

Abstract
Background
The TyrA protein family includes members that catalyze two dehydrogenase reactions in distinct pathways leading to L-tyrosine and a third reaction that is not part of tyrosine biosynthesis. Family members share a catalytic core region of about 30 kDa, where inhibitors operate competitively by acting as substrate mimics. This protein family typifies many that are challenging for bioinformatic analysis because of relatively modest sequence conservation and small size.
Results
Phylogenetic relationships of TyrA domains were evaluated in the context of combinatorial patterns of specificity for the two substrates, as well as the presence or absence of a variety of fusions. An interactive tool is provided for prediction of substrate specificity. Interactive alignments for a suite of catalytic-core TyrA domains of differing specificity are also provided to facilitate phylogenetic analysis. tyrA membership in apparent operons (or supraoperons) was examined, and patterns of conserved synteny in relationship to organismal positions on the 16S rRNA tree were ascertained for members of the domain Bacteria. A number of aromatic-pathway genes (hisHb, aroF, aroQ) have fused with tyrA, and it must be more than coincidental that the free-standing counterparts of all of the latter fused genes exhibit a distinct trace of syntenic association.
Conclusion
We propose that the ancestral TyrA dehydrogenase had broad specificity for both the cyclohexadienyl and pyridine nucleotide substrates. Indeed, TyrA proteins of this type persist today, but it is also common to find instances of narrowed substrate specificities, as well as of acquisition via gene fusion of additional catalytic domains or regulatory domains. In some clades a qualitative change associated with either narrowed substrate specificity or gene fusion has produced an evolutionary "jump" in the vertical genealogy of TyrA homologs. The evolutionary history of gene organizations that include tyrA can be deduced in genome assemblages of sufficiently close relatives, the most fruitful opportunities currently being in the Proteobacteria. The evolution of TyrA proteins within the broader context of how their regulation evolved and to what extent TyrA co-evolved with other genes as common members of aromatic-pathway regulons is now feasible as an emerging topic of ongoing inquiry.
Background
Dehydrogenases dedicated to L-tyrosine (TYR) biosynthesis comprise a family of TyrA homologs that have different specificities for the cyclohexadienyl substrate: ones specific for L-arogenate (AGN), ones specific for prephenate (PPA), and those that are able to use both [1,2]. Figure 1 illustrates the biochemical relationship of these specificities to divergent transformations beginning with chorismate (CHA) utilization and converging on TYR formation. Compounding this complexity, a given TyrA enzyme having any of the aforementioned cyclohexadienyl specificities may be specific for NAD+ or NADP+, or may use both. This is consistent with a growing appreciation [3,4] that different substrate specificities are often accommodated across a given protein family that nevertheless maintains a common scaffold of fundamental reaction chemistry. Even within the single category of broad TyrA specificity, there is a continuum ranging from examples where alternative substrates are accepted equally well to other cases where one substrate may be preferred by an order of magnitude or more. Table 1 provides a key to the nomenclature used to identify the various possible substrate-utilization combinations (both cyclohexadienyl and pyridine nucleotide) exhibited by TyrA proteins.
Figure 1.
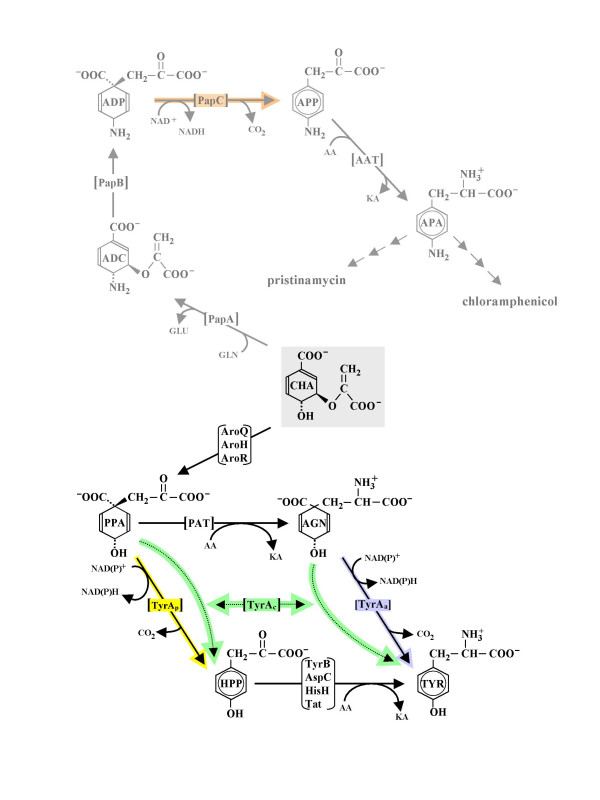
Composite of alternative biochemical routes from chorismate (CHA) to L-tyrosine (TYR) in nature. An antibiotic synthesis branch from CHA is also shown (dimmed). Here the intermediates shown to intervene between chorismate and pristinamycin or chloramphenicol are p-aminochorismate (ADC), p-aminoprephenate (ADP), p-aminophenylpyruvate (APP), and p-aminophenylalanine (APA). PPA may be transaminated by prephenate aminotransferase (PAT) to yield L-arogenate (AGN). The four TyrA homologs and the reactions they catalyze are colored differently. Arogenate dehydrogenase (TyrAa) converts AGN to TYR. Alternatively, prephenate dehydrogenase (TyrAp) converts PPA to 4-hydroxyphenylpyruvate (HPP) which is then transaminated to TYR via an homolog of TyrB, AspC, HisH, or Tat [49]. A broad-specificity cyclohexadienyl dehydrogenase (TyrAc) is competent to catalyze either the TyrAa or the TyrAp reaction. PapC converts the 4-amino analog of PPA to the 4-amino analog of HPP. AroQ, AroH, and AroR are distinct homologs known to exist in nature for performance of the chorismate mutase reaction. Other abbreviations: AA, amino acid donor, KA, keto-acid accepter.
