

Abstract
Cervical cancer (CESC) presents significant clinical challenges due to its complex tumor microenvironment (TME) and varied treatment responses. This study identified undifferentiated M0 macrophages as high-risk immune cells critically involved in CESC progression. Co-culture experiments further demonstrated that M0 macrophages significantly promoted HeLa cell proliferation, migration, and invasion, underscoring their pivotal role in modulating tumor cell behavior within the TME. A nine-gene prognostic model constructed from immune gene signatures highlighted CXCL8 as a key regulator of M0 macrophage behavior. Functional experiments demonstrated that CXCL8 knockdown in M0 macrophages inhibited their proliferation, shifted polarization toward an M1-dominant phenotype, and reduced tumor-promoting M2 polarization. Co-culture experiments with CXCL8-deficient M0 macrophages further revealed a suppression of HeLa cell proliferation, migration, and invasion. These findings position M0 macrophages as central regulators within the TME and suggest that targeting pathways like CXCL8 could provide novel therapeutic strategies for improving outcomes in CESC patients.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-024-81726-y.
Keywords: Cervical cancer (CESC), M0 macrophages, CXCL8, tumor microenvironment (TME), Single-cell RNA sequencing (scRNA-seq)
Subject terms: Cancer, Immunology, Oncology
Introduction
Cervical squamous cell carcinoma and endocervical adenocarcinoma (CESC) remains the fourth most common cancer in women globally and is a leading cause of cancer-related mortality, particularly in low- and middle-income countries1. Over 90% of CESC cases are associated with persistent infections by high-risk human papillomavirus (HPV) strains, which underscores the significant role of HPV in its etiology2. Although HPV vaccination has proven effective in reducing the incidence of HPV-associated cervical cancer, it does not benefit patients already diagnosed, especially those with advanced-stage disease3. The prognosis for late-stage CESC remains poor, with a median survival of approximately 16.9 months, even with current therapies4. Furthermore, HPV vaccines are ineffective for non-HPV-related CESC cases, necessitating alternative treatment strategies5.
Chemotherapy is a standard treatment for advanced CESC, yet its effectiveness is hampered by the emergence of drug resistance, which results in variable response rates between 29% and 63%6. This resistance, often driven by factors within the tumor microenvironment (TME), complicates efforts to improve patient outcomes7. The TME is a complex milieu consisting of various cellular components, such as fibroblasts, endothelial cells, and immune cells, along with non-cellular elements like cytokines and growth factors8. These components interact in ways that promote tumor progression, metastasis, and resistance to therapy9,10. Therefore, understanding how the TME influences CESC progression is essential for identifying new therapeutic targets.
Among the immune cells in the TME, macrophages are particularly notable for their plasticity. These cells can differentiate into either M1 macrophages, which promote anti-tumor responses, or M2 macrophages, which support tumor growth11,12. However, less attention has been paid to undifferentiated M0 macrophages, which have the potential to polarize towards either phenotype13. The role of M0 macrophages in CESC is not well understood, despite their capacity to influence the balance between tumor suppression and promotion14. Thus, investigating M0 macrophages’ functions could provide insights into new strategies for modulating the TME to improve treatment outcomes.
In our study, we employed bioinformatics analyses using single-cell RNA sequencing (scRNA-seq) and bulk RNA sequencing data to identify key immune cell populations associated with CESC. Our results indicated that M0 macrophages, along with activated mast cells, are high-risk immune cell types linked to poor patient outcomes15. We chose to focus on M0 macrophages due to their functional plasticity and significant impact on tumor dynamics. Through in vitro co-culture experiments with M0 macrophages and HeLa cells, we demonstrated that M0 macrophages significantly enhance HeLa cell proliferation, migration, and invasion. These findings highlight the pivotal role of M0 macrophages in modulating the TME, thus contributing to cancer progression.
To further elucidate the molecular mechanisms underlying the impact of M0 macrophages, we developed a prognostic model using immune gene signatures identified through univariate analysis and random survival forest algorithms. The analysis identified several key prognostic genes, including CXCL8, FTH1, and SOD216,17. Among these, CXCL8 stood out due to its strong association with M0 macrophage activity. Our experiments demonstrated that CXCL8 knockdown in M0 macrophages led to a reduction in their proliferation, along with a shift towards an M1-dominant phenotype. This polarization change resulted in decreased tumor-promoting behaviors in HeLa cells, including reduced proliferation, migration, and invasion.
Overall, our findings suggest that targeting CXCL8 in M0 macrophages may represent a promising strategy for improving CESC treatment. By influencing macrophage polarization and modulating the TME, CXCL8 could be a potential therapeutic target to enhance the efficacy of existing therapies and reduce tumor aggressiveness. Future studies should explore the use of CXCL8 inhibitors in preclinical models to validate their effectiveness and potential in combination therapies for overcoming drug resistance in CESC.
Results
Immune cell macrophages M0 as a high-risk factor in CESC
The flowchart related to the study is presented in Fig. 1. We first performed univariate Cox regression analysis based on immune cell expression levels to identify high-risk factors associated with cervical squamous cell carcinoma (CESC). The results, visualized using a forest plot, indicated that both activated mast cells and M0 macrophages were significant risk factors, while activated CD4 memory T cells and CD8 T cells served as protective factors (Fig. 2A). Although activated mast cells are potential high-risk factors, our interest in the plasticity of M0 macrophages guided subsequent analyses. M0 macrophages can polarize into either anti-tumor M1 or pro-tumor M2 phenotypes, making them pivotal in tumor immune regulation and progression18,19. In contrast, the functional dynamics of mast cells are less adaptable and lack the extensive plasticity of M0 macrophages20,21. Investigating M0 macrophages offers unique insights into tumor biology and potential therapeutic strategies targeting their polarization.
Fig. 1.

Flowchart of this study.
Fig. 2.

M0 macrophage promotes HeLa cell proliferation in vitro. (A) Univariate Cox regression analysis demonstrates that activated mast cells and macrophages M0 are significant risk factors. (B) Immunohistochemistry results show elevated expression of macrophage biomarker CD68, M0 biomarker CD14 and M2 biomarker CD206 in cancerous tissues compared to adjacent non-cancerous tissues, while M1 biomarker CD86 expression was decreased. (C) Schematic representation of the co-culture system: one group with macrophages M0 in the upper chamber and HeLa cells in the lower chamber, and the other group with only culture medium in the upper chamber and HeLa cells in the lower chamber. (D) Proliferative rate of HeLa cells was detected using Ki67 antibody immunofluorescence in each group. HeLa cell proliferation is significantly promoted by macrophages M0. (E) Migration of HeLa cells was assessed using the wound healing assay. Migration of HeLa cells is significantly promoted by macrophages M0. *P < 0.05. (F) Invasion of HeLa cells was evaluated using the transwell assay. Invasion of HeLa cells is promoted by macrophages M0. ***P < 0.001.
To validate the effects of M0 macrophages on tumor progression, we utilized an in vitro co-culture system with THP1-polarized M0 macrophages and HeLa cells. Immunohistochemistry results showed elevated expression of macrophage biomarker CD68, M0 biomarker CD14, and M2 biomarker CD206 in patient-derived cancerous tissues compared to adjacent non-cancerous tissues, while M1 biomarker CD86 expression was decreased. (Fig. 2B). M0 macrophages were cultured in the upper chamber while HeLa cells were placed in the lower chamber, with a control group using only culture medium in the upper chamber (Fig. 2C). Immunofluorescence staining with the Ki67 antibody demonstrated that the presence of M0 macrophages significantly enhanced HeLa cell proliferation (Fig. 2D). Additionally, wound healing assays showed increased cell migration (Fig. 2E), and transwell assays confirmed that M0 macrophages promoted HeLa cell invasion (Fig. 2F).
