

Abstract
Hydrogen sulfide (H2S) and other reactive sulfur species are important small molecules with biological significance. In addition to common reactive sulfur species like H2S, polysulfides, and persulfides, both carbonyl sulfide (COS) and carbon disulfide (CS2) have been postulated to be potential sources of reduced sulfur. To better understand this possible connection, we demonstrate that H2S can be converted to COS and CS2 by reaction with simple organic carbonate and thiocarbonate electrophiles, respectively.
Graphical Abstract
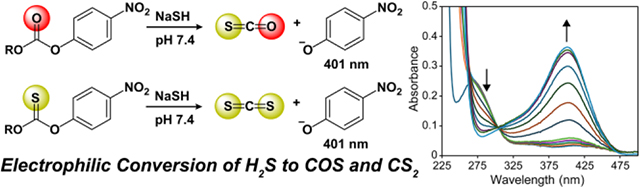
Reactive sulfur species (RSS) play important roles in synthetic chemistry and also in chemical biology, with common RSS oxidation states spanning from −2 to +6 in most organisms.1 The most common fully reduced RSS are thiols, hydrogen sulfide (H2S), and sulfides found in FeS clusters. Other small molecules like carbonyl sulfide (COS) and carbon disulfide (CS2) have been proposed to be potentially important biological molecules and also contain sulfur atoms in the −2 oxidation state. Focusing on COS, it has been previously proposed as a potential biomarker for various disease states including cystic fibrosis,2 acute rejection of organs,3 and liver disease.4 In addition, COS and CS2 may also play roles at the interface of H2S and NO chemistry by reacting with NO2− to generate different combinations of S/N hybrid species, NO, and sulfane sulfur motifs.5–6 These and other factors led to the proposition that COS may be involved in potential crosstalk through the gasotransmitters CO and H2S.7 In addition, Zn-based carbonic anhydrase (CA), which normally converts CO2 into bicarbonate, can also hydrolyze COS to H2S.8 We and others have leveraged this conversion to develop small molecule H2S donors that initially release COS as a precursor to H2S, which offers significant advantages over direct addition of sulfide salts like NaSH or Na2S.9–10
Common approaches to release COS include activatable organic thiocarbonates11 and thiocarbamates,9 both of which have been incorporated into responsive donors that respond to different triggers, including H2O2,12 enzyme activation,13 or reduction14 to undergo self-immolative cascade reactions to release COS, which is then converted to H2S by CA (Figure 1a). In addition, other COS-releasing platforms, including N-thiocarboxyanhydrides and perthiocarbonates have also been developed.15–16 Based on the utility of COS to H2S conversion, we were curious whether organic platforms could be designed to carry out the reverse reaction and convert H2S into COS (Figure 1b). To approach this question, we envisioned that addition of nucleophilic hydrosulfide (HS−),17 which is the most prevalent form of H2S at physiological pH (7.4),18 to carbonyl electrophiles could result in COS generation.
Figure 1.

(a) Example of prior work to develop distally activated COS/H2S donors. (b) General approach from this work to convert H2S to COS using activated carbonyl electrophiles.
To test this general approach, we treated 4-nitrophenyl chloroformate with 1 equiv. of NaSH in CD3OD, which resulted in immediate formation of a yellow solution corresponding to p-nitrophenol generation. This product identity was confirmed by 1H NMR spectroscopy (57% conversion), and the conversion could be improved to 96% conversion by using 2 equiv. of NaSH (Scheme 1, See SI for additional details). Under these unbuffered conditions, NaSH is likely acting as both nucleophile and a base based on the similar pKa values of H2S (7.0)18 and 4-nitrophenol (7.15),19 which is consistent with the increased conversion when excess NaSH is used.
Scheme 1.

Reaction of 4-nitrophenyl chloroformate with NaSH in CD3OD.
Next we investigated this reaction in pH 7.4 buffer to both determine the water compatibility of this reaction and to simplify the proton inventory. We monitored the initial rate of p-nitrophenol formation from 4-nitrophenyl chloroformate and observed rapid hydrolysis (rate ~ 7×10−5 M/s, t1/2 < 10 sec).20 To improve the stability of these compounds, we modified the electrophilicity of the carbonyl motif with the goal of improving water compatibility while maintaining reactivity toward HS−. We prepared an electronically varied series of aryl and alkyl carbonate derivates while maintaining the p-nitrophenol motif as a convenient UV-vis handle (p-nitrophenolate: λmax = 401 nm, ε = 14,400 ± 500 M−1 in pH 7.4 PBS).
To measure the reactivity of these electrophiles toward HS−, we treated 25 μM of each compound with 40 equiv. of NaSH in pH 7.4 PBS, measured initial rates of p-nitrophenolate formation, and compared reaction rates with NaSH with the background rate of hydrolysis. Using 4-nitrophenyl methyl carbonate (1a), we found the initial reaction rate with HS− was only 1.8 times faster than background hydrolysis.21 We found that CF3CH2 functionalized 1b reacted significantly faster NaSH (initial rate 9.05(8) × 10−9 M/s), which matched our expectation based on the pKa of the leaving group (MeOH = 15.54, CF3CH2OH = 12.46) (Figure 2). More importantly, the selectivity of 1b for NaSH over background hydrolysis was improved to 4.22:1. We also used this compound to definitively confirm COS release by GC after treating 25 μM of 1b with 40 equiv. of NaSH for 10 minutes and analyzing the headspace (see S21–S22).