Table 1.
Abbreviations used to designate substrate specificities of tyrA/TyrA homologs
| Abbreviationa | ||
| Gene | Gene Product | Description of specificityb |
| tyrAx | TyrAx | Specificity for cyclohexadienyl substrate is unknown |
| tyrAc | TyrAc | Broad-specificity cyclohexadienyl dehydrogenase (CDH) |
| tyrAp | TyrAp | Narrow-specificity prephenate dehydrogenase (PDH) |
| TyrAc_Δ | TyrAc_Δ | Broad-specificity cyclohexadienyl dehydrogenase having catalytic-core indels in correlation with an extra-core extension |
| tyrAa | TyrAa | Narrow-specificity arogenate dehydrogenase (ADH) |
| NAD tyrAa | NADTyrAa | TyrA homolog is AGN-specific and NAD+-specific |
| NADP tyrAa | NADPTyrAa | TyrA homolog is AGN-specific and NADP+-specific |
| NAD(P)tyrA a | NAD(P)TyrAa | TyrA homolog is AGN-specific but utilizes either NAD+ or NADP+ |
| xtyrAx | xTyrAx | Specificity for both the cyclohexadienyl and pyridine nucleotide substrates is unknown |
aAbbreviations in the upper-table (upper 5 rows) indicate the specificities for the cyclohexadienyl substrate. Abbreviations in the lower-table (lower 4 rows) indicate specificities for both cyclohexadienyl (right subscripts) and pyridine nucleotide substrates (left subscripts). Combinations not shown can be deduced from the examples given, e.g., a TyrA homolog specific for prephenate and NAD+ would be designated NADTyrAp.
bThe abbreviations CDH, PDH, and ADH (shown parenthetically) have been used frequently in the literature.
The TyrA family is typical of many protein families in that its members have a relatively small core domain that is not highly conserved. As such, substantial challenges for bioinformatic analysis are posed. Here we have not only carried out a labor-intensive manual analysis, but we have also developed tools intended to facilitate and refine follow-on studies of this protein family in the genome era. The approaches implemented in this study with the TYR segment of aromatic biosynthesis hopefully can serve as a template for forthcoming integrant analyses of other pathway segments of aromatic biosynthesis, and indeed for metabolic subsystems in general.
This manuscript contains three broad sections. First, the biochemical and enzymological complexity of the TyrA protein family is presented in terms of the diversity that exists in nature with respect to substrate specificity and the association of the core domain with other catalytic or regulatory domains. Secondly, the genomic colinear organization of tyrA genes with other genes is evaluated, i.e., tyrA is considered in its syntenic context. Thirdly, tyrA is evaluated in its context of regulation. These three sections are tied together in a framework of evolutionary perspective.
Results and discussion
Background of TyrA diversity
Our evolutionary analysis is limited by the amount of information that can be managed in a single study, with the focus fixed upon the domain Bacteria (due to the relative density of genome representation for Bacteria in the public databases). However, in order to show where future expansion of the analysis might lead, the selection of TyrA proteins in Fig. 2 are from all three domains of life, i.e., Bacteria, Archaea, and Eukarya (lower eukaryotes and higher plants). For practicality of presentation, numerous orphan (i.e., without close relatives) TyrA sequences are not shown, and not all members of a given group are necessarily included. The main purposes of the radial tree shown in Fig. 2 are: (i) to illustrate that TyrA proteins of major phylogenetic groupings are generally congruent with 16S rRNA groupings and (ii) to convey a snapshot visualization of the overall complexity of the TyrA protein family from the vantage point of its varied substrate specificities as well as its multiple fusion partners.
Figure 2.
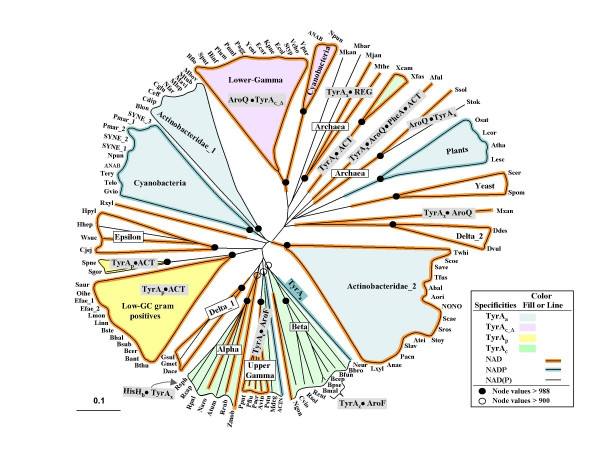
Phylogenetic tree for trimmed core domains of selected members of the TyrA Superfamily. Acronyms used for the various organisms are given in alphabetical order in Table 2. (A more extensive listing that includes organisms not shown in Fig. 2 and which also is hyperlinked to all of the individual GenBank records is given in Table S1. A similar table that also includes compilation of known and predicted substrate specificities is maintained at AroPath [73]. Lineages possessing experimentally established TyrAa, TyrAp, TyrAc or TyrAc_Δ proteins are indicated by fill colors specified in the legend. Three specificity patterns for the pyridine nucleotide substrate are shown by line colors (see figure box). Although the cyanobacteria are depicted as having NADP+-specific TyrA proteins, some of them can also accept NAD, albeit to a lesser degree. All proteins having an aspartate residue homologous to D-32 of the E. coli NADTyrAc_Δ domain are presumed specific for NAD+. Fusion of TyrA domains with other catalytic domains is indicated within grey boxes (AroQ•TyrA, TyrA•AroF, HisHb•TyrA, and TyrA•AroQ•PheA•ACT) using the convention of a bullet to represent the interdomain area. The boxes overlap any relevant lineages. TyrA proteins having carboxy-terminal fusions with regulatory domains (TyrA•ACT and TyrA•REG) are also shown. The distance scale bar at the bottom left represents substitutions per site.
