ferroptotic enzymes; glutathione reductase (GR), Ferroptosis suppressor protein 1 (FSP1), NAD(P)H Quinone Dehydrogenase 1 (NQO1), Dihydrofolate reductase (DHFR) and retinal dehydrogenases (RDH) and thioredoxin reductases (TR), which enable them to further propagate reduction reactions of multiple metabolites and proteins; retinol, retinal, tetrahydrobiopterin (BH4), dihydrobiopterin (BH2), α-tocopherol quinone (αTocQ), α-tocopherol quinol (αTocQH2), ascorbate, dehydroascorbate (DHA), glutathione (GSH), glutathione disulfide (GSSG), glutathione peroxidase 4 (GPX4), thioredoxin oxidised (Trx-S2), thioredoxin reduced (Trx-(SH)2), peroxiredoxin oxidised (Prx-S2), peroxiredoxin reduced (Prx-(SH)2), coenzyme Q10 (CoQ10), coenzyme Q10 reduced (CoQ10H2) and vitamin K (vit K), ultimately resulting in the prevention of lipid peroxidation. Figure created using Biorender.com
Glutathione reductase (GR)/GSH/GPX4 and ascorbate (vitamin C)
Glutathione reductase (GR) is a key enzyme that replenishes reduced glutathione from the oxidised form. GR contains several highly conserved domains, one of which binds NADPH (residues 198–238).156 GR transfers two electrons from NADPH to GSSG, which results in the formation of two molecules of GSH and NADP + . GSH, a tripeptide composed of glutamate, cysteine, and glycine, subsequently reduces and recycles both 1.) GPX4 and 2.) ascorbate, along with a range of other metabolites and enzymes.
GSH/GPX4
As an initial electron donor, NADPH provides reducing power for the reaction and subsequent activation of major ferroptotic defence enzyme, GPX4.152 GPX4 is a selenoenzyme that prevents ferroptosis by detoxifying lipid hydroperoxides in cell membranes.7,9,15 The reducing power of GPX4 enables the reduction of PLOOH (high ferroptosis risk) to benign phospholipid alcohols (PLOH). GPX4 activity depends on the availability of reduced glutathione (GSH).8 Cysteine is considered the rate limiting substrate for GSH biosynthesis and hence sustained GPX4 activity.157 Circulating cysteine in the blood exists as the oxidised di-sulfide, cystine.158 Once imported via the system Xc- cystine/glutamate antiporter, NADPH catalyses the two-electron reduction of cystine to cysteine via thioredoxin reductase 1 (TXNRD1).159 Hence the maintenance of active GPX4 depends on NADPH at two levels to reduce both cystine to cysteine and to reduce GSSG to GSH.
GPX4 is regulated at transcriptional, translational and post translational levels. At a translational level, selenocysteine incorporation in the GPX4 active site is required to facilitate its protective function.160 Selenocysteine is a selenium containing amino acid enabling an oxidoreductase property in half of all selenoproteins.161 Selenium was initially regarded as a toxin present in agricultural feed,162 however, the tissue-protective function of selenium in ‘factor 3’ was soon appreciated in a rat model of liver necrosis (later characterised as ferroptosis163) due to dietary vitamin E deficiency.164 Selenium enacts a protective role as the amino acid selenocysteine (Sec), a crucial amino acid giving rise to an oxidoreductase property in half of all selenoproteins.161 The first selenoprotein discovered in mammals was glutathione peroxidase 1 (GPX1) which was thought to explain selenium deficiency induced peroxidation of unsaturated lipids in membranes.165 However, it was later confirmed that only GPX4 harbors the unique capacity to detoxify membrane lipid peroxides.9,152 The importance of selenium in ferroptosis defence was recognised due to its incorporation in GPX4.160,166 Conrad’s group have extensively explored the essential role of sec in Gpx4 through attempts to rescue the embryonic lethal Gpx4-/-mice.166 They show that a selenocysteine (Sec) to serine replacement in GPX4 does not protect from early embryonic lethality,160 and animals where a Sec is substituted to Cys (Sec differs from cys only by the substitution of sulphur for Se) in GPX4 fail to survive past 3 weeks.160
At a post translational level, ubiquitination/deubiquitylation and acetylation/deacetylation can regulate GPX4 activity and/or stability.167–170 For example, OUT deubiquitinase 5 (OTUD5) can bind and stabilise GPX4 thus preventing ferroptosis vulnerability, but MTORC1 activation induces autophagy and degradation of OTUD5 and consequently GPX4 decay and increased ferroptosis.171 Supraphysiological levels of the essential metal copper may also a role in GPX4 breakdown. Cu2+ can directly bind to GPX4 and induce the formation of GPX4 aggregates. This might account for GPX4 autophagic degradation mediated by Tax1 binding protein 1 in Cu2+-treated cells.172 A caveat in this report in that Cu2+ is not reported in the cytoplasm of cells under physiological conditions, where copper is believed to be only in the Cu+ oxidation state. However, copper chelators have also been shown to decrease ferroptosis vulnerability against erastin and RSL3 in vitro. In addition, copper treatment accelerated ferroptosis-induced tumour suppression in a mouse model of pancreatic cancer, which was associated with decreased expression of GPX4.172 Copper induced GPX4 deficiency may be relevant to copper overload conditions like Wilson’s Disease, an autosomal recessive genetic disease (mutation of ATP7B) characterized by copper overload and degeneration in multiple organs including the liver and brain. Recent studies in a copper loaded rat model of Wilsons Disease demonstrated decreased GPX4 expression and increased oxidative stress and lipid peroxidation markers, thus implicating ferroptosis as a potential mechanism underlying the neurological symptoms of Wilsons disease.173 Further studies are required to investigate the presence of GPX4 deficiency in human diseases. We hypothesise that common physiological ferroptosis defence limitations are more likely to manifest as NADPH deficiency, subsequently reducing GPX4 recycling, however rare GPX4 mutations that impact function and/or stability are known to exacerbate human disease pathology,174,175 possibly due to altered ferroptosis vulnerability.
GSH/ascorbate (vitamin C)
Both ascorbate (vitamin C) and GSH are abundant and stable antioxidants capable of donating electrons and scavenging various species of ROS. Unlike GSH which is synthesised intracellularly, ascorbate is acquired solely through the diet in humans, with severe deficiency leading to scurvy.176 Vitamin C exists in several redox states, including ascorbic acid/ascorbate and its two-electron oxidized form dehydroascorbic acid (DHA). DHA is reduced spontaneously by glutathione or enzymatically in reactions using glutathione or NADPH.177 Vitamin C and GSH can also directly interact with each other to exert a protective effect: glutathione can reduce oxidised vitamin C products through the glutathione-ascorbic acid cycle, thus shielding vitamin C from oxidation.178–181
In the oxidative stress context, reduced Vitamin C (i.e., ascorbate; regenerated by GSH and NADPH) directly reduces the tocopheroxyl radical (Toc•) to produce reduced tocopherol (Toc), which allows Toc to exert an anti-oxidant effect in lipid environments.182 As a direct anti-oxidant, studies have suggested that GSH and ascorbate have a partial redundancy in defence; in cells (human myeloid HL-60) with depleted GSH, pre- loading with vitamin C protected cells from death induced by H2O2.183 Conversely, the pharmacological enhancement of GSH by glutathione monoethylester can delay the onset of scurvy in rodents,184 probably via increased ascorbate stabilisation. However, the greatest reduction of ROS occurs when both ascorbate and GSH are present.183 In the ferroptosis context, where ascorbate cannot compensate for a lack of cysteine, vitamin C has been positioned as a ferroptosis inducer,185 since vitamin C can also act as a pro-oxidant. Under conditions of high ascorbate, vitamin C catalyses the reduction of free transition metal ions, like iron, which can cause the formation of radicals.186 In the few studies investigating the role of vitamin C in the context of ferroptosis, the pro-oxidant role of vitamin C was shown to predominate over its antioxidant capacity.185,187,188 Vitamin C supplementation induced lipid peroxidation, ROS and cell death associated with an inactivation of GPX4 that was partially rescued by DFO.185,187 In addition, increased vitamin C import via upregulation of SVCT2 promoted the reduction of intracellular Fe3+ to Fe2+, which reacted with excessive Vitamin C to produce severe oxidative stress and trigger ferroptosis in melanoma.188
Ferroptosis suppressor protein 1 (FSP1), coenzyme-Q10, vitamin E and vitamin K
Apoptosis-inducing factor mitochondria-associated 2 (AIFM2) was initially identified as a pro-apoptotic gene,189 but it was later given another name ferroptosis suppressor protein 1 (FSP1) due to its newly appreciated role in ferroptosis defence.190 In the absence of functional GPX4, FSP1 defends against lipid peroxidation via 1) the NAD(P)H-dependent reduction of coenzyme-Q10 (ubiquinone) to the lipid peroxyl radical-quenching molecule, CoQ10-H2 (ubiquinol)190,191; and/or 2) the recruitment of endosomal sorting complexes required for transport (ESCRT)-III that repair oxidatively damaged sections of the plasma membrane.192 FSP1-mediated reduction of lipid peroxides is an alternative pathway to GPX4, but FSP1 and GPX4 are not redundant as their activities are differentially regulated and they act co-operatively.190,191 FSP1 can also prevent ferroptosis defence through the recycling of vitamin E and K.190,191,193,194 The investigation of FSP1 inhibitors recently lead to the discovery of a compound class of 3-phenylquinazolinones that induce phase separation of FSP1 into molecular condensates that renders cells vulnerable to ferroptosis inducers.20
Coenzyme-Q10
Coenzyme Q (CoQ) is a hydrophobic lipid consisting of a redox active benzoquinone ring fused to a polyprenoid tail of varying lengths of isoprenoid sidechains depending on the species (10 is the most common in humans, CoQ10).195 CoQ10 is ubiquitous in human tissue where it is manufactured at the mitochondrial inner membrane (IM).196,197 Due to a primary role as an electron carrier molecule in the electron transport chain to facilitate ATP production,198 CoQ10 is highly abundant in metabolically active tissue (ie., heart, liver, kidney and brain).195 However, reduced CoQ10 (ubiquinol) also acts as a potent antioxidant that traps lipid peroxyl radicals, consequently preventing ferroptosis.190,191 Oxidised CoQ10 in the cytosol is recycled by FSP1 using NAD(P)H acting as a glutathione independent system to suppress ferroptosis.190,191 Removal of CoQ10 from cells by blocking CoQ10 synthesis enzyme COQ2 lead to increased basal and RSL3 mediated lipid peroxidation.191
Vitamin E
Vitamin E is a lipid-soluble antioxidant that encompasses a group of compounds, including α-, β-, and γ-tocopherol (Toc) which have different chroman rings. α-tocopherol is the most biologically active and well-studied form in humans.199 Intracellular reduced α-tocopherol can act as a direct inhibitor of lipid peroxide propagation by donating one electron to an alkylperoxyl radical (LOO•) resulting in the production of a tocopheroxyl radical (Toc•) and LOOH.200 This disrupts the propagation step of lipid peroxidation, suppressing the further production of LOOH and consequent ferroptosis. In contrast, GPX4 suppresses ferroptosis by reductively converting LOOH to LOH.182 Reduced α-tocopherol can also suppress pro-ferroptotic lipoxygenase activity thus reducing the generation of doubly- and triply oxygenated (15-hydroperoxy)-di-acylated PE species.57,190
There are two key pathways cells use to regenerate reduced α-tocopherol: 1.) FSP1 uses NADPH to reduce CoQ10 to CoQ10-H2 which subsequently recycles oxidised α-tocopherol,190,191,193 and 2.) as previously discussed, reduced Vitamin C (regenerated by GSH and NADPH) directly reduces the tocopheroxyl radical (Toc•) to produces reduced tocopherol (Toc).182
In addition, overoxidation of α-tocopherol can yield a distinct chemical entity, α-tocopherol quinone, which exists in the oxidised state but can be reduced to α-tocopherol quinol, a highly active lipid peroxyl quencher.201
Vitamin K
Vitamin K is a fat-soluble antioxidant initially identified and characterised in 1934 for its key role in blood coagulation,202 and more recently has been shown to play a role in ferroptosis defence.203 Vitamin K is a term used for a range of compounds that share a common structure of a 2-methyl-1,4-naphthoquinone core, also known as menadione. K3 is regarded as the simplest form, containing only the core and serves as an intermediate in human metabolism and is not obtained through the diet.204 After intestinal absorption, dietary sourced vitamin Ks (i.e., phylloquinone (vitamin K1) and menaquinones (vitamin K2)) are transported into the blood by lipoproteins.205 Cellular uptake of vitamin K is mediated via lipoprotein receptors.
A screen of naturally abundant vitamin compounds in GPX4 knock out mouse embryonic fibroblasts identified three forms of vitamin K— phylloquinone, menaquinone-4 (MK-4), and menadione that could prevent cell death triggered by TAM-induced GPX4 deletion.206 In several human cell and mouse models vitamin K compounds enacted ferroptosis defence via inhibiting lipid peroxidation.203,206 FSP1 reduced via NADPH was identified as a vitamin K reductase that reduces vitamin K to its hydroquinone (VKH2) to support ferroptosis suppression.194 In addition to FSP1, vitamin K epoxide reductase complex subunit 1 like 1 (VKORC1L1) can also reduce vitamin K to generate vitamin K hydroquinone.207 VKORC1L1 was initially identified in CRISPR-Cas9 knockout screens as a ferroptosis suppressor.207 Currently, the physiological reductant of VKORC1L1 is unknown.
NAD(P)H quinone dehydrogenase 1 (NQO1)
NAD(P)H quinone dehydrogenase 1 (NQO1) is an intracellular, cytosolic enzyme which catalyses the two electron reduction of quinones and other compounds including quinones, nitroaromatic compounds, imidazoles, and iron ions.208,209 The enzymatic function is initiated by binding an FAD cofactor, which is reduced by NAD(P)H.210 In the context of ferroptosis defence, NQO1 can function in the plasma membrane to recycle forms of ubiquinone211 and vitamin E, including alpha-tocopherol quinone212 (discussed previously), and as a direct superoxide reductase at high levels.213,214
Genetic manipulation of NQO1 has produced varying results indicating a context specific effect on ferroptosis. Deletion of NQO1 in human bone osteosarcoma U2OS cells did not impact RSL3 sensitivity.191 However, when deleted in combination with FSP1, cells were more sensitive to RSL3 than cells only deficient for FSP1. NQO1 overexpression in FSP1 KO cells promoted minor protection to RSL3. In contrast, in neuronal SH-SY5Y cells, overexpression of NQO1 resulted in increased lipid peroxidation following treatment with RSL3 and erastin, while NQO1 knockdown protected cells against ferroptosis by lowering iron and lipid contents and increasing GPX4, xCT, and the GSH/GSSG system.215 The reason for altered iron homeostasis was not explored, however the authors hypothesised that it was due to decreased proliferation driven by degradation of c-fos. NQO1 has recently been shown to directly interact with unstructured DNA-binding domain of c-Fos, which inhibits its proteasome-mediated degradation. This induces CKS1 expression and control of cell cycle progression at the G2/M phase leading to cancer proliferation.216 Indeed, NQO1 is pleiotropic antioxidant enzyme and has also been shown to control the stability of multiple proteins including p53,217,218 p73,219 p33ING1b,220 and HIF-1α.221
Dihydrofolate reductase (DHFR), Tetrahydrobiopterin (BH4) and vitamin E
Dihydrofolate reductase (DHFR) regenerates tetrahydrobiopterin (BH4) while consuming NAD(P)H, which can act alone or in synergy with vitamin E as an endogenous radical trapping agent that protects lipid membranes from autoxidation.222 CRISPR-Cas9 screens identified BH4 as a metabolic modifier of lipid peroxidation upon GPX4 inhibition but not cysteine depletion.222 In response to erastin (but not RSL3), downregulation of BH4 via knockdown of the first-rate limiting enzyme of BH4 synthesis, GTP cyclohydrolase-1 (GCH1), increased lipid peroxidation and intracellular ferrous iron resulting in decreased in colorectal cancer cell viability.223 Supplementation of BH4 was sufficient to rescue erastin induced ferroptosis in GCH1 knockdown cells. Acting upstream of BH4, methotrexate synergizes with GPX4 inhibition to induce ferroptosis by reducing DHFR’s function.222
Retinol and derivatives
Retinol (vitamin A) is a lipid soluble micronutrient absorbed through dietary sources including retinyl esters and β-carotene. While retinoids participate in a variety of physiological functions including affecting the expression of genes that regulate cell proliferation, differentiation and death,224,225 the anti-ferroptotic function is proposed to be primarily due to a radical trapping capacity that directly interdicts lipid radicals.226,227 However, vitamin A also has a higher reactivity compared to endogenous esterified PUFAs towards lipid peroxidation via autoxidation (propagation) and can thus divert free radical chain reactions away from membrane phospholipids to prevent ferroptosis.228 Retinol is regenerated from retinal by retinal reductases, which require NADPH.
TRX (thioredoxin) and PRDX6
The thioredoxin system consists of thioredoxins (TRX) and thioredoxin reductase (TRXRD). TRXRD uses electrons from NADPH to reduce oxidised thioredoxin (TRX), which can subsequently reduce cystine to cysteine, the rate-limiting substrate for GSH biosynthesis and, in turn, regulate GPX4 activity.229,230 Indeed, overexpression of Trx-1 in mice reversed decreases of GPX4 induced by toxins used to model Parkinson’s disease (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine/1-methyl-4-phenylpyridinium (MPP + )).229 In vitro experiments confirmed that GPX4 deficiency and toxicity induced by MPP+ is rescued by ferrostatin-1 or by Trx-1 overexpression, implying a role of Trx-1 in ferroptosis defence.229 TRX was initially identified as an extracellular protein but is now known to localise intracellularly in the cytoplasm, mitochondria and nucleus.231,232 Together with NADPH, the TRX system reduces disulfide bonds in target proteins (i.e, peroxiredoxin family), restoring their activity and shielding them from oxidative damage.233
Peroxiredoxin 6 (PRDX6) is a member of the peroxiredoxin family of antioxidant enzymes that plays a crucial role in the repair of cell membrane lipid peroxidation.234 PRDX6 is a trifunctional enzyme that exhibits both peroxidase, phospholipase A2 (PLA2) and lysophosphatidylcholine acyl transferase (LPCAT) activities.235,236 The peroxidase activity enables PRDX6 to reduce peroxides and ROS by utilizing reducing equivalents from GSH and the thioredoxin system. The peroxidase activity can reduce a range of substrates with various implications; reduction of short chain hydroperoxides such as H2O2 would avert the formation of reactive oxygen (ROS) that are involved in biogenesis of lipid peroxidation, whereas the reduction of PLOOHs (i.e., phosphatidylcholine hydroperoxide (PCOOH)) would enable a lipid membrane repair process.236,237
NADPH – the key metabolite fuelling ferroptosis defence
NADPH is a substrate for both de novo lipid synthesis and for all key enzymes and metabolites that defend against ferroptosis, as the principal source of electrons (Fig. 3). NADPH also drives de novo synthesis of fatty acids, cholesterol, amino acids and nucleotides, as well as being used for nitric oxide signalling and ROS generation by NOX enzymes.238 To sustain these important roles in building biomass, signalling and cellular maintenance, the cell must constantly synthesise NADPH (Fig. 4).
Fig. 4.

Key metabolic pathways fuelling NADPH generation. 1.) The pentose phosphate pathway, shunts from glucose-6-phosphate (G6P) to regenerate two nicotinamide adenine dinucleotide phosphates (NADPH) in two dehydrogenase steps i. G6P to 6-phosphogluconate (6PG) via glucose 6-phosphate dehydrogenase (G6PD) and ii. 6PG to ribose 5-phosphate (Ru5P) via 6 phosphogluconate dehydrogenase (6PGD). 2.) Malic enzymes 1, 2 and 3. Malic enzymes located within cytoplasm (ME1) and mitochondria (ME2 and ME3) catalyse the oxidative decarboxylation of malate to pyruvate while concurrently generating NADPH from NADP. 3.) Isocitrate dehydrogenases (IDHs) catalyse oxidative decarboxylation to produce NADPH. IDH1 localizes to varying extents to the cytoplasm, and IDH2/3 localise to the mitochondria. 4.) One-carbon (1 C) and folate metabolism which involves a series of 1 C transformations that generate and consume redox equivalents including the oxidisation of 10-Formyltetrahydrofolate (10-formyl-THF) to carbon dioxide (CO2) by cytosolic (1)/mitochondrial (2) 10-formyltetrahydrofolate dehydrogenase (ALDH1L1/2). NADPH can also be produced by reversible conversions of 5,10-methylene-tetrahydrofolate (5,10-meTHF) to 10-formylTHF by cytosolic (1) and mitochondrial (2) Methylenetetrahydrofolate Dehydrogenase (MTHFD1/2 L). Figure created using Biorender.com
Metabolic pathways that produce NADPH
The oxidative pentose phosphate pathway is regarded as the major pathway for NADP+ reduction to NADPH and is a glucose-oxidising pathway, shunted from glucose-6-phosphate to produce ribose 5-phosphate via two dehydrogenase steps which regenerate two nicotinamide adenine dinucleotide phosphates (NADPH) (recently reviewed).239 The pentose phosphate pathway correlates with NADPH demand, which is enabled by NADP regulation of G6PD as a substrate and via an allosteric binding site on G6PD.240,241 Oxidative stress also imparts a higher NADPH demand and thus expression of several pentose phosphate genes (G6PD, 6PGD, TK and TALDO) are also upregulated by the nuclear respiratory factor 2 (NRF2) family of transcription factors.242,243 Genetic deficiencies in the pentose phosphate pathway occur commonly due to mutations in glucose-6-phopshate (discussed in more detail later).
NADPH can also be regenerated by cytosolic glycolytic and mitochondrial TCA cycle intermediates via Malic enzymes 1, 2 and 3. Malic enzymes, located in cytoplasm (ME1) and mitochondria (ME2 and ME3), catalyse the oxidative decarboxylation of malate to pyruvate while concurrently generating NADPH from NADP.244,245 Like the pentose phosphate pathway, MEs are upregulated in various cancer cell lines.246,247 Genomic deletion of ME2, which diminishes NADPH production, consequently induces higher levels of reactive oxygen species and cell death in pancreatic cancer cells.247
Isocitrate dehydrogenases (IDHs) are other enzymes that catalyse oxidative decarboxylation to produce NADPH. IDH1 and IDH2 share significant similarity and catalyse reversible reactions, whereas IDH3 catalyses an irreversible reaction with greater regulation (i.e., calcium, ADP and citrate), however all forms convert isocitrate to α-ketoglutarate while reducing NAD(P)+ to NAD(P)H.248–251 IDH1 localizes (variably) to the cytoplasm and IDH2/3 localise to the mitochondria.252 Examination of several gene expression databases from a range of cancer cell lines displayed co-expression of ME1 mRNA with G6PD and IDH1, indicating a coordination of metabolic pathways that produce NADPH.246
NADPH is also a product and substrate of several reactions in one-carbon (1 C) and folate metabolism, which involves a series of 1 C transformations that produce and consume redox equivalents.245 One of the only reactions that produces NADPH, and is not reversable, is the oxidation of 10-formyl-THF to CO2 by cytosolic (1)/mitochondrial (2) 10-formyltetrahydrofolate dehydrogenase (ALDH1L1/2).253 NADPH can also be produced by reversible conversions of 5,10-meTHF to 10-formylTHF, which is catalysed by methylenetetrahydrofolate dehydrogenases (MTHFDs).