Figure 2.
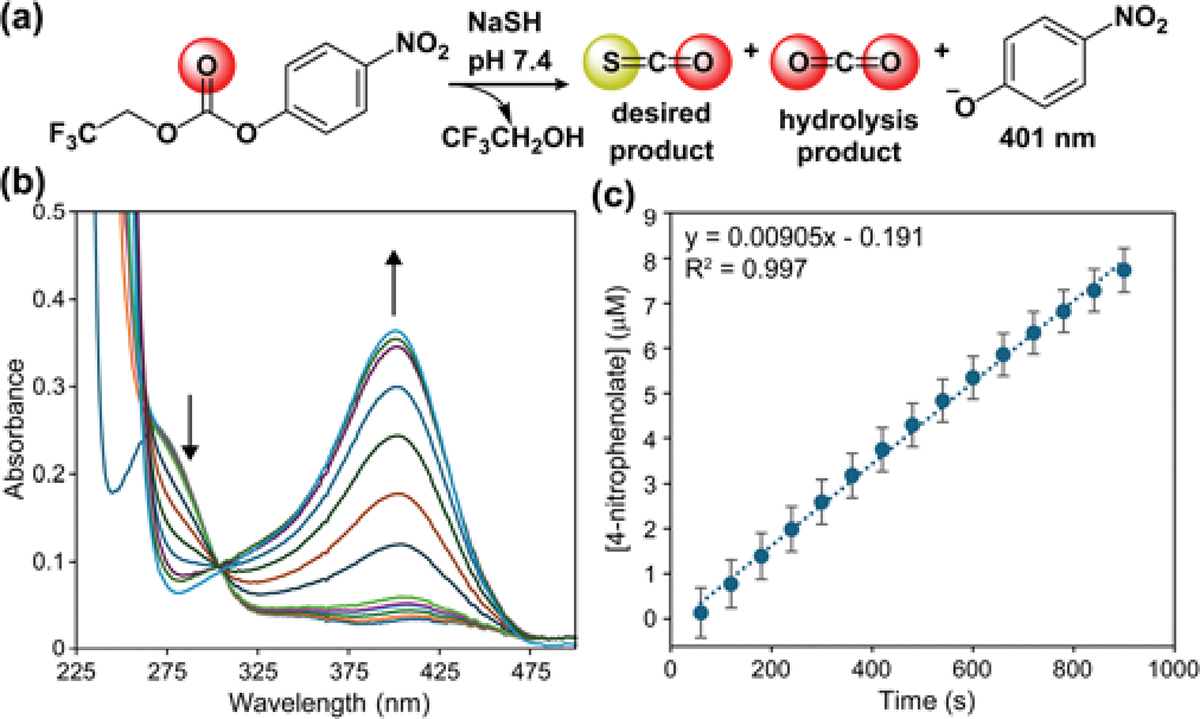
(a) Reaction scheme for conversion of 1b to COS by NaSH. (b) UV-vis spectra at various timepoints, and (c) initial rate data for the reaction. Condition: 25 μM 1b with 40 equiv. of NaSH in pH 7.4 PBS at 25 °C.
We further expanded the scope to include diaryl carbonates with electron withdrawing, neutral, or donating groups in bis(4-nitrophenyl) carbonate (1c), 4-nitrophenyl phenyl carbonate (1d), and 4-nitrophenyl 4′-methoxyphenyl carbonate (1e), respectively. Compounds 1c-1e followed the same general reactivity trends, with electron withdrawing groups facilitating with reaction with NaSH, with a change in initial rates of ~1 order of magnitude difference for each compound. Matching the trend observed for alkyl carbonates, we also observed improved selectivity for reaction with NaSH over background hydrolysis for electron withdrawn leaving groups, with 1c, 1d, and 1e showing selectivity ratios of 3.7:1, 3.0:1, and 1.4:1, respectively. Although the pKa values of the leaving group for 1c-1e were all lower than for CF3CH2OH (4-nitrophenol = 7.15, PhOH = 9.98, and 4-methoxyphenol = 10.21), only 1c reacted faster than 1b, which we attribute to steric influences in the accessibility of the carbonyl electrophile. Based on the pKa of H2S (7.0), about 72% is in the anionic HS− form at pH = 7.4 (Table 1). Our data show that organic carbonates 1a-e are significantly more reactive toward HS− (720 μM based on speciation) than water (~ 55 M), although clear differentiation from HO− (0.25 μM at pH = 7.4) is more difficult.22
Table 1.
Reaction parameters of the reaction of organic carbonates with NaSH

| |||||
|---|---|---|---|---|---|
|
| |||||
| Substrate | 1a | 1b | 1c | 1d | 1e |
|
| |||||
| R group | CH3 | CH2CF3 | 4-NO2-Ph | Ph | 4-OMe-Ph |
| pKa (ROH) | 15.54 | 12.46 | 7.15 | 9.98 | 10.21 |
| NaSHa | 4.8(4) | 90.5(8) | 230(50) | 49(8) | 6(2) |
| k (M−1s−1)b | 0.018(2) | 0.362(3) | 0.9(2) | 0.21(3) | 0.023(7) |
| Hydrolysisc | 2.7(1) | 21.5(1) | 60(20) | 16(2) | 4(1) |
| Ratio | 1.8:1 | 4.22:1 | 3.7:1 | 3.0:1 | 1.4:1 |
Initial rates (10−10 × M/s). Reaction monitored by UV-vis kinetics at 401 nm.