As an illustration of the detailed information that follows, note that the TyrA sequences from the beta Proteobacteria at five o'clock in Fig. 2 form a cohesive cluster (termed a 'congruency group'). In this clade there exists a proposed ancestral background of broad specificity where either AGN or PPA in combination with either NAD+ or NADP+ could be used. This profile of broad substrate use (which can be denoted as NAD(P)TyrAc; see Table 1) generally persists in the beta Proteobacteria. From this background, narrowed specificities for the AGN/NADP+ couple emerged once in the lineage represented by Nitrosomonas europaea (Fig. 2; dark blue line), narrowed specificity for NAD+ emerged once in species of Neisseria (orange line), and fusion of tyrAc with aroF (which encodes enolpyruvylshikimate-3-P synthase, the sixth enzyme in the common pathway of aromatic biosynthesis; see [5,6] for nomenclature used) occurred recently within the Burkholderia lineage. These character-state transformations appear to occur with relative ease, and independent emergence of the same character states can be seen elsewhere in the tree.
Phylogenetically congruent TyrA groupings
Multiple alignments of catalytic-core domains
A phylogenetic tree is only as good as the input alignment. An optimal multiple alignment of TyrA homologs requires a trimmed set of sequences that corresponds to the catalytic-core domain. Alignment of sequences with non-homologous N-terminal fusions (such as with chorismate mutase• (AroQ•), HisHb•, or plant transit peptides•; note the convention of using a bullet to indicate the fusion point of one domain with another domain) will make them appear to be more closely related than they actually are because residues in the non-homologous N-terminal regions find matches at random. Likewise, those TyrA sequences with C-terminal fusions (such as with •AroF, •ACT, or •REG) will appear to be anomalously close to one another. Even enzyme proteins that have much greater sequence conservation and amino-acid lengths than TyrA proteins cannot reasonably be expected to yield a protein tree that would be congruent over an extensive phylogenetic range with the overall 16S rRNA tree. However, if genome representation is sufficiently dense within a range of closely related organisms, 16S rRNA congruency with a given protein can be expected within that range of organisms provided that (i) the particular functional role has been retained and (ii) lateral gene transfer has not occurred to obscure the relationship. This expectation follows from the outcome of a detailed analysis of tryptophan-pathway proteins in Bacteria [7,8].
Congruency within major clades
TyrA sequences from higher-plant and yeast Eukarya form cohesive clusters. Genome representation among Archaea is still relatively limited. (Fig. 2 does reveal, however, that genes encoding TyrA proteins in Archaea have experienced various catalytic- and regulatory-domain fusions at least as frequently as those in Bacteria). Eventual expansion of both the tryptophan-pathway and tyrosine-pathway analyses to Archaea should be quite interesting.
The great majority of TyrA sequences available are from Bacteria, and one can see (by inspection of the major clades supported by high bootstrap values in Fig. 2) a qualitatively apparent congruence of TyrA-tree sub-sections with 16S rRNA expectations of vertical genealogy. Thus, all cyanobacteria possess a NADPTyrAa type of TyrA enzyme, and this is a very cohesive grouping. A few of the larger cyanobacterial genomes have a co-existing second enzyme of the TyrAc_Δ type (discussed in detail later). The low-GC gram-positive bacteria (Bacillus/Staphylococcus/Enterococcus/Listeria) exhibit the NADTyrAp pattern of specificity and also possess a C-terminal domain (ACT) of allosteric regulation. It is interesting that the TyrAp•ACT proteins of the Streptococcus lineage (at eight o'clock in Fig. 2) differ from the main low-GC clade in possessing broad specificity for pyridine nucleotides (as indicated with black line color). The most parsimonious evolutionary conclusion would be that in the low-GC gram-positive grouping, acquisition of the ACT domain and narrowed specificity for prephenate preceded narrowed specificity for NAD+. Thus, the latter event occurred after divergence of the Streptococcus lineage from the remainder of the low-GC clade. Members of the subclass taxon Actinobacteridae (mostly actinomycetes) possess AGN-specific TyrA enzymes (light blue fill color in Fig. 2), but they separate into two distinct groups that correlate either with broad specificity for pyridine nucleotides (Actinobacteridae_1) or a NAD+-specific pattern (Actinobacteridae_2). The Proteobacteria are discussed immediately below.