The functional importance of diverse pathways leading to NADPH production is likely influenced by contextual factors including cell type and proliferative state. In HEK293T cells, to assess relative contributions of key pathways, the cellular NADPH/NADP+ ratio was measured after knockdown of a range of enzymes that produce NADPH.245 Malic enzyme 1 (ME1), cytosolic or mitochondrial NADP-dependent isocitrate dehydrogenase (IDH1 and IDH2) knockdown did not materially change NADPH/NADP + , however glucose-6-phosphate dehydrogenase or either isozyme of methylene tetrahydrofolate dehydrogenase (MTHFD1, cytosolic, or MTHFD2, mitochondrial) knockdown significantly lowered NADPH.245
Other pathways involving NADPH
While depleting NADPH sensitises to models of ferroptosis and oxidative stress,254,255 under certain contexts NADPH can promote the generation of substrates (i.e., ROS via NOX) for ferroptosis (Fig. 5). NADPH can donate electrons to the centre of NOX catalytic subunits to generate O2- via the reduction of O2.256 Subsequently, SOD1 can convert NOX-generated O2- to H2O2. NOX enzymes are important for a several biological functions including host defence, cellular signalling, stress response and transcription and translation regulation. NOX-generated ROS can be triggered by external environmental factors (e.g., hypoxia) and internal signalling (e.g., cytokines, hormones such as angiotensin II, aldosterone, endothelin-1, platelet-derived growth factor, transforming growth factor β and tumor necrosis factor α.257–260 Different members of the NOX protein family localise to specific membranes i) NOX1, -2, and -5 localised to the plasma membrane, ii) NOX4 is localised to the ER, mitochondrial and nuclear membranes.256,261,262
Fig. 5.
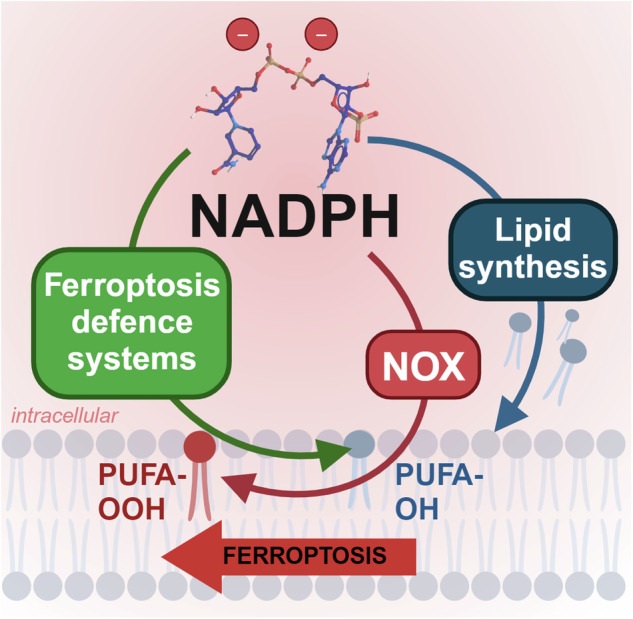
Dimorphic roles for NADPH in ferroptosis. NADPH promotes lipid synthesis for phospholipid production and is used by enzymes like heme-containing NADPH oxidases (NOXs) that transfer electrons from cytosolic NADPH to generate ROS, which promote lipid peroxidation (PUFA-OOH). Yet, NADPH is also recruited by anti-ferroptotic enzymes to prevent lipid peroxidation and to generate ferroptosis-resistant phospholipids (PUFA-OH). The recruitment of NADPH for ferroptosis-defence appears to be dominant in homeostasis, potentially to check the pro-ferroptosis pathways it fuels. Figure created using Biorender.com
Resources needed to synthesise NADPH
NADP + , the oxidised form of NADPH, is formed by the phosphorylation of nicotinamide adenine dinucleotide (NAD + ) via NAD+ kinases (NADKs).263 Thus, maintaining the NAD+ pool is essential to facilitate NADPH production (Fig. 6). NAD+, an abundant metabolite in the human body, serves several functions including; a coenzyme for oxidoreductases, a substrate for several enzymes (sirtuin family deacetylases, poly (ADP)-ribosyl polymerases and cADP-ribose synthases), and a redox carrier for bioenergetic processes including glycolysis, the TCA cycle and fatty acid oxidation.264 As a result, NAD+ dysregulation is shared by different diseases (i.e., cancer,265 metabolic diseases266 and neurodegeneration).267
Fig. 6.

The NADP(H) pool and metabolism of NAD +. Mammalian cells use dietary tryptophan to synthesise nicotinic acid adenine dinucleotide (NAD + ) via the kynurenine pathway. The Kynurenine pathway has two key branches with the main path preferentially converting kynurenine into 3-hydroxykynurenine (3-HK) and then 3-hydroxyanthranilic acid (4-HANA), 2-amino 3-carboxymuconate 6-semialdehyde (ACMS), and quinolinic acid (QA), which is converted to nicotinic acid mononucleotide (NAMN), a common intermediate of the Preiss-Handler pathway. NAMN is subsequently metabolised to nicotinic acid adenine dinucleotide (NAAD) a direct precursor to NAD + . Several enzymes including NADases, Poly (ADP-ribose) polymerases (PARPs), and Sirtuins (SIRTs) utilise NAD+ as a substrate and generate nicotinamide (NAM). The salvage pathway regenerates NAD+ from the precursor NAM which is first converted by Nicotinamide phosphoribosyltransferase (NAMPT) to nicotinamide mononucleotide (NMN) and subsequently to NAD+ by Nicotinamide mononucleotide adenylyl transferase 1-3 (NMNAT 1-3). NAD+ contributes to the NAD(H) and NADP(H) pool via several metabolic pathways and enzymes; TCA cycle, tricarboxylic acid cycle; ETC, electron transport chain; NNT, nicotinamide nucleotide transhydrogenase; NADK, NAD kinase; PPP, pentose phosphate pathway; IDP, isocitrate dehydrogenase; ME, malic enzyme; MTHFD1, Methylenetetrahydrofolate Dehydrogenase; NAPRT, nicotinate phosphoribosyltransferase. Figure created using Biorender.com
De novo NAD+ biosynthesis
Mammalian cells can use dietary tryptophan to synthesise NAD+ via the kynurenine pathway. Despite being well known as a precursor for serotonin, over 95% of tryptophan is diverted to the kynurenine pathway.268 The rate limiting step of the kynurenine pathway (converting tryptophan to kynurenine) is facilitated by tryptophan dioxygenase (TDO) and indoleamine 2,3-dioxygenase (IDO).269,270 TDO is predominantly expressed in the liver whereas IDO is more broadly expressed, particularly abundant in cells of the immune and central nervous system.271–273 The kynurenine pathway has two key branches with the main path preferentially converting kynurenine into 3-hydroxykynurenine and then 3-hydroxyanthranilic acid and quinolinic acid, the latter of which is converted to nicotinamide mononucleotide (NAMN) and then to NAD+.274 Alternatively, kynurenine can be converted into kynurenic acid or anthranilic acid, with the latter feeding back into the main pathway via 3-hydroxyanthranilic acid. In addition to de novo biosynthesis, most cellular NAD+ is recycled via salvage pathways from nicotinamide (NAM), a by-product of NAD+ degradation.275
NAD+ phosphorylation
The only way NADP+ is produced de novo is via phosphorylation of NAD+ by NAD+ kinases (NADKs) into NADP+ .276 This represents approximately 10% of the total NAD consumption.277 NADKs act via phosphorylating the 2’ position of the ribose ring connected to the adenine moiety. Since NADP+ and NADPH can not cross membranes, cells have subcellular localisation of NADKs; with NADK1 located in the cytosol and NADK2 in the mitochondria.278 NADP+ levels can be lowered via dephosphorylation by NADP phosphatase to produce NAD + .
Low NADPH as a signature of ferroptosis
Demonstrating a central role for NADPH in ferroptosis, NADPH was shown to be depleted during ferroptosis of 60 cell lines, and cellular NADP(H) abundance predicted vulnerability to ferroptosis inducers.10 NADPH depletion occurs due to an imbalance of NADPH synthesis and hydrolysis (e.g., for usage in antioxidant defences during ferroptosis). The NADPH/NADP+ ratio favours the reduced form in the cytosol in physiological conditions.263,279 However, various stresses, diseases and pathological states that decrease NADPH, thus withdrawing the foundation of ferroptosis defence, may render cells susceptible to ferroptosis. Hence, the level of NADPH could be considered a biomarker for ferroptosis sensitivity.280
Human Metazoan SpoT Homologue 1 (MESH1) was recently identified as a NADPH phosphatase with its upregulation consequently depleting NADPH, resulting in an impairment of glutathione regeneration and increased ferroptosis.255 Conversely, MESH1 removal preserved the NADPH pool in stressed cells and promoted their ferroptosis resistance. Higher levels of NADPH have also been shown to correlate with greater resistance to ferroptosis in cancer cells.10 Cancer cells are known for metabolic reprogramming that diverts carbon flux towards anabolic pathways such as the pentose phosphate pathway (PPP) to enable both rapid proliferation and generation of NADPH.239,281
Regulation of ferroptosis defence systems
Transcription factors can regulate gene expression programs that coordinate a parallel ferroptosis defence response through multiple pathways and organelles.282 The ER plays a crucial role in both lipid synthesis and the processing of transcripton factors that can initate or potentiate a ferroptosis defence gene expression program. Transcription factors in the ER including sterol regulatory element-binding proteins (SREBPs), regulate the expression of several enzymes involved in lipid metabolism (i.e., ACLY, ACACA, FASN, and SCD) and glucose metabolism including key proteins regulating the PPP (i.e., PKLR, PCK1, G6PC, and G6PD).283–286 SREBPs are tethered to the ER membrane and when activated are trafficked to the Golgi where they are proteolytically processed to release an active transcription factor that is subsequently imported into the nucleus to initiate trascription. SREBP1 has been reported to be regulated by PI3K-AKT-mTOR signalling,287,288 one of the most commonly altered signalling pathways in human cancers.289–291 As a result, oncogenic activation of the PI3K-AKT-mTOR signaling pathway induces protection against ferroptosis and pharmacological inhibition of this pathway could induce vulnerability to ferroptosis induction in cancer cells.292 A study looking at the impact of SREBF1 knockout, identified reduced expression of Stearoyl-CoA Desaturase 1 (SCD1, both mRNA and protein levels) as the most significantly impacted target. SCD1 catalyzes the rate-limiting step in MUFA synthesis, and since MUFAs are not vulnerable to peroxidation, protects against ferroptosis.292,293 Inhibition of SCD1 decreased CoQ10, an endogenous membrane antioxidant previously discussed, induced lipid oxidation and exacerbated ferroptosis sensitivity.294
The Nuclear factor erythroid 2–related factor 2 (NRF2)/NFE2L2-Kelch–like ECH-associated protein 1 (KEAP1) pathway is another well-known master regulator of cellular defence against ferroptosis, which also regulates NADPH generation and consumption.243,295,296 In unstressed conditions, NRF2 is minimally detected due to a very rapid half-life (less than 20 min).297 Keap1 acts as both an anchor that inhibits Nrf2 nuclear import and an adaptor that facilitates binding with Cullin 3-based E3 ligase, a protein-protein complex that ubiquitinates Nrf2 protein and leads to its rapid degradation through the proteasome system.298 Human Keap1 has 27 cysteine residues (25 cysteine residues in murine and rat) which are modified by both NRF2 activating compounds or oxidative/electrophilic stress.296,299,300 Oxidation, reduction, or alkylation of the sulfhydryl groups of cysteines in KEAP1 alter the conformation leading to the release of NRF2.
Unbound NRF2 translocates into the nucleus before heterodimerizing with musculoaponeurotic fibrosarcoma (Maf) protein and promoting the transcription of over 200 phase II and antioxidant genes.301–303 NRF2-regulated genes that are involved in ferroptosis defence include FSP1, GPX4, and xCT (full list available in ref. 304). In addition to several anti-ferroptotic proteins previously described to consume NADPH, NRF2 also regulates proteins that regenerate NADPH, including G6PD,296,305,306 PGD296,305 and ME1.296,305,307 Nrf2-regulated NADPH generation and consumption was investigated in Nrf2-null mice and Keap1-knockdown mice, with the latter having a higher concentration of hepatic NADPH.296 The authors indicated that NRF2 may also indirectly regulate the consumption of NADPH by downregulating genes involved in fatty acid synthesis and desaturation, concluding that Nrf2 protects against oxidative/electrophilic stress by helping with the production of NADPH.
The link between NADPH and ferroptosis in disease contexts
G6PD deficiency
Glucose-6-phosphate dehydrogenase (G6PD) deficiency is the most frequent human enzyme defect,308 causing hemolytic anemia upon exposure to certain stresses like infection, fava beans, aspirin etc. As the catalyst in the rate-limiting first step of the PPP which produces NADPH, G6PD deficiency disrupts a major metabolic pathway required to produce NADPH and power anti-ferroptotic defence. Complete G6PD deficiency is embryonically lethal in mice, but the human G6PD gene has over 200 variants, with the majority being missense mutations resulting in an unstable G6PD enzyme and G6PD deficiency.309 This helps explain why G6PD deficiencies due to these mutations predominantly affect red blood cells: mature red blood cells lack the ability to synthesise new proteins so they cannot replace mutant G6PD, which is more unstable and has a shorter half-life.310,311 The G6PD/NADPH pathway is the sole source of reduced glutathione in red blood cells. Red cells are put at risk of ferroptosis because they carry high concentrations of oxygen and iron, and must heavily rely upon the protection of G6PD/NADPH/glutathione. Comparatively, nonerythroid organs can compensate with increased G6PD synthesis and have metabolic changes consistent with mild G6PD deficiency.312 Certain medications and external sources of oxidative stress (i.e. infection) exploit inherent vulnerability in G6PD deficient individuals, primarily due to a decreased capacity to produce NADPH.313 The most common clinical presentations include acute haemolytic anaemia and neonatal jaundice, but studies have suggested an increased prevalence of diabetes mellitus and kidney disease.314 The avoidance of oxidative stress is one of the most beneficial management strategies to prevent haemolysis in patients with G6PD deficiency.308 While ferroptosis has not been explicitly studied in the context of G6PD deficiency, it is likely that individuals with G6PD deficiency and consequent NADPH depletion are more vulnerable to ferroptosis.
In contrast, various studies have stated a reduced incidence and mortality for specific cancers with hypomorphic mutations in G6PD.315–317 Metabolic re-wiring to upregulate the PPP and NADPH is characteristic of many cancers to boost oxidative stress and ferroptotic defence and provide metabolites for nucleotide and lipid synthesis. A recent pan-cancer study promoting a theoretical basis for developing G6PD inhibitors as anti-cancer drugs confirmed increased G6PD expression in hepatocellular carcinoma, glioma and breast cancer.281 In addition, a search for synthetic-lethal genes for neurofibromatosis Type II, a genetic condition characterised by benign tumors of the peripheral nervous system, using a genome-wide CRISPR/Cas9 screen identified ACSL3 and G6PD as two lethal partners; which was partly attributed to a diminished expression of genes associated with NADPH abundance.318 Pentose phosphate pathway metabolites are also enriched in metastasizing melanomas to generate NADPH for oxidative stress resistance.254 When G6PD activity is impaired in patient-derived melanomas, via mutation of the substrate binding site, mutant melanomas experience increased oxidative stress and decreased NADPH and GSH which suggests an increased metabolic vulnerability to ferroptosis when the PPP is impaired.254
The relationship between cholesterol synthesis and ferroptosis has recently been investigated in cancer cells.65,66 Enzymes and metabolites implicated in distal cholesterol biosynthesis have contrasting roles in regulating ferroptosis with 7-dehydrocholesterol, a cholesterol precursor synthesized by sterol C5-desaturase (SC5D) showing potent anti-ferroptotic activity. Interestingly DHCR7, the key enzyme converting 7-dehydrocholesterol to cholesterol, thus reducing 7-dehydrocholesterol, is also dependent on NADPH. In the oncogenic environment, 7-dehydrocholesterol conducts ferroptosis surveillance by using the conjugated diene to prevent phospholipid autoxidation, consequently protecting plasma and mitochondria membranes from phospholipid autoxidation and ferroptosis.65,66 However, in a non-oncogenic context, 7-dehydrocholesterol itself is extremely prone to free radical autoxidation resulting in the production of a dozen different toxic oxysterols.319 In a study before ferroptosis was coined, high concentrations of 7-DHC-derived oxysterols were cytotoxic to developing neurons by encouraging lipid peroxidation.320 Future studies are required to investigate the role of DHCR7 in preventing ferroptosis outside the oncogenic environment, as it may be another fundamental ferroptosis defence enzyme dependent on NADPH.
Ferroptosis in neurodegenerative contexts
Stroke, infarction and Ischemia-reperfusion damage
Ischemia is due to a restriction of blood flow that limits both the replenishment of oxygen and nutrients and the elimination of metabolic wastes from affected tissues. The subsequent reperfusion is needed to retain tissue function and viability, but while essential to prevent hypoxic damage, reperfusion paradoxically introduces a second oxygen chemical lesion that exacerbates oxidative stress.321 This can occur in various tissues and organs including the heart, kidney and brain due to a reduction of blood flow by a physical obstruction of a vessel or by a deleterious redistribution of blood flow away from a tissue or organ. In the heart, myocardial infarction can be caused by coronary atherosclerosis or the rupture of an artery plaque, which can trigger thrombosis and artery occlusion.322 In the kidney, interruption of renal blood flow is the leading cause of perioperative acute kidney injury which can occur in several clinical settings including major surgeries, sepsis, trauma and transplantation.323 In the brain, reperfusion damage is common post-stroke (e.g. after clot retrieval or thrombolytic treatment).
Several studies suggest that ferroptosis as a key mechanism involved in the onset and progression of ischemic-reperfusion injury in a range of organs.324,325 Ferroptosis inhibitors liproxstatin-1326 and ferrostatin-1327 reduce cell death and infarct size while maintaining mitochondrial integrity in ischaemic-reperfusion heart injury models. Ferroptosis has also been implicated as a significant cell death pathway in ischemic reperfusion-induced acute kidney injury, particularly in renal tubular cell death, which is also alleviated with ferrostatin328 and liproxstatin.329 Interestingly, one model describes NADPH abundance as a gradient that defines the risk of ferroptosis and dictates progression of synchronized cell death in renal tubules.330 Briefly, an initiating cell under oxidative stress undergoes NADPH-depleting necrosis while it is linked to the cytoplasm of neighbouring cells via tight junctions and gap junctions that subsequently recruits NADPH from the adjacent cells. These neighbouring cells are therefore at risk for ferroptosis due to a depletion of NADPH. In ischemic stroke models, a model which are strongly associated with ferroptosis, iron elevation associated with ischemic damage is mitigated by ferroptosis inhibitors liproxstatin-1 and ferrostatin-1,331 selenium supplementation to enhance GPX4 activity,332,333 ceruloplasmin to facilitate iron export,334,335 or genetic mutations that decrease labile intracellular iron; tau-knockout mice,336,337 which increase iron export and NCOA4 deletion, prevent ferritinophagy.334
According to studies that suggest excessive lipid peroxidation and cell death begins in the ischemic period, pre-treatment with ferroptosis inhibitors DFO and ferrostatin-1 reduced cytotoxicity and reversed a depletion of total GSH and NADPH.338 This could imply that the abundance of NADPH promotes resistance to lipid peroxidation, indicating a role for NADPH in the defence against ferroptosis during ischemia. Indeed, NADPH supplementation increased ATP and the reduced form of glutathione, lowering intracellular oxidative stress and protecting neurons against ischemia/reperfusion-induced injury.333
Conversely, NADPH may also be utilized by NADPH oxidases (NOXs) to produce ROS in certain contexts.339 NADPH oxidase (Nox) 2 and 4 are the major sources of O2− and H2O2 in the heart and are upregulated in response to ischemia-reperfusion.340 Suppression of either can reduce ROS and IR injury, but synergistic inhibition of both Nox2 and Nox4 exacerbates myocardial I/R injury.340 In a model of intermittent hypoxia, (ROS) generated by NADPH oxidases (specifically Nox2) created a signal necessary to increase HIF-1α synthesis and stability for metabolic adaptation.341 Thus, NADPH abundance plays a paradoxical role in both preventing and contributing to ferroptosis in ischemic reperfusion injuries.
Clearly the ability to re-wire central carbon metabolism in ischemia, and inevitably alter NAD(P)H levels, plays a key role in pathogenic hypoxia adaptation, however the direct implications and timing of NAD(P)H in ischemic-reperfusion induced ferroptosis requires further characterisation. Utilising novel tracer methods to measure metabolic flux during hypoxia will shed light on the contribution of NADPH to ischemia reperfusion-induced ferroptosis. While NADPH production is often attributed to PPP flux, to accurately characterise NADPH metabolism, studies should combine several tracers (deuterium (2H) tracer methods) to quantitatively analyse NADPH production from all potential pathways including the PPP, folate metabolism and malic enzymes.342 Future studies should subsequently investigate the function of NADPH produced during hypoxia and deliberate whether and how it contributes to ferroptosis initiation or defence.
Alzheimer’s disease (AD)
AD is the most common dementia with late-onset sporadic AD accounting for 95% while familial AD caused by autosomal dominant mutations accounts for less than 1% of all diagnosed cases.343 The accumulation of amyloid-β (Aβ) plaques and neurofibrillary tangles composed of hyperphosphorylated tau are pathological hallmarks of AD. However, high clearance anti-amyloid therapies have recently been approved by the FDA, show modest efficacy in slowing cognitive deterioration, as well as serious adverse effects.344,345 Studies have also identified a potential ferroptosis signature in AD models consisting of disrupted iron homeostasis, decreased GSH and increased markers of lipid peroxidation and oxidative stress (recently reviewed346).