Conditions: 25 μM 1, 1000 μM NaSH, pH 7.4 PBS
Calculated from concentrations of 1 and NaSH.
Conditions: 25 μM 1, pH 7.4 PBS.
Building from HS−-mediated COS release, we next investigated whether thiocarbonate analogues could undergo a similar reaction to generate CS2. In general, small molecules that release CS2 are much less common than those that release COS.23 Ford and co-workers demonstrated acid-mediated CS2 release from hindered dithiocarbamates.24 In contrast, general strategies used for triggered COS release do not appear to work for CS2 release, with caged dithiocarbamates releasing H2S and an isothiocyanate rather than CS2.25 To determine whether electron poor thiocarbonates could release CS2, we treated 4-nitrophenyl chlorothioformate with 2 equiv. of NBu4SH in CD3CN (for better solubility than NaSH and to better monitor HS− concentration through NBu4+ integration) and observed quantitative formation of 4-nitrophenolate ion by 1H NMR spectroscopy.26 In addition, the 13C{1H} NMR spectrum showed a new peak at 193.6 ppm that matched the chemical shift of authentic CS2 (Figure 3). CS2 was not quantified directly by 13C NMR spectroscopy, but no other C=S containing resonances were observed.
Figure 3.
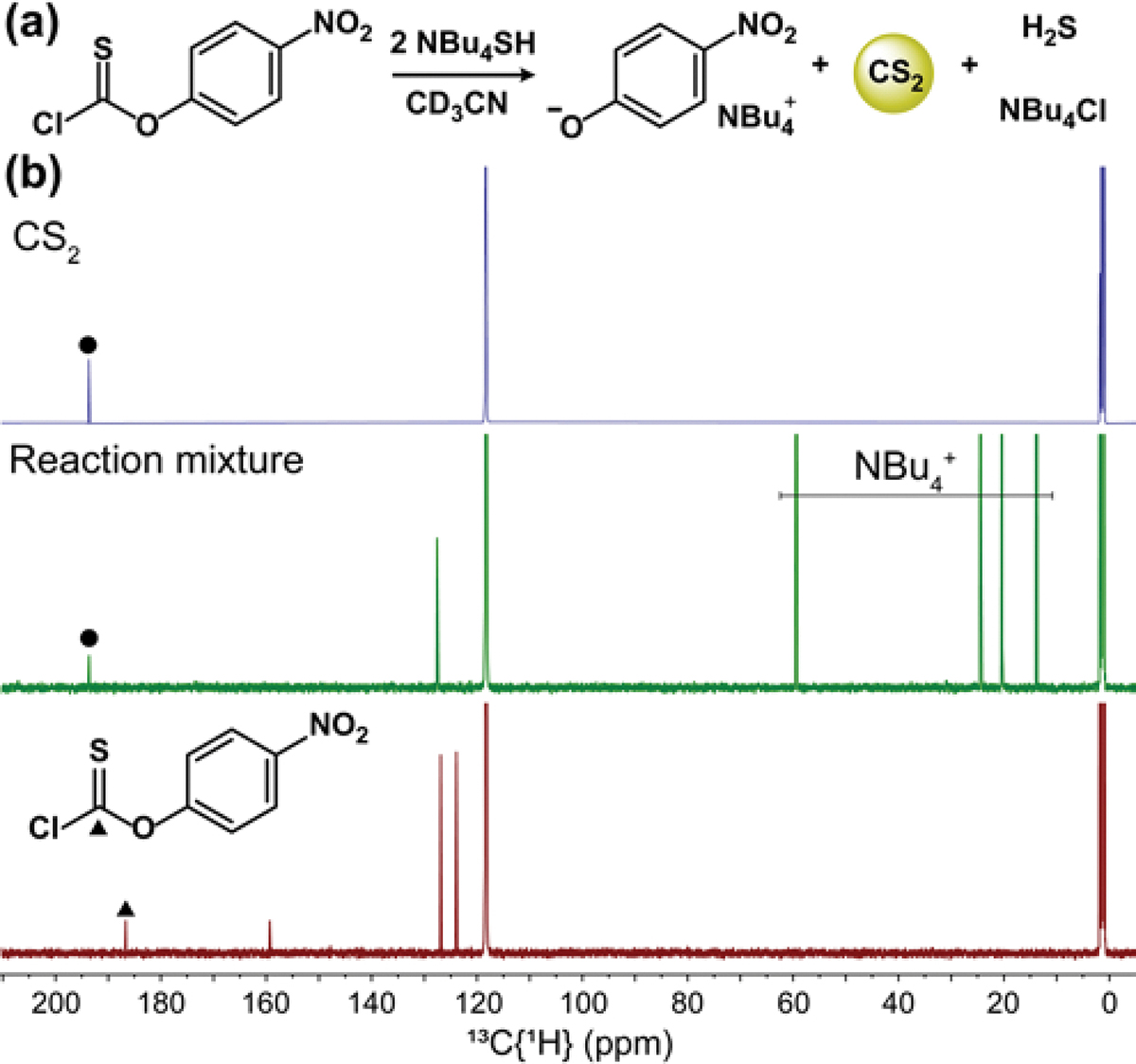
(a) Reaction for CS2 release from chlorothioformates. (b) Stacked 13C{H} NMR spectra in CD3CN of CS2 formation from chlorothioformate treated with 2 equiv. of NBu4SH, and the parent 4-nitrophenyl chlorothioformate.