Proteobacteria
By far the greatest genomic density available is for Proteobacteria, the group of Bacteria that includes purple bacteria and their relatives. The various divisions of Proteobacteria, as currently named, lack hierarchical equivalence. For example, the epsilon and delta divisions branch from much deeper positions on the phylogenetic tree than do the alpha Proteobacteria. As genome representation expands for epsilon and delta Proteobacteria, it is probable that these will subdivide to newly named groupings of approximate hierarchical equivalence with alpha Proteobacteria. The most recently diverged Proteobacteria are the beta and gamma divisions. From the combination of our previous analysis of tryptophan biosynthesis [7,8], TYR biosynthesis (this paper), and other segments of aromatic biosynthesis (unpublished data), we find it useful to separate "upper-gamma" Proteobacteria from "lower-gamma" Proteobacteria (an "enteric lineage" with Shewanella oneidensis as approximately the most divergent member). This separation is because the beta Proteobacteria and the upper-gamma Proteobacteria exhibit a smooth continuity of relatively few evolutionary events with respect to aromatic biosynthesis, in striking contrast to extraordinarily dynamic evolutionary events in the lower-gamma Proteobacteria. As a consequence, the lower-gamma Proteobacteria are much more distinct (in terms of aromatic biosynthesis) from the upper-gamma Proteobacteria than the upper-gamma are from the beta Proteobacteria.
Figure 2 shows that alpha, beta and epsilon divisions of Proteobacteria form phylogenetically coherent clusters with respect to their TyrA proteins. Although delta Proteobacteria fall into two well-separated groupings denoted as Delta_1 and Delta_2, this should not be surprising since these groupings diverge at a deep level on the 16S rRNA tree where genome representation is poor. In addition, the Myxococcus xanthus TyrA sequence, currently an orphan (three o'clock in Fig. 2), represents a third divergent lineage in delta Proetobacteria. In contrast to delta Proteobacteria, genomic representation for the gamma Proteobacteria is relatively good. Nevertheless their TyrA sequences separate into several well-spaced groupings, albeit for entirely different reasons. In this case, the split seen between two clades of these fairly close relatives (upper-gamma and lower-gamma) is attributed to particularly dynamic evolutionary events compressed into a relatively short time span in the lower-gamma Proteobacteria. (We refer to such a dynamic divergence as an evolutionary jump; see the next section.) Note that the allocation of upper-gamma and lower-gamma Proteobacteria to separate TyrA congruency groups is not the same as being incongruent. It is quite possible that as new genomes come on line, new and intermediate TyrA sequences may result in the merging of the foregoing two congruency groups (currently tyrosine congruency group 1 (TyrCG-1) and tyrosine congruency group 2 (TyrCG-2)).
Comparison of tryptophan and tyrosine congruency groups
Although the true extent of lateral gene transfer (LGT) at present must be described as intensely controversial, there is little doubt that any given organism is mosaic with respect to some unknown fraction of its gene repertoire. Our "accounting" system for keeping track of proteins that are faithful to the vertical genealogy is to formulate congruency groupings that are defined by congruence of given protein-tree clusters to a section of the 16S rRNA tree. Ultimately this information will reveal which organisms are "pure" with respect to the vertical inheritance of a given pathway or pathway segment. Our congruency groups are intended to be fluid, in that with the continued availability of new sequences, a previous orphan sequence may very well become the seed for a new congruency group. On the other hand, previously separate congruency groups have the potential to merge. (See Methods for more information.) The present tyrosine congruency groups are listed on the AroPath website [9].
Seven tryptophan congruency groups in Bacteria were previously formulated [8] based upon the correspondence of cohesive clusters in trees of Trp-protein concatenates with sections of 16S rRNA trees. The information input for formulation of tryptophan congruency groups is of greater quality than for tyrosine congruency groups because seven-protein concatenates could be used for the former. On the other hand, the broad information input supporting tyrosine congruency groups in this study is more comprehensive because of greater genome availability. Tryptophan congruency group 1 (TrpCG-1) corresponds perfectly with the organisms represented in TyrCG-1, these being the lower-gamma Proteobacteria (enteric lineage). The upper-gamma Proteobacteria (TyrCG-2) and the beta Proteobacteria (tyrosine congruency group 3; TyrCG-3) are represented by different tyrosine congruency groups. In contrast, the membership of tryptophan congruency group 2 (TrpCG-2) includes both the upper-gamma Proteobacteria and the beta Proteobacteria. The latter merging probably reflects the advantage conferred by the greater information content of the concatenated sequences used to define tryptophan congruency groups.
Species of Xylella and Xanthomonas are usually referred to as gamma Proteobacteria. They probably represent an outlying deeply branching lineage, although trees based on concatenated strings of proteins [10] or 16S rRNA [11] position them with beta Proteobacteria. In any event, Trp-protein concatenate trees placed Xylella and Xanthomonas within TrpCG-2, which contains both upper-gamma and beta Proteobacteria. In contrast, the TyrA domains from Xylella and Xanthomonas were well separated (at about two o'clock in Fig. 2) from those of any other organism. This might simply be due to the limited resolving power of a single protein in combination with too few close relatives. (Note that single Trp-protein trees sometimes failed to achieve the congruency-group placements that were resolved by seven-protein Trp concatenates [8]). An additional clue may be relevant. The TyrA proteins from the Xylella/Xanthomonas genera possess an ACT domain, which has not been observed in any other proteobacterial TyrA proteins thus far. In view of this, origin by LGT seems to be a distinct possibility, but with the important caveat that no likely genome donors are yet obvious on the criterion of sequence similarity. Perhaps more likely is the following possible explanation that postulates a basis for accelerated divergence. The TyrA domains of Xanthomonas/Xylella proteins have an indel structuring (insertions and/or deletions) that places them within the TyrAc_Δ specificity subclass (see below). We suggest (see below) that such indel structuring reflects interaction of the core TyrA domain with an extra-domain extension. Thus, selection for amino acid changes accomplishing a new domain-domain interaction could account for accelerated divergence of the Xanthomonas/Xylella sequences on the TyrA tree (Fig. 2).