Before ferroptosis had been defined, iron accumulation was observed in AD brains and was implicated as a contributing factor to disease progression.347 Elevated CSF ferritin, a biomarker of brain iron, is associated with the Alzheimer’s major risk allele, APOE e4 and AD progression (cognitive decline and brain atrophy).348–350 An association with iron and AD progression was later confirmed in quantitative susceptibility mapping (QSM) –magnetic resonance imaging351 and directly in post-mortem brains.352,353 Unbiased ‘omics analyses have also identified iron homeostasis as a biological process affected in AD.354,355 Excessive lipid peroxidation is more direct evidence of ferroptosis. Indeed, multiple biomarkers of lipid peroxidation (including F2-IsoPs, 4-HNE, malondialdehyde, and protein-bound acrolein) are elevated in CSF, post-mortem AD brain samples and animal models of AD.356,357 Substituting dietary PUFAs with deuterium-reinforced PUFAs, which are more resistant to lipid peroxidation, supresses lipid peroxidation (cortex and hippocampus), and improves cognition in a model of oxidative stress-related cognitive impairment that exhibits AD-like pathologies.357
Recent studies report a decrease in GSH in the hippocampus, frontal cortex, and cingulate cortex of AD subjects358–362 and a positive correlation with impairment in Mini-Mental State Examination scores.360 Low GSH suggests that the AD brain is unable to maintain a strong defence against excessive lipid-peroxidation, potentially enabling ferroptosis. In a clinical study, levels of GSH in the left hippocampus of patients with mild cognitive impairment and AD were inversely correlated with iron.358 Oxidised GSH may serve as a proxy for a depletion of NADPH within the cytoplasm, however further studies are needed to unpack the metabolic dysregulation involved. An indirect way of assessing NADPH in AD is to measure the pentose phosphate pathway. Limitations of phosphometabolite detection have prevented a comprehensive metabolite assessment, however studies have looked at the key enzyme G6PDH. Evidence in AD patients is conflicting and restricted by small sample sizes, with a decrease in synaptosomal G6PDH activity observed in the frontal cortex361 and hippocampus,363 and an increase in the inferior temporal cortex364 and cerebral hemisphere.365 To add to the complexity, an upregulation of PPP to produce NADPH may not necessarily increase the availability of NADPH for ferroptosis defence systems since studies report a decrease in several NADPH-dependent antioxidant systems (GPX, CAT, and PRDX) and an increase in NAPH-dependent NOX activity.361,366,367 Moreover, an elevated NADPH/NADP+ ratio significantly reduces G6PD activity, thus proteomic studies cannot delineate a directionality of PPP regulation and subsequent NADPH generation.368,369
Other hallmarks of AD progression may also implicate an impairment of NADPH production due to depleted NAD + . Mitochondrial impairment is well-documented in AD pathogenesis and may be caused by complex I impairment and/or impaired lysosomal-dependent mitophagy.370 The mitochondrial electron transport chain contains several iron-containing proteins, and iron chelation can decrease mitochondrial energy production. Thus, we highlight caution when using iron chelation as an anti-ferroptotic strategy in diseases with underlying mitochondrial pathologies.86,88,89,371 As complex I is crucial to reduce NADH to NAD + , dysfunctional complex I or mitochondrial impairment may lead to a loss of NAD + . Supplementation with NAD+ precursors has been explored as a treatment for cognitive decline (recently reviewed372). Across multiple models of cognitive decline (Alzheimer’s disease, vascular dementia, diabetes, stroke, and traumatic brain injury), the majority report cognitive benefits,373,374 however others have reported null or adverse effects. In 2021 a systematic metanalysis in investigating NAD + , its derivatives, and their association with cognitive function restricted to the AD context revealed that NAD+ improves learning and memory.374 Subsequent studies concurred, showing cognitive benefits in a range of rodent AD models including APP/PS1-mutant mice,373,375 and intracerebroventricular injection of Aβ1–42.376
There are no clinical studies investigating NADPH and limited comprehensive and well controlled human studies on the impact of supplementation with NAD+ precursors on cognitive function in AD. In 1996, 17 Alzheimer’s disease patients were treated with NADH disodium salt (10 mg/day) which benefited cognitive function based on the MMSE.377 However, an attempt to repeat these findings in 2000 included non-Alzheimer’s dementia in their study and showed no effect.378 The inconsistency suggests that NAD+ precursor supplementation might be selectively beneficial for AD among dementias, where there is a metabolic dysregulation that depletes NADPH resources, potentially an energetic failure, leading to increased ferroptosis susceptibility.
Parkinson’s disease
Parkinson’s disease (PD) is characterised by α-synuclein aggregation in cells of the midbrain dopaminergic neurons and cortical neurons. Chronic and progressive neurodegeneration of dopaminergic neurons results in the motor symptoms of PD including tremors, rigidity and bradykinesia. Early studies demonstrated iron elevation, excessive oxidative stress and damaged lipids in the most severely affected subpopulation of melanized neurons located in the substantia nigra pars compacta,379–381 implicating a potential role of ferroptosis. In addition, epigenetic modifications involving hypermethylation and consequently downregulation of system xc- has been shown to be associated with PD.382
Excess iron in PD brains has been attributed to various mechanisms including iron-loaded neurotoxic microglia,383 α-synuclein stabilisation of DMT1,384 α-synuclein binding with iron385 and amplified IRP1 activity resulting in a reduction of ferritin concentrations and increase in TfR1 expression,3 decreased transferrin and ceruloplasmin, which facilitates iron export from ferroportin and decreased APP, which stabilizes ferroportin at the cell surface.91,386,387 Aberrant α-synuclein oligomer incorporation into membranes of human iPSC-derived neurons with SNCA triplication, led to dysregulated membrane conductance, abnormal calcium influx and lipid peroxidation.388 Erastin exacerbated α-synuclein oligomer induced toxicity in human iPSC-derived neurons with SNCA (α-synuclein gene) triplication, which was reduced by three classes of ferroptosis inhibitor- deuterated PUFAs, iron chelator deferoxamine, and ferrostatin-1.388 Similarly, ferroptosis evasion occurred in neurons depleted of α-synuclein, which was attributed to a reduction in ether-linked phospholipids that are essential for ferroptosis.389
In addition to iron accumulation and excessive lipid peroxidation, further ferroptosis vulnerability in PD is demonstrated by a depleted glutathione levels, decreased system xc- and diminished coenzyme Q10.390 In a mouse model of PD, GPX4 levels were decreased in midbrain dopaminergic neurons.391 The proposed mechanism suggested that iron-induced dopamine oxidation modified GPX4 leading to its degradation. Conditional knockdown of Gpx4 in substantia nigra was also shown to accelerate the onset of parkinsonism in SNCAA53T/Gpx4+/fl double transgenic mice.391 Several features of PD pathology parallel the ferroptosis pathway, opening therapeutic opportunities of targeting this cell death pathway discussed below.
Mitochondrial dysfunction, specifically a reduction in Complex I activity has also been characterised as a hallmark of PD.392 Disruption of mitochondrial complex I via deletion of Ndufs2 specifically from dopaminergic neuron downregulation in mice induced a Warburg-like metabolic shift (upregulation of genes associated with glycolysis and downregulation of those genes associated with OXPHOS) that enabled neuronal survival but triggered progressive, axon-first, levodopa-responsive parkinsonism393 as observed in humans.394 In rodents, the complex I inhibitor rotenone, has also been able to induce parkinsonism clinical, pathological, and biochemical characteristics.395,396 Mechanisms of toxicity in rotenone models of PD rule out a depletion of ATP, since glycolysis inhibitors deplete ATP to a similar magnitude as rotenone but do not cause toxicity.396 Instead, excessive oxidative stress and dopaminergic neuronal loss is blocked by α-tocopherol, a anti-ferroptotic nutrient. Thus, it is likely that excess oxidative stress exerts a tax on NADPH in neurons. In humans, dietary vitamin E has emerged as a protective factor of PD, where increased Vitamin E dietary consumption was inversely associated with PD occurrence irrespective of age and gender.397
Huntington’s disease
Huntington’s disease (HD) is an autosomal dominant progressive and ultimately fatal neurodegenerative disease caused by an expanded CAG repeat in the huntingtin gene.398 The toxic gain of function has been attributed to a polyglutamine strand of variable length at the N-terminus which leads to misfolding and protein aggregate formations. Mutant huntingtin aggregates affect a variety of cellular functions, however the precise mechanism leading to neuronal cell death is poorly understood.399 Ferroptosis has been implicated due to the classic ferroptotic signature of increased iron accumulation and lipid peroxidation with decreased GSH and blunted NRF2 response.400–404 Anti-ferroptotic compounds have also shown efficacy; ferrostatin-1 prevents the degeneration of medium spiny neurons in rat corticostriatal brain slices overexpressing the huntingtin exon 1 fragment with a pathogenic repeat,405 iron chelator deferoxamine (DFO) benefited striatum pathology and motor phenotype in R6/2 HD mouse,406 and deuterium-reinforced linoleic acid, all lowered lipid peroxidation and alleviated cognitive decline in a mouse model of HD (Q140 knock in).407
Like AD and PD, HD is also characterised by perturbed mitochondria. Energy metabolism defects were initially implicated due to presentations of weight loss at early stages in patients with HD.408,409 Using localized proton nuclear magnetic resonance spectroscopy, bioenergetic dysregulation was further evidenced by elevations of lactate in the occipital cortex of symptomatic HD patients that correlated with illness duration.410–412 Increased lactate may occur due to decreased electron transport chain activity and/or elevated glycolysis. FDG-PET studies have revealed hypometabolism in areas impacted early in disease progression (i.e., caudate, putamen, and cerebral cortex) in HD subjects with symptomatic disease,413,414 and in non-symptomatic known HD gene carriers.414–418 A reduction of glucose uptake and flux through glycolysis and may possibly reduce PPP flux consequently limiting NADPH production and GSH recycling needed for antioxidant protection.419 In contrast, hypermetabolism was observed in the cerebellum and some thalamic nuclei, areas effected later in disease progression.417 It has been hypothesised that the increase in glucose metabolism is an attempt to prevent mutant huntingtin toxicity,420 potentially via increased metabolic flux to increase NADPH production to fuel anti-oxidant defence. Mitochondrial abnormalities were first observed in post mortem cortical tissue from HD patients in 1978421 and have been confirmed in multiple HD mouse models.422 Prolonged energy impairment due to Complex II inhibition via 3-nitropropionic acid administration has also been used as model for HD.421,423–425
Multiple sclerosis
Multiple sclerosis (MS) is an autoimmune condition characterised by degeneration of the myelin sheath surrounding axons impacting the brain and spinal cord. Degeneration in the deep grey matter has been associated with clinical disabilities including visual abnormalities, weakness, spasticity, urinary dysfunction, and cognitive symptoms. Iron elevation has been observed in deep grey matter from both MS patient autopsies which showed global neurodegeneration associated with an accumulation of oxidised phospholipids and DNA in neurons, oligodendrocytes and axons426 and in living MS patients via MRI.427 Iron is a necessary factor for several processes that enable myelination by oligodendrites (i.e., ATP, cholesterol, lipid synthesis and myelin basic protein function).428 As a result, oligodendrocytes are the brain cells with the highest iron levels.429 However in MS, iron accumulation in microglia and other macrophages suggests a pathological role of iron, implicating ferroptosis vulnerability.427 Indeed, Luoqian et al. found that ACSL4 was altered in a genomic database of MS patients, and that a ferroptosis signature of accrued lipid ROS and mitochondrial shrinkage were observed in the experimental autoimmune encephalitis mouse model, one of the most commonly used models for MS.430 Both liproxstatin-1 and ACSL4 repression (AAV8-Acsl4-KD) improved behavioural phenotypes, reduced inflammation and prevented neuronal cell death. Ferroptosis was proposed to cause neurodegeneration through hyperactivity of T-cells, since RSL3-treated neurons injected into naive recipients significantly exacerbated experimental autoimmune encephalitis pathogenesis.430 This study highlights a role for ferroptosis in driving immune-mediated neurodegeneration in MS.430,431
Mitochondrial abnormalities including morphological changes (swelling), fragmentation and impaired trafficking are also observed in the autoimmune encephalitis mouse model.432,433 In vivo imaging of the mouse spinal cord revealed that depolarisation of both the axonal mitochondria and the axons themselves correlated with neurological function and that mitochondrial abnormalities were most severe in regions of perivascular inflammatory cells.433 Mitochondrial dysfunction, including decreased activities of ETC complexes I, III434 and IV,435,436 has also been widely reported in MS patient neurons. It has been suggested that a bioenergetic insufficiency impacts ion homeostasis, inducing Ca2+ mediated axonal degeneration.434 Remyelination exerts an enormous energy demand, with estimations suggesting that 1 gram of myelin demands 3.3 × 1023 ATP molecules.437 Thus, oligodendrocytes have a high mitochondrial oxidative phosphorylation (OXPHOS) rate before and during myelination.438 Consequently, bioenergetic insufficiency can promote neurodegeneration in MS.
Common vulnerabilities and predisposing risk factors for ferroptosis in neurodegeneration
Energy stress, mitochondrial abnormalities and excessive oxidative stress parallels ferroptosis signatures in multiple forms of neurodegeneration with diverse genetic origins (AD, PD, HD and MS).439 Ultimately, imported glucose must either flux down through glycolysis and oxidative phosphorylation to produce ATP, or be shunted via the PPP to produce NADPH for antioxidant recycling and/or macromolecule synthesis. This creates a metabolic intersection to either create energy (ATP) or fuel ferroptosis defence (NADPH) from imported glucose. A recent study showed that neurons actively preserve low glycolytic flux via proteolytic destabilization of 6-phosphofructo-2-kinase–fructose-2,6-bisphosphatase-3 (PFKFB3; a key glycolysis promoting enzyme), to increase the PPP and therefore boost antioxidant defence to prevent oxidative stress-induced mitochondrial impairment.440 Overexpression of Pfkfb3 in genetically engineered mouse neurons resulted in an accumulation of anomalous mitochondria, complex I disassembly, bioenergetic deficiency, mitochondrial redox stress and decreased GSH, which translated to accelerated cognitive decline. Importantly, behavioural abnormalities were ameliorated with neuron-specific genetic ablation of mitochondrial redox stress (via expression of mitochondrial matrix-tagged catalase) or brain NAD+ restoration (via supplementation of NAD+ precursor, nicotinamide mononucleotide).440 Ferroptosis was not investigated in this mouse model, however, we hypothesise that the decreased GSH and excessive oxidative stress would promote ferroptosis.
Consequently, we propose that energy stress creates an inherent vulnerability for ferroptosis in neurodegenerative diseases. Several consequences of energy stress may render neurons more susceptible to ferroptosis, including decreased glucose flux through the PPP to produce NADPH, reduced ATP required to synthesise key anti-ferroptosis defence substrate GSH, and/or excessive oxidative stress production via compromised mitochondria. Mitochondrial reactive oxygen species can also mediate PFKFB3 stabilisation,441 thus mitochondrial impairment may exacerbate both energy stress and impair PPP flux, compounding to increase vulnerability to ferroptosis.
Interaction of different regulated cell death pathways in neurodegeneration
Despite ferroptosis being classified as a distinct programmed cell death pathway compared to other regulated death mechanisms (i.e., apoptosis and autophagic death), there are several common proteins and cross-signalling pathways. Some common signalling increases the vulnerability of multiple regulated cell death programs; for example, p53 increases ferroptosis through targeting SLC7A11A, spermidine, N1-acetyltransferase 1 or glutaminase 2, but can also increase apoptosis by directly binding to BAX to increase mitochondrial membrane permeabilization, or combine with anti-apoptotic mitochondrial proteins (i.e., Bcl-2 and Bcl-XL) to induce apoptosis.442–444 In contrast, the level of a common protein can differentially regulate different cell death programs; for example, increased BAP1 is associated with increased ferroptosis, but decreased BAP1 can cause apoptosis.445–447 Erastin, a common ferroptosis inducer, is also capable of binding VDAC which can influence vulnerability to apoptosis at high doses.448–452 While in vitro, a tightly controlled environment with cell-death specific inducers can create the illusion of clean single cell death mechanisms, human diseases are likely to comprise of multiple regulated cell death pathways due to the complexity of interacting signalling pathways. Understanding the interaction and interrelation of regulated cell death pathways will be important in understanding the pathogenesis of neurodegeneration.
Therapeutic strategies and clinical research strategies
Most therapeutic strategies that inadvertently or intentionally target ferroptosis can be classified into three broad categories 1. Radical trapping agents (RTAs) 2. Iron modulation and 3. Glutathione-dependent redox support. By virtue of reducing the fuel or increasing the defence of ferroptosis, these therapeutic strategies also reduce the cellular demand on NADPH generation. Similarly, therapeutic strategies that target NADPH synthesis (i.e. NAD precursors including NAM, NMN and NR376,453–457) may also boost cellular defence against ferroptosis. While increasing ferroptosis sensitivity has been exploited in the oncogenic context,458–460 this review will focus on pharmacological approaches that increase ferroptosis defence.
RTAs
RTAs are recognised as a pivotal approach to inhibit phospholipid peroxidation. RTAs can be further classified as endogenous (vitamin E isoforms,461 reduced coenzyme Q10,462 BH4463 and Vitamin K isoforms203; discussed previously) or synthetic (Ferrostatin 1,1 liproxstatin-1,9 liproxstatin-2,464 CuATSM,465 UAMC-3203,466 SRS11-92, SRS9-11, SRS16-86, UAMC-2418, CFI-4061, CFI-4082,405,467 Phenothiazine,468 2-{1-[4-(4-methylpiperazin-1-yl)phenyl]ethyl}-10H-phenothiazine,4-{4-[1-(10H-phenothiazin-2-yl)vinyl]phenyl}morpholine,469,470 3-CF3-8-tBu-PNX471; recently reviewed472). Endogenous RTAs were identified as protective in GPX4 deficient mice preceding the coining of ‘ferroptosis’ by Conrad’s group who showed that cell death could be abolished by alpha-tocopherol.71 Whereas development of synthetic RTAs to target ferroptosis began with ferrostatin-1, which was identified by Stockwell’s group through high-throughput screening of a small molecule library encompassing varied drug-like soluble compounds in the study first defining ferroptosis.1 Ferrostatin-1’s RTA capacity was later attributed to the amine group and necessary N-cyclohexyl moiety that enabled anchoring in lipid membranes.465 Ferrostatin-1 was first modified to UAMC-2418 by replacing the labile ester with amide or sulfonamide and adding a benzyl ring to NH2.467 Modification of ferrostatin-1 to improve stability however solubility became an issue. UAMC-3203 included additional solubility enhancing groups showing improved solubility and efficacy and a lack of toxicity in mice.466,473,474 At a similar time to ferrostatin-1 development, liproxstatin-1 was identified from a small molecule screening in TAM-inducible gpx4−/− mouse embryonic fibroblasts.9 The RTA capacity to block peroxyl radicals was later attributed to the liproxstatin’s quinaxoline ring.475 Conrad and coworkers since created liproxstatin-2, which has improved pharmacokinetic properties and efficacy (chemical structure not disclosed).464 Despite efficacy in inhibiting ferroptosis, non-RTA ferroptosis inhibitors are often perceived as more druggable due to their specific protein targets.472
Iron modulation
Since iron can contribute to lipid peroxidation both enzymatically and non-enzymatically,476 iron chelation has emerged as a common anti-ferroptotic therapeutic strategy. Iron chelation involves the use of ligands that bind iron before being excreted from the body via stool or urine to reduce tissue iron.477,478 Common iron chelators used to prevent ferroptosis include deferoxamine, deferiprone and deferasirox, which have previously been developed and tested for iron overload disorders such as hemochromatosis, beta-thalassemia and sickle cell disease.421,479,480 Deferoxamine chelates ferric iron at a 1:1 stoichiometry enabled by a chain of 3 hydroxamic acids terminating in a free amino acid group.481 However, the hydrophilic structure of deferoxamine results in poor absorption and rapid metabolism which forces frequent parenteral administration in patients (over 8 to 12 h per day), thus imposing a significant treatment burden.479,482 Deferiprone overcame treatment burdens and was the first orally active iron chelating drug, however was associated with gastrointestinal side effects.483 Deferasirox on the other hand can coordinate iron to for a 2:1 stoichochemistry, enabled by a triazolyl nitrogen and two phenolic oxygens as donor groups. Deferasirox can also conveniently be taken orally, but has been associated with hepatic, gastrointestinal and renal toxicities.484
MRI studies have confirmed the potential of deferiprone to cross the blood brain barrier and remove iron from the brain, leading to the hypothesis that iron chelation may slow down the progression of neurodegenerative diseases associated with elevated iron.485 Indeed, in a preliminary study, deferiprone treatment for 12 months (30 mg per kilogram per day) in patients with early-stage PD experienced a decrease in both substantia nigra iron deposits and motor-scale indicators of disease progression.486 However, in a follow up multi-centre phase 2 trial in participants with early Parkinson’s disease, deferiprone was associated with worse scores in measures of parkinsonism than those with placebo over a period of 36 weeks.487 The contradictory results of iron chelation in PD may be explained by the difference in dopaminergic medications. In the preliminary study, subjects remained on stabilised L-DOPA regimes, however in the phase 2 multicentre trial, subjects were not treated with L-DOPA. Iron is a cofactor for tyrosine hydroxylase, an enzyme required for dopamine serotonin synthesis.488 Thus, iron chelation may have limited dopamine synthesis, confounding any potential benefit of iron chelation in preventing ferroptosis. However, iron also plays a key role in energy metabolism; in the form of iron sulfur clusters, iron enables electron transport to drive oxidative phosphorylation and energy production. As previously discussed, decreased energy production is a hallmark of several neurodegenerative diseases thus caution is advised in using iron-chelation. As a double edge sword, iron chelation may prevent labile iron catalysing ferroptosis, but may also disrupt energy production. Ramifications of reducing iron-overload via iron chelation in a highly metabolically active tissue was evidenced in a limb ischemic reperfusion mouse model.89 Despite showing hallmarks of ferroptosis (excess iron and lipid peroxidation), mice treated with deferiprone experienced an exacerbated ischemic reperfusion injury to hindlimb muscle. Recently, iron chelation in an Alzheimer’s disease clinical trial similarly exacerbated disease pathology raising questions about the suitability of iron chelation in complex in vivo systems, particularly when energy demand is high, or energy impairment is characteristic of the condition {Ayton, 2024 #1578}.
Glutathione-dependent redox support
N-acetylcysteine (NAC) has been shown in several in vitro studies to inhibit ferroptosis by targeting cysteine and GSH metabolism.489–493 NAC has also been clinically shown to improve neurodegeneration-related symptoms in PD patients.494 However poor bioavailability and blood-brain barrier permeation have been implicated in the inconsistency of several clinical trial results. To overcome these limitations of NAC, an amide derivative N-Acetylcysteine amide (NACA) has been created to advance the lipophilicity, membrane permeability, and antioxidant property to increase stability and blood brain barrier penetration.495,496 Selenium supplementation is another approach to increase glutathione-mediated ferroptosis defence.163,336,497,498 There is currently a large phase II randomised, double-blind, placebo-controlled trial of sodium selenate as a disease-modifying therapy for behavioural variant frontotemporal dementia.499 This clinical trial is based on a proposed mechanism that sodium selenate acts as a specific agonist for PP2A, one of the implicated phosphatases in regulating tau protein phosphorylation.500 As a result, sodium selenate-treated transgenic TAU441 mice had significantly reduced phospho- and total tau levels (hippocampus and amygdala), and demonstrated improvements in spatial learning and memory.500 However, this does not rule out a complimentary GPX4 boosting affect.