Much like the oxygen congener, the parent chlorothioformate showed limited stability in water (Figure S23), but we prepared thiocarbonate analogues 2a-2e to both improve water stability and also allow for more direct comparisons between carbonates and thiocarbonates. Electron deficient 4-nitrophenyl methyl thiocarbonate (2a) showed significantly higher selectivity for HS− than 1a (see Table 2), with a 60-fold selectivity for reaction with NaSH over background hydrolysis. Similar improved selectivity was observed for trifluoroethyl derivative 2b. The most electrophilic thiocarbonate, bis(4-nitrophenyl) thiocarbonate (2c), not only the reacted faster than any of the carbonates or thiocarbonates, but also showed the highest selectivity over hydrolysis (690:1). The diaryl thiocarbonates with neutral or donating groups (2d and 2e) reacted with NaSH much more slowly than the corresponding carbonates 2a-c, with 4-nitrophenyl phenyl carbonate (2d) showing the slowest overall reactivity. Overall, with the exceptions of 2a and 2d, the series of thiocarbonates followed the same reactivity and selectivity trends as 1a-e.
Table 2.
Initial Rates of Organic Thiocarbonates with NaSH

| |||||
|---|---|---|---|---|---|
|
| |||||
| Substrate | 2a | 2b | 2c | 2d | 2e |
|
| |||||
| R identity | CH3 | CH2CF3 | 4-NO2-Ph | Ph | 4-OMe-Ph |
| pKa (ROH) | 15.54 | 12.46 | 7.15 | 9.98 | 10.21 |
| NaSHa | 124(6) | 500(30) | 820(40) | 3.7(0.4) | 16.5(0.4) |
| k (M−1s−1)b | 0.51(2) | 2.1(1) | 3.3(2) | 0.015(2) | 0.07(2) |
| Hydrolysisc | 2.06(4) | 15.9(7) | 1.2(3) | 0.8(3) | 9.4(5) |
| Ratio | 59.8:1 | 32:1 | 690 | 5:1 | 1.8:1 |
Initial rates (10−10 × M/s). Reaction monitored by UV-vis kinetics at 401 nm.
Conditions: 25 μM 2, 1000 μM NaSH, pH 7.4 PBS
Calculated from concentrations of 2 and NaSH.
Conditions: 25 μM 2, pH 7.4 PBS.
One key observation in comparing the carbonate and thiocarbonate derivatives is the higher selectivity of the thiocarbonates for attack by HS− over background hydrolysis, which is particularly apparent for 2a and 1a. Thiocarbonate 2a exhibits higher water stability than 1a, yet in stark contrast, the rate of 4-nitrophenolate release in the presence of NaSH for 2a is 26 times faster than for 1a. Considering that nucleophilic addition of HS− is faster with electron withdrawing and sterically unhindered substrates, we hypothesize that formation of the initial tetrahedral intermediate is likely the rate limiting step of the reaction with HS−. It is possible that there are changes in subsequent steps of the mechanism between the HS− and hydrolysis reactions, particularly between differently electronically-substituted compounds where the degree of resonance donation from the alkoxy group may impact the electrophilicity of the thiocarbonyl.
In conclusion, we have demonstrated that electrophilic organic carbonates and thiocarbonates can react with HS− to produce COS and CS2, respectively. In general, the thiocarbonates react faster with hydrosulfide than the respective carbonates and are also more stable in water, which we attribute to differences in electrophilic character on the carbonyl / thiocarbonyl carbon. We found that the selectivity for COS and CS2 formation over background hydrolysis can be modified by electronic substitution of the (thio)carbonate precursor. In general, this approach for interconversion between HS− and COS/CS2 may open new pathways for developing constructs for shuttling between different RSS while maintaining fully-reduced sulfur incorporation. In addition, if carried out under conditions to limit the hydrolysis of COS, this method could likely be adapted as a simple approach to deliver COS on demand from organic precursors for synthetic applications.
Experimental Section
General.
The reagents 4-nitrophenyl chloroformate, p-substituted phenols, thiophosgene, 1c, and anhydrous NaSH were purchased from TCI, Thermo Fischer Scientific, Oakwood, and Strem. DMSO, CDCl3, and CD3CN were purchased from Sigma-Aldrich and Cambridge Isotope Laboratories. CD3CN was dried over CaH2, distilled, and freeze-pump-thawed. NBu4SH,27 4-nitrophenyl chlorothioformate,28 and 1a29 were prepared as previously reported. PBS buffer (pH 7.4) was prepared with 140 mM NaCl, 3 mM KCl, and 10 mM phosphate with PBS tablets in milliQ water. UV-Vis experiments were performed using an Agilent Cary 60 spectrophotometer. NMR spectra were acquired using a Bruker 500 MHz instrument and processed with MestRe Nova software version 14.0.0. GC measurements were made on an Agilent 8890A GC with a PFPD and SPME headspace autosampler. Note: carbonyl sulfide, carbon disulfide, and hydrosulfide salts are acutely toxic. These reagents were used inside a fume hood or glovebox in order to minimize exposure. Air-tight screw-cap cuvettes were used for UV-vis experiments. Finally, all reactions in which hydrosulfide salts were utilized were quenched using an aqueous solution of zinc acetate, sodium citrate, and sodium hydroxide. This quenching procedure converts all hydrosulfide salts into zinc sulfide which is insoluble and substantially less toxic than the precursor salts.