Cohesive tryptophan congruency groups of the alpha Proteobacteria (tryptophan congruency group 3; TrpCG-3) and the cyanobacteria (tryptophan congruency group 4; TrpCG-4) match up well with the corresponding tyrosine congruency groups (tyrosine congruency group 4 (TyrCG-4) and tyrosine congruency group 8 (TyrCG-8), respectively). The TyrA proteins of epsilon Proteobacteria define a cohesive tyrosine congruency group (tyrosine congruency group 5; TyrCG-5), whereas the Trp-protein concatenates of epsilon Proteobacteria did not exhibit a coherent congruency group, due at least in part to LGT [8]. The delta Proteobacteria separate into two distinct tyrosine congruency groups: Delta_1 (tyrosine congruency group 6; TyrCG-6) and Delta_2 (tyrosine congruency group 7; TyrCG-7), as shown in Fig. 2. It is likely that corresponding tryptophan congruency groups exist (work in progress), but at the time of the Xie et al. study [8] only Trp-pathway protein concatenates for Desulfovibrio vulgaris (Delta_2) and Geobacter sulfurreducens (Delta_1) were available, and they were provisionally listed as "orphans". In the present work TyrA sequences from Deinococcus radiodurans and Thermus thermophilus are the sole members of tyrosine congruency group 12 (TyrCG-12). At the time of the Trp-pathway work, the genome of Thermus was unavailable and the Deinococcus concatenate was listed as an orphan. It is expected that the Deinococcus and Thermus concatenates will now seed a new tryptophan congruency group.
Whereas tryptophan congruency group 5 (TrpCG-5) is defined by cohesive concatenates from actinomycete bacteria, the TyrA proteins from the same organisms separated into two distinct congruency groups. It is intriguing that this partitioning into two congruency groups correlates with narrowed specificity for NAD+ (indicating an evolutionary jump) in one of the groups. The latter group (tyrosine congruency group 11; TyrCG-11) is denoted Actinobacteridae_2 in Fig. 2, whereas tyrosine congruency group 10 (TyrCG-10) is displayed as Actinobacteridae_1. The opposite scenario whereby a single tyrosine congruency group corresponds to split tryptophan congruency groups applies in the case of low-GC gram-positive bacteria. Whereas TyrA proteins form a single congruency group in these organisms (tyrosine congruency group 9; TyrCG-9), a small cluster of Trp-pathway concatenates from Bacillus subtilis, B. stearothermophilus, and B. halodurans (tryptophan congruency group 6; TrpCG-6) separate distinctly from the remaining organisms (tryptophan congruency group 7; TrpCG-7). The latter evolutionary jump reflects a dynamic scenario of tryptophan-pathway evolutionary events that include loss of one gene from the trp operon, insertion of the trp operon into a 6-gene aro operon to produce a supraoperon, and acquisition of the TRAP (tryptophan-activated RNA-binding protein) mechanism of regulation by an RNA-binding protein [7].
Tyrosine congruency groups and tryptophan congruency groups are maintained and updated at the AroPath website [12].
Distribution in nature of TyrA specificity subclasses for the cyclohexadienyl substrate
Four qualitative classes of specificity for the cyclohexadienyl substrate populate the TyrA superfamily of homologs (Fig. 1). These include PPA-specific (TyrAp), AGN-specific (TyrAa), the broad-specificity cyclohexadienyl (TyrAc) dehydrogenases and a fourth class represented by an enzyme of antibiotic biosynthesis (PapC) that converts 4-amino-4-deoxy-prephenate to 4-amino-phenylpyruvate [13]. Representatives of each specificity class have been studied at molecular and genetic levels. TyrA family members sharing a given substrate specificity do not necessarily cluster tightly together, and assignment of substrate specificity to experimentally uncharacterized TyrA homologs is uncertain unless they exhibit very high amino acid identities with experimentally characterized TyrA proteins. In some cases we do not accept older literature reports without more recent verification. For example, the yeast Saccharomyces cerevisiae TyrAx was characterized as a TyrAp protein [14] long before it was recognized [15] that PPA preparations were often contaminated with AGN (an unknown compound at that time).
Our collection of curated TyrA sequences at AroPath (see Table 3) contains trimmed sequences that comprise catalytic-core domains. This collection was divided into two groups based on whether the sequences contained the relatively short N-terminal pyridine-nucleotide discriminator segment or the longer C-terminal cyclohexadienyl-substrate core segment. The sequences in the latter group were assembled into subgroups representing established substrate specificities (TyrAa, TyrAp and TyrAc) and were aligned separately to obtain overall consensus sequences for cyclohexadienyl-substrate core segments. The TyrAc group members from the lower-gamma assemblage of Proteobacteria (as well as from a few other lineages) were so distinctive that a fourth group (TyrAc_Δ) was defined. This latter group is, in fact, the most divergent of the four. Figure 3 shows a comparison of the four consensus sequences, with invariant anchor residues shaded yellow and residues conserved across all groups shaded in gray. Residues within each group that are >50% conserved are shown in capital letters. In pairwise BLAST (Basic Local Alignment Tool) [16]comparisons, TyrAa and TyrAc consensus sequences are most similar (47% identity), followed by the TyrAc/TyrAp pair (40% identity), with TyrAa and TyrAp exhibiting 34% identity. TyrAc_Δ is quite distinct from the other three groupings, exhibiting only 27% identity with TyrAc, 23% identity with TyrAc, and 18% identity with TyrAp.
Table 3.