Drug repurposing to discover novel ferroptosis therapeutics
Drug repurposing offers a potentially expedited path to clinical trials at lower costs and reduced safety risks.501 Recently, 1176 FDA approved drugs were screened using erastin for ferroptosis inhibitors as neuroprotective agents.502 89 drugs showed anti-ferroptotic activity and the top 26 drugs with EC50 values below 10 μM were further investigated to categorise mechanistic activity. Most of the drugs scavenged free radicals (n = 25) while a subset chelated Fe2+(n = 6) and inhibited 15-LOX (n = 6). 11 of the top 26 drugs (lumateperone tosylate, eltrombopag olamine, osimertinib, isoetharine mesylate salt, tizoxanide, indacaterol, clofazimine, indacaterol, valrubicin and fenoldopam mesylate) were newly identified and hold promise for further characterisation in bona fide ferroptosis disease models. Four of the top 26 drugs were antipsychotics (lumateperone, promethazine, thioridazine and olanzapine), with EC50s < 4 µM. Lumateparone was the most potent of the 26. This is curious because recent work has identified an elevation of iron in the brain in schizophrenia503,504 and bipolar disorder.505
Ferroptosis has been implicated in a wide range of diseases as previously discussed and the list will continue to grow with ongoing clinical trials investigating biomarkers of ferroptosis in other contexts (e.g., air pollution exposure NCT05753332/ NCT05758129, Myelodysplastic syndromes NCT05924074, Vitiligo NCT06261086, Lead exposure NCT05950386, Lymphedema NCT06237907 and epilepsy NCT05269901). 16 Clinical trials associated with ferroptosis were identified on clinicaltrials.gov using a filter of key search term ‘ferroptosis’ (Table 1).
Table 1.
Clinical trials associated with ferroptosis
| Clinical trial ID | Status | Title | Study objectives | Key Outcomes |
|---|---|---|---|---|
| NCT05753332 | Active, Not recruiting | Association Between Short-term PM2.5 Exposures and Nrf2 Dependent Ferroptosis Pathway | Discover of the effects of air pollution (PM2.5) on the Nrf2- dependent ferroptosis pathway | To be confirmed (TBC) |
| NCT05758129 | Active, Not recruiting | Association Between PM2.5 Exposure and Ferroptosis in Seizures Patients | Discover the possible effects of PM2.5 exposures on the Nrf2- dependent ferroptosis pathway in seizure patients | TBC |
| NCT05493800 | Active, Not recruiting |
Evaluate the Safety and Efficacy for Oral Mucositis Prevention of MIT-001 in Auto HSCT (Capella) ClinicalTrials.gov ID NCT05493800 |
Evaluate the efficacy and safety for the prevention of oral mucositis and PK of MIT-001 (a ferroptosis inhibitor506) for lymphoma or multiple myeloma patients receiving conditioning chemotherapy for autologous hematopoietic stem cell transplantation(auto-HSCT). | TBC |
| NCT05924074 | Not yet recruiting | Ferroptosis Study in SF3B1-mutant Myelodysplastic Syndromes (FerMDS) (FerMDS) | Ferroptosis will be analyzed using flow cytometry (labelling of peroxidized lipids with C11-BODIPY) from bone marrow samples from patients with Myelodysplastic syndromes (MDS): clonal diseases of hematopoietic stem cells characterized by dysplastic and inefficient hematopoiesis related to excessive progenitor cell death. | TBC |
| NCT06491394 | Not yet recruiting | Lactoferrin Effect on Kidney and Heart of Rhabdomyolysis Rats | To investigate the possible beneficial effect of Lactoferrin administration on the kidney and the heart of glycerol-induced rhabdomyolysis in rats and its relation to ferroptosis and AMPK/Nrf2/ HO-1 signalling pathway. | TBC |
| NCT06218524 | Not yet recruiting | The Effectiveness of HP and TMZ Synergism on Adult Recurrence GBM | To investigate the potential of haloperidol (an antipsychotic drug) to reverse ferroptosis resistance triggered by Temozolomide in patients who suffer from recurrent glioblastoma. | TBC |
| NCT04211636 | Not yet recruiting | Autoimmunity And Immune Deficiency After Spinal Cord Injury: Association With Rehabilitation Outcomes | To systematically analyze humoral autoantibody responses and their interaction with post-spinal cord injury immune-deficiency and infections as well as their association with the clinical course of rehabilitation and markers of ferroptosis in plasma and CSF. | TBC |
| NCT06261086 | Not yet recruiting | Evaluation of Pyroptosis-related Indicators in the Pathogenesis of Vitiligo: Across-sectional Comparative Study | To investigate necroptosis, pyroptosis, and ferroptosis markers in serum to delineate a role of cell death pathways in the pathogenesis of Vitiligo, an acquired pigmentary disorder on skin and/or mucosae, which is characterized by death of melanocytes. | TBC |
| NCT05950386 | Recruiting | Effects of Lead Exposure on Ferroptosis Pathway | Investigate the effects of chronic lead exposure on mRNA levels of genes associated with iron metabolism (FTH1, FPN1, DMT1) and the Nrf2-dependent ferroptosis pathway (Nrf2, SLC7A11, GPX4) in blood samples from lead acid battery factory workers. | TBC |
| NCT06237907 | Recruiting | Pyroptosis and Ferroptosis in the Pathophysiology of Lymphedema | Compare the differences in the expression of cell death through apoptosis and iron-dependent cell death in the subcutaneous adipose tissue after the reduction of edema symptoms following lymphedema surgery in patients | TBC |
| NCT06102993 | Recruiting | Ferroptosis in Patients With COPD With/Without Risk of Cardiovascular Events. Pathophysiological Implications, Diagnostics and Prognoses. FerrEPOC Study. (FerrEPOC) | To investigate a small group of circulating proteins in blood previously identified using Differentially Expressed Genes (DEGs) related to ferroptosis that overlap with the DEGs of COPD and the DEGs of atherosclerosis to evaluate the relationship between these molecules and clinical variables of COPD and their potential utility in identifying the risk of exacerbations, admissions, and cardiovascular events in COPD. | TBC |
| NCT06048367 | Recruiting | Carbon Nanoparticle-Loaded Iron [CNSI-Fe(II)] in the Treatment of Advanced Solid Tumor (CNSI-Fe(II)) | evaluate the safety, tolerability, pharmacokinetics (PK) profile, dose and preliminary efficacy of intratumoral injection of Carbon Nanoparticle-Loaded Iron [CNSI-Fe(II)] (to induce ferroptosis507) in patients with advanced solid tumors. | TBC |
| NCT06151548 | Recruiting | Effect of Krill Oil Supplementation on Red Blood Cell Physiology Against Changes in Markers of Iron Metabolism. | To investigate whether supplementation with krill oil may have a beneficial effect on athletes by limiting lipid peroxidation and inhibiting ferroptosis which in consequence may lead to red blood cell membrane protection. | TBC |
| NCT05410665 | Unknown | The Roles of IL-9/E-cadherin and Ferroptosis in Intestinal Mucosal Barrier Injury in Sepsis | Evaluate the roles of IL-9/E-cadherin and ferroptosis in the intestinal mucosal barrier injury of sepsis. | TBC |
| NCT05269901 | Completed | Association Between Ferroptosis and Epilepsy | Obtain peripheral blood from 20 newly epileptic diagnosed untreated school-aged children (6 –12 years) and 20 age-matched healthy controls to investigate three glutathione peroxidase 4 (GPX4) dependent ferroptosis pathway biomarkers: Nrf2, SLC7A11, and GPX4. | mRNA expression levels of Nrf2, SLC7A11, and GPX4 were significantly reduced in peripheral blood from patients with newly diagnosed and untreated seizures suggesting that the Nrf2-mediated ferroptosis pathway might be associated with the occurrence of seizures in a clinical setting.508 |
| NCT04378075 | Completed | A Study to Evaluate Efficacy and Safety of Vatiquinone for Treating Mitochondrial Disease in Participants With Refractory Epilepsy (MIT-E) | To investigate the efficacy and Safety Study of Vatiquinone (ferroptosis inhibitor509) for the Treatment of Mitochondrial Disease Subjects With Refractory Epilepsy | Not yet posted to Clintrials.gov; press release by PTC Therapeutics June 2023 reported failure to achieve primary endpoint but significantly reduced seizure incidence in the subset of patients with Leigh syndrome.510 |
Conclusion, future directions and open questions
Since ferroptosis was coined in 2012, the field has grown rapid momentum. Over the last decade, the development of a range of ferroptosis induces and rescuers, (oxidised) lipidomic techniques and CRISPR knock out screens has enabled the field to create a sound understanding of the biochemical pathways underpinning ferroptosis initiation and defence. This review focuses on key ferroptotic defence enzymes and metabolites; TR, GR, FSP1, DHFR, NQO1 and retinal reductases, and identified a common thread: a dependence on a reducing equivalent for redox recycling. NADPH is the major reducing agent of the cell, and its abundance is dependent on the complex interaction between metabolic pathways that control fundamental life processes including energy production, molecule synthesis and antioxidant defence. This leads us to pose whether NADPH or downstream protein/metabolites form the foundation of ferroptosis defence in disease?
Parallel emerging evidence in ischemic reperfusion and neurodegenerative diseases support the presence of both ferroptosis and metabolic dysregulation centred around NADPH and metabolite resources required to synthesise and recycle NADPH. Ferroptosis research has been recurrently linked to neurodegeneration. Still, while a wealth of literature suggests a ferroptotic component in neuronal cell death, we lack a granular understanding of how ferroptosis can be activated in the presence of its main guardian – glutathione peroxidase 4 (GPX4). Ferroptosis is classically induced by impaired cystine/cysteine transport (leading to GSH loss) or direct GPX4 inhibition. Yet, it is unclear if this occurs in any common chronic disease. This disconnect between how ferroptosis is modelled within the laboratory, and the pathophysiology of chronic disease has been a barrier to translation of ferroptosis concepts and therapeutics into the realm of clinical research. Given the foundational role of NAPDH in ferroptosis defence, this leads us to question whether ferroptosis may be unleashed by metabolic dysregulation leading to altered carbon flux and failure to supply enough NADPH to regenerate GSH and other ferroptosis suppressors.
The translation of ferroptosis theories into therapies is currently limited by a lack of understanding of physiological triggers in the context of human disease. While in-vitro and genetic animal models have demonstrated molecular pathways downstream of ferroptosis induction, the next frontier for the ferroptosis field will involve understanding the causes of ferroptosis in human diseases. It is likely that induction of ferroptosis in disease will occur through the erosion of ferroptosis defences that are ineffectually restored by disrupted or fatigued metabolic pathways. By understanding the metabolic pathway disruption and other pathophysiological determinants that dismantle ferroptosis defence, this may expose druggable targets to restore metabolic flux and recover ferroptosis defences in disease contexts.
Acknowledgements
This research was funded by the National Health and Medical Research Council (GNT2008359, GNT1194028). For the purposes of open access, the authors have applied a CC BY public copyright licence to any Author Accepted Manuscript version arising from this submission. The Florey Institute of Neuroscience and Mental Health acknowledges support from the Victorian Government, in particular, funding from the Operational Infrastructure Support Grant.
Author contributions
FA. supervised, coordinated and participated in the literature search, in the writing and figure generation; P.M.T.N and DL participated in the literature search and in the writing; S.A has supervised and participated in the writing and revision of the paper; A.B. has supervised and participated in the writing and revision of the paper. All authors have read and approved the article.
Competing interests
Ashley Bush is an Associate Editor of Signal Transduction and Targeted Therapy, but he has not been involved in the process of the manuscript handling. All the other authors declare no conflict of interest.
Contributor Information
Ashley I. Bush, Email: Ashley.bush@florey.edu.au
Scott Ayton, Email: scott.ayton@florey.edu.au.
References
- 1.Dixon, S. J. et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell149, 1060–1072 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dixon, S. J. & Stockwell, B. R. The role of iron and reactive oxygen species in cell death. Nat. Chem. Biol.10, 9–17 (2014). [DOI] [PubMed] [Google Scholar]
- 3.Feng, S. et al. The mechanism of ferroptosis and its related diseases. Mol. Biomed.4, 33 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yang, W. S. et al. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc. Natl. Acad. Sci. USA113, E4966–E4975 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ayala, A., Muñoz, M. F. & Argüelles, S. Lipid Peroxidation: Production, Metabolism, and Signaling Mechanisms of Malondialdehyde and 4-Hydroxy-2-Nonenal. Oxid. Med. Cell. Longev.2014, 360438 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Galy, B., Conrad, M. & Muckenthaler, M. Mechanisms controlling cellular and systemic iron homeostasis. Nat. Rev. Mol. Cell. Biol.25, 133–155 (2024). [DOI] [PubMed]
- 7.Seibt, T. M., Proneth, B. & Conrad, M. Role of GPX4 in ferroptosis and its pharmacological implication. Free Radic. Biol. Med.133, 144–152 (2019). [DOI] [PubMed] [Google Scholar]
- 8.Weaver, K. & Skouta, R. The Selenoprotein Glutathione Peroxidase 4: From Molecular Mechanisms to Novel Therapeutic Opportunities. Biomedicines. 10, 891 (2022). [DOI] [PMC free article] [PubMed]
- 9.Friedmann Angeli, J. P. et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol.16, 1180–1191 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shimada, K., Hayano, M., Pagano, N. C. & Stockwell, B. R. Cell-Line Selectivity Improves the Predictive Power of Pharmacogenomic Analyses and Helps Identify NADPH as Biomarker for Ferroptosis Sensitivity. Cell Chem. Biol.23, 225–235 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim, S. E. et al. Ultrasmall nanoparticles induce ferroptosis in nutrient-deprived cancer cells and suppress tumour growth. Nat. Nanotechnol.11, 977–985 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Doll, S. et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol.13, 91–98 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Drew, R. & Miners, J. O. The effects of buthionine sulphoximine (BSO) on glutathione depletion and xenobiotic biotransformation. Biochem. Pharmacol.33, 2989–2994, (1984). [DOI] [PubMed] [Google Scholar]
- 14.Dixon, S. J. et al. Pharmacological inhibition of cystine–glutamate exchange induces endoplasmic reticulum stress and ferroptosis. elife3, e02523 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gaschler, M. M. et al. FINO(2) initiates ferroptosis through GPX4 inactivation and iron oxidation. Nat. Chem. Biol.14, 507–515 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nakamura, T. et al. A tangible method to assess native ferroptosis suppressor activity. Cell Rep. Methods.4, 100710 (2024). [DOI] [PMC free article] [PubMed]
- 17.Shintoku, R. et al. Lipoxygenase‐mediated generation of lipid peroxides enhances ferroptosis induced by erastin and RSL3. Cancer Sci.108, 2187–2194 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bae, C. et al. Induction of ferroptosis using functionalized iron-based nanoparticles for anti-cancer therapy. Mater. Today Bio.17, 100457 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Belaidi, A. A. et al. Apolipoprotein E potently inhibits ferroptosis by blocking ferritinophagy. Mol. Psychiatry.29, 211–220 (2024). [DOI] [PMC free article] [PubMed]
- 20.Nakamura, T. et al. Phase separation of FSP1 promotes ferroptosis. Nature619, 371–377 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bassi, M. T. et al. Identification and characterisation of human xCT that co-expresses, with 4F2 heavy chain, the amino acid transport activity system xc. Pflug. Arch.442, 286–296 (2001). [DOI] [PubMed] [Google Scholar]
- 22.McCullagh, E. A. & Featherstone, D. E. Behavioral characterization of system xc-mutant mice. Behav. Brain Res.265, 1–11 (2014). [DOI] [PubMed] [Google Scholar]
- 23.De Bundel, D. et al. Loss of system x(c)- does not induce oxidative stress but decreases extracellular glutamate in hippocampus and influences spatial working memory and limbic seizure susceptibility. J. Neurosci.31, 5792–5803 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang, H. et al. Characterization of ferroptosis in murine models of hemochromatosis. Hepatology66, 449–465 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jyotsana, N., Ta, K. T. & DelGiorno, K. E. The Role of Cystine/Glutamate Antiporter SLC7A11/xCT in the Pathophysiology of Cancer. Front. Oncol. 12, 858462 (2022). [DOI] [PMC free article] [PubMed]
- 26.Qiu, B. et al. Phospholipids with two polyunsaturated fatty acyl tails promote ferroptosis. Cell187, 1177–1190.e1118 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.von Krusenstiern, A. N. et al. Identification of essential sites of lipid peroxidation in ferroptosis. Nat. Chem. Biol.19, 719–730 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Alborzinia, H. et al. Golgi stress mediates redox imbalance and ferroptosis in human cells. Commun. Biol.1, 210 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zou, Y. et al. Plasticity of ether lipids promotes ferroptosis susceptibility and evasion. Nature585, 603–608 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dierge, E. et al. Peroxidation of n-3 and n-6 polyunsaturated fatty acids in the acidic tumor environment leads to ferroptosis-mediated anticancer effects. Cell Metab.33, 1701–1715.e1705 (2021). [DOI] [PubMed] [Google Scholar]
- 31.Minami, J. K. et al. CDKN2A deletion remodels lipid metabolism to prime glioblastoma for ferroptosis. Cancer Cell41, 1048–1060.e1049 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Halliwell, B. Understanding mechanisms of antioxidant action in health and disease. Nat. Rev. Mol. Cell Biol.25, 13–33 (2024). [DOI] [PubMed] [Google Scholar]
- 33.Anandhan, A. et al. Breakdown of an Ironclad Defense System: The Critical Role of NRF2 in Mediating Ferroptosis. Cell Chem. Biol.27, 436–447 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rodencal, J. et al. Sensitization of cancer cells to ferroptosis coincident with cell cycle arrest. Cell Chem. Biol.31, 234–248.e213 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee, H. et al. Cell cycle arrest induces lipid droplet formation and confers ferroptosis resistance. Nat. Commun.15, 79 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gryzik, M. et al. NCOA4-mediated ferritinophagy promotes ferroptosis induced by erastin, but not by RSL3 in HeLa cells. Biochim. Biophys. Acta Mol. Cell Res.1868, 118913 (2021). [DOI] [PubMed] [Google Scholar]
- 37.Jin, L. et al. STING promotes ferroptosis through NCOA4-dependent ferritinophagy in acute kidney injury. Free Radic. Biol. Med.208, 348–360 (2023). [DOI] [PubMed] [Google Scholar]
- 38.Liu, J. et al. Autophagy-Dependent Ferroptosis: Machinery and Regulation. Cell Chem. Biol.27, 420–435 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Swanda, R. V. et al. Lysosomal cystine governs ferroptosis sensitivity in cancer via cysteine stress response. Mol. Cell.83, 3347–3359.e3349 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Armenta, D. A. et al. Ferroptosis inhibition by lysosome-dependent catabolism of extracellular protein. Cell Chem. Biol.29, 1588–1600.e1587 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Magtanong, L. et al. Context-dependent regulation of ferroptosis sensitivity. Cell Chem. Biol.29, 1409–1418.e1406 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Riegman, M. et al. Ferroptosis occurs through an osmotic mechanism and propagates independently of cell rupture. Na. Cell Biol.22, 1042–1048 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dolma, S., Lessnick, S. L., Hahn, W. C. & Stockwell, B. R. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer cell3, 285–296, (2003). [DOI] [PubMed] [Google Scholar]
- 44.Co, H. K. C., Wu, C.-C., Lee, Y.-C. & Chen, S. h. Emergence of large-scale cell death through ferroptotic trigger waves. Nature631, 654–662 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gelens, L., Anderson, G. A. & Ferrell, J. E. Jr Spatial trigger waves: positive feedback gets you a long way. Mol. Biol. Cell.25, 3486–3493 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zaikin, A. & Zhabotinsky, A. Concentration wave propagation in two-dimensional liquid-phase self-oscillating system. Nature225, 535–537, (1970). [DOI] [PubMed] [Google Scholar]
- 47.Lorent, J. H. et al. Plasma membranes are asymmetric in lipid unsaturation, packing and protein shape. Nat. Chem. Biol.16, 644–652 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wiernicki, B. et al. Excessive phospholipid peroxidation distinguishes ferroptosis from other cell death modes including pyroptosis. Cell Death Dis.11, 922 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wagner, B. A., Buettner, G. R. & Burns, C. P. Free Radical-Mediated Lipid Peroxidation in Cells: Oxidizability Is a Function of Cell Lipid bis-Allylic Hydrogen Content. Biochemistry33, 4449–4453, (1994). [DOI] [PubMed] [Google Scholar]
- 50.Gardner, H. W. Oxygen radical chemistry of polyunsaturated fatty acids. Free Radic. Biol. Med.7, 65–86 (1989). [DOI] [PubMed] [Google Scholar]
- 51.Barclay, L. R. C. 1992 Syntex Award Lecture Model biomembranes: quantitative studies of peroxidation, antioxidant action, partitioning, and oxidative stress. Can. J. Chem.71, 1–16 (1993). [Google Scholar]
- 52.Catalá, A. Lipid peroxidation of membrane phospholipids generates hydroxy-alkenals and oxidized phospholipids active in physiological and/or pathological conditions. Chem. Phys. Lipids157, 1–11 (2009). [DOI] [PubMed] [Google Scholar]
- 53.Dennis, E. A. Diversity of group types, regulation, and function of phospholipase A2. J. Biol. Chem.269, 13057–13060, (1994). [PubMed] [Google Scholar]
- 54.Shindou, H. & Shimizu, T. Acyl-CoA:Lysophospholipid Acyltransferases. J. Biol. Chem.284, 1–5 (2009). [DOI] [PubMed] [Google Scholar]
- 55.Dixon, S. J. et al. Human Haploid Cell Genetics Reveals Roles for Lipid Metabolism Genes in Nonapoptotic Cell Death. ACS Chem. Biol.10, 1604–1609 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liang, D. et al. Ferroptosis surveillance independent of GPX4 and differentially regulated by sex hormones. Cell186, 2748–2764.e2722 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kagan, V. E. et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat. Chem. Biol.13, 81–90 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang, Q. et al. The structural basis for the phospholipid remodeling by lysophosphatidylcholine acyltransferase 3. Nat. Commun.12, 6869 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Beharier, O. et al. PLA2G6 guards placental trophoblasts against ferroptotic injury. Proc. Natl. Acad. Sci.117, 27319–27328 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sun, W.-Y. et al. Phospholipase iPLA2β averts ferroptosis by eliminating a redox lipid death signal. Nat. Chem. Biol.17, 465–476 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang, Q. et al. PAFAH2 suppresses synchronized ferroptosis to ameliorate acute kidney injury. Nat. Chem. Biol. 20, 835–846 (2024). [DOI] [PubMed]