General Procedure for UV-vis Kinetic Studies.
In an O2-free glovebox, screw-cap UV-Vis cuvettes were filled with 3.00 mL of 7.4 pH PBS buffer. A 2.5 mM solution of the desired carbonate or thiocarbonate were prepared in DMSO. A vial containing NaSH (3.4 mg, 0.061 mmol) was evacuated and backfilled with N2 on the Schlenk line, after which 500 μL of degassed 7.4 pH PBS was added to generate a 0.12 M solution. To measure the background hydrolysis reactions, 30 μL of a 2.5 mM carbonate or thiocarbonate solution was injected into the cuvette, which was then inverted several times prior to analysis by UV-vis spectroscopy. For reactions with NaSH, 25 μL of 0.12 M NaSH solution (40 equiv.) was injected within 2 min of carbonate or thiocarbonate addition to provide kinetics that were readily measurable under the reaction conditions while staying within the buffering capacity of the solution. The reaction was monitored at 401 nm by UV-vis spectroscopy at specific time intervals (~1 min) depending on the rate of the reaction.
Synthesis of 1b. 4-Nitrophenyl chloroformate (2.017 g, 10.00 mmol) was dissolved in 40 mL of CH2Cl2 in a round bottom flask with a stir bar. 2,2,2-Trifluoroethanol (755 μL, 10.0 mmol) and NEt3 were dissolved in 10 mL of CH2Cl2 in a 20 mL scintillation vial. Both solutions were cooled to 0 °C and the trifluoroethoxide solution was slowly added to the stirring solution of 4-nitrophenyl chloroformate. The reaction mixture was stirred at 0 °C for 20 minutes and then allowed to warm to room temperature. After 30 minutes, an aliquot of the reaction mixture was concentrated under vacuum and analyzed by 1H and 19F NMR spectroscopy to confirm conversion. The reaction mixture was then filtered through celite and the celite was washed with CH2Cl2 (3 × 10 mL). The crude product was concentrated onto 9 g of silica and dry loaded onto a column and purified by column chromatograph (2%–20% EtOAc in hexanes) to afford the desired product as a clear liquid (1.722 g, 65% yield). 1H NMR (500 MHz, CDCl3) δ: 8.31 (d, J = 9.2 Hz, 2H), 7.42 (d, J = 9.2 Hz, 2H), 4.65 (q, JH-F = 8.0 Hz, 2H). 13C{1H} NMR (126 MHz, CDCl3) δ: 155.1, 151.7, 146.0, 125.6, 122.4 (q, JC-F = 277 Hz), 121.8, 64.4 (q, JC-F = 37.6 Hz). 19F NMR (471 MHz, CD3Cl) δ: −74.1 (t, JF-H = 7.9 Hz, 3F). HRMS-ESI m/z [M]+ calcd for [C9H6O5NF3], 265.0198; found, 265.0190.
Synthesis of 1d. Phenyl chloroformate (112.7 mg, 0.7189 mmol) was dissolved in 2 mL of CH2Cl2 in a 20 mL scintillation vial with a stir bar and stirred at 0 °C. 4-Nitrophenol (100 mg, 0.719 mmol) was dissolved in 2 mL of CH2Cl2. NEt3 (100 μL, 0.717 mmol) was added to the 4-nitrophenol solution, which turned a bright yellow. The 4-nitrophenolate solution was added slowly over 2 minutes to the phenyl chloroformate solution. The solution was allowed to stir at 0 °C for 15 minutes and was then warmed to room temperature over the course of 30 minutes. An aliquot of the reaction was concentrated under vacuum and analyzed by 1H NMR spectroscopy. The crude reaction mixture was purified by recrystallization by layering hexanes at −30 °C to afford the product as a white solid (60.8 mg, 33% yield). 1H NMR (500 MHz, CDCl3) δ: 8.32 (d, J = 9.2 Hz, 2H), 7.49 (d, J = 9.2 Hz, 2H), 7.47–7.43 (m, 2H), 7.32 (t, J = 7.4 Hz, 1H), 7.28 (d, J = 7.6 Hz, 2H). 13C{1H} NMR (126 MHz, CDCl3) δ: 155.4, 151.2, 150.8, 145.7, 129.9, 126.9, 125.6, 121.9, 120.9. The NMR data matched prior reports of this compound.30
