

Abstract
We have performed restriction mapping of DNA molecules using restriction endonucleases in nanochannels with diameters of 100-200 nm. The location of the restriction reaction within the device is controlled by electrophoresis and diffusion of Mg2+ and EDTA. We have successfully used the restriction enzymes SmaI, SacI, and PacI, and have been able to measure the positions of restriction sites with a precision of ≈1.5 kbp in 1 min using single DNA molecules.
Restriction mapping with endonucleases is a central method in molecular biology (1, 2). It is based on the measurement of fragment lengths after digestion, while possibly maintaining the respective order. We present here an approach to restriction mapping that is based on stretching DNA in nanofluidic channels, in which DNA is linearized to a length-independent fraction of its contour length (3, 4).
To date, the most powerful method for constructing restriction maps of long DNA molecules (100 kbp and above) is the one developed by Schwartz and coworkers (5, 6). Their technique consists of stretching the DNA on a surface to establish a one-to-one mapping between spatial and genomic position, initiating the restriction by exposing the DNA to restriction enzymes and Mg2+, and optically observing the lengths of the resulting fragments. An elegant study of the same basic approach showing separation of specific binding and induced cutting was published by Taylor et al. (7).
A fundamental principle in determining the error of a measurement, and a common strategy to reduce the experimental error, is to take multiple, statistically independent measurements of the same quantity. Note that the fixing of the molecule to the surface in Schwartz's approach prevents any fluctuations, and hence multiple molecules have to be observed to obtain statistically independent measurements of the same cut position. Moreover, genomic-length DNA molecules stretched on surfaces often exhibit inhomogeneous stretching, including breaks, and thus averaging over multiple molecules becomes imperative (6, 8). In contrast, a molecule stretched inside a nanochannel is not subject to any forces other than those causing lateral confinement and thus is able to thermally relax and fluctuate around an equilibrium conformation. The evaluation of a single molecule is thus sufficient if complete digestion is achieved. A detailed description of the statistics and dynamics of DNA molecules in nanochannels is published in ref. 9.
The main challenge in employing the concept of stretching and mapping DNA inside a closed fluidic system is to separate the steps of stretching and cutting. We have solved this problem by introducing the DNA electrophoretically into nanochannels and controlling the concentration of the enzyme cofactor Mg2+ in the device shown in Fig. 1. It consists of a microfluidic “loading” channel containing the DNA to be analyzed, the restriction enzyme, and EDTA, and a microfluidic “exit” channel containing Mg2+ and the restriction enzyme. The two microfluidic channels are linked by 10 nanofluidic channels of ≈100 nm in diameter. We expect that, in the absence of an applied voltage along the nanochannels, a Mg2+ gradient will be established as the result of Mg2+ and EDTA diffusion, and chelation of Mg2+ by EDTA. With an applied voltage, we expect the higher mobility of the Mg2+ ions to result in a constant Mg2+ concentration equal to that on the “exit” side. Note that the particular shape of the Mg2+ concentration is not crucial to the functioning of the device, only the facts that there is a concentration sufficient for restriction in the active region, i.e., the nanochannels, and a negligible concentration in the “loading” microchannel.
Fig. 1.

Schematic of the device used in the experiments. Fluidic components were first fabricated on a fused silica substrate and later sealed with a fused silica coverslip. We linked two microfluidic channels (a and b, 1 μm × 100 μm cross-section) with 10 nanochannels of ≈100 nm × 100 nm (c). The solution in the “loading” microchannel (b) contained DNA and EDTA, and the “exit” microchannel (a) contained Mg2+. Both channels contained restriction enzyme. Both DNA and Mg2+ were moved through the device by electrophoresis using four electrodes. The voltage applied across the length of the nanochannels is marked ΔVn (≈2 V), and the voltage across the microchannels is ΔVμ (≈2 V). During DNA imaging, no voltages were applied.
We present here restriction mapping of DNA molecules inside nanochannels controlled by this Mg2+-gradient, discuss the attainable resolution, and discuss how the cuts become visible.