- 62.Dong, L., Li, Y. & Wu, H. Platelet activating-factor acetylhydrolase II: A member of phospholipase A2 family that hydrolyzes oxidized phospholipids. Chem. Phys. Lipids239, 105103 (2021). [DOI] [PubMed] [Google Scholar]
- 63.Magtanong, L. et al. Exogenous Monounsaturated Fatty Acids Promote a Ferroptosis-Resistant Cell State. Cell Chem. Biol.26, 420–432.e429 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mann, J. et al. Ferroptosis inhibition by oleic acid mitigates iron-overload-induced injury. Cell Chem. Biol.31, 249–264 (2024). [DOI] [PMC free article] [PubMed]
- 65.Freitas, F. P. et al. 7-Dehydrocholesterol is an endogenous suppressor of ferroptosis. Nature626, 401–410 (2024). [DOI] [PubMed] [Google Scholar]
- 66.Li, Y. et al. 7-Dehydrocholesterol dictates ferroptosis sensitivity. Nature626, 411–418 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Imam, M. U. et al. Antioxidants Mediate Both Iron Homeostasis and Oxidative Stress. Nutrients.9, 671 (2017). [DOI] [PMC free article] [PubMed]
- 68.Levi, S. & Rovida, E. The role of iron in mitochondrial function. Biochim. Biophys. Acta Gen. l Subj.1790, 629–636 (2009). [DOI] [PubMed] [Google Scholar]
- 69.Conrad, M. & Pratt, D. A. The chemical basis of ferroptosis. Nat. Chem. Biol.15, 1137–1147 (2019). [DOI] [PubMed] [Google Scholar]
- 70.Shah, R., Shchepinov, M. S. & Pratt, D. A. Resolving the Role of Lipoxygenases in the Initiation and Execution of Ferroptosis. ACS Cent. Sci.4, 387–396 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Seiler, A. et al. Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-mediated cell death. Cell Metab.8, 237–248 (2008). [DOI] [PubMed] [Google Scholar]
- 72.Saraev, D. D. & Pratt, D. A. Reactions of lipid hydroperoxides and how they may contribute to ferroptosis sensitivity. Curr. Opin. Chem. Biol.81, 102478 (2024). [DOI] [PubMed] [Google Scholar]
- 73.Lu, B. et al. Identification of PRDX6 as a regulator of ferroptosis. Acta Pharmacol. Sin.40, 1334–1342 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Fujii, J. & Yamada, K. i. Defense systems to avoid ferroptosis caused by lipid peroxidation-mediated membrane damage. Free Radic. Res.57, 353–372 (2023). [DOI] [PubMed] [Google Scholar]
- 75.Vermot, A., Petit-Härtlein, I., Smith, S. M. E. & Fieschi, F. NADPH Oxidases (NOX): An Overview from Discovery, Molecular Mechanisms to Physiology and Pathology. Antioxidants10, 890 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zou, Y. et al. Cytochrome P450 oxidoreductase contributes tophospholipid peroxidation in ferroptosis. Nat. Chem. Biol.16, 302–309 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Arosio, P. et al. The importance of eukaryotic ferritins in iron handling and cytoprotection. Biochem. J.472, 1–15 (2015). [DOI] [PubMed] [Google Scholar]
- 78.Pham, C. G. et al. Ferritin heavy chain upregulation by NF-kappaB inhibits TNFalpha-induced apoptosis by suppressing reactive oxygen species. Cell119, 529–542 (2004). [DOI] [PubMed] [Google Scholar]
- 79.Eid, R. et al. Identification of human ferritin, heavy polypeptide 1 (FTH1) and yeast RGI1 (YER067W) as pro-survival sequences that counteract the effects of Bax and copper in Saccharomyces cerevisiae. Exp. Cell Res.342, 52–61 (2016). [DOI] [PubMed] [Google Scholar]
- 80.Arosio, P. & Levi, S. Cytosolic and mitochondrial ferritins in the regulation of cellular iron homeostasis and oxidative damage. Biochim. Biophys. Acta1800, 783–792, (2010). [DOI] [PubMed] [Google Scholar]
- 81.Bou-Abdallah, F. The iron redox and hydrolysis chemistry of the ferritins. Biochim. Biophys. Acta1800, 719–731 (2010). [DOI] [PubMed] [Google Scholar]
- 82.Theil, E. C. Ferritin protein nanocages-the story. Nanotechnol. Percept.8, 7–16 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kato, J. et al. Iron/IRP-1-dependent regulation of mRNA expression for transferrin receptor, DMT1 and ferritin during human erythroid differentiation. Exp. Hematol.35, 879–887 (2007). [DOI] [PubMed] [Google Scholar]
- 84.Hentze, M. W., Muckenthaler, M. U., Galy, B. & Camaschella, C. Two to tango: regulation of Mammalian iron metabolism. Cell142, 24–38 (2010). [DOI] [PubMed] [Google Scholar]
- 85.Pantopoulos, K. Iron metabolism and the IRE/IRP regulatory system: an update. Ann. N. Y. Acad. Sci.1012, 1–13 (2004). [DOI] [PubMed] [Google Scholar]
- 86.Alves, F. M. et al. Age-Related Changes in Skeletal Muscle Iron Homeostasis. J. Gerontol. A Biol. Sci. Med. Sci.78, 16–24 (2022). [DOI] [PubMed] [Google Scholar]
- 87.Quiles del Rey, M. & Mancias, J. D. NCOA4-Mediated Ferritinophagy: A Potential Link to Neurodegeneration. Front. Neurosci. 13, 238 (2019). [DOI] [PMC free article] [PubMed]
- 88.Alves, F. M. et al. Iron overload and impaired iron handling contribute to the dystrophic pathology in models of Duchenne muscular dystrophy. J. Cachexia Sarcopenia Muscle13, 1541–1553 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Alves, F. M. et al. Iron accumulation in skeletal muscles of old mice is associated with impaired regeneration after ischaemia–reperfusion damage. J. Cachexia, Sarcopenia Muscle12, 476–492 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kaur, D. et al. Genetic or pharmacological iron chelation prevents MPTP-induced neurotoxicity in vivo: a novel therapy for Parkinson’s disease. Neuron37, 899–909 (2003). [DOI] [PubMed] [Google Scholar]
- 91.Lei, P. et al. Tau deficiency induces parkinsonism with dementia by impairing APP-mediated iron export. Nat. Med.18, 291–295 (2012). [DOI] [PubMed] [Google Scholar]
- 92.Hou, W. et al. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy12, 1425–1428 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Gao, M. et al. Ferroptosis is an autophagic cell death process. Cell Res26, 1021–1032 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wang, Y. Q. et al. The Protective Role of Mitochondrial Ferritin on Erastin-Induced Ferroptosis. Front. Aging Neurosci.8, 308 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Chen, X., Yu, C., Kang, R. & Tang, D. Iron Metabolism in Ferroptosis. Front. Cell Dev. Biol.8, 590226 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Escobar, A., Gaete, V. & Núñez, M. T. Effect of ascorbate in the reduction of transferrin-associated iron in endocytic vesicles. J. Bioenerg. Biomembr.24, 227–233, (1992). [DOI] [PubMed] [Google Scholar]
- 97.Lane, D. J. R., Chikhani, S., Richardson, V. & Richardson, D. R. Transferrin iron uptake is stimulated by ascorbate via an intracellular reductive mechanism. Biochim. Biophys. Acta1833, 1527–1541, (2013). [DOI] [PubMed] [Google Scholar]
- 98.Levina, A. & Lay, P. A. Transferrin Cycle and Clinical Roles of Citrate and Ascorbate in Improved Iron Metabolism. ACS Chem. Biol.14, 893–900 (2019). [DOI] [PubMed] [Google Scholar]
- 99.Gao, M. et al. Glutaminolysis and Transferrin Regulate Ferroptosis. Mol. Cell59, 298–308 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Feng, H. et al. Transferrin Receptor Is a Specific Ferroptosis Marker. Cell Rep.30, 3411–3423.e3417 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Yi, L. et al. TFRC upregulation promotes ferroptosis in CVB3 infection via nucleus recruitment of Sp1. Cell Death Dis.13, 592 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Geng, N. et al. Knockdown of ferroportin accelerates erastin-induced ferroptosis in neuroblastoma cells. Eur. Rev. Med. Pharmacol. Sci.22, 3826–3836 (2018). [DOI] [PubMed] [Google Scholar]
- 103.Li, Y. et al. Erastin induces ferroptosis via ferroportin-mediated iron accumulation in endometriosis. Hum. Reprod.36, 951–964 (2021). [DOI] [PubMed] [Google Scholar]
- 104.Collins, J. F., Wessling-Resnick, M. & Knutson, M. D. Hepcidin regulation of iron transport. J. Nutr.138, 2284–2288, (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Feder, J. N. et al. A novel MHC class I-like gene is mutated in patients with hereditary haemochromatosis. Nat. Genet.13, 399–408 (1996). [DOI] [PubMed] [Google Scholar]
- 106.Lebrón, J. A. & Bjorkman, P. J. The transferrin receptor binding site on HFE, the class I MHC-related protein mutated in hereditary hemochromatosis. J. Mol. Biol.289, 1109–1118 (1999). [DOI] [PubMed] [Google Scholar]
- 107.Lebrón, J. A., West, A. P. Jr. & Bjorkman, P. J. The hemochromatosis protein HFE competes with transferrin for binding to the transferrin receptor. J. Mol. Biol.294, 239–245 (1999). [DOI] [PubMed] [Google Scholar]
- 108.Malyszko, J. Hemojuvelin: the hepcidin story continues. Kidney Blood Press Res.32, 71–76, (2009). [DOI] [PubMed] [Google Scholar]
- 109.Milet, J. et al. Common variants in the BMP2, BMP4, and HJV genes of the hepcidin regulation pathway modulate HFE hemochromatosis penetrance. Am. J. Hum. Genet.81, 799–807 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Biasiotto, G. et al. Identification of new mutations of the HFE, hepcidin, and transferrin receptor 2 genes by denaturing HPLC analysis of individuals with biochemical indications of iron overload. Clin. Chem.49, 1981–1988 (2003). [DOI] [PubMed] [Google Scholar]
- 111.Worthen, C. A. & Enns, C. A. The role of hepatic transferrin receptor 2 in the regulation of iron homeostasis in the body. Front. Pharmacol.5, 34 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Weber, F. P. Hæmochromatosis, with Diabetes Mellitus, Hepatic Cirrhosis and Chronic Ascites. Proc. R. Soc. Med.24, 478 (1931). [PMC free article] [PubMed] [Google Scholar]
- 113.Schumacher, H. R. Jr HEMOCHROMATOSIS AND ARTHRITIS. Arthritis Rheum.7, 41–50 (1964). [DOI] [PubMed] [Google Scholar]
- 114.Cecchetti, G. et al. Cardiac alterations in 36 consecutive patients with idiopathic haemochromatosis: polygraphic and echocardiographic evaluation. Eur. Heart J.12, 224–230 (1991). [DOI] [PubMed] [Google Scholar]
- 115.Ong, S. Y. et al. Reduction of body iron in HFE-related haemochromatosis and moderate iron overload (Mi-Iron): a multicentre, participant-blinded, randomised controlled trial. Lancet Haematol.4, e607–e614 (2017). [DOI] [PubMed] [Google Scholar]
- 116.Alves, F. M. et al. Disruption of Hfe leads to skeletal muscle iron loading and reduction of hemoproteins involved in oxidative metabolism in a mouse model of hereditary hemochromatosis. Biochim. Biophys. Acta Gen. Subj.1866, 130082 (2022). [DOI] [PubMed] [Google Scholar]
- 117.Truman-Rosentsvit, M. et al. Ferritin is secreted via 2 distinct nonclassical vesicular pathways. Blood131, 342–352 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Brown, C. W. et al. Prominin2 Drives Ferroptosis Resistance by Stimulating Iron Export. Dev. Cell.51, 575–586.e574 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Zhang, C. et al. Ferroptosis in cancer therapy: a novel approach to reversing drug resistance. Mol. Cancer21, 47 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Meunier, E. & Neyrolles, O. Die another way: Ferroptosis drives tuberculosis pathology. J. Exp. Med.216, 471–473 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Zhang, Y., Qian, D., Bai, X. & Sun, S. Ferroptosis Is a Potential Therapeutic Target for Pulmonary Infectious Diseases. Cell. Microbiol.2023, 3875897 (2023). [Google Scholar]
- 122.Xiao, L. et al. Ferroptosis: A mixed blessing for infectious diseases. Front. Pharmacol.13, 992734 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Pieracci, F. M. & Barie, P. S. Iron and the risk of infection. Surg. Infect. (Larchmt.).6, S41–S46 (2005). [DOI] [PubMed] [Google Scholar]
- 124.Otto, B. R., Verweij-van Vught, A. M. & MacLaren, D. M. Transferrins and heme-compounds as iron sources for pathogenic bacteria. Crit. Rev. Microbiol.18, 217–233, (1992). [DOI] [PubMed] [Google Scholar]
- 125.Chu, B. C. et al. Siderophore uptake in bacteria and the battle for iron with the host; a bird’s eye view. Biometals23, 601–611 (2010). [DOI] [PubMed] [Google Scholar]
- 126.Dahl, S. L. et al. Lipocalin-2 Functions as Inhibitor of Innate Resistance to Mycobacterium tuberculosis. Front. Immunol. 9, 2717 (2018). [DOI] [PMC free article] [PubMed]
- 127.Smith, K. D. Iron metabolism at the host pathogen interface: lipocalin 2 and the pathogen-associated iroA gene cluster. Int. J. Biochem. Cell Biol.39, 1776–1780, (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Mayeur, S., Spahis, S., Pouliot, Y. & Levy, E. Lactoferrin, a Pleiotropic Protein in Health and Disease. Antioxid. Redox Signal.24, 813–836, (2016). [DOI] [PubMed] [Google Scholar]
- 129.Ganz, T. Iron and infection. Int. J. Hematol.107, 7–15 (2018). [DOI] [PubMed] [Google Scholar]
- 130.Sia, A. K., Allred, B. E. & Raymond, K. N. Siderocalins: Siderophore binding proteins evolved for primary pathogen host defense. Curr. Opin. Chem. Biol.17, 150–157, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Shields-Cutler, R. R. et al. Human Metabolome-derived Cofactors Are Required for the Antibacterial Activity of Siderocalin in Urine. J. Biol. Chem.291, 25901–25910 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Flo, T. H. et al. Lipocalin 2 mediates an innate immune response to bacterial infection by sequestrating iron. Nature432, 917–921 (2004). [DOI] [PubMed] [Google Scholar]
- 133.Wu, H. et al. Lipocalin 2 is protective against E. coli pneumonia. Respir. Res11, 96 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Nicolas, G. et al. The gene encoding the iron regulatory peptide hepcidin is regulated by anemia, hypoxia, and inflammation. J. Clin. Invest.110, 1037–1044 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Faux, N. G. et al. An anemia of Alzheimer’s disease. Mol. Psychiatry19, 1227–1234 (2014). [DOI] [PubMed] [Google Scholar]
- 136.Pichler, I. et al. Serum iron levels and the risk of Parkinson disease: a Mendelian randomization study. PLoS Med10, e1001462 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Zierfuss, B. et al. Iron in multiple sclerosis - Neuropathology, immunology, and real-world considerations. Mult. Scler. Relat. Disord.78, 104934 (2023). [DOI] [PubMed] [Google Scholar]
- 138.Gardenghi, S. et al. Distinct roles for hepcidin and interleukin-6 in the recovery from anemia in mice injected with heat-killed Brucella abortus. Blood123, 1137–1145 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Deschemin, J. C. & Vaulont, S. Role of hepcidin in the setting of hypoferremia during acute inflammation. PLoS One8, e61050 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Brissot, P. et al. Haemochromatosis. Nat. Rev. Dis. Prim.4, 18016 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Centis, F. et al. The importance of erythroid expansion in determining the extent of apoptosis in erythroid precursors in patients with beta-thalassemia major. Blood96, 3624–3629 (2000). [PubMed] [Google Scholar]
- 142.Srole, D. N. & Ganz, T. Erythroferrone structure, function, and physiology: Iron homeostasis and beyond. J. Cell Physiol.236, 4888–4901 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Kautz, L. et al. Identification of erythroferrone as an erythroid regulator of iron metabolism. Nat. Genet.46, 678–684 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Amaral, E. P. et al. A major role for ferroptosis in Mycobacterium tuberculosis–induced cell death and tissue necrosis. J. Exp. Med.216, 556–570 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Qiang, L. et al. A mycobacterial effector promotes ferroptosis-dependent pathogenicity and dissemination. Nat. Commun.14, 1430 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Gan, B. Ferroptosis hijacking by Mycobacterium tuberculosis. Nat. Commun.14, 1431 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Chen, X., Kang, R., Kroemer, G. & Tang, D. Ferroptosis in infection, inflammation, and immunity. J. Exp. Med. 218, e20210518 (2021). [DOI] [PMC free article] [PubMed]
- 148.Youssef, L. A. et al. Increased erythrophagocytosis induces ferroptosis in red pulp macrophages in a mouse model of transfusion. Blood131, 2581–2593 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Matsushita, M. et al. T cell lipid peroxidation induces ferroptosis and prevents immunity to infection. J. Exp. Med.212, 555–568 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Han, L. et al. PPARG-mediated ferroptosis in dendritic cells limits antitumor immunity. Biochem. Biophys. Res. Commun.576, 33–39 (2021). [DOI] [PubMed] [Google Scholar]
- 151.Kong, R. et al. IFNγ-mediated repression of system xc− drives vulnerability to induced ferroptosis in hepatocellular carcinoma cells. J. Leukoc. Biol.110, 301–314 (2021). [DOI] [PubMed] [Google Scholar]
- 152.Yang, WanS. et al. Regulation of Ferroptotic Cancer Cell Death by GPX4. Cell156, 317–331 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Wang, D. & DuBois, R. N. The Role of Prostaglandin E(2) in Tumor-Associated Immunosuppression. Trends Mol. Med.22, 1–3 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Wiernicki, B. et al. Cancer cells dying from ferroptosis impede dendritic cell-mediated anti-tumor immunity. Nat. Commun.13, 3676 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Friedmann Angeli, J. P., Krysko, D. V. & Conrad, M. Ferroptosis at the crossroads of cancer-acquired drug resistance and immune evasion. Nat. Rev. Cancer19, 405–414 (2019). [DOI] [PubMed] [Google Scholar]
- 156.Couto, N., Wood, J. & Barber, J. The role of glutathione reductase and related enzymes on cellular redox homoeostasis network. Free Radic. Biol. Med.95, 27–42 (2016). [DOI] [PubMed] [Google Scholar]
- 157.Zhang, Y. et al. mTORC1 couples cyst(e)ine availability with GPX4 protein synthesis and ferroptosis regulation. Nat. Commun.12, 1589 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Parker, J. L. et al. Molecular basis for redox control by the human cystine/glutamate antiporter system xc−. Nat. Commun.12, 7147 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Liu, X. et al. NADPH debt drives redox bankruptcy: SLC7A11/xCT-mediated cystine uptake as a double-edged sword in cellular redox regulation. Genes Dis.8, 731–745 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Ingold, I. et al. Selenium Utilization by GPX4 Is Required to Prevent Hydroperoxide-Induced Ferroptosis. Cell172, 409–422.e421 (2018). [DOI] [PubMed] [Google Scholar]
- 161.Ingold, I. & Conrad, M. Selenium and iron, two elemental rivals in the ferroptotic death process. Oncotarget9, 22241–22242 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Franke, K. W. & Tully, W. C. A New Toxicant Occurring Naturally in Certain Samples of Plant Foodstuffs*: V. Low Hatchability Due to Deformities in Chicks. Poult. Sci.14, 273–279 (1935). [Google Scholar]
- 163.Conrad, M. & Proneth, B. Selenium: Tracing Another Essential Element of Ferroptotic Cell Death. Cell Chem. Biol.27, 409–419 (2020). [DOI] [PubMed] [Google Scholar]
- 164.Schwarz, K. & Foltz, C. M. Factor 3 Activity of Selenium Compounds. J. Biol. Chem.233, 245–251, (1958). [PubMed] [Google Scholar]
- 165.Flohe, L., Gunzler, W. & Schock, H. Glutathione peroxidase: a selenoenzyme. FEBS lett.32, 132–134, (1973). [DOI] [PubMed] [Google Scholar]
- 166.Ingold, I. et al. Expression of a Catalytically Inactive Mutant Form of Glutathione Peroxidase 4 (Gpx4) Confers a Dominant-negative Effect in Male Fertility. J. Biol. Chem.290, 14668–14678 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Wei, X., Yi, X., Zhu, X. H. & Jiang, D. S. Posttranslational Modifications in Ferroptosis. Oxid. Med. Cell Longev.2020, 8832043 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Reinke, E. N. et al. Translational regulation of GPx-1 and GPx-4 by the mTOR pathway. PLoS One9, e93472 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Yang, L. et al. Broad Spectrum Deubiquitinase Inhibition Induces Both Apoptosis and Ferroptosis in Cancer Cells. Front Oncol.10, 949 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Conrad, M. & Friedmann Angeli, J. P. Glutathione peroxidase 4 (Gpx4) and ferroptosis: what’s so special about it? Mol. Cell Oncol.2, e995047 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Chu, L.-K. et al. Autophagy of OTUD5 destabilizes GPX4 to confer ferroptosis-dependent kidney injury. Nat. Commun.14, 8393 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Xue, Q. et al. Copper-dependent autophagic degradation of GPX4 drives ferroptosis. Autophagy19, 1982–1996 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Wang, X. et al. Ferulic Acid Activates SIRT1-Mediated Ferroptosis Signaling Pathway to Improve Cognition Dysfunction in Wilson’s Disease. Neuropsychiatr. Dis. Treat.19, 2681–2696 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Barbosa, P., Abo El-Magd, N. F., Hesketh, J. & Bermano, G. The Role of rs713041 Glutathione Peroxidase 4 (GPX4) Single Nucleotide Polymorphism on Disease Susceptibility in Humans: A Systematic Review and Meta-Analysis. Int. J. Mol. Sci. 23, 15762 (2022). [DOI] [PMC free article] [PubMed]
- 175.Bermano, G. et al. Evidence that a polymorphism within the 3′UTR of glutathione peroxidase 4 is functional and is associated with susceptibility to colorectal cancer. Genes Nutr.2, 225–232 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Carpenter, K. J. The discovery of vitamin C. Ann. Nutr. Metab.61, 259–264 (2012). [DOI] [PubMed] [Google Scholar]
- 177.Linster, C. L. & Van Schaftingen, E. Vitamin C. Biosynthesis, recycling and degradation in mammals. Febs j.274, 1–22 (2007). [DOI] [PubMed] [Google Scholar]
- 178.Ortwerth, B. & Olesen, P. Glutathione inhibits the glycation and crosslinking of lens proteins by ascorbic acid. Exp. Eye Res.47, 737–750, (1988). [DOI] [PubMed] [Google Scholar]
- 179.Winkler, B. S., Orselli, S. M. & Rex, T. S. The redox couple between glutathione and ascorbic acid: a chemical and physiological perspective. Free Radic. Biol. Med.17, 333–349, (1994). [DOI] [PubMed] [Google Scholar]
- 180.Sasaki, H. et al. A protective role for glutathione-dependent reduction of dehydroascorbic acid in lens epithelium. Invest. Ophthalmol. Vis. Sci.36, 1804–1817 (1995). [PubMed] [Google Scholar]
- 181.Foyer, C. H. & Noctor, G. Ascorbate and glutathione: the heart of the redox hub. Plant Physiol.155, 2–18 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Ursini, F. & Maiorino, M. Lipid peroxidation and ferroptosis: The role of GSH and GPx4. Free Radic. Biol. Med.152, 175–185 (2020). [DOI] [PubMed] [Google Scholar]
- 183.Guaiquil, V. H., Vera, J. C. & Golde, D. W. Mechanism of Vitamin C Inhibition of Cell Death Induced by Oxidative Stress in Glutathione-depleted HL-60 Cells. J. Biol. Chem.276, 40955–40961, (2001). [DOI] [PubMed] [Google Scholar]
- 184.Mårtensson, J., Han, J., Griffith, O. W. & Meister, A. Glutathione ester delays the onset of scurvy in ascorbate-deficient guinea pigs. Proc. Natl. Acad. Sci. USA90, 317–321 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Wu, K. et al. Ascorbic acid induces ferroptosis via STAT3/GPX4 signaling in oropharyngeal cancer. Free Radic. Res.58, 117–129 (2024). [DOI] [PubMed] [Google Scholar]
- 186.Kaźmierczak-Barańska, J., Boguszewska, K., Adamus-Grabicka, A. & Karwowski, B. T. Two Faces of Vitamin C—Antioxidative and Pro-Oxidative Agent. Nutrients12, 1501 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Wang, X. et al. Vitamin C induces ferroptosis in anaplastic thyroid cancer cells by ferritinophagy activation. Biochem. Biophys. Res. Commun.551, 46–53 (2021). [DOI] [PubMed] [Google Scholar]
- 188.Yang, R. et al. Blue light promotes vitamin C-mediated ferroptosis of melanoma through specifically upregulating transporter SVCT2 and generating Fe2+. Biomaterials299, 122186 (2023). [DOI] [PubMed] [Google Scholar]
- 189.Wu, M. et al. AMID, an Apoptosis-inducing Factor-homologous Mitochondrion-associated Protein, Induces Caspase-independent Apoptosis. J. Biol. Chem.277, 25617–25623 (2002). [DOI] [PubMed] [Google Scholar]