To deepen our understanding of the specific genes expressed by immune cells in cancerous tissues and to develop a prognostic model for the further exploration of key prognostic genes in M0 macrophages related to CESC in subsequent analyses, we utilized single-cell RNA sequencing (scRNA-seq) technology. This approach enables a comprehensive examination of the tumor microenvironment at the cellular level, revealing distinct gene expression patterns that may play critical roles in the progression of CESC.
Quality control and PCA analysis of single-cell data
We integrated single-cell RNA sequencing (scRNA-seq) data from the GSE208653 dataset, which included three samples. Quality control filtering was applied (nFeature_RNA > 50 and percent.mt < 25) (Supplementary Fig. 1A-B), and principal component analysis (PCA) confirmed the absence of significant batch effects (Supplementary Fig. 1D). Using an ElbowPlot, we determined that 15 principal components were optimal for downstream analysis (Supplementary Fig. 1E), leading to the identification of 22 distinct subclusters through t-SNE clustering (Supplementary Fig. 2A). This approach allowed us to explore the heterogeneity within the tumor microenvironment and provided a deeper understanding of M0 macrophages’ specific contributions.
Immune cell cluster annotation and interaction analysis
Further annotation of the 22 subclusters was conducted using the R package SingleR, classifying them into nine major cell types, including T cells, epithelial cells, neutrophils, macrophages, monocytes, B cells, fibroblasts, NK cells, and endothelial cells (Supplementary Fig. 2B). We analyzed ligand-receptor interactions using the CellChat package, revealing intricate communication networks, particularly between endothelial cells and macrophages (Supplementary Fig. 3B). Differential expression analysis using thresholds of |avg_log2FC| > 2 and p_val_adj < 0.05 identified 315 marker genes associated with immune cell types (Supplementary Fig. 4A-F). These comprehensive analyses of immune cell interactions and marker genes laid the foundation for constructing a prognostic model to predict patient outcomes in CESC.
Identification of prognostic genes and prognostic model construction
Utilizing bulk RNA-seq data from the TCGA database, we performed univariate Cox regression on the 315 marker genes to identify prognostic indicators, resulting in 14 genes with significant associations (p < 0.01) (Fig. 3A). LASSO regression further narrowed this set to nine key genes (Fig. 3B-D). Patients were then randomly divided into training and validation cohorts (3:1 ratio), and a LASSO-derived risk score formula was developed to categorize patients into high-risk and low-risk groups. Kaplan-Meier survival analysis showed significantly lower overall survival in the high-risk group (Fig. 3E-F), with ROC curve analysis confirming the predictive performance of the model (Fig. 3G-H). Although the model was constructed using a comprehensive set of immune cell marker genes, we focused on specific genes to validate the model’s reliability.
Fig. 3.
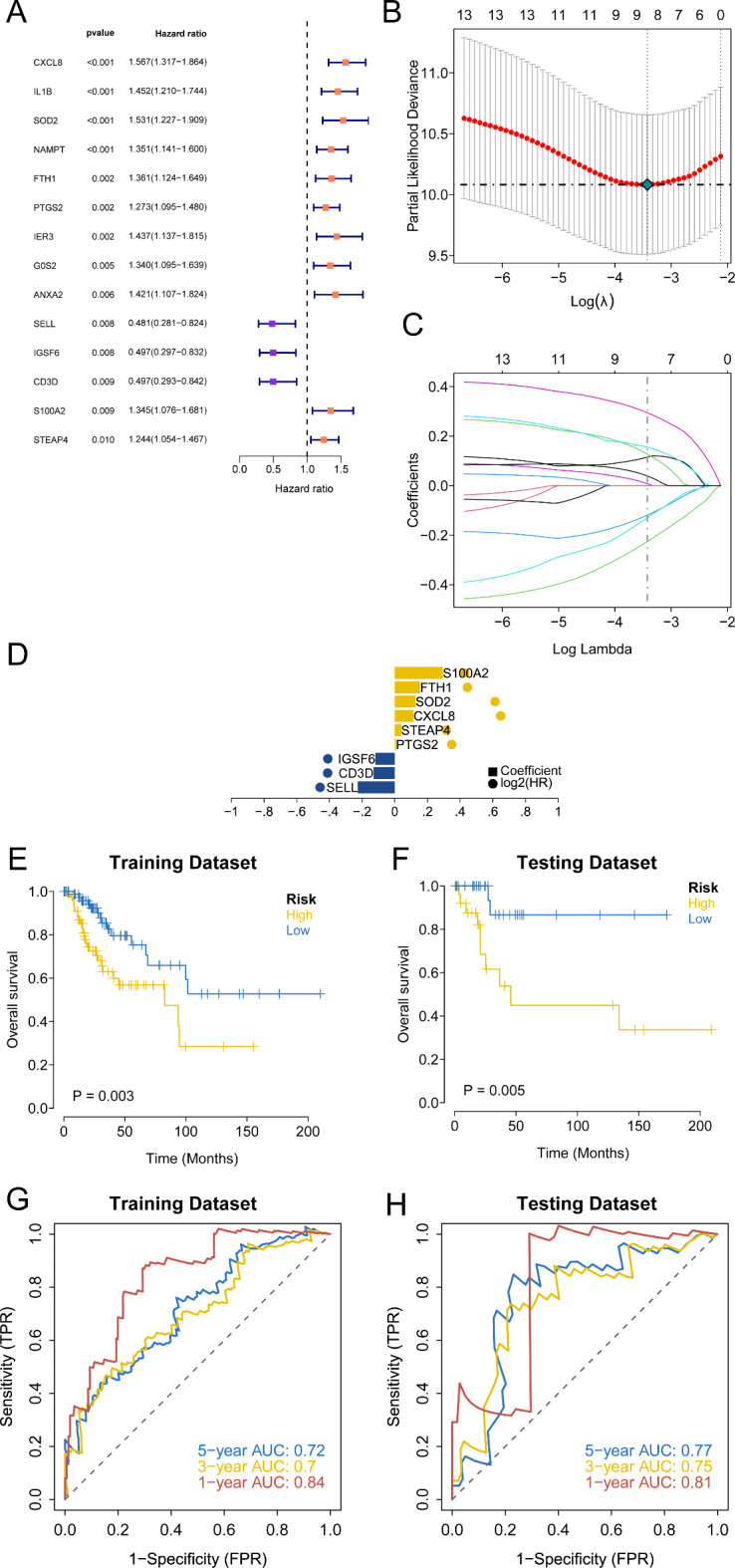
Identification of the key prognostic potential of marker genes associated with immune cells. (A) The forest figure for Univariate Cox regression analysis of the differentially expressed marker genes associated with immune cells. (B-D) The LASSO regression model selecting key genes from the identified prognostic markers. (E, F) Kaplan-Meier survival curve showing survival probability of high-risk score or low-risk score subgroups in both the training and validation sets. (G, H) ROC curves assessing the model’s predictive accuracy in both the training and validation sets.
External dataset validation of prognostic model robustness
Processed CESC patient data with survival information (GSE52903) and immunotherapy dataset IMOV were downloaded from the GEO database. Using the prognostic model, clinical stratification of CESC patients in these datasets was performed. Kaplan-Meier analysis evaluated survival differences between the high-risk and low-risk groups, assessing the model’s stability. The results demonstrated that in external validation sets, the overall survival (OS) of the high-risk group was significantly lower than that of the low-risk group (Supplementary Fig. 5A-B).
Experimental validation of key high-risk genes to confirm prognostic model significance
To confirm the robustness of our prognostic model, we selected key representative genes for further experimental validation. From the nine genes identified through LASSO regression, random survival forest analysis highlighted four genes with the highest predictive importance (importance score > 0.3) (Fig. 4A). A Venn diagram was used to illustrate the overlap between the high-risk genes (FTH1, SOD2, and CXCL8) and key prognostic genes (FTH1, SOD2, CXCL8, and SELL), with FTH1, SOD2, and CXCL8 being identified in both categories (Fig. 4B). These three genes were chosen as representative key high-risk genes to assess the model’s predictive accuracy.
Fig. 4.