Methods
Device Fabrication. Devices were fabricated on polished quartz wafers. Microchannels ≈1 μm deep and 100 μm wide were patterned by standard optical lithography and reactive ion etching using a CF4/H2 chemistry (10). We used 1.4-μm-thick AZ5214-E photoresist (Clariant, Somerville, NJ), exposed by using a mask aligner. The ratio of CF4 to H2 during the etching was 26.5-6, at a pressure of 35 mtorr (1 torr = 133 Pa) and a power density of 1 W/cm2. The microchannels were separated from each other by either 100 or 200 μm. Holes for accessing the microfluidic system were drilled by using dental sandblasting tool (Danville Engineering, San Ramon, CA). Nanochannels were prepared by focused ion milling after first coating with a 5-nm Au film (11). The gold was removed by using aqua regia, and the device was sealed with a quartz coverslip via quartz-quartz bonding (4).
Microscopy. We imaged DNA molecules and the Mg2+-sensitive dye using a 1.4-numerical aperture oil-immersion objective (Nikon) and an intensified charge-coupled device camera (Roper Scientific) under illumination with the 488 nm-line from an Ar/Kr ion laser. Images were collected by using 10-ms exposures spaced by ≈250 ms. The laser beam was shut between the frames of acquisition.
Magnesium Imaging. The “exit” side was filled with a solution containing 10 mM Tris, 1 mM DTT, and 100 μg/ml POP6 (Applied Biosystems). The “loading” side was filled with an identical solution except for the addition of 5 mM EDTA. Magnesium-acetate (5 mM) was placed into one reservoir on the “exit” side, and the Mg2+ was brought into the microchannel by applying 10 V across both microchannels, with no voltage across the nanochannels. During image acquisition, the microchannel voltage was 2 V, and the nanochannel voltage was 1 V. Images were acquired at a frame rate of 1 Hz.
Restriction Protocols. DNA was dyed with the bis-intercalating dye TOTO-1 (Molecular Probes) at a ratio of 1 dye molecule per 20 bp. λ-DNA was obtained from New England Biolabs and used at a concentration of 0.5 μg/ml or 1 μg/ml. The basic buffer for restriction using SmaI, SacI, and PacI consisted of 20 mM Tris-acetate, 50 mM potassium-acetate, 1 mM DTT, 100 μg/ml BSA, and 100 μg/ml POP6. The restriction enzymes (New England Biolabs) were used with concentrations of ≈3 nM for SmaI, ≈3 nM for SacI, and ≈13 nM for PacI. We chose these concentrations because of the specific binding constant for SmaI of ≈109 M-1 (12). The solution that was filled into the “entry”-side also contained 5 mM EDTA. After filling of the device and inspection under the microscope, Mg2+ was brought into the “exit”-side microchannel by the method described in the Mg2+ imaging. During the restriction experiment, 2 V was applied over each microchannel. The potential of the two microfluidic systems was offset by 1 V for 100-μm-long nanochannels and 2 V for 200-μm-long nanochannels. Sealed chips could be used up to 5 days.
Preparation of PAC DNA. The PAC DNA was isolated from Escherichia coli strains PRCI 21 168-F5 by using standard protocols (2, 13). The DNA was digested with NotI (New England Biolabs), followed by pulsed-field gel electrophoresis separation. The 61-kb band was cut out and eluted by dialysis against 0.5× TBE buffer. The DNA sample was concentrated to ≈10 μg/ml with centrifugal filters (Microcon YM-100, Millipore).
Results and Discussion
We have imaged the Mg2+ concentration inside nanochannels (Fig. 2) using a Mg2+-sensitive dye (Magnesium Green, Molecular Probes). The dye has a roughly linear response at low Mg2+ levels and a well defined level of fluorescence in the absence of Mg2+. Without any voltage, the predicted Mg2+ gradient was observed, while a constant intensity was established when a voltage was applied along the nanochannels. To avoid a poisoning of the “loading” channel with Mg2+, we applied a voltage across the microchannel that carries free positive ions away from the entrance to the nanochannels. We also imaged the entrance region of the nanochannels with and without voltages and found no observable leakage of free Mg2+ into the “entry” microchannel. We further calculated the Mg2+ concentration in our channel geometry and found qualitative agreement with our experimental observations (data not shown).
Fig. 2.

Images of 100-μm-long nanochannels containing a Mg2+-sensitive dye. (Scale bar: 5 μm.) The image in a was recorded without voltages, and b was acquired with 1 V over the nanochannel. Magnesium entered the nanochannels from the left, and the images were averaged for 40 s. c is the ratio of images a/b taken along the nanochannels.