- 190.Doll, S. et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature575, 693–698 (2019). [DOI] [PubMed] [Google Scholar]
- 191.Bersuker, K. et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature575, 688–692 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Dai, E. et al. AIFM2 blocks ferroptosis independent of ubiquinol metabolism. Biochem. Biophys. Res. Commun.523, 966–971 (2020). [DOI] [PubMed] [Google Scholar]
- 193.Kajarabille, N. & Latunde-Dada, G. O. Programmed Cell-Death by Ferroptosis: Antioxidants as Mitigators. Int. J. Mol. Sci.20, 4968 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Jin, D.-Y. et al. A genome-wide CRISPR-Cas9 knockout screen identifies FSP1 as the warfarin-resistant vitamin K reductase. Nat. Commun.14, 828 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Bhagavan, H. N. & Chopra, R. K. Coenzyme Q10: absorption, tissue uptake, metabolism and pharmacokinetics. Free Radic. Res.40, 445–453, (2006). [DOI] [PubMed] [Google Scholar]
- 196.Wang, Y. & Hekimi, S. Molecular genetics of ubiquinone biosynthesis in animals. Crit. Rev. Biochem. Mol. Biol.48, 69–88 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Allan, C. M. et al. Identification of Coq11, a New Coenzyme Q Biosynthetic Protein in the CoQ-Synthome in Saccharomyces cerevisiae*. J. Biol. Chem.290, 7517–7534 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Schniertshauer, D. et al. Accelerated Regeneration of ATP Level after Irradiation in Human Skin Fibroblasts by Coenzyme Q10. Photochem. Photobiol.92, 488–494 (2016). [DOI] [PubMed] [Google Scholar]
- 199.Robinson, I., de Serna, D. G., Gutierrez, A. & Schade, D. S. Vitamin E in Humans: An Explanation of Clinical Trial Failure. Endocr. Pract.12, 576–582, (2006). [DOI] [PubMed] [Google Scholar]
- 200.Traber, M. G. & Head, B. Vitamin E: How much is enough, too much and why! Free Radic. Biol. Med.177, 212–225 (2021). [DOI] [PubMed] [Google Scholar]
- 201.Neuzil, J., Witting, P. K. & Stocker, R. Alpha-tocopheryl hydroquinone is an efficient multifunctional inhibitor of radical-initiated oxidation of low density lipoprotein lipids. Proc. Natl. Acad. Sci. USA94, 7885–7890 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Dam, H. Hæmorrhages in Chicks Reared on Artificial Diets: a New Deficiency Disease. Nature133, 909–910 (1934). [Google Scholar]
- 203.Kolbrink, B. et al. Vitamin K1 inhibits ferroptosis and counteracts a detrimental effect of phenprocoumon in experimental acute kidney injury. Cell. Mol. Life Sci.79, 387 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Shearer, M. J. & Newman, P. Recent trends in the metabolism and cell biology of vitamin K with special reference to vitamin K cycling and MK-4 biosynthesis. J. Lipid Res.55, 345–362, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.EFSA Panel on Dietetic Products, N. et al. Dietary reference values for vitamin K. EFSA J.15, e04780 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Mishima, E. et al. A non-canonical vitamin K cycle is a potent ferroptosis suppressor. Nature608, 778–783 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Yang, X. et al. Regulation of VKORC1L1 is critical for p53-mediated tumor suppression through vitamin K metabolism. Cell Metab.35, 1474–1490.e1478 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Ernster, L., Danielson, L. & Ljunggren, M. DT diaphorase I. Purification from the soluble fraction of rat-liver cytoplasm, and properties. Biochim. Biophys. Acta58, 171–188, (1962). [DOI] [PubMed] [Google Scholar]
- 209.Pey, A. L., Megarity, C. F. & Timson, D. J. NAD(P)H quinone oxidoreductase (NQO1): an enzyme which needs just enough mobility, in just the right places. Biosci. Rep. 39, BSR20180459 (2019). [DOI] [PMC free article] [PubMed]
- 210.Hosoda, S., Nakamura, W. & Hayashi, K. Properties and reaction mechanism of DT diaphorase from rat liver. J. Biol. Chem.249, 6416–6423 (1974). [PubMed] [Google Scholar]
- 211.Beyer, R. E. et al. The role of DT-diaphorase in the maintenance of the reduced antioxidant form of coenzyme Q in membrane systems. Proc. Natl. Acad. Sci. USA93, 2528–2532 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Siegel, D. et al. The reduction of α-tocopherolquinone by human NAD (P) H: quinone oxidoreductase: the role of α-tocopherolhydroquinone as a cellular antioxidant. Mol. Pharmacol.52, 300–305 (1997). [DOI] [PubMed] [Google Scholar]
- 213.Siegel, D. et al. NAD (P) H: quinone oxidoreductase 1: role as a superoxide scavenger. Mol. Pharmacol.65, 1238–1247 (2004). [DOI] [PubMed] [Google Scholar]
- 214.Zhu, H. et al. The highly expressed and inducible endogenous NAD (P) H: quinone oxidoreductase 1 in cardiovascular cells acts as a potential superoxide scavenger. Cardiovasc. Toxicol.7, 202–211 (2007). [DOI] [PubMed] [Google Scholar]
- 215.Lee, J. & Hyun, D. H. NAD(P)H-quinone oxidoreductase 1 induces complicated effects on mitochondrial dysfunction and ferroptosis in an expression level-dependent manner. Biosci. Trends.6, 153–164 (2024). [DOI] [PubMed]
- 216.Oh, E. T. et al. NQO1 regulates cell cycle progression at the G2/M phase. Theranostics13, 873–895 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Dinkova-Kostova, A. T. & Talalay, P. NAD(P)H:quinone acceptor oxidoreductase 1 (NQO1), a multifunctional antioxidant enzyme and exceptionally versatile cytoprotector. Arch. Biochem. Biophys.501, 116–123 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Asher, G. et al. NQO1 stabilizes p53 through a distinct pathway. Proc. Natl. Acad. Sci. USA99, 3099–3104 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Asher, G., Tsvetkov, P., Kahana, C. & Shaul, Y. A mechanism of ubiquitin-independent proteasomal degradation of the tumor suppressors p53 and p73. Genes Dev.19, 316–321 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Garate, M. et al. NAD(P)H quinone oxidoreductase 1 inhibits the proteasomal degradation of the tumour suppressor p33(ING1b). EMBO Rep.9, 576–581 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Oh, E. T. et al. NQO1 inhibits proteasome-mediated degradation of HIF-1α. Nat. Commun.7, 13593 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Soula, M. et al. Metabolic determinants of cancer cell sensitivity to canonical ferroptosis inducers. Nat. Chem. Biol.16, 1351–1360 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Hu, Q. et al. Blockade of GCH1/BH4 Axis Activates Ferritinophagy to Mitigate the Resistance of Colorectal Cancer to Erastin-Induced Ferroptosis. Front. Cell Dev. Biol.10, 810327 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Dhokia, V. & Macip, S. A master of all trades – linking retinoids to different signalling pathways through the multi-purpose receptor STRA6. Cell Death Discov.7, 358 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Malaspina, A. & Michael‐Titus, A. T. Is the modulation of retinoid and retinoid‐associated signaling a future therapeutic strategy in neurological trauma and neurodegeneration? J. Neurochem.104, 584–595 (2008). [DOI] [PubMed] [Google Scholar]
- 226.Jakaria, M., Belaidi, A. A., Bush, A. I. & Ayton, S. Vitamin A metabolites inhibit ferroptosis. Biomed. Pharmacother.164, 114930 (2023). [DOI] [PubMed] [Google Scholar]
- 227.Lai, X. et al. Retinoic acid protects against lipopolysaccharide-induced ferroptotic liver injury and iron disorders by regulating Nrf2/HO-1 and RARβ signaling. Free Radic. Biol. Med.205, 202–213 (2023). [DOI] [PubMed] [Google Scholar]
- 228.Do, Q., Zhang, R., Hooper, G. & Xu, L. Differential Contributions of Distinct Free Radical Peroxidation Mechanisms to the Induction of Ferroptosis. JACS Au3, 1100–1117 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Bai, L. et al. Thioredoxin-1 Rescues MPP(+)/MPTP-Induced Ferroptosis by Increasing Glutathione Peroxidase 4. Mol. Neurobiol.58, 3187–3197 (2021). [DOI] [PubMed] [Google Scholar]
- 230.Maiorino, M., Conrad, M. & Ursini, F. GPx4, Lipid Peroxidation, and Cell Death: Discoveries, Rediscoveries, and Open Issues. Antioxid. Redox Signal.29, 61–74 (2017). [DOI] [PubMed] [Google Scholar]
- 231.Arai, R. J. et al. Nitric oxide induces thioredoxin-1 nuclear translocation: Possible association with the p21Ras survival pathway. Biochem. Biophys. Res. Commun.348, 1254–1260 (2006). [DOI] [PubMed] [Google Scholar]
- 232.Léveillard, T. & Aït-Ali, N. Cell Signaling with Extracellular Thioredoxin and Thioredoxin-Like Proteins: Insight into Their Mechanisms of Action. Oxid. Med. Cell. Longev.2017, 8475125 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Vlamis-Gardikas, A. & Holmgren, A. Thioredoxin and glutaredoxin isoforms. Methods Enzymol.347, 286–296 (2002). [DOI] [PubMed] [Google Scholar]
- 234.Fisher, A. B. The phospholipase A(2) activity of peroxiredoxin 6. J. Lipid Res.59, 1132–1147 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Fisher, A. B. et al. A novel lysophosphatidylcholine acyl transferase activity is expressed by peroxiredoxin 6. J. Lipid Res.57, 587–596 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Fisher, A. B. Peroxiredoxin 6: A Bifunctional Enzyme with Glutathione Peroxidase and Phospholipase A2 Activities. Antioxid. Redox Signal.15, 831–844 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Fisher, A. B. et al. Phospholipid Hydroperoxides Are Substrates for Non-selenium Glutathione Peroxidase. J. Biol. Chem.274, 21326–21334 (1999). [DOI] [PubMed] [Google Scholar]
- 238.Ju, H.-Q. et al. NADPH homeostasis in cancer: functions, mechanisms and therapeutic implications. Signal Transduc. Target. Ther.5, 231 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.TeSlaa, T., Ralser, M., Fan, J. & Rabinowitz, J. D. The pentose phosphate pathway in health and disease. Nat. Metab.5, 1275–1289 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Garcia, A. A. et al. Stabilization of glucose-6-phosphate dehydrogenase oligomers enhances catalytic activity and stability of clinical variants. J. Biol. Chem.298, 101610 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Wrigley, N. G., Heather, J. V., Bonsignore, A. & De Flora, A. Human erythrocyte glucose 6-phosphate dehydrogenase: electron microscope studies on structure and interconversion of tetramers, dimers and monomers. J. Mol. Biol.68, 483–499, (1972). [DOI] [PubMed] [Google Scholar]
- 242.Kitamura, H. & Motohashi, H. NRF2 addiction in cancer cells. Cancer Sci.109, 900–911 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Mitsuishi, Y. et al. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell22, 66–79 (2012). [DOI] [PubMed] [Google Scholar]
- 244.Jiang, P. et al. Reciprocal regulation of p53 and malic enzymes modulates metabolism and senescence. Nature493, 689–693 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Fan, J. et al. Quantitative flux analysis reveals folate-dependent NADPH production. Nature510, 298–302 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Simmen, F. A., Alhallak, I. & Simmen, R. C. M. Malic enzyme 1 (ME1) in the biology of cancer: it is not just intermediary metabolism. J. Mol. Endocrinol.65, R77–r90 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Dey, P. et al. Genomic deletion of malic enzyme 2 confers collateral lethality in pancreatic cancer. Nature542, 119–123 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Zeng, Y. et al. Isocitrate dehydrogenase from bovine heart: primary structure of subunit 3/4. Biochem. J.310, 507–516 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Gabriel, J. L., Zervos, P. R. & Plaut, G. W. Activity of purified NAD-specific isocitrate dehydrogenase at modulator and substrate concentrations approximating conditions in mitochondria. Metabolism35, 661–667, (1986). [DOI] [PubMed] [Google Scholar]
- 250.Al-Khallaf, H. Isocitrate dehydrogenases in physiology and cancer: biochemical and molecular insight. Cell Biosci.7, 37 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Stoddard, B. L., Dean, A. & Koshland, D. E. Jr. Structure of isocitrate dehydrogenase with isocitrate, nicotinamide adenine dinucleotide phosphate, and calcium at 2.5-.ANG. resolution: A pseudo-Michaelis ternary complex. Biochemistry32, 9310–9316 (1993). [DOI] [PubMed] [Google Scholar]
- 252.Pirozzi, C. J. & Yan, H. The implications of IDH mutations for cancer development and therapy. Nat. Rev. Clin. Oncol.18, 645–661 (2021). [DOI] [PubMed] [Google Scholar]
- 253.Ducker, G. S. & Rabinowitz, J. D. One-Carbon Metabolism in Health and Disease. Cell Metab.25, 27–42 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Aurora, A. B. et al. Loss of glucose 6-phosphate dehydrogenase function increases oxidative stress and glutaminolysis in metastasizing melanoma cells. Proc. Natl. Acad. Sci. USA119, e2120617119 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Ding, C.-K. C. et al. MESH1 is a cytosolic NADPH phosphatase that regulates ferroptosis. Nat. Metab.2, 270–277 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Bedard, K. & Krause, K. H. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol. Rev.87, 245–313 (2007). [DOI] [PubMed] [Google Scholar]
- 257.Oskarsson, H. J. & Heistad, D. D. Oxidative stress produced by angiotensin too. Implications for hypertension and vascular injury. Circulation95, 557–559, (1997). [DOI] [PubMed] [Google Scholar]
- 258.Sahoo, S., Meijles, D. N. & Pagano, P. J. NADPH oxidases: key modulators in aging and age-related cardiovascular diseases? Clin. Sci.130, 317–335 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Touyz, R. M. et al. p47phox associates with the cytoskeleton through cortactin in human vascular smooth muscle cells: role in NAD(P)H oxidase regulation by angiotensin II. Arterioscler. Thromb. Vasc. Biol.25, 512–518 (2005). [DOI] [PubMed] [Google Scholar]
- 260.Tarafdar, A. & Pula, G. The Role of NADPH Oxidases and Oxidative Stress in Neurodegenerative Disorders. Int. J. Mol. Sci.19, 3824 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Sarkar, S. Mechanism of Gene-Environment Interactions Driving Glial Activation in Parkinson’s Diseases. Curr. Environ. Health Rep.8, 203–211 (2021). [DOI] [PubMed] [Google Scholar]
- 262.Sorce, S. & Krause, K. H. NOX enzymes in the central nervous system: from signaling to disease. Antioxid. Redox Signal.11, 2481–2504, (2009). [DOI] [PubMed] [Google Scholar]
- 263.Pollak, N., Dölle, C. & Ziegler, M. The power to reduce: pyridine nucleotides-small molecules with a multitude of functions. Biochem J.402, 205–218, (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Xiao, W., Wang, R. S., Handy, D. E. & Loscalzo, J. NAD(H) and NADP(H) Redox Couples and Cellular Energy Metabolism. Antioxid. Redox Signal.28, 251–272 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Navas, L. E. & Carnero, A. NAD+ metabolism, stemness, the immune response, and cancer. Signal Transduct. Target. Ther.6, 2 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Liu, Y. et al. Association of NAD+ levels with metabolic disease in a community-based study. Front. Endocrinol. 14, 1164788 (2023). [DOI] [PMC free article] [PubMed]
- 267.Hou, Y. et al. NAD supplementation reduces neuroinflammation and cell senescence in a transgenic mouse model of Alzheimer disease via cGAS-STING. Proc. Natl Acad. Sci.118, e2011226118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Bender, D. A. Effects of a dietary excess of leucine on the metabolism of tryptophan in the rat: a mechanism for the pellagragenic action of leucine. Br. J. Nutr.50, 25–32 (1983). [DOI] [PubMed] [Google Scholar]
- 269.Dolšak, A., Gobec, S. & Sova, M. Indoleamine and tryptophan 2,3-dioxygenases as important future therapeutic targets. Pharmacol. Ther.221, 107746 (2021). [DOI] [PubMed] [Google Scholar]
- 270.Sugimoto, H. et al. Crystal structure of human indoleamine 2,3-dioxygenase: catalytic mechanism of O2 incorporation by a heme-containing dioxygenase. Proc. Natl. Acad. Sci. USA103, 2611–2616 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Knox, W. E. & Mehler, A. H. The conversion of tryptophan to kynurenine in liver. I. The coupled tryptophan peroxidase-oxidase system forming formylkynurenine. J. Biol. Chem.187, 419–430, (1950). [PubMed] [Google Scholar]
- 272.Fujigaki, S. et al. Lipopolysaccharide induction of indoleamine 2,3-dioxygenase is mediated dominantly by an IFN-γ-independent mechanism. Eur. J. Immunol.31, 2313–2318 (2001). [DOI] [PubMed] [Google Scholar]
- 273.Lanz, T. V. et al. Mouse mesenchymal stem cells suppress antigen-specific TH cell immunity independent of indoleamine 2, 3-dioxygenase 1 (IDO1). Stem Cells Dev.19, 657–668 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Savitz, J. The kynurenine pathway: a finger in every pie. Mol. Psychiatry25, 131–147 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Braidy, N. et al. Role of Nicotinamide Adenine Dinucleotide and Related Precursors as Therapeutic Targets for Age-Related Degenerative Diseases: Rationale, Biochemistry, Pharmacokinetics, and Outcomes. Antioxid. Redox Signal.30, 251–294 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Oka, S. I., Titus, A. S., Zablocki, D. & Sadoshima, J. Molecular properties and regulation of NAD(+) kinase (NADK). Redox Biol.59, 102561 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Liu, L. et al. Quantitative Analysis of NAD Synthesis-Breakdown Fluxes. Cell Metab.27, 1067–1080.e1065 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Murata, K. Polyphosphate-dependent nicotinamide adenine dinucleotide (NAD) kinase: A novel missing link in human mitochondria. Proc. Jpn. Acad. Ser. B. Phys. Biol. Sci.97, 479–498 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Ying, W. NAD+/NADH and NADP+/NADPH in cellular functions and cell death: regulation and biological consequences. Antioxid. Redox Signal.10, 179–206 (2008). [DOI] [PubMed] [Google Scholar]
- 280.Nguyen, K. T. et al. The MARCHF6 E3 ubiquitin ligase acts as an NADPH sensor for the regulation of ferroptosis. Nat. Cell Biol.24, 1239–1251 (2022). [DOI] [PubMed] [Google Scholar]
- 281.Liu, B. et al. Pan-cancer analysis of G6PD carcinogenesis in human tumors. Carcinogenesis44, 525–534 (2023). [DOI] [PubMed] [Google Scholar]
- 282.Dixon, S. J. & Olzmann, J. A. The cell biology of ferroptosis. Nat. Rev. Mol. Cell Biol.25, 424–442 (2024). [DOI] [PubMed] [Google Scholar]
- 283.Shimano, H. & Sato, R. SREBP-regulated lipid metabolism: convergent physiology - divergent pathophysiology. Nat. Rev. Endocrinol.13, 710–730 (2017). [DOI] [PubMed] [Google Scholar]
- 284.Horton, J. D., Goldstein, J. L. & Brown, M. S. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Invest.109, 1125–1131 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Düvel, K. et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol. Cell.39, 171–183 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Ruiz, R. et al. Sterol regulatory element-binding protein-1 (SREBP-1) is required to regulate glycogen synthesis and gluconeogenic gene expression in mouse liver. J. Biol. Chem.289, 5510–5517 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Ricoult, S. J., Yecies, J. L., Ben-Sahra, I. & Manning, B. D. Oncogenic PI3K and K-Ras stimulate de novo lipid synthesis through mTORC1 and SREBP. Oncogene35, 1250–1260, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Yecies, J. L. et al. Akt stimulates hepatic SREBP1c and lipogenesis through parallel mTORC1-dependent and independent pathways. Cell Metab.14, 21–32 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Zhang, Y. et al. A Pan-Cancer Proteogenomic Atlas of PI3K/AKT/mTOR Pathway Alterations. Cancer Cell31, 820–832.e823 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Fruman, D. A. et al. The PI3K Pathway in Human Disease. Cell170, 605–635 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Saxton, R. A. & Sabatini, D. M. mTOR Signaling in Growth, Metabolism, and Disease. Cell169, 361–371 (2017). [DOI] [PubMed] [Google Scholar]
- 292.Yi, J. et al. Oncogenic activation of PI3K-AKT-mTOR signaling suppresses ferroptosis via SREBP-mediated lipogenesis. Proc. Natl. Acad. Sci. USA117, 31189–31197 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Sen, U., Coleman, C. & Sen, T. Stearoyl coenzyme A desaturase-1: multitasker in cancer, metabolism, and ferroptosis. Trends Cancer9, 480–489 (2023). [DOI] [PubMed] [Google Scholar]
- 294.Tesfay, L. et al. Stearoyl-CoA Desaturase 1 Protects Ovarian Cancer Cells from Ferroptotic Cell Death. Cancer Res.79, 5355–5366 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Bensaad, K. et al. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell126, 107–120 (2006). [DOI] [PubMed] [Google Scholar]
- 296.Wu, K. C., Cui, J. Y. & Klaassen, C. D. Beneficial role of Nrf2 in regulating NADPH generation and consumption. Toxicol. Sci.123, 590–600 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Kobayashi, A. et al. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol. Cell Biol.24, 7130–7139 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Cullinan, S. B. et al. The Keap1-BTB Protein Is an Adaptor That Bridges Nrf2 to a Cul3-Based E3 Ligase: Oxidative Stress Sensing by a Cul3-Keap1 Ligase. Mol. Cell. Biol.24, 8477–8486 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Kobayashi, M. & Yamamoto, M. Nrf2-Keap1 regulation of cellular defense mechanisms against electrophiles and reactive oxygen species. Adv. Enzym. Regul.46, 113–140, (2006). [DOI] [PubMed] [Google Scholar]
- 300.Hong, F., Sekhar, K. R., Freeman, M. L. & Liebler, D. C. Specific Patterns of Electrophile Adduction Trigger Keap1 Ubiquitination and Nrf2 Activation. J. Biol. Chem.280, 31768–31775, (2005). [DOI] [PubMed] [Google Scholar]
- 301.Itoh, K. et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun.236, 313–322 (1997). [DOI] [PubMed] [Google Scholar]
- 302.Dodson, M. et al. Modulating NRF2 in Disease: Timing Is Everything. Annu. Rev. Pharmacol. Toxicol.59, 555–575 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Song, X. & Long, D. Nrf2 and Ferroptosis: A New Research Direction for Neurodegenerative Diseases. Front. Neurosci. 14, 267 (2020). [DOI] [PMC free article] [PubMed]
- 304.Abdalkader, M. et al. Targeting Nrf2 to Suppress Ferroptosis and Mitochondrial Dysfunction in Neurodegeneration. Front. Neurosci. 12, 466 (2018). [DOI] [PMC free article] [PubMed]
- 305.Thimmulappa, R. K. et al. Identification of Nrf2-regulated genes induced by the chemopreventive agent sulforaphane by oligonucleotide microarray. Cancer Res.62, 5196–5203 (2002). [PubMed] [Google Scholar]
- 306.Lee, J.-M. et al. Identification of the NF-E2-related factor-2-dependent genes conferring protection against oxidative stress in primary cortical astrocytes using oligonucleotide microarray analysis. J. Biol. Chem.278, 12029–12038 (2003). [DOI] [PubMed] [Google Scholar]
- 307.Kwak, M.-K. et al. Modulation of gene expression by cancer chemopreventive dithiolethiones through the Keap1-Nrf2 pathway: identification of novel gene clusters for cell survival. J. Biol. Chem.278, 8135–8145 (2003). [DOI] [PubMed] [Google Scholar]
- 308.Cappellini, M. D. & Fiorelli, G. Glucose-6-phosphate dehydrogenase deficiency. Lancet371, 64–74 (2008). [DOI] [PubMed] [Google Scholar]
- 309.Luzzatto, L., Ally, M. & Notaro, R. Glucose-6-phosphate dehydrogenase deficiency. Blood136, 1225–1240 (2020). [DOI] [PubMed] [Google Scholar]
- 310.Beutler, E. The molecular biology of G6PD variants and other red cell enzyme defects. Annu. Rev. Med.43, 47–59 (1992). [DOI] [PubMed] [Google Scholar]
- 311.Mandas, A. et al. Glucose-6-phosphate-dehydrogenase deficiency as a risk factor in proliferative disorder development. Nat Preced.6, 2928–35 (2009).