Synthesis of 1e. 4-Methoxyphenol (124.2 mg, 1.001 mmol) and NEt3 (139.4 μL, 1.000 mmol) were combined in 4 mL of CH2Cl2 in a scintillation vial. 4-Nitrophenyl chloroformate (201.8 mg, 1.001 mmol) was dissolved in 4 mL of CH2Cl2 with a stir bar in a scintillation vial. Both solutions were allowed to cool to 0 °C, and the 4-methoxy phenolate solution was slowly added to the stirring solution of 4-chloroformate. The reaction mixture was stirred at 0 °C for 15 minutes, warmed to room temperature, and stirred for an additional 30 minutes. The CH2Cl2 was removed under vacuum and the residue was reconstituted in ethyl acetate. Unreacted 4-methoxyphenol and 4-nitrophenol byproduct were removed by aqueous washing with 5% NaOH (aq) (3 × 10 mL). During these washings, the aqueous layer was initially a bright yellow color and changed to nearly clear. The organic layer was further washed with 3 M HCl (3 × 10 mL) to remove residual NEt3. The product was purified by recrystallization at –30 °C (ethyl acetate and hexanes layering) to provide the white crystalline product (113.5 mg, 39% yield). 1H NMR (500 MHz, CDCl3) δ: 8.31 (d, J = 9.2 Hz, 2H), 7.48 (d, J = 9.2 Hz, 2H), 7.19 (d, J = 9.1 Hz, 2H), 6.93 (d, J = 9.1 Hz, 2H), 3.82 (s, 3H). The NMR data matched prior reports of the same compound.31
Synthesis of 2a. 4-Nitrophenyl chlorothioformate (93.9 mg, 0.431 mmol) was dissolved in 2.2 mL of CH2Cl2 in a scintillation vial, after which methanol (17.4 μL, 0.431 mmol), and then NEt3 (60.1 μL, 0.431 mmol) were added. Monitoring the reaction by TLC showed that most starting material was consumed after 10 min. The crude reaction mixture was purified by column chromatography (2–20% ethyl acetate in hexanes) to afford the desired product as a yellow oil (13.5 mg, 15% yield). 1H NMR (500 MHz, CDCl3) δ: 8.32 (d, J = 9.1 Hz, 2H), 7.29 (d, J = 9.1 Hz, 2H), 4.19 (s, 3H), which matched the previously reported spectrum.32
Synthesis of 2b. 4-Nitrophenyl chlorothioformate (108.4 mg, 0.4981 mmol) was dissolved in 2.5 mL of CH2Cl2 in scintillation vial, after which 2,2,2-trifluoroethanol (38 μL, 0.50 mmol) and then NEt3 (70 μL, 0.50 mmol) were added. After 5 min, TLC showed complete consumption of starting material. The crude reaction mixture was purified by column chromatography (2–20% ethyl acetate in hexanes) to afford the desired product as a clear liquid (81.2 mg, 58% yield). 1H NMR (500 MHz, CDCl3) δ: 8.34 (d, J = 9.1 Hz, 2H), 7.33 (d, J = 9.1 Hz, 2H), 4.89 (q, JH-F = 8.0 Hz, 2H). 13C{1H} NMR (126 MHz, CDCl3) δ: 192.5, 157.4, 146.5, 125.7, 123.2, 122.3 (q, JC-F = 278 Hz), 68.7 (q, JC-F = 37.2 Hz). 19F NMR (471 MHz, CD3Cl) δ: −73.2 (t, JF-H = 7.8 Hz, 3F). HRMS-ESI m/z [M]+ calcd for [C9H6O4NSF3], 280.9970; found, 280.9957.
Synthesis of 2c. Prepared by modifying an existing procedure.33 4-Nitrophenol (500.8 mg, 3.600 mmol) was dissolved in 20 mL of dry THF in a 100 mL round bottom flask and cooled to 0 °C. Triethyl amine (251 μL, 1.80 mmol) was added to the flask, after which thiophosgene (85%, 162 μL, 1.80 mmol) was added dropwise. The resultant reaction mixture stirred at 0 °C for 25 minutes, during which time a precipitate formed. The precipitate was filtered, washed with THF (3 × 5 mL), and dried to afford the pure product as an orange solid (514 mg, 89% yield). 1H NMR (500 MHz, CDCl3) δ: 8.37 (d, J = 9.1 Hz, 4H), 7.41 (d, J = 9.2 Hz, 4H), which matched the previously reported spectrum.