Evaluating Prognostic Model Significance by Assessing the Impact of FTH1, SOD2, and CXCL8 Knockdowns on HeLa Cell Proliferation and Migration. (A) Displaying the importance scores of the four genes identified as significant markers by the random survival forest analysis. (B) Venn diagram illustrating the overlap between high-risk genes (FTH1, SOD2, and CXCL8) and key prognostic genes (FTH1, SOD2, CXCL8, and SELL), showing the intersection which includes FTH1, SOD2, and CXCL8. (C) Immunohistochemical staining reveals that the expression levels of FTH1, SOD2 and CXCL8 are significantly higher in the cancerous tissues of patients compared to the adjacent non-cancerous tissues. (D) After individually knocking down CXCL8, FTH1, or SOD2 using siRNA, the expression level of CXCL8, FTH1, or SOD2 was detected by qRT-PCR analysis. (E) MTT assay results show that the proliferation of HeLa cells is inhibited after knocking down FTH1, SOD2 or CXCL8 respectively. **P < 0.01, ***P < 0.001. (F) Scratch assay results indicate that the migration of HeLa cells is inhibited after knocking down FTH1, SOD2 or CXCL8 respectively. ****P < 0.0001. (G) Transwell assay results demonstrate that the invasion of HeLa cells is inhibited after knocking down FTH1, SOD2 or CXCL8 respectively. ****P < 0.0001.
Immunohistochemical staining of CESC patient tissue samples demonstrated significantly higher expression of FTH1, SOD2, and CXCL8 in cancerous tissues compared to adjacent non-cancerous tissues (Fig. 4C). To further assess their functional relevance, we performed siRNA-mediated knockdown in HeLa cells (Fig. 4D). The MTT assay showed reduced cell proliferation following gene knockdown (Fig. 4E). Additionally, scratch and transwell assays demonstrated that knocking down FTH1, SOD2, or CXCL8 led to decreased cell migration (Fig. 4F) and invasion (Fig. 4G), respectively. These findings underscore the accuracy of our prognostic model in reflecting patient outcomes in CESC.
Multi-omics study to explore the clinical predictive value of the prognostic model
To further validate the robustness of the prognostic model and explore its potential clinical applications, we extended our analysis to external datasets and multi-omics approaches. While external dataset validation confirmed that the risk groups accurately reflect patient survival outcomes, multi-omics analysis provided insights into risk score correlations with drug sensitivity and pathway enrichment, bridging prognosis with actionable therapeutic strategies. Our study utilized drug sensitivity data from the GDSC database and the R package ‘pRRophetic’ to predict chemotherapy sensitivity for each tumor sample and evaluate correlations with risk scores. The results highlighted significant associations between risk scores and sensitivity to multiple chemotherapy agents, including ATRA, AUY922, and AS601245 (Fig. 5A). Among these, AUY922, an HSP90 inhibitor, emerged as a particularly effective option for CESC patients with high expression of high-risk model genes (Fig. 5A). Its potential to target tumors with distinct molecular profiles underscores its translational relevance, and we therefore selected AUY922 for further in vitro experimental validation.
Fig. 5.

Multi-omics study to explore the clinical predictive value of the model. (A) Illustrating the correlation between risk scores and sensitivity to chemotherapy drugs. (B) GSVA analysis results highlight differential pathway enrichment between high-risk and low-risk groups. (C) GSEA results further illustrate key signaling pathways differentially enriched between different risk groups. (D) Showing the molecular interaction networks of pathways associated with high-risk and low-risk groups.
Subsequent biological experiments using the MTT assay showed that after knocking down high-risk group genes CXCL8, FTH1, or SOD2 in HeLa cells and adding different concentrations of the AUY922 inhibitor, the IC50 values in HeLa cells increased (Supplementary Fig. 6A). When AUY922 was directly added to HeLa cells, their proliferation was significantly inhibited (Supplementary Fig. 6B). In an in vitro co-culture system (Supplementary Fig. 6C), AUY922 was added to macrophages M0 in the upper chamber for 48 h without HeLa cells in the lower chamber. Then, the medium was replaced with normal medium in macrophages M0, and HeLa cells were seeded in a new lower chamber to form a co-culture system. The proliferation of HeLa cells in the lower chamber was significantly inhibited (Supplementary Fig. 6D).
Interestingly, when CXCL8 was knocked down in macrophages M0 (Supplementary Fig. 6E), the IC50 value of the AUY922 inhibitor increased. Conversely, when CXCL8 (IL8) was added to macrophages M0, the IC50 value of the AUY922 inhibitor decreased, suggesting that low expression of CXCL8 in macrophages reduces sensitivity to the AUY922 inhibitor (Supplementary Fig. 6F).
Next, we explored specific signaling pathways related to high and low-risk models to understand the molecular mechanisms by which risk scores affect tumor progression. GSVA results showed that differential pathways between groups were mainly enriched in the P53_PATHWAY, TGF_BETA_SIGNALING, and XENOBIOTIC_METABOLISM pathways (Fig. 5B). GSEA results indicated pathways involved in chemokine signaling, IL-17 signaling, and T cell receptor signaling (Fig. 5C). The molecular interaction networks of these pathways are illustrated (Fig. 5D).
Disease risk and independent prognostic analysis
Samples were divided into high-risk and low-risk groups based on the median risk score. Regression analysis results presented in a nomogram demonstrated that the risk score significantly contributed to the predictive accuracy of the nomogram model (Supplementary Fig. 7A). Additionally, predictions for the three-year and five-year overall survival of CESC patients were analyzed (Supplementary Fig. 7B-D). Both univariate and multivariate Cox regression analyses indicated that the risk score is an independent prognostic factor for CESC patients (Supplementary Fig. 7E-F).
Motif analysis and miRNA network aalysis
The prognostic model genes were analyzed as part of a gene set to discover shared regulatory mechanisms involving multiple transcription factors. Enrichment analysis of these transcription factors using cumulative recovery curves revealed significant motifs (Supplementary Fig. 8A). The motif annotated as cisbp__M3356 showed enrichment with a normalized enrichment score (NES) of 6.63, involving 4 model genes. We displayed all motifs and corresponding transcription factors enriched in the model genes (Supplementary Fig. 8B). Using the miRcode database, reverse prediction identified 79 miRNAs and 259 mRNA-miRNA interaction pairs for the 9 model genes, visualized with Cytoscape (Supplementary Fig. 8C).
Correlation analysis between prognostic model genes and tumor regulatory genes
We obtained CESC-related disease genes from the GeneCards database. Analysis of tumor gene expression differences between groups revealed significant expression differences for TP53, MET, PTEN, and PALB2 among patients and healthy controls (Supplementary Fig. 9A). The expression levels of the nine model genes were significantly correlated with several tumor-related genes. Notably, S100A2 was positively correlated with EGFR (r = 0.528), and CD3D was negatively correlated with AKT1 (r=-0.324) (Supplementary Fig. 9B). In cervical cancer, both EGFR (epidermal growth factor receptor) and AKT1 play significant roles in promoting tumor growth and survival22,23. EGFR, when activated, can stimulate the PI3K/AKT signaling pathway, leading to the activation of AKT124. This pathway is crucial for various cellular processes, including proliferation, survival, and metabolism25,26. The positive correlation between S100A2 and EGFR suggests their potential interaction in specific cellular environments and implies that elevated S100A2 expression may promote CESC progression.
Additionally, differential expression analysis revealed that CD3D, CXCL8, FTH1, IGSF6, S100A2, SELL, and STEAP4 were significantly upregulated in tumor tissues compared to controls (Supplementary Fig. 10A-I). Specifically, S100A2 expression was notably higher in cervical cancer (Supplementary Fig. 10F). Immunohistochemical staining of pathological sections from CESC patients confirmed significantly higher expression of S100A2 in cancerous tissues compared to adjacent non-cancerous tissues (Supplementary Fig. 11A). In vitro knockdown of S100A2 in HeLa cells (Supplementary Fig. 11B) resulted in inhibited proliferation, migration, and invasion of these cells (Supplementary Fig. 11 C-E). These findings suggest that lower expression of the high-risk gene S100A2 inhibits the progression of HeLa cells, thereby supporting the prognostic model.