- 312.D’Alessandro, A. et al. Hematologic and systemic metabolic alterations due to Mediterranean class II G6PD deficiency in mice. JCI Insight. 6, e147056 (2021). [DOI] [PMC free article] [PubMed]
- 313.Beutler, E. Glucose-6-phosphate dehydrogenase deficiency: a historical perspective. Blood111, 16–24 (2008). [DOI] [PubMed] [Google Scholar]
- 314.Spencer, N. Y. & Stanton, R. C. Glucose 6-phosphate dehydrogenase and the kidney. Curr. Opin. Nephrol. Hypertens.26, 43–49 (2017). [DOI] [PubMed] [Google Scholar]
- 315.Pes, G. M., Bassotti, G. & Dore, M. P. Colorectal Cancer Mortality in Relation to Glucose - 6 - Phosphate Dehydrogenase Deficiency and Consanguinity in Sardinia: A Spatial Correlation Analysis. Asian Pac. J. Cancer Prev.18, 2403–2407 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Pes, G. M. et al. Glucose-6-phosphate dehydrogenase deficiency reduces susceptibility to cancer of endodermal origin. Acta Oncol.58, 1205–1211 (2019). [DOI] [PubMed] [Google Scholar]
- 317.Dore, M. P. et al. Glucose-6-phosphate dehydrogenase deficiency and risk of colorectal cancer in Northern Sardinia: A retrospective observational study. Med. (Baltim.)95, e5254 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Kyrkou, A. et al. G6PD and ACSL3 are synthetic lethal partners of NF2 in Schwann cells. Nat. Commun.15, 5115 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Xu, L., Korade, Z. & Porter, N. A. Oxysterols from free radical chain oxidation of 7-dehydrocholesterol: product and mechanistic studies. J. Am. Chem. Soc.132, 2222–2232, (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Korade, Z., Xu, L., Shelton, R. & Porter, N. A. Biological activities of 7-dehydrocholesterol-derived oxysterols: implications for Smith-Lemli-Opitz syndrome. J. Lipid Res.51, 3259–3269, (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Kalogeris, T., Baines, C. P., Krenz, M. & Korthuis, R. J. Cell biology of ischemia/reperfusion injury. Int. Rev. Cell Mol. Biol.298, 229–317 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Heusch, G. Myocardial ischaemia–reperfusion injury and cardioprotection in perspective. Nat. Rev. Cardiol.17, 773–789 (2020). [DOI] [PubMed] [Google Scholar]
- 323.Bonventre, J. V. & Weinberg, J. M. Recent advances in the pathophysiology of ischemic acute renal failure. J. Am. Soc. Nephrol.14, 2199–2210, (2003). [DOI] [PubMed] [Google Scholar]
- 324.Zhou, L. et al. Ferroptosis-A New Dawn in the Treatment of Organ Ischemia-Reperfusion Injury. Cells. 11, 3653 (2022). [DOI] [PMC free article] [PubMed]
- 325.Yellon, D. M. & Hausenloy, D. J. Myocardial reperfusion injury. N. Engl. J. Med.357, 1121–1135, (2007). [DOI] [PubMed] [Google Scholar]
- 326.Feng, Y. et al. Liproxstatin-1 protects the mouse myocardium against ischemia/reperfusion injury by decreasing VDAC1 levels and restoring GPX4 levels. Biochem. Biophys. Res. Commun. 10, 606–611 (2019). [DOI] [PMC free article] [PubMed]
- 327.Li, W. et al. Ferroptotic cell death and TLR4/Trif signaling initiate neutrophil recruitment after heart transplantation. J. Clin. Invest.129, 2293–2304 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.Linkermann, A. et al. Synchronized renal tubular cell death involves ferroptosis. Proc. Natl. Acad. Sci. USA111, 16836–16841 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329.Shi, Z. et al. Liproxstatin-1 Alleviated Ischemia/Reperfusion-Induced Acute Kidney Injury via Inhibiting Ferroptosis. Antioxidants13, 182 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.Tonnus, W. & Linkermann, A. Death is my Heir”—Ferroptosis Connects Cancer Pharmacogenomics and Ischemia-Reperfusion Injury. Cell Chem. Biol.23, 202–203 (2016). [DOI] [PubMed] [Google Scholar]
- 331.Enomoto, M. et al. Clinical Effects of Early Edaravone Use in Acute Ischemic Stroke Patients Treated by Endovascular Reperfusion Therapy. Stroke50, 652–658 (2019). [DOI] [PubMed] [Google Scholar]
- 332.Wang, X. X. et al. NADPH is superior to NADH or edaravone in ameliorating metabolic disturbance and brain injury in ischemic stroke. Acta Pharm. Sin.43, 529–540 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333.Li, M. et al. Reduced Nicotinamide Adenine Dinucleotide Phosphate, a Pentose Phosphate Pathway Product, Might Be a Novel Drug Candidate for Ischemic Stroke. Stroke47, 187–195 (2016). [DOI] [PubMed] [Google Scholar]
- 334.Tuo, Q. Z. et al. Tau-mediated iron export prevents ferroptotic damage after ischemic stroke. Mol. Psychiatry22, 1520–1530 (2017). [DOI] [PubMed] [Google Scholar]
- 335.Li, C. et al. Nuclear receptor coactivator 4-mediated ferritinophagy contributes to cerebral ischemia-induced ferroptosis in ischemic stroke. Pharmacol. Res.174, 105933 (2021). [DOI] [PubMed] [Google Scholar]
- 336.Alim, I. et al. Selenium Drives a Transcriptional Adaptive Program to Block Ferroptosis and Treat Stroke. Cell177, 1262–1279.e1225 (2019). [DOI] [PubMed] [Google Scholar]
- 337.Tuo, Q. Z. et al. Characterization of Selenium Compounds for Anti-ferroptotic Activity in Neuronal Cells and After Cerebral Ischemia-Reperfusion Injury. Neurotherapeutics18, 2682–2691 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338.Ma, X. H. et al. ALOX15-launched PUFA-phospholipids peroxidation increases the susceptibility of ferroptosis in ischemia-induced myocardial damage. Signal Transduct. Target. Ther.7, 288 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339.Chen, H., Song, Y. S. & Chan, P. H. Inhibition of NADPH oxidase is neuroprotective after ischemia—reperfusion. J. Cereb. Blood Flow. Metab.29, 1262–1272 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 340.Matsushima, S., Tsutsui, H. & Sadoshima, J. Physiological and pathological functions of NADPH oxidases during myocardial ischemia–reperfusion. Trends Cardiovasc. Med.24, 202–205, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341.Nanduri, J. et al. HIF-1α activation by intermittent hypoxia requires NADPH oxidase stimulation by xanthine oxidase. PLoS One10, e0119762 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342.Liu, L. et al. Malic enzyme tracers reveal hypoxia-induced switch in adipocyte NADPH pathway usage. Nat. Chem. Biol.12, 345–352 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343.Masters, C. L. et al. Alzheimer’s disease. Nat. Rev. Dis. Prim.1, 15056 (2015). [DOI] [PubMed] [Google Scholar]
- 344.Ayton, S. & Bush, A. I. β-amyloid: The known unknowns. Ageing Res. Rev.65, 101212 (2021). [DOI] [PubMed] [Google Scholar]
- 345.Alves, F., Kalinowski, P. & Ayton, S. Accelerated Brain Volume Loss Caused by Anti-β-Amyloid Drugs: A Systematic Review and Meta-analysis. Neurology100, e2114–e2124 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346.Lane, D. J. R., Alves, F., Ayton, S. J. & Bush, A. I. Striking a NRF2: The Rusty and Rancid Vulnerabilities Toward Ferroptosis in Alzheimer’s Disease. Antioxid. Redox Signal.39, 141–161 (2023). [DOI] [PubMed] [Google Scholar]
- 347.Smith, M. A., Harris, P. L., Sayre, L. M. & Perry, G. Iron accumulation in Alzheimer disease is a source of redox-generated free radicals. Proc. Natl. Acad. Sci. USA94, 9866–9868 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 348.Ayton, S. et al. Ferritin levels in the cerebrospinal fluid predict Alzheimer’s disease outcomes and are regulated by APOE. Nat. Commun.6, 6760 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.Ayton, S., Diouf, I. & Bush, A. I. Evidence that iron accelerates Alzheimer’s pathology: a CSF biomarker study. J. Neurol. Neurosurg. Psychiatry89, 456 (2018). [DOI] [PubMed] [Google Scholar]
- 350.Diouf, I., Fazlollahi, A., Bush, A. I. & Ayton, S. Cerebrospinal fluid ferritin levels predict brain hypometabolism in people with underlying β-amyloid pathology. Neurobiol. Dis.124, 335–339 (2019). [DOI] [PubMed] [Google Scholar]
- 351.Ayton, S. et al. Cerebral quantitative susceptibility mapping predicts amyloid-β-related cognitive decline. Brain140, 2112–2119 (2017). [DOI] [PubMed] [Google Scholar]
- 352.Ayton, S. et al. Regional brain iron associated with deterioration in Alzheimer’s disease: A large cohort study and theoretical significance. Alzheimer’s Dement17, 1244–1256 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 353.Ayton, S. et al. Brain iron is associated with accelerated cognitive decline in people with Alzheimer pathology. Mol. Psychiatry25, 2932–2941 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354.Bai, B. et al. Deep Multilayer Brain Proteomics Identifies Molecular Networks in Alzheimer Disease Progression. Neuron105, 975–991.e977 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355.Zhou, Y. et al. Human and mouse single-nucleus transcriptomics reveal TREM2-dependent and TREM2-independent cellular responses in Alzheimer’s disease. Nat. Med.26, 131–142 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 356.Bradley-Whitman, M. A. & Lovell, M. A. Biomarkers of lipid peroxidation in Alzheimer disease (AD): an update. Arch. Toxicol.89, 1035–1044, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 357.Elharram, A. et al. Deuterium-reinforced polyunsaturated fatty acids improve cognition in a mouse model of sporadic Alzheimer’s disease. FEBS J.284, 4083–4095 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 358.Mandal, P. K. et al. Hippocampal glutathione depletion with enhanced iron level in patients with mild cognitive impairment and Alzheimer’s disease compared with healthy elderly participants. Brain Commun. 4, fcac215 (2022). [DOI] [PMC free article] [PubMed]
- 359.Mandal, P. K. et al. Quantitation of Brain and Blood Glutathione and Iron in Healthy Age Groups Using Biophysical and In Vivo MR Spectroscopy: Potential Clinical Application. ACS Chem. Neurosci.14, 2375–2384 (2023). [DOI] [PubMed] [Google Scholar]
- 360.Mandal, P. K., Saharan, S., Tripathi, M. & Murari, G. Brain glutathione levels-a novel biomarker for mild cognitive impairment and Alzheimer’s disease. Biol. Psychiatry78, 702–710, (2015). [DOI] [PubMed] [Google Scholar]
- 361.Ansari, M. A. & Scheff, S. W. Oxidative stress in the progression of Alzheimer disease in the frontal cortex. J. Neuropathol. Exp. Neurol.69, 155–167, (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 362.Gu, M. et al. Mitochondrial function, GSH and iron in neurodegeneration and Lewy body diseases. J. Neurol. Sci.158, 24–29 (1998). [DOI] [PubMed] [Google Scholar]
- 363.Bigl, M. et al. Activities of key glycolytic enzymes in the brains of patients with Alzheimer’s disease. J. Neural Transm.106, 499–511 (1999). [DOI] [PubMed] [Google Scholar]
- 364.Palmer, A. M. The activity of the pentose phosphate pathway is increased in response to oxidative stress in Alzheimer’s disease. J. Neural Transm. (Vienna).106, 317–328 (1999). [DOI] [PubMed] [Google Scholar]
- 365.Martins, R. N., Harper, C. G., Stokes, G. B. & Masters, C. L. Increased cerebral glucose-6-phosphate dehydrogenase activity in Alzheimer’s disease may reflect oxidative stress. J. Neurochem.46, 1042–1045, (1986). [DOI] [PubMed] [Google Scholar]
- 366.Youssef, P. et al. Evidence supporting oxidative stress in a moderately affected area of the brain in Alzheimer’s disease. Sci. Rep.8, 11553 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 367.Baldeiras, I. et al. Oxidative damage and progression to Alzheimer’s disease in patients with mild cognitive impairment. J. Alzheimers Dis.21, 1165–1177 (2010). [DOI] [PubMed] [Google Scholar]
- 368.Yan, X. et al. Metabolic Dysregulation Contributes to the Progression of Alzheimer’s Disease. Front. Neurosci. 14, 530219 (2020). [DOI] [PMC free article] [PubMed]
- 369.Holten, D., Procsal, D. & Chang, H.-L. Regulation of pentose phosphate pathway dehydrogenases by NADP+/NADPH ratios. Biochem. Biophys. Res. Commun.68, 436–441, (1976). [DOI] [PubMed] [Google Scholar]
- 370.Askenazi, M. et al. Compilation of reported protein changes in the brain in Alzheimer’s disease. Nat. Commun.14, 4466 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371.Fryknäs, M. et al. Iron chelators target both proliferating and quiescent cancer cells. Sci. Rep.6, 38343 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 372.Campbell, J. M. Supplementation with NAD(+) and Its Precursors to Prevent Cognitive Decline across Disease Contexts. Nutrients. 14, 3231 (2022). [DOI] [PMC free article] [PubMed]
- 373.Hou, Y. et al. NAD(+) supplementation reduces neuroinflammation and cell senescence in a transgenic mouse model of Alzheimer’s disease via cGAS-STING. Proc. Natl. Acad. Sci. USA. 118, e2011226118 (2021). [DOI] [PMC free article] [PubMed]
- 374.Hosseini, L. et al. Protective Effects of Nicotinamide Adenine Dinucleotide and Related Precursors in Alzheimer’s Disease: A Systematic Review of Preclinical Studies. J. Mol. Neurosci.71, 1425–1435 (2021). [DOI] [PubMed] [Google Scholar]
- 375.Yao, Z., Yang, W., Gao, Z. & Jia, P. Nicotinamide mononucleotide inhibits JNK activation to reverse Alzheimer disease. Neurosci. Lett.647, 133–140 (2017). [DOI] [PubMed] [Google Scholar]
- 376.Rehman, I. U. et al. Nicotinamide Ameliorates Amyloid Beta-Induced Oxidative Stress-Mediated Neuroinflammation and Neurodegeneration in Adult Mouse Brain. Biomedicines. 9, 408 (2021). [DOI] [PMC free article] [PubMed]
- 377.Birkmayer, J. G. Coenzyme nicotinamide adenine dinucleotide: new therapeutic approach for improving dementia of the Alzheimer type. Ann. Clin. Lab. Sci.26, 1–9 (1996). [PubMed] [Google Scholar]
- 378.Rainer, M. et al. No evidence for cognitive improvement from oral nicotinamide adenine dinucleotide (NADH) in dementia. J. Neural Transm. (Vienna).107, 1475–1481 (2000). [DOI] [PubMed] [Google Scholar]
- 379.Hirsch, E. C. & Faucheux, B. A. Iron metabolism and Parkinson’s disease. Mov. Disord.13, 39–45 (1998). [PubMed] [Google Scholar]
- 380.Jenner, P. & Olanow, C. W. Understanding cell death in Parkinson’s disease. Ann. Neurol.44, S72–S84 (1998). [DOI] [PubMed] [Google Scholar]
- 381.Foley, P. & Riederer, P. Influence of neurotoxins and oxidative stress on the onset and progression of Parkinson’s disease. J. Neurol.247, Ii82–Ii94 (2000). [DOI] [PubMed] [Google Scholar]
- 382.Vallerga, C. L. et al. Analysis of DNA methylation associates the cystine-glutamate antiporter SLC7A11 with risk of Parkinson’s disease. Nat. Commun.11, 1238 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 383.Ryan, S. K. et al. Microglia ferroptosis is regulated by SEC24B and contributes to neurodegeneration. Nat. Neurosci.26, 12–26 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 384.Bi, M. et al. α-Synuclein Regulates Iron Homeostasis via Preventing Parkin-Mediated DMT1 Ubiquitylation in Parkinson’s Disease Models. ACS Chem. Neurosci.11, 1682–1691 (2020). [DOI] [PubMed] [Google Scholar]
- 385.Peng, Y. et al. Binding of α-synuclein with Fe(III) and with Fe(II) and biological implications of the resultant complexes. J. Inorg. Biochem.104, 365–370 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 386.Ayton, S. et al. Transferrin protects against Parkinsonian neurotoxicity and is deficient in Parkinson’s substantia nigra. Signal Transduct. Target Ther.1, 16015 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 387.Ayton, S. et al. Parkinson’s disease iron deposition caused by nitric oxide-induced loss of β-amyloid precursor protein. J. Neurosci.35, 3591–3597 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 388.Angelova, P. R. et al. Alpha synuclein aggregation drives ferroptosis: an interplay of iron, calcium and lipid peroxidation. Cell Death Differ.27, 2781–2796 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 389.Mahoney-Sanchez, L. et al. Alpha synuclein determines ferroptosis sensitivity in dopaminergic neurons via modulation of ether-phospholipid membrane composition. Cell Rep.40, 111231 (2022). [DOI] [PubMed] [Google Scholar]
- 390.Mahoney-Sánchez, L. et al. Ferroptosis and its potential role in the physiopathology of Parkinson’s Disease. Prog. Neurobiol.196, 101890 (2021). [DOI] [PubMed] [Google Scholar]
- 391.Sun, J. et al. Midbrain dopamine oxidation links ubiquitination of glutathione peroxidase 4 to ferroptosis of dopaminergic neurons. J. Clin. Invest. 133, e165228 (2023). [DOI] [PMC free article] [PubMed]
- 392.Schapira, A. H. Mitochondrial complex I deficiency in Parkinson’s disease. Adv. Neurol.60, 288–291, (1993). [PubMed] [Google Scholar]
- 393.González-Rodríguez, P. et al. Disruption of mitochondrial complex I induces progressive parkinsonism. Nature599, 650–656 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 394.Kordower, J. H. & Burke, R. E. Disease Modification for Parkinson’s Disease: Axonal Regeneration and Trophic Factors. Mov. Disord.33, 678–683 (2018). [DOI] [PubMed] [Google Scholar]
- 395.Van Laar, A. D. et al. Transient exposure to rotenone causes degeneration and progressive parkinsonian motor deficits, neuroinflammation, and synucleinopathy. npj Parkinson Dis.9, 121 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 396.Sherer, T. B. et al. Mechanism of toxicity in rotenone models of Parkinson’s disease. J. Neurosci.23, 10756–10764 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 397.Schirinzi, T. et al. Dietary Vitamin E as a Protective Factor for Parkinson’s Disease: Clinical and Experimental Evidence. Front. Neurol.10, 148 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 398.Ross, C. A. & Tabrizi, S. J. Huntington’s disease: from molecular pathogenesis to clinical treatment. Lancet Neurol.10, 83–98 (2011). [DOI] [PubMed] [Google Scholar]
- 399.Walker, F. O. Huntington’s disease. Lancet369, 218–228 (2007). [DOI] [PubMed] [Google Scholar]
- 400.Agrawal, S., Fox, J., Thyagarajan, B. & Fox, J. H. Brain mitochondrial iron accumulates in Huntington’s disease, mediates mitochondrial dysfunction, and can be removed pharmacologically. Free Radic. Biol. Med.120, 317–329 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 401.Klepac, N. et al. Oxidative stress parameters in plasma of Huntington’s disease patients, asymptomatic Huntington’s disease gene carriers and healthy subjects. J. Neurol.254, 1676–1683 (2007). [DOI] [PubMed] [Google Scholar]
- 402.Lee, J. et al. Modulation of lipid peroxidation and mitochondrial function improves neuropathology in Huntington’s disease mice. Acta Neuropathol.121, 487–498 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 403.Rosas, H. D. et al. Alterations in Brain Transition Metals in Huntington Disease: An Evolving and Intricate Story. Arch. Neurol.69, 887–893 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 404.Quinti, L. et al. KEAP1-modifying small molecule reveals muted NRF2 signaling responses in neural stem cells from Huntington’s disease patients. Proc. Natl. Acad. Sci.114, E4676–E4685 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 405.Skouta, R. et al. Ferrostatins Inhibit Oxidative Lipid Damage and Cell Death in Diverse Disease Models. J. Am. Chem. Soc.136, 4551–4556 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 406.Chen, J. et al. Iron Accumulates in Huntington’s Disease Neurons: Protection by Deferoxamine. PLOS ONE8, e77023 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 407.Hatami, A. et al. Deuterium-reinforced linoleic acid lowers lipid peroxidation and mitigates cognitive impairment in the Q140 knock in mouse model of Huntington’s disease. FEBS J.285, 3002–3012 (2018). [DOI] [PubMed] [Google Scholar]
- 408.Djoussé, L. et al. Weight loss in early stage of Huntington’s disease. Neurology59, 1325–1330 (2002). [DOI] [PubMed] [Google Scholar]
- 409.Mahant, N., McCusker, E. A., Byth, K. & Graham, S. Huntington’s disease: clinical correlates of disability and progression. Neurology61, 1085–1092, (2003). [DOI] [PubMed] [Google Scholar]
- 410.Jenkins, B. G., Koroshetz, W. J., Beal, M. F. & Rosen, B. R. Evidence for impairment of energy metabolism in vivo in Huntington’s disease using localized 1H NMR spectroscopy. Neurology43, 2689–2695, (1993). [DOI] [PubMed] [Google Scholar]