To perform real-time observations of restriction in nanochannels, the reaction has to be completed within seconds, to avoid photobleaching. This can be achieved by adjusting the enzyme concentration so that all recognition sites are occupied before they enter the channel. In that case, the rate-limiting step is the release of the DNA from the enzyme after cutting, or ≈1 s (14). Note that only a subset of restriction enzymes is able to bind to the correct sequence in the absence of divalent metal ions. Some of these are SmaI and EcoRI, for which the specific and nonspecific binding constants have been determined (12, 15). Kinetic tests of λ-DNA restriction by SmaI using a moving Mg2+ front inside a microchannel has confirmed that the cutting occurs on a time scale shorter than 4 s with our combination of buffer and enzyme concentrations. We thus expect that in the case of SmaI, fully decorated DNA molecules enter the channel, and complete digestion of all accessible recognition sites occurs within a few seconds. If an enzyme does not exhibit specific binding in the absence of divalent metal ions, the enzyme needs to diffuse along the DNA molecule or bind to the DNA from solution before cutting. These additional steps will lead to somewhat longer times between entry of the DNA into the channel and cutting. However, the rate should rise with rising enzyme concentration.
We have successfully performed restriction mapping of λ-DNA (48.5 kbp) using the enzymes SmaI and SacI in nanochannels where the DNA was stretched to ≈40% and 30% of its full length, respectively (Fig. 3). Molecules were observed for ≈1 min, and both typical frames as well as time traces are shown. The latter is a stack of lines, each of which shows the intensity along the molecule for a single 10-ms frame. SmaI cuts λ-DNA into fragments of 19.4, 12.2, 8.3, and 8.6 kbp, in that order. All four fragments were clearly observed. SacI cuts λ-DNA into three fragments 22.6, 0.9, and 24.8 kbp long. Only the longer two fragments could be clearly observed.
Fig. 3.

Time-resolved restriction mapping of λ-DNA in nanochannels. (a) Restriction of three λ-DNA (48.5 kbp) molecules by using SmaI in channels of ≈120 nm × 120 nm cross-section. The DNA is stretched to ≈40% of its contour length. (Left) Individual 10-ms frames. (Right) Time traces, in which each line corresponds to intensity along the nanochannel in a single frame. From the known DNA sequence, we expect fragments of 19.4, 12.2, 8.3, and 8.6 kbp, in that order. (b) Restriction of three λ-DNA molecules by using SacI in channels of 140 nm × 180 nm cross-section. (Left) Individual 10-ms frames. (Right) Time traces. The DNA is expected to stretch to ≈25-30% of its contour length in channels of these dimensions. We expect fragments of 22.6, 0.9, and 24.8 kbp. The smallest 0.9-kbp segment is in general not visible. (c) Cutting 61-kbp DNA with PacI. The top panels are time traces of cutting in roughly 120 nm wide nano-channels, where we expect a stretch of 40%. (Right) Pulsed-field gel electrophoresis separation of the digestion product of an unpurified 61-kbp DNA and cloning vector. Lane 1,λ-DNA ladder; lane 2, long-range PFGE ladder; lanes 3 and 4, digestion product after Pacl.
Fig. 3. demonstrates that cuts become visible because restriction fragments diffuse in the channels. We do not observe an obvious recoil of the ends at the cutting positions, probably because the thermalized polymer has a constant stretching independent of its length. Hence, the time of cutting can be inferred with a precision equal to the diffusion time of the fragment through a length corresponding to the resolution of the microscope. Note that the mild bleaching of DNA during the observation time in Fig. 3 may be either photobleaching or bleaching due to high Mg2+ concentrations. Thus, observation times longer than 3 min were not possible. However, one could easily extend the observation periods using lower Mg2+ concentrations and lower read-out times, possibly in connection with the use of an oxygen-scavenging system.
Although most cuts were observed within a few seconds, some late cutting events, at half a minute or later, were observed. These late cuts could either be due to random photoinduced cutting (16) or unspecific enzyme action. Because the photocutting rate is expected to have a square dependence on exposure time, we excluded cuts that occurred at 20 s or later from the further analysis. We performed negative controls with insufficient Mg2+ and found no early cutting events, and very few late ones. Typically, molecules stayed intact for 90 s or until near-complete photobleaching.