Relationship between prognostic model and immune microenvironment
The TME comprising tumor-associated fibroblasts, immune cells, extracellular matrix, various growth factors, inflammatory factors, unique physicochemical characteristics, and cancer cells, significantly influences tumor diagnosis, survival outcomes, and clinical treatment sensitivity. By analyzing the relationship between risk scores and tumor immune infiltration, we explored the potential molecular mechanisms by which risk scores affect CESC progression. The proportion of immune cells differed significantly between low-risk and high-risk groups (Fig. 6A). Notably, macrophages M0 and activated dendritic cells were significantly elevated, while naive B cells and CD8 T cells were significantly reduced in the high-risk group compared to the low-risk group (Fig. 6B).
Fig. 6.

Identification of the key prognostic potential of immune cells. (A) Proportions of various immune cells in High-Risk and Low-Risk groups. (B) Showing the abundance of infiltrating immune cell populations at different risk-score.
CXCL8 as a key prognostic gene expressed in macrophages: implications for tumor progression
Notebly, among the nine risk model genes identified through LASSO regression, CXCL8, SOD2, and FTH1 were notable for their expression in macrophages (Fig. 7A-B). Furthermore, Kaplan-Meier survival analysis revealed that while all three genes exhibited significant prognostic value, CXCL8 was uniquely associated with poorer overall survival in patients with high expression levels (Supplementary Fig. 12A-C). This distinct correlation, combined with its expression pattern in macrophages, highlights CXCL8 as a particularly compelling candidate for experimental validation in subsequent experiment (Supplementary Fig. 12D).
Fig. 7.

Expression of key prognostic genes across various cell types. (A) Displaying the differential expression of the 9 key genes in various cell types. (B) Dot plot of 9 key genes for each cell type according to the previous research.
CXCL8 silencing impairs M0 macrophage proliferation and enhances the M1/M2 polarization balance
We investigated the role of CXCL8 in regulating M0 macrophage proliferation and polarization. First, we knocked down CXCL8 in M0 macrophages, and the results showed that this knockdown inhibited M0 cell proliferation (Fig. 8A). MTT assays and Ki67 antibody staining indicated that the proliferation of CXCL8-deficient M0 macrophages during polarization into M1 and M2 macrophages was significantly reduced (Fig. 8B-C). Interestingly, upon polarization, the expression of the M1 marker CD86 increased in CXCL8-deficient M0 macrophages, whereas the expression of the M2 marker CD206 decreased ( Fig. 8D-E). Conversely, when exogenous CXCL8 (IL8) was added to M0 macrophages prior to polarization, CD86 expression decreased while CD206 expression increased ( Fig. 8D-E). These results indicate that the deletion of CXCL8 can inhibit the proliferation of macrophages M0 and simultaneously increase the subsequent M1/M2 polarization ratio.
Fig. 8.

The absence of CXCL8 inhibits macrophage proliferation and promotes an increase in the M1/M2 ratio. (A) MTT assay results show that knocking down CXCL8 in macrophages M0 inhibited their proliferation, whereas adding extra CXCL8 promoted proliferation. (B, C) MTT assay and Ki67 immunofluorescence staining indicated decreased proliferation ability in CXCL8-deficient macrophages M0 polarized into M1 and macrophages M2. (D) WB analysis shows that the expression of the macrophage biomarker CD68 decreased in CXCL8-deficient macrophages M0; the M1 biomarker CD86 increased and the M2 biomarker CD206 decreased after polarization. (E) Flow cytometry analysis confirmed that CXCL8-deficient macrophages M0 exhibited decreased CD68 expression. Additionally, an increase in CD86 was observed after polarization into macrophages M1, while a decrease in CD206 was noted following polarization into macrophages M2.
The role of CXCL8 in mediating M0 macrophage-tumor cell interactions
To better clarify the role of CXCL8 in shaping the tumor immune microenvironment, we used a co-culture system with THP1-derived M0 macrophages and HeLa cells (Fig. 9A). After knocking down CXCL8 in M0 macrophages and subsequently co-culturing them with HeLa cells, we found that, compared to the control group of M0 macrophages co-cultured with HeLa cells, HeLa cell proliferation, migration, and invasion were significantly inhibited, as shown by Ki67 immunofluorescence, scratch assays, and Transwell assays (Fig. 9B-D). Conversely, after adding exogenous CXCL8 (IL8) to the M0 macrophage culture medium for 48 h and then replacing it with regular medium before co-culturing with HeLa cells, we observed an enhancement in HeLa cell proliferation, migration, and invasion compared to the control group (Fig. 9B-D). These findings underscore the crucial role of CXCL8 in regulating M0 macrophage-mediated tumor progression. By affecting M0 macrophage proliferation and polarization, as well as their interaction with tumor cells, CXCL8 acts as a pivotal link between the tumor immune microenvironment and cancer cell behavior, reinforcing its potential as a therapeutic target in CESC.
Fig. 9.

The expression of CXCL8 in M0 macrophages influences the proliferation, migration, and invasion of co-cultured HeLa cells. (A) Schematic representation of the co-culture system used to study the effect of CXCL8 expression in macrophages M0 on HeLa cells. THP1-polarized macrophages M0 were co-cultured with HeLa cells. (B) Ki67 immunofluorescence staining indicates that the proliferation of HeLa cells is inhibited when CXCL8 was knocked down in macrophages M0, whereas the addition of exogenous CXCL8 promotes HeLa cell proliferation. (C) Scratch assay results demonstrates that the migration of HeLa cells is inhibited by the deletion of CXCL8 in macrophages M0 and enhanced by the addition of exogenous CXCL8. (D) Transwell assay results show that the invasion of HeLa cells is suppressed when CXCL8 was knocked down in macrophages M0 and promoted when exogenous CXCL8 was added.
Discussion
The tumor microenvironment (TME) is an intricate network comprising diverse cellular and molecular components, including immune cells, stromal cells, blood vessels, cytokines, chemokines, and various signaling molecules within tumor tissues25–28. This dynamic environment not only supports tumor cell survival but also actively influences tumor growth, metastasis, and therapeutic resistance. In cervical squamous cell carcinoma (CESC), immune cells such as tumor-associated macrophages (TAMs), dendritic cells (DCs), regulatory T cells (Tregs), natural killer (NK) cells, and γδ T cells play essential roles in tumor progression through complex interactions that facilitate immune evasion, chronic inflammation, and modulation of immune responses29–31. These interactions make immune cells a focal point in understanding the pathophysiology of CESC and exploring novel therapeutic strategies.
Macrophages within the CESC immune microenvironment are primarily undifferentiated M0 macrophages, which possess the unique potential to polarize into either pro-inflammatory M1 or tumor-promoting M2 macrophages depending on the microenvironmental stimuli32. M1 macrophages secrete pro-inflammatory cytokines, such as TNF-α, IL-12, IL-1β, and IL-18, which contribute to anti-tumor immune responses by inhibiting angiogenesis and promoting tumor cell apoptosis33,34. Conversely, M2 macrophages are associated with tumor-supportive functions, secreting anti-inflammatory cytokines like IL-10 and TGF-β that facilitate tumor cell proliferation, invasion, and immune suppression35,36. While current research has extensively examined M2 macrophages in various cancers, the specific role and influence of undifferentiated M0 macrophages in the CESC tumor microenvironment remain less explored37,38. Understanding how these cells contribute to tumor dynamics and how their differentiation pathways can be modulated may open new avenues for cancer treatment.