- 411.Reynolds, N. C. Jr., Prost, R. W. & Mark, L. P. Heterogeneity in 1H-MRS profiles of presymptomatic and early manifest Huntington’s disease. Brain Res.1031, 82–89 (2005). [DOI] [PubMed] [Google Scholar]
- 412.Koroshetz, W. J., Jenkins, B. G., Rosen, B. R. & Beal, M. F. Energy metabolism defects in Huntington’s disease and effects of coenzyme Q10. Ann. Neurol.41, 160–165, (1997). [DOI] [PubMed] [Google Scholar]
- 413.Kuwert, T. et al. Cortical and subcortical glucose consumption measured by PET in patients with Huntington’s disease. Brain113, 1405–1423 (1990). [DOI] [PubMed] [Google Scholar]
- 414.Antonini, A. et al. Striatal glucose metabolism and dopamine D2 receptor binding in asymptomatic gene carriers and patients with Huntington’s disease. Brain119, 2085–2095 (1996). [DOI] [PubMed] [Google Scholar]
- 415.Kuwert, T. et al. Striatal glucose consumption in chorea-free subjects at risk of Huntington’s disease. J. Neurol.241, 31–36 (1993). [DOI] [PubMed] [Google Scholar]
- 416.Ciarmiello, A. et al. Brain white-matter volume loss and glucose hypometabolism precede the clinical symptoms of Huntington’s disease. J. Nucl. Med.47, 215–222 (2006). [PubMed] [Google Scholar]
- 417.Feigin, A. et al. Metabolic network abnormalities in early Huntington’s disease: an [(18)F]FDG PET study. J. Nucl. Med.42, 1591–1595 (2001). [PubMed] [Google Scholar]
- 418.Feigin, A. et al. Thalamic metabolism and symptom onset in preclinical Huntington’s disease. Brain130, 2858–2867 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 419.Dubinsky, J. M. Towards an Understanding of Energy Impairment in Huntington’s Disease Brain. J. Huntingt. Dis.6, 267–302 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 420.Browne, S. E. Mitochondria and Huntington’s Disease Pathogenesis. Ann. N. Y. Acad. Sci.1147, 358–382 (2008). [DOI] [PubMed] [Google Scholar]
- 421.Zuccato, C., Valenza, M. & Cattaneo, E. Molecular mechanisms and potential therapeutical targets in Huntington’s disease. Physiol. Rev.90, 905–981, (2010). [DOI] [PubMed] [Google Scholar]
- 422.Chen, C. M. Mitochondrial dysfunction, metabolic deficits, and increased oxidative stress in Huntington’s disease. Chang Gung Med. J.34, 135–152, (2011). [PubMed] [Google Scholar]
- 423.Borlongan, C. V., Koutouzis, T. K. & Sanberg, P. R. 3-Nitropropionic acid animal model and Huntington’s disease. Neurosci. Biobehav. Rev.21, 289–293, (1997). [DOI] [PubMed] [Google Scholar]
- 424.Guyot, M. C. et al. Quantifiable bradykinesia, gait abnormalities and Huntington’s disease-like striatal lesions in rats chronically treated with 3-nitropropionic acid. Neuroscience79, 45–56 (1997). [DOI] [PubMed] [Google Scholar]
- 425.Vis, J. C. et al. 3-Nitropropionic acid induces a spectrum of Huntington’s disease-like neuropathology in rat striatum. Neuropathol. Appl. Neurobiol.25, 513–521 (1999). [DOI] [PubMed] [Google Scholar]
- 426.Haider, L. et al. Multiple sclerosis deep grey matter: the relation between demyelination, neurodegeneration, inflammation and iron. J. Neurol. Neurosurg. Psychiatry85, 1386 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 427.Stephenson, E. et al. Iron in multiple sclerosis: roles in neurodegeneration and repair. Nat. Rev. Neurol.10, 459–468 (2014). [DOI] [PubMed] [Google Scholar]
- 428.Badaracco, M. E., Siri, M. V. & Pasquini, J. M. Oligodendrogenesis: the role of iron. Biofactors36, 98–102 (2010). [DOI] [PubMed] [Google Scholar]
- 429.Cheli, V. T., Correale, J., Paez, P. M. & Pasquini, J. M. Iron Metabolism in Oligodendrocytes and Astrocytes, Implications for Myelination and Remyelination. ASN Neuro12, 1759091420962681 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 430.Luoqian, J. et al. Ferroptosis promotes T-cell activation-induced neurodegeneration in multiple sclerosis. Cell. Mol. Immunol.19, 913–924 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 431.White, A. R. Ferroptosis drives immune-mediated neurodegeneration in multiple sclerosis. Cell. Mol. Immunol.20, 112–113 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 432.Feng, J. et al. Curcumin inhibits mitochondrial injury and apoptosis from the early stage in EAE mice. Oxid. Med. Cell. Longev.2014, 728751 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 433.Sadeghian, M. et al. Mitochondrial dysfunction is an important cause of neurological deficits in an inflammatory model of multiple sclerosis. Sci. Rep.6, 33249 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 434.Dutta, R. et al. Mitochondrial dysfunction as a cause of axonal degeneration in multiple sclerosis patients. Ann. Neurol.59, 478–489 (2006). [DOI] [PubMed] [Google Scholar]
- 435.Mahad, D., Ziabreva, I., Lassmann, H. & Turnbull, D. Mitochondrial defects in acute multiple sclerosis lesions. Brain131, 1722–1735, (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 436.Campbell, G. R. et al. Mitochondrial DNA deletions and neurodegeneration in multiple sclerosis. Ann. Neurol.69, 481–492 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 437.Meyer, N. & Rinholm, J. E. Mitochondria in Myelinating Oligodendrocytes: Slow and Out of Breath? Metabolites11, 359 (2021). [DOI] [PMC free article] [PubMed]
- 438.Atkinson, K. C., Osunde, M. & Tiwari-Woodruff, S. K. The complexities of investigating mitochondria dynamics in multiple sclerosis and mouse models of MS. Front. Neurosci.17, 1144896 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 439.Wilson, D. M. et al. Hallmarks of neurodegenerative diseases. Cell186, 693–714 (2023). [DOI] [PubMed] [Google Scholar]
- 440.Jimenez-Blasco, D. et al. Weak neuronal glycolysis sustains cognition and organismal fitness. Nat. Metab.6, 1253–1267 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 441.Lopez-Fabuel, I. et al. Aberrant upregulation of the glycolytic enzyme PFKFB3 in CLN7 neuronal ceroid lipofuscinosis. Nat. Commun.13, 536 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 442.Maiuri, M. C. et al. Functional and physical interaction between Bcl‐XL and a BH3‐like domain in Beclin‐1. EMBO J.26, 2527–2539 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 443.Wirawan, E. et al. Caspase-mediated cleavage of Beclin-1 inactivates Beclin-1-induced autophagy and enhances apoptosis by promoting the release of proapoptotic factors from mitochondria. Cell Death Dis.1, e18–e18 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 444.Song, X. et al. AMPK-mediated BECN1 phosphorylation promotes ferroptosis by directly blocking system Xc–activity. Curr. Biol.28, 2388–2399.e2385 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 445.Bononi, A. et al. BAP1 regulates IP3R3-mediated Ca2+ flux to mitochondria suppressing cell transformation. Nature546, 549–553 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 446.He, M. et al. Intrinsic apoptosis shapes the tumor spectrum linked to inactivation of the deubiquitinase BAP1. Science364, 283–285 (2019). [DOI] [PubMed] [Google Scholar]
- 447.Zhang, Y., Zhuang, L. & Gan, B. BAP1 suppresses tumor development by inducing ferroptosis upon SLC7A11 repression. Mol. Cell Oncol.6, 1536845 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 448.Maldonado, E. N. et al. Voltage-dependent anion channels modulate mitochondrial metabolism in cancer cells: regulation by free tubulin and erastin. J. Biol. Chem.288, 11920–11929 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 449.Veenman, L., Shandalov, Y. & Gavish, M. VDAC activation by the 18 kDa translocator protein (TSPO), implications for apoptosis. J. Bioenerg. Biomembr.40, 199–205 (2008). [DOI] [PubMed] [Google Scholar]
- 450.Veenman, L., Papadopoulos, V. & Gavish, M. Channel-like functions of the 18-kDa translocator protein (TSPO): regulation of apoptosis and steroidogenesis as part of the host-defense response. Curr. Pharm. Des.13, 2385–2405, (2007). [DOI] [PubMed] [Google Scholar]
- 451.Yagoda, N. et al. RAS–RAF–MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature447, 865–869 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 452.Tan, W. & Colombini, M. VDAC closure increases calcium ion flux. Biochim. Biophys. Acta Biomembr.1768, 2510–2515 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 453.Lautrup, S., Sinclair, D. A., Mattson, M. P. & Fang, E. F. NAD+ in Brain Aging and Neurodegenerative Disorders. Cell Metab.30, 630–655 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 454.Iqbal, T. & Nakagawa, T. The therapeutic perspective of NAD+ precursors in age-related diseases. Biochem. Biophys. Res. Commun.702, 149590 (2024). [DOI] [PubMed] [Google Scholar]
- 455.Cantó, C., Menzies, KeirJ. & Auwerx, J. NAD+ Metabolism and the Control of Energy Homeostasis: A Balancing Act between Mitochondria and the Nucleus. Cell Metab.22, 31–53 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 456.Belenky, P., Bogan, K. L. & Brenner, C. NAD+ metabolism in health and disease. Trends Biochem. Sci.32, 12–19, (2007). [DOI] [PubMed] [Google Scholar]
- 457.Zhao, Y. et al. NAD+ improves cognitive function and reduces neuroinflammation by ameliorating mitochondrial damage and decreasing ROS production in chronic cerebral hypoperfusion models through Sirt1/PGC-1α pathway. J. Neuroinflammation.18, 207 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 458.Zhou, Q. et al. Ferroptosis in cancer: From molecular mechanisms to therapeutic strategies. Signal Transduct. Target. Ther.9, 55 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 459.Nie, Q. et al. Induction and application of ferroptosis in cancer therapy. Cancer Cell Int22, 12 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 460.Chen, Z. et al. Ferroptosis as a potential target for cancer therapy. Cell Death Dis.14, 460 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 461.Li, B. et al. Besting Vitamin E: Sidechain Substitution is Key to the Reactivity of Naphthyridinol Antioxidants in Lipid Bilayers. J. Am. Chem. Soc.135, 1394–1405 (2013). [DOI] [PubMed] [Google Scholar]
- 462.Shimada, K. et al. Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat. Chem. Biol.12, 497–503 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 463.Fanet, H. et al. Tetrahydrobioterin (BH4) pathway: from metabolism to neuropsychiatry. Curr. Neuropharmacol.19, 591–609 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 464.Alli, A. A. et al. Kidney tubular epithelial cell ferroptosis links glomerular injury to tubulointerstitial pathology in lupus nephritis. Clin. Immunol.248, 109213 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 465.Zilka, O., Poon, J.-F. & Pratt, D. A. Radical-trapping antioxidant activity of copper and nickel bis (thiosemicarbazone) complexes underlies their potency as inhibitors of ferroptotic cell death. J. Am. Chem. Soc.143, 19043–19057 (2021). [DOI] [PubMed] [Google Scholar]
- 466.Devisscher, L. et al. Discovery of novel, drug-like ferroptosis inhibitors with in vivo efficacy. J. Med. Chem.61, 10126–10140 (2018). [DOI] [PubMed] [Google Scholar]
- 467.Hofmans, S. et al. Novel ferroptosis inhibitors with improved potency and ADME properties. J. Med. l Chem.59, 2041–2053 (2016). [DOI] [PubMed] [Google Scholar]
- 468.Shah, R., Margison, K. & Pratt, D. A. The potency of diarylamine radical-trapping antioxidants as inhibitors of ferroptosis underscores the role of autoxidation in the mechanism of cell death. ACS Chem. Biol.12, 2538–2545 (2017). [DOI] [PubMed] [Google Scholar]
- 469.Yang, W. et al. Structure-activity relationship studies of phenothiazine derivatives as a new class of ferroptosis inhibitors together with the therapeutic effect in an ischemic stroke model. Eur. J. Med. Chem.209, 112842 (2021). [DOI] [PubMed] [Google Scholar]
- 470.You, J. et al. Discovery of 2-vinyl-10H-phenothiazine derivatives as a class of ferroptosis inhibitors with minimal human Ether-a-go-go related gene (hERG) activity for the treatment of DOX-induced cardiomyopathy. Bioorg. Med. Chem. Lett.74, 128911 (2022). [DOI] [PubMed] [Google Scholar]
- 471.Farmer, L. A. et al. Intrinsic and extrinsic limitations to the design and optimization of inhibitors of lipid peroxidation and associated cell death. J. Am. Chem. Soc.144, 14706–14721 (2022). [DOI] [PubMed] [Google Scholar]
- 472.Scarpellini, C. et al. Beyond ferrostatin-1: a comprehensive review of ferroptosis inhibitors. Trends Pharmacol. Sci.44, 902–916 (2023). [DOI] [PubMed] [Google Scholar]
- 473.Van Coillie, S. et al. Targeting ferroptosis protects against experimental (multi) organ dysfunction and death. Nat. Commun.13, 1046 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 474.Jin, T. et al. UAMC-3203 or/and deferoxamine improve post-resuscitation myocardial dysfunction through suppressing ferroptosis in a rat model of cardiac arrest. Shock57, 344–350 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 475.Zilka, O. et al. On the mechanism of cytoprotection by ferrostatin-1 and liproxstatin-1 and the role of lipid peroxidation in ferroptotic cell death. ACS Cent. Sci.3, 232–243 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 476.Stoyanovsky, D. et al. Iron catalysis of lipid peroxidation in ferroptosis: Regulated enzymatic or random free radical reaction? Free Radic. Biol. Med.133, 153–161 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 477.Buss, J. L. et al. Iron chelators in cancer chemotherapy. Curr. Top. Med. Chem.4, 1623–1635 (2004). [DOI] [PubMed] [Google Scholar]
- 478.Hider, R. C. & Hoffbrand, A. V. The Role of Deferiprone in Iron Chelation. N. Engl. J. Med.379, 2140–2150 (2018). [DOI] [PubMed] [Google Scholar]
- 479.Treadwell, M. J. et al. Barriers to adherence of deferoxamine usage in sickle cell disease. Pediatr. Blood Cancer44, 500–507 (2005). [DOI] [PubMed] [Google Scholar]
- 480.Tam, F. T. et al. Iron Chelator Research: Past, Present, and Future. Curr. Med. Chem.10, 983–995 (2003). [DOI] [PubMed] [Google Scholar]
- 481.Hershko, C. Iron chelators in medicine. Mol. Asp. Med.13, 113–165 (1992). [DOI] [PubMed] [Google Scholar]
- 482.Elalfy, M. S. et al. Cardiac events and cardiac T2* in Egyptian children and young adults with beta-thalassemia major taking deferoxamine. Hematol. Oncol. Stem Cell Ther.3, 174–178 (2010). [DOI] [PubMed] [Google Scholar]
- 483.Kontoghiorghes, G., Pattichis, K., Neocleous, K. & Kolnagou, A. The design and development of deferiprone (L1) and other iron chelators for clinical use: targeting methods and application prospects. Curr. Med. Chem.11, 2161–2183, (2004). [DOI] [PubMed] [Google Scholar]
- 484.Ribeiro, L. B. et al. The challenges of handling deferasirox in sickle cell disease patients older than 40 years. Hematology24, 596–600 (2019). [DOI] [PubMed] [Google Scholar]
- 485.Barnham, K. Metal-based Neurodegeneration: From Molecular Mechanisms to Therapeutic Strategies. ChemMedChem1, 742–743 (2006). By Robert R. Crichton and Roberta J. Ward. [Google Scholar]
- 486.Devos, D. et al. Targeting chelatable iron as a therapeutic modality in Parkinson’s disease. Antioxid. Redox Signal.21, 195–210 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 487.Devos, D. et al. Trial of Deferiprone in Parkinson’s Disease. N. Engl. J. Med.387, 2045–2055 (2022). [DOI] [PubMed] [Google Scholar]
- 488.Kim, J. & Wessling-Resnick, M. Iron and mechanisms of emotional behavior. J. Nutr. Biochem.25, 1101–1107 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 489.Li, Q. et al. NAC alleviative ferroptosis in diabetic nephropathy via maintaining mitochondrial redox homeostasis through activating SIRT3-SOD2/Gpx4 pathway. Free Radic. Biol. Med.187, 158–170 (2022). [DOI] [PubMed] [Google Scholar]
- 490.Li, J. et al. Targeting Molecular Mediators of Ferroptosis and Oxidative Stress for Neurological Disorders. Oxid. Med. Cell Longev.2022, 3999083 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 491.Karuppagounder, S. S. et al. N-acetylcysteine targets 5 lipoxygenase-derived, toxic lipids and can synergize with prostaglandin E(2) to inhibit ferroptosis and improve outcomes following hemorrhagic stroke in mice. Ann. Neurol.84, 854–872 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 492.Huang, J. et al. The role of ferroptosis and endoplasmic reticulum stress in intermittent hypoxia-induced myocardial injury. Sleep. Breath.27, 1005–1011 (2023). [DOI] [PubMed] [Google Scholar]
- 493.Hu, M. et al. Suppression of uterine and placental ferroptosis by N-acetylcysteine in a rat model of polycystic ovary syndrome. Mol. Hum. Reprod. 27, gaab067 (2021). [DOI] [PubMed]
- 494.Monti, D. A. et al. N-Acetyl Cysteine May Support Dopamine Neurons in Parkinson’s Disease: Preliminary Clinical and Cell Line Data. PLoS One11, e0157602 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 495.Sunitha, K. et al. N-Acetylcysteine amide: a derivative to fulfill the promises of N-Acetylcysteine. Free Radic. Res.47, 357–367 (2013). [DOI] [PubMed] [Google Scholar]
- 496.Ates, B., Abraham, L. & Ercal, N. Antioxidant and free radical scavenging properties of N-acetylcysteine amide (NACA) and comparison with N-acetylcysteine (NAC). Free Radic. Res.42, 372–377, (2008). [DOI] [PubMed] [Google Scholar]
- 497.Belavgeni, A. et al. Exquisite sensitivity of adrenocortical carcinomas to induction of ferroptosis. Proc. Natl. Acad. Sci. USA116, 22269–22274 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 498.Vande Voorde, J. et al. Improving the metabolic fidelity of cancer models with a physiological cell culture medium. Sci. Adv.5, eaau7314 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 499.Vivash, L. et al. A study protocol for a phase II randomised, double-blind, placebo-controlled trial of sodium selenate as a disease-modifying treatment for behavioural variant frontotemporal dementia. BMJ Open10, e040100 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 500.Corcoran, N. M. et al. Sodium selenate specifically activates PP2A phosphatase, dephosphorylates tau and reverses memory deficits in an Alzheimer’s disease model. J. Clin. Neurosci.17, 1025–1033 (2010). [DOI] [PubMed] [Google Scholar]
- 501.Pushpakom, S. et al. Drug repurposing: progress, challenges and recommendations. Nat. Rev. Drug Dis.18, 41–58 (2019). [DOI] [PubMed] [Google Scholar]
- 502.Tan, Q. et al. Identifying eleven new ferroptosis inhibitors as neuroprotective agents from FDA-approved drugs. Bioorg. Chem.146, 107261 (2024). [DOI] [PubMed] [Google Scholar]
- 503.Ravanfar, P. et al. In Vivo 7-Tesla MRI Investigation of Brain Iron and Its Metabolic Correlates in Chronic Schizophrenia. Schizophrenia (Heidelb.)8, 86 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 504.Lotan, A. et al. Perturbed iron biology in the prefrontal cortex of people with schizophrenia. Mol. Psychiatry28, 2058–2070 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 505.Lei, P. et al. Lithium suppression of tau induces brain iron accumulation and neurodegeneration. Mol. Psychiatry22, 396–406 (2017). [DOI] [PubMed] [Google Scholar]
- 506.Tong, X. et al. Targeting cell death pathways for cancer therapy: recent developments in necroptosis, pyroptosis, ferroptosis, and cuproptosis research. J. Hematol. Oncol.15, 174 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 507.Xie, P. et al. Carbon nanoparticles-Fe(II) complex for efficient theranostics of xenografted colonic tumor. Cancer Nanotechnol. 14, 38 (2023).
- 508.Xie, R. et al. Quercetin alleviates kainic acid-induced seizure by inhibiting the Nrf2-mediated ferroptosis pathway. Free Radic. Biol. Med.191, 212–226 (2022). [DOI] [PubMed] [Google Scholar]
- 509.Zhou, Y. et al. Ferroptosis and Its Potential Role in the Nervous System Diseases. J. Inflamm. Res.15, 1555–1574 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 510.Kayser, E. B. et al. Evaluating the efficacy of vatiquinone in preclinical models of mitochondrial disease. Res. Sq.10.21203/rs.3.rs-4202689/v1 (2024).