We have analyzed the size of the restriction fragments by fitting the intensity along the molecules to (4)
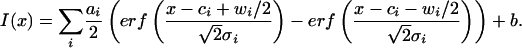 |
[1] |
Here, I is the intensity, the sum runs over all fragments, erf is the error function, w is the width of a segment, c its center, σ is the sharpness of the molecules end, and b is a constant describing the background. Each experimental data frame was fit individually and thus, if frames are spaced by a time longer than the relaxation time for Brownian fluctuations, the measured lengths are statistically independent. The exact length of a fragment can hence in principle be determined from a single molecule by averaging over those independent measurements.
For small molecules and short exposure times (10 ms), we have found that σ can become comparable to w, and hence w does not serve as a good measure of the amount of DNA in a single restriction fragment. Therefore, we determined the relative lengths of restriction fragments by integrating the fitted function and using the proportionality between total fluorescence intensity and length (17). The absolute number of base pairs was determined by first calculating the relative lengths of the restriction fragments and then multiplying it by the known length of the DNA. In principle, for well controlled nanochannels, the length of the DNA can be determined directly from the observed length of the whole DNA and the known channel dimensions.
Fig. 4 is a histogram of calculated cut positions of SmaI on λ-DNA, constructed from all molecules with two or three observed cuts. There are three known restriction sites, and the positions derived from the histogram correspond to the positions in the sequence (Table 1). We believe that the incomplete digestion is due to the staining of the DNA with an intercalating dye, which is known to interfere with sequence-recognizing enzymes (18).§ Furthermore, the recognition sequence for SmaI contains a known preferred binding site of the intercalating dye TOTO-1 (19). We find that Fig. 4 shows no signs of photoinduced or unspecific enzymatic cutting. Note also that cuts by SmaI on λ-DNA appear within, at most, a few seconds (Fig. 3).
Fig. 4.

The absolute cut positions from 29 molecules with two and three cuts. The line is a fit to the histogram by using the sum of three Gaussian distributions.
Table 1. Cutting positions of Smal on λ-DNA in kbp derived from molecules with two and three cuts.
| Sequence | Histogram | Weighted average |
|---|---|---|
| 19.4 | 19.3 ± 1.2 | 19.9 ± 1.3 |
| 31.6 | 32.1 ± 1.0 | 32.8 ± 1.3 |
| 39.9 | 40.6 ± 2.0 | 40.7 ± 1.7 |
The histogram values were obtained from a three-Gaussian fit to the histogram in Fig. 4. The weighted averages were calculated from the average values for the individual molecules, by forming the weighted mean of the the individual-molecule averages with the observation times for the individual molecules as weights. The errors are the statistical variances, and 29 molecules were used for the statistical analysis.
We noticed that some of the experimental values for the fragment lengths in Table 2 differ from the sequence data by more than the expected error of the measurement (the error of the mean was ≈0.3 kbp in this experiment). In particular, the 8.6-kbp fragment appears systematically underestimated, whereas the 12.2-kbp fragment was systematically overestimated. This misestimation probably stems from the combination of optical system response and the functional relationship used for fitting. In particular, the imaging system does not have a purely Gaussian response, but it also a slower-decaying component. The slowly decaying component causes a differently shaped molecule fluorescence profile depending on the length of the fragment. Shoulders that do not follow the error function model become particularly prominent for long molecules. These shoulders then lead to a greater apparent width of the longer restriction fragments and reduced apparent length for shorter molecules. Note also that for the same reason, the uncertainty for the position of the third cut is larger than that of the others.
Table 2. Fragment lengths of Smal on λ-DNA in kbp derived from molecules with two and three cuts.
| Sequence | Histogram | Weighted average |
|---|---|---|
| 19.4 | 19.3 (−0.1) | 19.9 (+0.5) |
| 12.2 | 12.8 (+0.6) | 12.9 (+0.7) |
| 8.3 | 8.5 (+0.2) | 8.0 (−0.3) |
| 8.6 | 7.9 (−0.7) | 7.8 (−0.8) |
The histogram values were obtained from a three-Gaussian fit to the histogram in Fig. 4. The weighted averages were calculated from the average values for the individual molecules, by forming the weighted mean of the the individual-molecule averages with the observation times for the individual molecules as weights. We have indicated the difference between the experimental and the sequencing values in brackets.
We confirmed that our method of restriction mapping is also applicable to PacI, a known rare cutter often used to establish landmark sites for verification of shotgun sequencing of genomic DNA. We tested this enzyme using a 61-kbp-long PAC insert, known to contain a single PacI site. Time traces of molecules digested with PacI are presented in Fig. 3.