Curated TyrA amino-acid sequence files at AroPath [35]
| Complete TyrA sequences |
| Catalytic-core domainsa |
| Pyridine-nucleotide discriminator segmentsb |
| NAD+-specific |
| NADP+-specific |
| Broad specificity |
| Cyclohexadienyl-substrate core segments |
| Arogenate-specific (TyrAa) |
| Prephenate-specific (TyrAp) |
| Broad specificity |
| TyrAc |
| TyrAc_Δ |
| Pseudogene TyrA sequences |
aTrimmed free of N-terminal or C-terminal extensions, including any fusions with regulatory domains or other catalytic domains.
bHigh-glycine βαβ Rossmann fold at the N-terminus.
Figure 3.
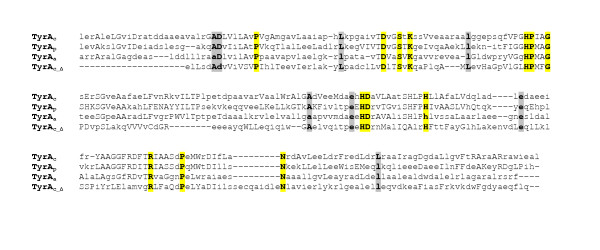
Multiple alignment of the HMM consensus sequences obtained for different substrate-specificity groupings within cyclohexadienyl-substrate core segments (see Table 3). Invariant anchor residues are highlighted in yellow, conserved residues in grey. These consensus sequences will change continuously as corrections and refinements are made. The version shown was current as of April, 2005.
Cyclohexadienyl dehydrogenases
Many TyrA proteins (at least in the domain Bacteria) are of the TyrAc subclass. The cyclohexadienyl dehydrogenases commonly accept PPA or AGN about equally well, but various degrees of preference for one of the alternative substrates are also observed. Detailed molecular and genetic studies of TyrAc proteins from Pseudomonas aeruginosa, [17], P. stutzeri [1], and Zymomonas mobilis [18] have been carried out. The distinct variety of TyrAc mentioned above, which has been denoted TyrAc_Δ exhibits a number of indels (mostly deletions) within the catalytic-core region when its consensus sequence is aligned with those of the other TyrA classes (Fig. 3). It is intriguing that the indel structuring of TyrAc_Δ correlates with the presence of an extra-core extension. This extension is often AroQ, but not always. For example, in the genera Nostoc and Anabaena it appears to be a degraded, catalytically inactive AroQ, whereas in Xanthomonas or Xylella it is an ACT domain. Since the one large clade of TyrAc_Δ proteins that has so far been studied prefers PPA over AGN by well over an order of magnitude, an evolutionary relationship of indel insertions to the narrowing of substrate preference for PPA might exist. If so, however, this cannot be the only molecular change to accomplish favored utilization of PPA over AGN since a number of TyrAc proteins, (e.g., TyrAc from Neisseria gonorrhoeae), also exhibits an overwhelming preference for PPA, even though this class lacks the indel structuring.
Arogenate dehydrogenases
The TyrAa class of specificity is currently represented by higher plants and at least three widely spaced bacterial lineages: cyanobacteria, actinomycetes and Nitrosomonas europaea. This discontinuity of phylogenetic spacing is consistent with a fundamental evolutionary scenario [19] whereby the ancestral dehydrogenase was a broad-specificity TyrAc that evolved narrowed substrate specificity (to yield either TyrAp or TyrAa) independently on multiple occasions in modern lineages. The ubiquitous presence of TyrAa in cyanobacteria has been heavily documented [20]. Nitrosomonas europaea currently (as of March, 2005) has no sufficiently close genome relatives that have been sequenced. The first BLAST hit returned from a NADPTyrAa query from N. europaea (March,2005) is the protein from Ralstonia solanacearum (48% identity), which is known to possess broad specificity for both of its substrates (i.e., NAD(P)TyrAc) [21,22].
The TyrA sequences of Actinobacteria separate into two distinct groupings on the protein tree (Fig. 2). Coryneform bacteria in one sub-cluster have been rigorously characterized as the NAD(P)TyrAa substrate specificity type. On the other hand, a variety of Streptomyces species have been shown [23,24] to possess NADTyrAa, and TyrA proteins of these organisms populate the second Actinobacteria sub-cluster of Fig. 2. Figure 4 shows sequence alignments of the N-terminal pyridine-nucleotide discriminator regions of currently available actinomycetes. The conserved 'D' residue (highlighted in yellow) in the upper group is a reliable indicator of NAD+ specificity, in part because NADP+ is repelled by the negative charge at this position. The asparagine residue (highlighted in blue) in the corresponding position in members of the lower group indicates NAD(P)+ specificity as discussed by Bonner et al. [25]. Rubrobacter xylanophilus is the most distant representative of the Actinobacteria, being the sole member of the subclass taxon Rubrobacteridae, and its protein (denoted Rxyl) appears as an orphan in Fig. 2.
Figure 4.

Alignment of the N-terminal glycine-rich P-loop of TyrA•ACT proteins from the Class Actinobacteria. These are specific for L-arogenate as substrate, but fall into two groups with respect to the pyridine nucleotide co-substrate. The top NAD+-specific group possesses an aspartate (D) at position 32 (E. coli numbering), whereas the bottom NAD+/NADP+ group possesses an asparagine at the homologous position. Residue numbers are shown at the left. The species in the middle are color coded to match the hierarchical taxon positions obtained from NCBI. The variable loop of the Wierenga fingerprint [26], which in E. coli contains five residues (22–26), contains the minimal two residues in all of the Actinobacteria shown. The organisms on the right are color coded according to the taxonomic position indicated on the left (NCBI). The Rubrobacter xylanophilus TyrAa sequence is an orphan in the tree displayed in Fig. 2, as consistent with its outlying position in the taxonomy scheme.