Synthesis of 2d. 4-Nitrophenyl chlorothioformate (54.5 mg, 0.250 mmol) was dissolved in 1 mL of CH2Cl2 in a 20 mL scintillation with a stir-bar. Phenol (23.5 mg, 0.250 mmol) and NEt3 (38 μL, 0.27 mmol) were dissolved in 1 mL of CH2Cl2, and the phenolate solution was slowly added to the stirring solution of 4-nitrophenyl chlorothioformate over 1 minute. After 5.5 h, the crude reaction mixture was directly loaded onto a SiO2 column and purified by flash chromatography (30% ethyl acetate in hexanes) to afford a white solid (43.1 mg, 63% yield). 1H NMR (500 MHz, CDCl3) δ: 8.35 (d, J = 9.1 Hz, 2H), 7.50–7.45 (m, 2H), 7.41 (d, J = 9.1 Hz, 2H), 7.38–7.34 (m, 1H), 7.24–7.20 (m, 2H). 13C{1H} NMR (126 MHz, CDCl3) δ: 193.5, 157.7, 153.5, 146.3, 130.0, 127.3, 125.6, 123.4, 121.8. The NMR spectra matched prior reports of the compound.34
Synthesis of 2e. Compound 2c (100 mg, 0.312 mmol) was dissolved in 2 mL of CH2Cl2 in a 20 mL scintillation vial with a stir-bar. 4-Methoxyphenol (37.2 mg, 0.300 mmol) was dissolved in 2 mL of CH2Cl2. The solutions were cooled to 0 °C, and NEt3 (42 μL, 0.30 mmol) was added to the 4-methoxyphenol solution. The 4-methoxyphenolate solution was added slowly over about 2 min to the stirring solution of the 2c. The reaction mixture was stirred at 0 °C for 15 min, then allowed to warm to room temperature over the course of 15 minutes. The reaction mixture was washed with H2O (2 × 20 mL), concentrated, and recrystallized twice by layering with hexanes at −30 °C. Remaining 4-methoxylphenol was removed by washing with 5% NaOH to afford the pure product as a white solid (45.4 mg, 50% yield). 1H NMR (500 MHz, CDCl3) δ: 8.35 (d, J = 9.1 Hz, 2H), 7.40 (d, J = 9.1 Hz, 2H), 7.13 (d, J = 9.0 Hz, 2H), 6.96 (d, J = 9.2 Hz, 2H), 3.83 (s, 3H). 13C{1H} NMR (126 MHz, CDCl3) δ: 194.1, 158.3, 157.7, 147.2, 146.3, 125.6, 123.4, 122.5, 114.9, 55.8. HRMS-ESI m/z [M]+ calcd for [C14H11O5NS], 305.0358; found, 305.0357.
Supplementary Material
ACKNOWLEDGMENT
We thank the NIH (R01GM113030) for support of this research.
Footnotes
ASSOCIATED CONTENT
Supporting Information Statement
Supporting Information. Calibration curves, NMR spectra, kinetic data, GC data (pdf).
Data Availability Statement
The data underlying this study are available in the published article and its Supporting Information.
References
- 1.Lau N; Pluth MD, Reactive sulfur species (RSS): persulfides, polysulfides, potential, and problems. Curr. Opin. Chem. Biol. 2019, 49, 1–8. [DOI] [PubMed] [Google Scholar]
- 2.Kamboures MA; Blake DR; Cooper DM; Newcomb RL; Barker M; Larson JK; Meinardi S; Nussbaum E; Rowland FS, Breath sulfides and pulmonary function in cystic fibrosis. Proc Natl Acad Sci U S A 2005, 102 (44), 15762–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Studer SM; Orens JB; Rosas I; Krishnan JA; Cope KA; Yang S; Conte JV; Becker PB; Risby TH, Patterns and significance of exhaled-breath biomarkers in lung transplant recipients with acute allograft rejection. J. Heart Lung Transpl. 2001, 20 (11), 1158–66. [DOI] [PubMed] [Google Scholar]
- 4.Sehnert SS; Jiang L; Burdick JF; Risby TH, Breath biomarkers for detection of human liver diseases: preliminary study. Biomarkers 2002, 7 (2), 174–87. [DOI] [PubMed] [Google Scholar]
- 5.Kolliyedath G; Sahana T; Johnson SM; Kundu S, Synergistic Activation of Nitrite and Thiocarbonyl Compounds Affords NO and Sulfane Sulfur via (Per)thionitrite (SNO−/SSNO−). Angew. Chem. Int. Ed. 2023, 62 (50), e202313187. [DOI] [PubMed] [Google Scholar]
- 6.Davis AG; Pluth MD, Experimental Insights into the Formation, Reactivity, and Crosstalk of Thionitrite (SNO−) and Perthionitrite (SSNO−) Angew. Chem. Int. Ed. 2024, 10.1002/anie.202413092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Steiger AK; Zhao Y; Pluth MD, Emerging Roles of Carbonyl Sulfide in Chemical Biology: Sulfide Transporter or Gasotransmitter? Antioxid. Redox Signal 2018, 28 (16), 1516–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Haritos VS; Dojchinov G, Carbonic anhydrase metabolism is a key factor in the toxicity of CO2 and COS but not CS2 toward the flour beetle Tribolium castaneum [Coleoptera: Tenebrionidae]. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 2005, 140 (1), 139–147. [DOI] [PubMed] [Google Scholar]
- 9.Levinn CM; Cerda MM; Pluth MD, Development and Application of Carbonyl Sulfide-Based Donors for H2S Delivery. Acc. Chem. Res. 2019, 52 (9), 2723–2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Powell CR; Dillon KM; Matson JB, A review of hydrogen sulfide (H2S) donors: Chemistry and potential therapeutic applications. Biochem. Pharmacol. 2018, 149, 110–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhao Y; Henthorn HA; Pluth MD, Kinetic Insights into Hydrogen Sulfide Delivery from Caged-Carbonyl Sulfide Isomeric Donor Platforms. J. Am. Chem. Soc. 2017, 139 (45), 16365–16376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhao Y; Pluth MD, Hydrogen Sulfide Donors Activated by Reactive Oxygen Species. Angew. Chem. Int. Ed. 2016, 55 (47), 14638–14642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Levinn CM; Steiger AK; Pluth MD, Esterase-Triggered Self-Immolative Thiocarbamates Provide Insights into COS Cytotoxicity. ACS Chem. Biol. 2019, 14 (2), 170–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Steiger AK; Pardue S; Kevil CG; Pluth MD, Self-Immolative Thiocarbamates Provide Access to Triggered H2S Donors and Analyte Replacement Fluorescent Probes. J. Am. Chem. Soc. 2016, 138 (23), 7256–7259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Powell CR; Foster JC; Okyere B; Theus MH; Matson JB, Therapeutic Delivery of H2S via COS: Small Molecule and Polymeric Donors with Benign Byproducts. J. Am. Chem. Soc. 2016, 138 (41), 13477–13480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Khodade VS; Pharoah BM; Paolocci N; Toscano JP, Alkylamine-Substituted Perthiocarbamates: Dual Precursors to Hydropersulfide and Carbonyl Sulfide with Cardioprotective Actions. J. Am. Chem. Soc. 2020, 142 (9), 4309–4316. [DOI] [PubMed] [Google Scholar]
- 17.Swain CG; Scott CB, Quantitative Correlation of Relative Rates. Comparison of Hydroxide Ion with Other Nucleophilic Reagents toward Alkyl Halides, Esters, Epoxides and Acyl Halides1. J. Am. Chem. Soc. 1953, 75 (1), 141–147. [Google Scholar]
- 18.Li Q; Lancaster JR Jr., Chemical foundations of hydrogen sulfide biology. Nitric Oxide 2013, 35, 21–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Abdollahi M; Mohammadirad A, Nitrophenol, 4. In Encyclopedia of Toxicology (Third Edition), Wexler P, Ed. Academic Press: Oxford, 2014; pp 575–577. [Google Scholar]
- 20. This reaction occurs too quickly to monitor accurately in solution. See Figure S23.