In our experiments using SmaI, the cutting typically occurs at between 1 and 4 s after the DNA entered the channel, whereas the time increases to 10 s or so for SacI or KpnI. We can compare those cutting times to the relaxation times that were measured for varying channel widths and DNA lengths by Reisner et al. (9), who found that 48.5-kbp-long λ-DNA in 100-nm-wide channels relaxes within 1 s or so. We thus expect the DNA to be partially or fully thermally relaxed at the time the cut occurs, and we expect no significant recoil of the ends due to tension along the DNA backbone. The gaps must thus arise mainly from diffusive motion of the fragments. The average relative diffusion coefficient of two fragments resulting from a cut using SacI was estimated from a fit to the graph of mean squared distance between the centers of mass versus time. We find a constant of 0.5 μm2/s for the 22- to 26-kbp-long fragments. This value has to be taken as an upper limit, because only molecules in which both segments did not touch after the initial cutting, and to which good curve fitting was possible for >20 s, were included. This restriction of data created average separations that overestimate the true ensemble. It appears reasonable to assume that the diffusion constant of DNA in nanochannels scales roughly linear with the length of the molecule. We thus anticipate that gap formation due to diffusion will be observable for 100- or 200-kbp-long DNA molecules within practicable times. We have not observed a passing-through of the long segments through each other, or of the short segments through the long one, but cannot exclude that the short segments exchanged their positions. We have explained the mechanism of stretching in the papers by Tegenfeldt et al. (4) and Reisner et al. (9), and in summary the stretching is reliant on semiflexibility, entropic effects, and self-exclusion. The self-exclusion places a large energy penalty on placing two polymer strands next to each other, and we expect that interpenetration by Brownian motion is unlikely.
We note that the method for optical restriction mapping is also expected to be applicable to molecules beyond that length for the following reasons. The relaxation time of DNA confined to nanochannels is expected to scale with the square of the molecule length (9). Hence, very long molecules, which typically enter the nanochannels in an overstretched state, will not be thermally relaxed at the time of cutting. The resulting restriction fragments themselves will have a relaxation time that is only dependent on their own length and thus will relax much faster, opening visible gaps in the DNA and enabling observation of the cutting. We thus anticipate that the technique will be universally applicable, as long as the relaxation times of the fragments make time averaging feasible.
We return at this point to a comparison of our method to that of Schwartz et al. (5, 6). These authors have achieved a resolution of 1.1 kbp using 20-150 molecules (20). Alternatively, the error was given as 2.9%. We thus can see that the resolution attained by using a single molecule in our method is comparable. Note also that the error in our method is expected to scale with the square root of the fragment size. Thus, the technique should perform significantly better on long fragments. Note that a significant amount of noise in our device may stem from nonuniform channels, which can be reduced (4). Two key advantages of our technique are the easy integration into microfluidic systems able of handling microliters of fluid and the capability of single-molecule mapping. Isolated and purified DNA samples can be analyzed within 1 h, including chip wetting, sample loading, and mounting in a microscope.
In conclusion, we have demonstrated a method for the construction of ordered restriction maps using single DNA molecules stretched in nanofluidic channels, and integrated control of the enzymatic cofactor magnesium. We have used the method for the mapping of λ-DNA and have shown a resolution of 1.5 kbp within 1 min. We believe the presented technology could be used to study the site-selective interaction between DNA and a range of metal-ion-induced enzymes.
Acknowledgments
This work was supported by Defense Advanced Research Planning Agency Grant MDA972-00-1-0031, National Institutes of Health Grants HG01506 and E21F46G1, National Science Foundation Nanobiology Technology Center Grant BSCECS9876771, State of New Jersey Grant NJCST 99-100-082-2042-007, and U.S. Genomics.
Author contributions: R.R., Y.-M.W., E.C.C., and R.H.A. designed research; R.R., M.L., and S.F.L. performed research; R.R., M.L., and E.C.C. contributed new reagents/analytic tools; R.R. and S.F.L. analyzed data; and R.R., Y.-M.W., E.C.C., and R.H.A. wrote the paper.
Footnotes
We have observed that Mg2+ concentrations of 2 mM or more lead to a quenching of fluorescence from bis-intercalating DNA dyes. We believe this is due to removal of dye from the DNA. Thus, at sufficiently high Mg2+ concentrations, intercalating dyes such as TOTO-1 or Sybr Green do not interfere with sequence-specific enzymes, because the dyes are not effectively blocking the interaction anymore.