Using the CIBERSORT algorithm to analyze immune cell composition, we identified M0 macrophages as high-risk immune cells associated with poor prognosis in CESC patients. Further cell communication analysis revealed strong interactions between M0 macrophages and both endothelial and epithelial cells within the tumor microenvironment (TME). These interactions suggest that M0 macrophages may actively contribute to tumor progression and immune modulation. This finding emphasizes the pivotal role of M0 macrophages as key players in the TME, whose interactions with other cell types potentially drive tumor aggressiveness and influence the immune landscape of CESC.
To explore the tumor-promoting potential of M0 macrophages in greater depth, we conducted in vitro co-culture experiments with HeLa cells, a commonly used model for CESC. Our results demonstrated that M0 macrophages significantly enhanced the proliferation, migration, and invasion capabilities of HeLa cells, underscoring their tumor-promoting characteristics. This observation highlights the relevance of M0 macrophages as modulators within the tumor microenvironment (TME), directly influencing tumor cell behavior and contributing to disease progression in CESC. These findings indicate that M0 macrophages, even in their undifferentiated state, play an active role in shaping the TME. This discovery opens a relatively unexplored area regarding their regulatory mechanisms, warranting further molecular investigation.
To further investigate the key genes involved in M0 macrophage-related mechanisms, we first identified an immune gene signature (IGS) and constructed a prognostic model39,40. This model was designed to identify high-risk genes associated with CESC progression. Through this approach, we identified nine prognosis-related genes, among which FTH1, SOD2, and CXCL8 were selected for experimental validation. The primary purpose of these validations was to confirm the model’s robustness and effectiveness in predicting CESC progression, independent of their roles in the immune microenvironment or their direct association with M0 macrophages. This validation step demonstrated that the deletion of FTH1, SOD2, and CXCL8 significantly suppressed HeLa cell proliferation, migration, and invasion, thereby confirming the prognostic reliability of the model.
With the validated model, we performed immune infiltration analysis, which revealed significantly higher levels of M0 macrophage infiltration in high-risk CESC patients compared to low-risk groups. This finding suggested that M0 macrophages play a critical role in shaping the immune microenvironment and promoting tumor aggressiveness. The combination of model validation and immune infiltration analysis further reinforced the biological significance of M0 macrophages in CESC progression.
Building on these results, we applied single-cell RNA sequencing (scRNA-seq) to investigate the expression patterns of the model genes within specific immune cell populations, particularly macrophages. This analysis identified FTH1, SOD2, and CXCL8 as highly expressed in macrophages, with CXCL8 showing a significant association with CESC prognosis. Consequently, we focused on CXCL8 as a high-risk gene expressed in M0 macrophages for further exploration. Our subsequent experiments aimed to elucidate the role of CXCL8 in regulating M0 macrophage behavior and its broader impact on tumor progression in CESC.
While previous studies have documented the involvement of CXCL8 in cervical cancer, there is limited research specifically examining its impact on M0 macrophages. To address this gap, our study focused on elucidating the pivotal role of CXCL8 in influencing M0 macrophage behavior within the CESC tumor microenvironment. In particular, we demonstrated that the knockdown of CXCL8 in M0 macrophages not only inhibited their proliferation but also altered their polarization, causing a shift from a pro-tumorigenic M2 phenotype towards an anti-tumorigenic M1 phenotype. This shift significantly reduced the proliferation, migration, and invasion of HeLa cells in co-culture, suggesting that CXCL8 plays a crucial role in regulating M0 macrophage behavior and its interactions with tumor cells. These findings underscore the importance of understanding immune cell dynamics and their interactions with tumor cells in the context of CESC.
Future research should focus on investigating the role of M0 macrophages as high-risk immune cells in CESC through rigorous in vitro and in vivo experimentation. A comprehensive analysis of key prognostic genes expressed in M0 macrophages, particularly CXCL8, is essential for understanding how their proliferation and polarization are regulated, as this may significantly influence tumor progression and the immune microenvironment. Additionally, exploring the molecular mechanisms by which CXCL8 influences M0 macrophages could provide critical insights into their interactions with tumor cells and highlight the potential of M0 macrophages as therapeutic targets. This knowledge can inform the development of small molecule inhibitors specifically targeting CXCL8 signaling in M0 macrophages. Such inhibitors could disrupt pro-tumorigenic pathways, inhibit tumor cell proliferation, and enhance the effectiveness of existing treatments. By selectively modulating M0 macrophages, we might improve patient outcomes and pave the way for innovative therapeutic strategies that harness the immune system to combat tumor progression.
Materials and methods
Data source and acquisition
The single-cell data files of GSE208653 were downloaded from the Gene Expression Omnibus (GEO) public database. Three samples with complete expression profiles were selected for analysis. Additionally, the Series Matrix File of GSE52903 was obtained, annotated with platform GPL6244, comprising 55 CESC patient data with complete expression profiles and survival information. Immunotherapy data were sourced using the R package “IMvigor210CoreBiology” (v. 2.0.0), retaining 348 samples with complete prognostic information and survival time greater than zero. The The Cancer Genome Atlas (TCGA) database (https://portal.gdc.cancer.gov/), as the largest cancer gene information repository, provided processed mRNA expression data for CESC, including normal (n = 3) and tumor groups (n = 306).
CIBERSORT analysis
The CIBERSORT41 method is widely used to evaluate immune cell types in the microenvironment. Based on the principle of support vector regression, it performs deconvolution analysis on the expression matrix of immune cell subtypes. CIBERSORT includes 547 biomarkers42–47, distinguishing 22 human immune cell phenotypes, such as T cells, B cells, plasma cells, and myeloid subpopulations. In this study, the CIBERSORT algorithm was applied to patient data to infer the relative proportions of these 22 immune-infiltrating cells.
Single-cell analysis
The expression profiles were imported using the Seurat package48, and low-expression genes were filtered out (nFeature_RNA > 50 & percent.mt < 25). The data underwent normalization, scaling, PCA, and analysis. The optimal number of principal components (15) was determined using an ElbowPlot. t-SNE analysis was performed to visualize the positional relationships among clusters. Cluster annotation was done using the celldex package, linking clusters to cells crucial in tumor development. Differentially expressed genes in immune cells were identified with criteria of |avg_log2FC|>2 and p_val_adj < 0.05.
Ligand-receptor Interaction Analysis (CellChat)
CellChat49 is a tool designed to quantitatively infer and analyze intercellular communication networks from single-cell data. It employs network analysis and pattern recognition to predict the main signaling inputs and outputs of cells and how these cells and signals coordinate their functions. In this analysis, the normalized single-cell expression profiles were used as input data, with cell subtypes from the single-cell analysis serving as cell information. The interactions were quantified by interaction strength (weights) and frequency (count), allowing us to observe the activity and impact of each cell type in the disease context.
Prognostic model construction and validation
Differential genes related to immune cells were selected for further analysis using LASSO regression to construct a prognostic model. Each patient’s risk score was calculated using a formula based on the expression values of specific genes, weighted by their LASSO regression coefficients. Patients were divided into low-risk and high-risk groups using the median risk score as a cutoff. Kaplan-Meier analysis evaluated survival differences between groups, compared using the log-rank test. LASSO regression and stratified analysis were used to test the risk score’s prognostic value. The model’s predictive a CESC uracy was assessed using ROC curves.
Random survival forest analysis
We used the randomForestSRC package for feature selection. Additionally, we utilized the random survival forest algorithm to rank the importance of prognostic-related genes (nrep = 1000, indicating 1000 iterations in the Monte Carlo simulation). Genes with a relative importance > 0.3 were identified as the final marker genes.
Drug sensitivity analysis
Based on the largest pharmacogenomics database (GDSC, https://www.cancerrxgene.org/), we used the R package “pRRophetic”50 to predict the chemotherapy sensitivity of each tumor sample. The regression method provided IC50 estimates for specific chemotherapy drugs, validated through 10-fold cross-validation with the GDSC training set. Default parameters were used, including “combat” for batch effect removal and averaging for duplicate gene expression values.