A similar relationship of phylogenetic separation associated with narrowed specificity for pyridine-nucleotide substrate exists for the low-GC gram-positive bacteria (eight o'clock in Fig. 2). Here the major clade is NAD+-specific, whereas species of Streptococcus have retained the ancestral breadth of specificity for NAD+/NADP+. Alignments of the pyridine-nucleotide discriminator regions of these latter two groups match up extremely well with the upper alignment of Fig. 4 where residue 32 of the Wierenga fingerprint [26] is 'D' and with the lower alignment where residue 32 is 'N' (data not shown).
Recently, a plant tyrAa from Arabidopsis thaliana has been reported to consist of two near-identical domains that are fused [27]. The gene encoding this 68-kDa protein co-exists in the genome with a single-domain paralog [28] that encodes a predicted 37-kDa protein, somewhat larger than the catalytic-core domain of TyrAa from Synechocystis. TyrAa (known to be located in higher-plant chloroplasts [2]) may have originated from cyanobacteria via endosymbiosis. If so, however, the plant TyrAa sequences have diverged sufficiently that they no longer share a specific phylogenetic grouping with the cyanobacterial TyrA sequences. This is in marked contrast with the phylogenetic coherence of the tryptophan synthase subunit proteins (TrpEa and TrpEb_1) from cyanobacteria and higher plants [29].
Prephenate dehydrogenases
TyrAp is conspicuously represented by a large clade of low-GC gram-positive organisms, of which Bacillus subtilis TyrAp is the best studied [30]. Thus far, all TyrAp proteins are fused to a C-terminal ACT domain, and therefore no "minimal" TyrAp proteins that consist only of a catalytic core are available as yet. At the level of physiological function, it should be added that those cyclohexadienyl dehydrogenases that exhibit a very substantial preference for prephenate are for all practical purposes prephenate dehydrogenases, even though they carry a formal designation of TyrAc or TyrAc_Δ. These include most, if not all, of the AroQ•TyrAc_Δ enzymes of the enteric lineage (lower-gamma in Fig. 2). The TyrAc protein from Neisseria gonorrhoeae (and by inference, the closely related N. meningitides) is also a well-studied example of overwhelming preference for prephenate [21].
PapC dehydrogenases
PapC participates in the formation of p-aminophenylalanine as a step in the synthesis of at least two antibiotics (see Fig. 1). It is so far represented by only a few sequences. The PapC specificity is strongly indicated by absence of the otherwise invariant residue H197 (E. coli numbering) that is associated with recognition of a 4-hydroxy moiety in the cyclohexadienyl substrates of the aforementioned dehydrogenases. This moiety, of course, differs in being a 4-amino substituent in the substrate used by the PapC dehydrogenase (Fig. 1). See Bonner et al. [25] for a more detailed overview.
The "redundant" trp/aro supraoperon of Nostoc/Anabaena
All cyanobacteria possess a highly conserved tyrAa gene, as well as a complete suite of tryptophan-pathway genes that are dispersed (unlinked) in the genome. The large-genome cyanobacterial lineage consisting of the Nostoc and Anabaena genera possess in addition a unique and seemingly redundant trp/aro supraoperon consisting of most of the aforementioned genes [31]. These include a second tyrA gene (curated as tyrAc_Δ), six trp-pathway genes (all except trpC), and genes encoding the first two common-pathway steps of aromatic amino acid biosynthesis. All of these supraoperonic genes appear to be redundant in that they are represented by homologs (paralogs or xenologs) elsewhere in the Nostoc and Anabaena genomes at scattered loci. The closest BLAST hits for the Nostoc/Anabaena TyrAc_Δ proteins are not the co-existing TyrAa homologs present in their own genomes (and universally present in cyanobacteria). Rather the closest BLAST hits are to the TyrAc_Δ domains of the AroQ•TyrAc_Δ fusions in the enteric lineage. Since the enteric proteins are NAD+-specific and strongly prefer prephenate, it is likely that the "extra" cyanobacterial proteins are also NADTyrAc_Δ proteins. Indeed, this would be consistent with enzymological evidence provided in the literature for both Nostoc and Anabaena [20].
Concerning the evolutionary origin of the redundant block of linked genes found in the Nostoc and Anabaena genomes, at least two possibilities await further illumination. (i) These genes might have been acquired by a common ancestor of Nostoc and Anabaena via lateral gene transfer. This is consistent with the observation that biosynthetic-pathway operons are generally absent in the cyanobacteria, and all of the linked genes could have been recruited in a single event. However, at present no candidate donor genomes are known that possess this supraoperon combination of genes. If the TyrAc_Δ proteins of Nostoc/Anabaena and the enteric lineage are possibly related by LGT, it is of interest that the N-terminal extension of TyrAc_Δ from Nostoc/Anabaena resembles a degraded AroQ domain of AroQ•TyrAc_Δ from enterics. In both cases the N-terminal residues may compensate for indel deletions within the catalytic core region of TyrAc_Δ. Subsequently, AroQ function may have evolved in one lineage (or have been lost in the other). This possibility of domain-domain interaction is consistent with the established interdependence of the AroQ• and •TyrAc_Δ domains from E. coli [32]. Alternatively, tyrAa and tyrAc_Δ (and the duplicated trp and aro genes present in the supraoperon) might be ancient paralogs within the cyanobacterial lineage. If so, at a time following divergence of heterocystous cyanobacteria from the unicellular cyanobacteria, the latter may have lost the clustered block of aromatic-pathway genes in a single event of reductive evolution. The supraoperonic genes might be related to a specialized function associated with "developmental" physiological processes that typify the filamentous, heterocyst-forming cyanobacteria. This might be reminiscent of the nature of the phenazine-pigment operon of Pseudomonas aeruginosa. Here unique phenazine-pathway genes are combined with a redundant gene of common-pathway aromatic biosynthesis and two redundant (and fused) genes of tryptophan biosynthesis. This accomplishes the linkage of specific phenazine biosynthesis with a supply of 2-amino-2-deoxy-isochorismate, the branchpoint of divergence toward phenazine and tryptophan [33,34]. This complexity in which multiple paralogs are differentially deployed is consistent with the large genome sizes of Anabaena (7.2 MB) and Nostoc (9.2 MB), compared with the much smaller unicellular genomes of Prochlorococcus marinus (1.7 MB), Synechococcus sp. WH8102 (2.4 MB), and Synechocystis sp. PCC6803 (3.6 MB).