- 21. Reactions were performed under oxygen-free conditions.
- 22. Less than 1% of the solution is DMSO.
- 23.Yu B; Yuan Z; Yang X; Wang B, Prodrugs of Persulfides, Sulfur Dioxide, and Carbon Disulfide: Important Tools for Studying Sulfur Signaling at Various Oxidation States. Antioxid. Redox Signal 2020, 33 (14), 1046–1059. [DOI] [PubMed] [Google Scholar]
- 24.DeMartino AW; Souza ML; Ford PC, Uncaging carbon disulfide. Delivery platforms for potential pharmacological applications: a mechanistic approach. Chem. Sci. 2017, 8 (10), 7186–7196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Antioxid Redox SignalZhao Y; Henthorn HA; Pluth MD, Kinetic Insights into Hydrogen Sulfide Delivery from Caged-Carbonyl Sulfide Isomeric Donor Platforms. J. Am. Chem. Soc. 2017, 139 (45), 16365–16376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. One equivalent was serves as a nucleophile, and the second equivalent serves as a base to form the 4-nitrophenolate ion, the colorimetric indicator.
- 27.Hartle MD; Meininger DJ; Zakharov LN; Tonzetich ZJ; Pluth MD, NBu4SH provides a convenient source of HS− soluble in organic solution for H2S and anion-binding research. Dalton Trans. 2015, 44 (46), 19782–19785. [DOI] [PubMed] [Google Scholar]
- 28.Hagooly Y; Sasson R; Welch MJ; Rozen S, Preparation of Alkyl and Aryl Chlorodifluoromethyl Ethers Using BrF3. Eur. J. Org. Chem. 2008, 2008 (17), 2875–2880. [Google Scholar]
- 29.Sonkaria S; Bourcher G; Florez-Alvarez J; Said B; Hussain S; Ostler Elizabeth L.; Gul S; Thomas Emrys W.; Resmini M; Gallacher G; Brocklehurst K, Evidence for ‘lock and key’ character in an anti-phosphonate hydrolytic antibody catalytic site augmented by non-reaction centre recognition: variation in substrate selectivity between an anti-phosphonate antibody, an anti-phosphate antibody and two hydrolytic enzymes. Biochem. J. 2004, 381 (1), 125–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Um I-H; Kim EY; Park H-R; Jeon S-E, Aminolyses of 4-Nitrophenyl Phenyl Carbonate and Thionocarbonate: Effect of Modification of Electrophilic Center from CO to CS on Reactivity and Mechanism. J. Org. Chem. 2006, 71 (6), 2302–2306. [DOI] [PubMed] [Google Scholar]
- 31.Freer R; McKillop A, Synthesis of Symmetrical and Unsymmetrical Ureas Using Unsymmetrical Diaryl Carbonates. Synth. Commun. 1996, 26 (2), 331–349. [Google Scholar]
- 32.Castro EA; Cubillos M; Santos JG; Téllez J, Kinetics and Mechanism of the Pyridinolysis of Alkyl Aryl Thionocarbonates. J. Org. Chem. 1997, 62 (8), 2512–2517. [DOI] [PubMed] [Google Scholar]
- 33.Castro EA; Santos JG; Téllez J; Umaña MI, Structure–Reactivity Correlations in the Aminolysis and Pyridinolysis of Bis(phenyl) and Bis(4-nitrophenyl) Thionocarbonates. J. Org. Chem. 1997, 62 (19), 6568–6574. [Google Scholar]
- 34.Baroudi A; Alicea J; Flack P; Kirincich J; Alabugin IV, Radical O→C Transposition: A Metal-Free Process for Conversion of Phenols into Benzoates and Benzamides. J. Org. Chem. 2011, 76 (6), 1521–1537. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this study are available in the published article and its Supporting Information.