GSVA analysis (Gene Set Variation Analysis)
Gene Set Variation Analysis (GSVA)51 is a non-parametric, unsupervised method used to evaluate gene set enrichment in transcriptomics data. GSVA scores gene sets of interest, converting gene-level changes into pathway-level changes to infer biological functions in samples. In this study, gene sets were downloaded from the Molecular Signatures Database, and the GSVA algorithm was applied to each gene set to score and assess potential biological function changes across different samples.
GSEA analysis
Patients were divided into high-risk and low-risk groups based on the model’s risk scores. GSEA52 was performed to analyze differences in signaling pathways between these groups. Background gene sets were downloaded from the MsigDB database, and subtype pathway differential expression analysis was conducted. Significantly enriched gene sets (adjusted p-value < 0.05) were ranked based on consistency scores. GSEA analysis is commonly used to investigate the association between tumor subtypes and their biological significance.
Nomogram model construction
A nomogram was constructed based on regression analysis, incorporating gene expression levels and clinical symptoms. Using a scaled line, the nomogram visually represents the relationships between variables in the predictive model. A multifactor regression model was developed, assigning scores to each influencing factor based on their contribution to the outcome variable (regression coefficients). The individual scores were summed to obtain a total score, which was then used to calculate the predicted value.
Regulatory network analysis of model genes
In this study, we used the R package “RcisTarget”40 to predict transcription factors. RcisTarget performs all computations based on motifs. The normalized enrichment score (NES) of motifs depends on the total number of motifs in the database. In addition to annotated motifs, further annotations were inferred based on motif similarity and gene sequences. The first step in estimating motif overexpression in the gene set is calculating the area under the curve (AUC) for each motif-gene set pair. The NES for each motif is then calculated based on the AUC distribution across all motifs in the gene set. We used the rcistarget.hg19.motifdb.cisbpont.500 bp database for gene-motif rankings.
miRNA network construction
miRNAs (MicroRNAs) are small non-coding RNAs that regulate gene expression by promoting mRNA degradation or inhibiting mRNA translation. We analyzed whether key miRNAs regulate the transcription or degradation of some risk genes within our key genes. Relevant miRNAs for key genes were obtained from the miRcode database53. The miRNA network of these genes was visualized using Cytoscape software.
Statistical analysis
Survival curves were generated using the Kaplan-Meier method and compared with the log-rank test. Multivariate analysis was performed using the Cox proportional hazards model. All statistical analyses were conducted with R software (version 4.2). All statistical tests were two-sided, with p-values less than 0.05 considered statistically significant.
Immunohistochemical analysis
All histological sections were obatained from Department of Pathology in the afflicted hospital of Guizhou Medical University. These sections contained samples for 75 cases of CESC tissues and paired adjacent normal tissues, and 28 cases of unpaired CESC tissues. All samples were clinically and pathologically diagnosed. The Human Ethics Committee of Guizhou Medical University approved the collection of all samples from the affiliated hospital. All participants provided informed consent before any treatment such as radiotherapy or chemotherapy. These tissue samples had previously been analyzed through immunohistochemistry (IHC) staining. This analysis involved the use of CXCL8 antibody at a dilution of 1:200 (IPB3915, Baijia, China).
Cell culture
The HeLa cell line (derived from human cervical adenocarcinoma) was obtained from the Cell Bank of the Chinese Academy of Sciences in Shanghai, China. HeLa cells were cultured in DMEM medium with 10% fetal bovine serum (164210, Procell, China). siRNAs for S100A2, FTH1, SOD2, and CXCL8 (synthesized by Shanghai Kei Lei Biological Technology Co., Ltd.) were transfected into macrophages M0 using Lipofectamine Lipo3000 (L3000015, Thermo Fisher Scientific, USA). siRNA sequences are listed in Supplementary Table 1.
The THP-1 human monocytic cell line (ATCC, USA) was cultured in RPMI-1640 medium (C11875500BT, Gibco, USA) with 10% fetal bovine serum (164210, Procell, China). For macrophage differentiation, THP-1 monocytes were treated with PMA (100 ng/ml) for 48 h to derive macrophages M0, followed by LPS (100 ng/ml) and IFN-γ (20 ng/ml) for macrophages M1, or IL-4 (20 ng/ml) for macrophages M2. CXCL8-siRNA was transfected into macrophages M0 using Lipofectamine Lipo3000 according to the manufacturer’s instructions. In co-culture experiments, macrophages M0 and HeLa cells were co-cultured in a 6-well Transwell system (TCS021024, JETBIOFIL, China).
Quantitative real-time polymerase chain reaction (qRT-PCR)
Total RNA was isolated from cell lines using TRIzol reagent (CP_20211115002, Invitrogen, USA) in aCESCordance with the manufacturer’s protocol. RNA concentration were determined using a Nanodrop 300 spectrophotometer (ALLSHENG, China). For cDNA synthesis, 2 µg of total RNA was reverse-transcribed using Hifair® Lyo Multiplex One Step RT-qPCR Kit (11831ES60, Yeasen, China) following the manufacturer’s protocol. Quantitative real-time PCR (qRT-PCR) was conducted with SYBR Green Hieff Unicon® Universal TaqMan multiplex qPCR master mix (11211ES03, Yeasen, China) on a Real-Time PCR System (BIORAD, USA).The thermal cycling conditions were: initial denaturation at 95 °C for 15 min, followed by 32 cycles of 95 °C for 10 s, annealing at 58 °C for 25 s, and extension at 72 °C for 15 s. β-actin served as the internal control. Relative gene expression levels were calculated using the 2 − ΔΔCt method. The primers utilized for qRT-PCR are detailed in Supplementary Table 2.
Western blot
Total protein was lysed with a potent precooled lysate containing 2% PMSF protease inhibitor. After centrifugation, the protein supernatant was absorbed and quantified using the bicinchoninic acid method. SDS-PAGE electrophoresis and semi-wet protein transfer on PVDF membrane were performed. CXCL8 (IPB3915, Baijia, China), CD68 (HA601115, HUABIO, China), CD86 (AF1447, Beyotime, China), CD206 (AG2660, Beyotime, China), and GAPDH (60004-1-Ig, Proteintech, China) with dilution of 1:1000 were added overnight at 4 ℃. After washing thrice, a secondary antibody (a dilution 1:2000) was added and incubated for two hours at room temperature. A high-sensitivity ECL exposure solution was added and developed using an imager.
Flow cytometry
M0 macrophage cells across all experimental groups were digested using trypsin, and the reaction was halted by adding cell culture medium. After centrifugation, the cells were collected and washed twice with PBS. The cells were subsequently stained for 1 h at room temperature using antibodies specific to CD68 (HA601115, HUABIO, China), CD86 (AF1447, Beyotime, China), and CD206 (AG2660, Beyotime, China). Following three PBS washes, the cells were incubated with a FITC-conjugated secondary antibody (33106ES60,Yeasen, China) in darkness at room temperature for 1 h. This preparation was then analyzed using flow cytometry.
MTT assay
Each well of a 96-well plate was seeded with 2 × 10^4 THP-1 cells, and each experimental group comprised six replicate wells. The cells were treated with 100 ng/ml PMA to induce polarization into macrophages M0 over a 48-hour period. Subsequently, THP-1 M0 cells were transfected using RNATransMate transfection reagent with either siCXCL8 or siNC. Cells were either polarized into macrophages M1 or macrophages M2. Following another 48-hour incubation, the culture medium was discarded and each well was supplemented with 100 µl of 0.5 mg/ml MTT solution. The plates were then incubated at 37 °C for 4 h. Post incubation, the supernatant was removed and 100 µl of DMSO was added to each well. The plates were agitated in the dark at 100 rpm for 10 min to ensure complete dissolution of the formazan crystals. The absorbance was measured at 490 nm using a microplate reader.