References
- 1.Pingoud, A. & Jeltsch, A. (2001) Nucleic Acids Res. 29, 3705-3727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sambrook, J. & Russel, D. W. (1989) Molecular Cloning: A Laboratory Manual (Cold Spring Harbor Lab. Press, Cold Spring Harbor, NY).
- 3.Li, W. L., Tegenfeldt, J. O., Chen, L., Austin, R. H., Chou, S. Y., Kohl, P. A., Krotine, J. & Sturm, J. C. (2003) Nanotechnology 14, 578-583. [Google Scholar]
- 4.Tegenfeldt, J. O., Prinz, C., Cao, H., Chou, S., Reisner, W. W., Riehn, R., Wang, Y. M., Cox, E. C., Sturm, J. C., Silberzahn, P. & Austin, R. H. (2004) Proc. Natl. Acad. Sci. USA 101, 10979-10983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schwartz, D. C., Li, X., Hernandez, L. I., Ramnarain, S. P., Huff, E. J. & Wang, Y.-K. (1993) Science 262, 110-114. [DOI] [PubMed] [Google Scholar]
- 6.Lin, J., Qi, R., Aston, C., Jing, J., Anantharaman, T. S., Mishra, B., White, O., Daly, M. J., Minton, K. W., Venter, J. C. & Schwartz, D. C. (1999) Science 285, 1558-1562. [DOI] [PubMed] [Google Scholar]
- 7.Taylor, J. R., Fang, M. M. & Nie, S. (2000) Anal. Chem. 72, 1979-1986. [DOI] [PubMed] [Google Scholar]
- 8.Bensimon, D., Simon, A. J., Croquette, V. & Bensimon, A. (1995) Phys. Rev. Lett. 74, 4754-4757. [DOI] [PubMed] [Google Scholar]
- 9.Reisner, W. W., Morton, K., Riehn, R., Wang, Y. M., Yu, Z., Rosen, M., Sturm, J., Chou, S. Y., Frey, E. & Austin, R. H. (2005) Phys. Rev. Lett. 94, 196101. [DOI] [PubMed] [Google Scholar]
- 10.Madou, M. J. (2002) Fundamentals of Microfabrication (CRC, Boca Raton, FL)
- 11.Wang, Y. M., Tegenfeldt, J. O., Reisner, W. W., Riehn, R., Guan, X.-J., Guo, L., Golding, I., Cox, E. C., Sturm, J. C. & Austin, R. H. (2005) Proc. Natl. Acad. Sci. USA 102, 9796-9801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Withers, B. E. & Dunbar, J. C. (1995) J. Biol. Chem. 270, 6496-6504. [DOI] [PubMed] [Google Scholar]
- 13.Osoegawa, K., Tateno, M., Woon, P. Y., Frengen, E., Mammoser, A. G., Catanese, J. J., Hayashizaki, Y. & de Jong, P. J. (2000) Genome Res. 10, 116-128. [PMC free article] [PubMed] [Google Scholar]
- 14.Wright, D. J., Jack, W. E. & Modrich, P. (1999) J. Biol. Chem. 274, 31896-31902. [DOI] [PubMed] [Google Scholar]
- 15.Terry, B., Jack, W., Rubin, R. & Modrich, P. (1983) J. Biol. Chem. 258, 9820-9825. [PubMed] [Google Scholar]
- 16.Åkerman, B. & Tuite, E. (1996) Nucleic Acids Res. 24, 1080-1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goodwin, P. M., Johnson, M. E., Martin, J. C., Ambrose, W. P., Marronce, B. L., Jett, J. H. & Keller, R. A. (1993) Nucleic Acids Res. 21, 803-806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Meng, X., Cai, W. & Schwartz, D. C. (1996) J. Biomol. Struct. Dyn. 13, 945-951. [DOI] [PubMed] [Google Scholar]
- 19.Hansen, L. F., Jensen, L. K. & Jacobsen, J. P. (1996) Nucleic Acids Res. 24, 859-867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cai, W., Jing, J., Irvin, B., Ohler, L., Rose, E., Shizuya, H., Kim, U.-J., Simon, M., Anantharaman, T., Mishra, B. & Schwartz, D. C. (1998) Proc. Natl. Acad. Sci. USA 95, 3390-3395. [DOI] [PMC free article] [PubMed] [Google Scholar]