Immunofluorescence
THP-1 cells were seeded into a 12-well plate equipped with coverslips and induced to differentiate into macrophages M0 by the addition of 100 ng/ml PMA for 48 h. Following this, siCXCL8 and siNC were transfected into the THP-1 M0 cells using RNATransMate transfection reagent, and the cells were subsequently induced to polarize into either macrophages M1 or macrophages M2. After incubating for 48 h, the cell culture medium was removed, and the cells were washed three times with PBS. The cells were then fixed with paraformaldehyde at room temperature for 15 min, and the fixative was removed. After three washes with PBS, the cells were permeabilized with 0.1% PBST at room temperature for 10 min. Following the removal of the permeabilization solution and three PBS washes, the cells were blocked with 5% BSA for 1 h. Ki67 antibody (AF1738, Beyotime, China) was added at a dilution of 1:200 and incubated overnight at 4 °C. After three washes with PBS, the cells were incubated with a secondary antibody (33106ES60,Yeasen, China) at room temperature for 1 h. Finally, the slides were mounted using a DAPI-containing antifade mounting medium.
Wound healing assay
THP-1 cells were plated in 6-well plates and differentiated into macrophages M0. The monolayers were then disrupted with a vertical scratch using a 200 µL pipette tip, introducing a wound across the M0 macrophage cells in each experimental group. Following the scratch, floating cells were removed by washing, and the wells were filled with serum-free medium. Wound healing was assessed at 0 and 24 h using an inverted optical microscope (Axiovert 200; Carl Zeiss, Germany). The migration of macrophages M0 towards the wound was evaluated by measuring the area of wound closure during this interval.
Transwell assay
HeLa cells were suspended in serum-free DMEM to a concentration of 1 × 10^6 cells/mL. Then, 200 µL of this suspension was added to the upper compartment of a Matrigel-pre-coated transwell insert (JETBIOFIL, China). In the lower section, macrophages M0 from various experimental groups were seeded in 600 µL of DMEM supplemented with 10% FBS. Following the incubation period, cells in the upper chamber were fixed using 4% paraformaldehyde for 20 min and then stained with 0.5% crystal violet for 15 min, with gentle washing afterward to remove non-invasive cells. The cells that migrated through the Matrigel to the underside of the membrane were subsequently observed under an orthotopic light microscope (Axioscope5; Carl Zeiss).
Electronic supplementary material
Below is the link to the electronic supplementary material.
Author contributions
Xiyan Zhao, Wei Pan, and Tengxiang Chen conceptualized and designed the experiments and drafted the manuscript. Zhirui Zeng, Tuo Zhang, and Jigang Pan prepared Figures 1–6 and Supplementary Figures 1–3, while Li Yang, Ziwen Xiao, and Yushi Yang prepared Figures 7–9 and Supplementary Figures 4–12. All authors reviewed and approved the final version of the manuscript.
Funding
This study is funded by the Continuous Support Fund for Excellent Scientific Research Platform of Colleges and Universities in Guizhou Province (QJJ (2022) 020 to Tengxiang Chen), Discipline Leading Talents Project of the Affiliated Hospital of Guizhou Medical University (No.gyfyxkyc-2023-01 to Tengxiang Chen) and Postdoctoral Research Funding of the Affiliated Hospital of Guizhou Medical University (BSH-Q-2022-1 to Xiyan Zhao).
Data availability
In this study, we analyzed publicly available datasets from the following sources: GEO (https://www.ncbi.nlm.nih.gov/), TCGA (https://portal.gdc.cancer.gov/), and IMvigor210CoreBiologies (http://research-pub.gene.com/IMvigor210CoreBiologies).
Declarations
Ethics approval and consent to participate
The experiments involving cervical cancer samples in this study were approved by the Human Research Ethics Review Committee of Guizhou Medical University (approval number: 2024-77). Forty cervical tissue samples were obtained and processed in accordance with the principles of the Declaration of Helsinki, and all participants provided informed consent.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors are contributed equally to this work: Xiyan Zhao and Li Yang.
Contributor Information
Tengxiang Chen, Email: txch@gmc.edu.cn.
Ziwen Xiao, Email: xzwe@hotmail.com.
Wei Pan, Email: 313831139@qq.com.
References
- 1.Hao, Z. et al. (ed AghaKouchak, A.) Global integrated drought monitoring and prediction system (GIDMaPS) data sets. Figshare10.6084/m9.Figshare.853801 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Small, W. Jr et al. Cervical cancer: a global health crisis. Cancer123 (13), 2404–2412. 10.1002/cncr.30667 (2017). [DOI] [PubMed] [Google Scholar]
- 3.Arbyn, M. et al. Prophylactic vaccination against human papillomaviruses to prevent cervical cancer and its precursors. Cochrane Database Syst. Reviews no. 510.1002/14651858.CD009069.pub3 (2019). [DOI] [PMC free article] [PubMed]
- 4.Cohen, P. A. et al. Cerv. cancer Lancet ;393(10167):169–182. 10.1016/S0140-6736(18)32470-X. (2019). [DOI] [PubMed] [Google Scholar]
- 5.Lin, Z. et al. Decoding the TME and molecular mechanism: unraveling cervical cancer subpopulations and prognostic signatures through scRNA-Seq and bulk RNA-seq analyses. Front. Immunol.10.3389/fimmu.2024.1351287 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tewari, K. S. et al. Improved survival with bevacizumab in advanced cervical cancer. N Engl. J. Med.370 (8), 734–743. 10.1056/NEJMoa1309748 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hanahan, D. & Weinberg, R. A. Hallmarks of cancer: The next generation. Cell, vol. 144, no. 5, pp. 646–674. (2011). 10.1016/j.cell.2011.02.013 [DOI] [PubMed]
- 8.Coussens, L. M., Werb, Z. & Inflammation and cancer. Nature, vol. 420, no. 6917, pp. 860–867. (2002). 10.1038/nature01322 [DOI] [PMC free article] [PubMed]
- 9.Joyce, J. A. & Fearon, D. T. T cell exclusion, immune privilege, and the TME. Science, vol. 348, no. 6230, pp. 74–80. (2015). 10.1126/science.aaa6204 [DOI] [PubMed]
- 10.Quail, D. F. & Joyce, J. A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med.19 (11), 1423–1437. 10.1038/nm.3394 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Galon, J. et al. Cancer classification using the immunoscore: a worldwide task force. J. Translational Med.10 (1), 205. 10.1186/1479-5876-10-205 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fridman, W. H. et al. The immune contexture in human tumours: impact on clinical outcome. Nat. Rev. Cancer. 12 (4), 298–306. 10.1038/nrc3245 (2012). [DOI] [PubMed] [Google Scholar]
- 13.Vinay, D. S. et al. Immune evasion in cancer: mechanistic basis and therapeutic strategies. Sem. Cancer Biol.3510.1016/j.semcancer.2015.03.004 (2015). pp. S185-S198. [DOI] [PubMed]
- 14.Mantovani, A. et al. Macrophage plasticity and polarization in tissue repair and remodelling. J. Pathol.229 (2), 176–185. 10.1002/path.4133 (2013). [DOI] [PubMed] [Google Scholar]
- 15.Junttila, M. R. & de Sauvage, F. J. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature, vol. 501, no. 7467, pp. 346–354. (2013). 10.1038/nature12626 [DOI] [PubMed]
- 16.Polyak, K. et al. Tumor heterogeneity and precision oncology. Breast Cancer Res.17 (1), 112. 10.1186/s13058-015-0628-0 (2015).26286584 [Google Scholar]
- 17.Newman, A. M. et al. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods. 12 (5), 453–457. 10.1038/nmeth.3337 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Walther, M. et al. Efficient polymeric nanoparticles for RNAi in macrophages reveal complex effects on polarization markers upon knockdown of STAT3/STAT6. Eur. J. Pharm. Biopharm.197, 114232. 10.1016/j.ejpb.2024.114232 (2024). [DOI] [PubMed] [Google Scholar]
- 19.Huang, L. et al. EFEMP2 indicates assembly of M0 macrophage and more malignant phenotypes of glioma. Aging (Albany NY). 12 (9), 8397–8412. 10.18632/aging.103147 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Galli, S. J. et al. Phenotypic and functional plasticity of cells of innate immunity: macrophages, mast cells and neutrophils. Nat. Immunol.12 (11), 1035–1044. 10.1038/ni.2109 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mekori, Y. A. et al. Integrating innate and adaptive immune cells: mast cells as crossroads between regulatory and effector B and T cells. Eur. J. Pharmacol.778, 84–89. 10.1016/j.ejphar.2015.03.087 (2015). [DOI] [PubMed] [Google Scholar]
- 22.Patel, A. P. et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science, vol. 344, no. 6190, pp. 1396–1401. (2014). 10.1126/science.1254257 [DOI] [PMC free article] [PubMed]
- 23.Tirosh, I. et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science, vol. 352, no. 6282, pp. 189–196. (2016). 10.1126/science.aad0501 [DOI] [PMC free article] [PubMed]
- 24.Engelman, J. A. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat. Rev. Cancer. 9 (8), 550–562. 10.1038/nrc2664 (2009). [DOI] [PubMed] [Google Scholar]
- 25.Villani, A. C. et al. Single-cell RNA-seq reveals new types of human blood dendritic cells, monocytes, and progenitors. Science, vol. 356, no. 6335, pp. 1–9. (2017). 10.1126/science.aah4573 [DOI] [PMC free article] [PubMed]
- 26.Waugh, D. J. & Wilson, C. The interleukin-8 pathway in cancer. Clin. Cancer Res.14 (21), 6735–6741. 10.1158/1078-0432.CCR-07-4843 (2008). [DOI] [PubMed] [Google Scholar]
- 27.Cancer Genome Atlas Research Network. Integrated genomic and molecular characterization of cervical cancer. Nature543 (7645), 378–384. 10.1038/nature21386 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Alzahrani, A. S. PI3K/Akt/mTOR inhibitors in cancer: at the bench and bedside. Sem. Cancer Biol.59, 125–132. 10.1016/j.semcancer.2019.07.009 (2019). [DOI] [PubMed] [Google Scholar]
- 29.Gajewski, T. F., Schreiber, H. & Fu, Y. X. Innate and adaptive immune cells in the TME. Nat. Immunol.14 (10), 1014–1022. 10.1038/ni.2703 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Binnewies, M. et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med.24 (5), 541–550. 10.1038/s41591-018-0014-x (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Joyce, J. A. & Pollard, J. W. Microenvironmental regulation of metastasis. Nat. Rev. Cancer. 9 (4), 239–252. 10.1038/nrc2618 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pollard, J. W. Trophic macrophages in development and disease. Nat. Rev. Immunol.9 (4), 259–270. 10.1038/nri2528 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ostrand-Rosenberg, S. & Sinha, P. Myeloid-derived suppressor cells: linking inflammation and cancer. J. Immunol.182 (8), 4499–4506. 10.4049/jimmunol.0802740 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Biswas, S. K. & Mantovani, A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat. Immunol.11 (10), 889–896. 10.1038/ni.1937 (2010). [DOI] [PubMed] [Google Scholar]
- 35.Ruffell, B. & Coussens, L. M. Macrophages and therapeutic resistance in cancer. Cancer Cell.27 (4), 462–472. 10.1016/j.ccell.2015.02.015 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.DeNardo, D. G. et al. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov. 1 (1), 54–67. 10.1158/2159-8274.CD-10-0028 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Papalexi, E. & Satija, R. scRNA-seq to explore immune cell heterogeneity. Nat. Rev. Immunol.18 (1), 35–45. 10.1038/nri.2017.76 (2018). [DOI] [PubMed] [Google Scholar]
- 38.Mantovani, A., Allavena, P., Sica, A. & Balkwill, F. Cancer-related inflammation. Nature454 (7203), 436–444. 10.1038/nature07205 (2008). [DOI] [PubMed] [Google Scholar]
- 39.Fridman, W. H., Pages, F., Sautes-Fridman, C. & Galon, J. The immune contexture in human tumours: impact on clinical outcome. Nat. Rev. Cancer. 12 (4), 298–306. 10.1038/nrc3245 (2012). [DOI] [PubMed] [Google Scholar]
- 40.Aibar, S. et al. SCENIC: single-cell regulatory network inference and clustering. Nat. Methods. 14 (11), 1083–1086. 10.1038/nmeth.4463 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Newman, A. M. et al. Profiling Tumor infiltrating Immune cells with CIBERSORT. Methods Mol. Biol.1711, 243–259. 10.1007/978-1-4939-7493-1_12 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ma, H. et al. Review Progress in Cervical Cancer Biomarkers. Clinical and 51, 4, p. 89. (2024). 10.31083/j.ceog5104089
- 43.Dubey, H. et al. Evaluation of HE4 as a prognostic biomarker in uterine cervical cancer. Cancer Treatment and Research Communications, X, X, p. 100672. (2022). 10.1016/j.ctarc.2022.100672 [DOI] [PubMed]
- 44.Mallik, S. et al. A Linear Regression and Deep Learning Approach for Detecting Reliable Genetic Alterations in Cancer Using DNA Methylation and Gene Expression Data. Genes, vol. 11, no. 8, p. 931. (2020). 10.3390/genes11080931 [DOI] [PMC free article] [PubMed]
- 45.Seth, S. et al. Dimensionality Reduction and Louvain Agglomerative Hierarchical Clustering for Cluster-Specified Frequent Biomarker Discovery in Single-Cell Sequencing Data.Front. Genet., 13, 10.3389/fgene.2022.828479. (2022). [DOI] [PMC free article] [PubMed]
- 46.Seth, S. et al. Identifying Genetic Signatures from Single-Cell RNA Sequencing Data by Matrix Imputation and Reduced Set Gene Clustering. Mathematics, vol. 11, no. 20, p. 4315. (2023). 10.3390/math11204315
- 47.Zhou et al. Prediction of prognosis and chemotherapeutic sensitivity based on cuproptosis-Associated lncRNAs in cervical squamous cell carcinoma and Endocervical Adenocarcinoma. Genes, 14, 7, p. 1381. (2023). 10.3390/genes14071381 [DOI] [PMC free article] [PubMed]
- 48.Jin, S. et al. Integrated analysis of multimodal single-cell data. Cell184 (13), 3573–3587. .e29 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jin, S. et al. Inference and analysis of cell-cell communication using CellChat. Nat. Commun.12 (1), 1088. 10.1038/s41467-021-21246-9 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Geeleher, P. et al. pRRophetic: an R package for prediction of clinical chemotherapeutic response from tumor gene expression levels. PLoS One. 9 (9). 10.1371/journal.pone.0107468 (2014). [DOI] [PMC free article] [PubMed]
- 51.Hänzelmann, S. et al. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. ;14:7. 10.1186/1471-2105-14-7. (2013). [DOI] [PMC free article] [PubMed]
- 52.Yu, G. et al. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS16 (5), 284–287. 10.1089/omi.2011.0118 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen, Y. et al. miRDB: an online database for prediction of functional microRNA targets. Nucleic Acids Res.48(D1)10.1093/nar/gkz757 (2020). [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Citations
- Seth, S. et al. Dimensionality Reduction and Louvain Agglomerative Hierarchical Clustering for Cluster-Specified Frequent Biomarker Discovery in Single-Cell Sequencing Data.Front. Genet., 13, 10.3389/fgene.2022.828479. (2022). [DOI] [PMC free article] [PubMed]
- Hänzelmann, S. et al. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. ;14:7. 10.1186/1471-2105-14-7. (2013). [DOI] [PMC free article] [PubMed]
Supplementary Materials
Data Availability Statement
In this study, we analyzed publicly available datasets from the following sources: GEO (https://www.ncbi.nlm.nih.gov/), TCGA (https://portal.gdc.cancer.gov/), and IMvigor210CoreBiologies (http://research-pub.gene.com/IMvigor210CoreBiologies).