Profile hidden Markov models (HMMs) to distinguish specificity subfamilies for cyclohexadienyl substrate
The limited information thus far available about specific molecular roles of particular TyrA amino acid residues has been summarized recently [25]. The catalytic-core domains of known TyrAa, TyrAp, TyrAc, and TyrAc_Δ proteins were selected from our files of TyrA catalytic-core domains [35], and a new subset of sequences was prepared that lacked the pyridine nucleotide discriminator segment, a glycine-rich βαβ region at the N terminus. Although the glycine-rich βαβ region is not the only segment that contacts pyridine nucleotide substrate, it is the sole region that discriminates between NAD+ and NADP+. The resulting trimmed sequence is defined as the "cyclohexadienyl-substrate core segment". No distinctive motifs were found that, in isolation, would be a clear predictive indicator of specificity for cyclohexadienyl substrate. Similar substrate specificity profiles probably can be dictated by alternative patterns of interplay between different residue combinations.
Because of the rapid accumulation of incorrectly annotated TyrA entries in GenBank and other databases, partly due to the complications of misnaming that are associated with gene fusions and partly to a failure to assimilate published substrate specificities, the use of BLAST does not return reliable annotations with respect to substrate specificity. Even the HMMs used in Pfam [36] and Interpro [37] were not helpful in this case because the HMM deployed in those databases was broadly but incorrectly defined as 'prephenate dehydrogenase (NADP+) activity' for all TyrA dehydrogenases (accession number PF02153 in Pfam and entry IPR003099 in Interpro). However, Profile HMM is known to be well suited for modeling a particular sequence family of interest and for finding additional remote homologs [38]. It is reputed to outperform methods that rely only upon pair-wise alignment of homologous residues in predicting protein function [39]. Therefore, profile HMMs were constructed using our multiple sequence alignments of each curated TyrA specificity subfamily, using the HMMER package [38].
The profile HMMs obtained are only tentatively reliable for prediction of substrate specificity. To facilitate ongoing and future functional annotations, we have made our profile HMMs available as a working resource for "specificity prediction" at AroPath [40]. Users can match query sequences against the four profile HMMs to predict the subfamily to which a query sequence belongs. It is anticipated that future experimental data relevant to substrate specificity will facilitate refinement of the prediction program. For example, at present the program predicts that the TyrA sequences from organisms such as Helicobacter pylori and Saccharomyces cerevisiae belong to the TyrAa grouping, and it will be interesting to see whether this holds up to experimental confirmation. It is additionally fascinating that (i) the dehydrogenase from Archaeoglobus fulgidus is predicted to belong to the indel-containing TyrAc_Δ grouping and (ii) that it possesses a possible cooperatively interacting extra-core domain extension (an AroQ fusion), just as occurs for the large clade of enteric bacteria. If this is relevant, it is even more fascinating that the Archaeoglobus aroQ is fused at the C-terminal side of tyrAc_Δ, rather than at the N terminus as is the case with enteric bacteria.
Users at AroPath [41] can enter query sequences into interactive multiple sequence alignments with any of the four sets of "cyclohexadienyl-substrate core segments" sequences that were used to train the profile HMMs. An ongoing effort is in process to extend the predictor capability to include the pyridine nucleotide substrate as well. One can also align query sequences of interest with either an assemblage of the complete set of curator-approved TyrA catalytic-core TyrA sequences or with any desired subset of seed sequences.
The catalytic-core domain of TyrA proteins
The simplest set of fully functional TyrA proteins consists only of the catalytic-core domain (about 180 amino acids) [1] and includes the well-characterized TyrAc enzymes from Neisseria gonorrhoeae [21] and Zymomonas mobilis [18], as well as TyrAa from a cyanobacterium [25]. In addition the catalytic-core domain from Pseudomonas stutzeri has been engineered for study from a tyrAc•aroF fusion [1]. These model core proteins are roughly as divergent from one another on the TyrA protein tree as are the organisms that contain them (Fig. 2). In view of the possibility raised in this paper about inter-domain interactions, the single-domain TyrA proteins are undoubtedly the simplest sources for study of the fundamental properties of the catalytic-core domain.
Xie et al. [1] suggested that in the set of catalytic-core TyrA proteins, inhibitors bind at the catalytic site and exhibit classical competitive inhibition with respect to the particular cyclohexadienyl substrates that can be accepted by a given organism. This model predicts that the specificity for the sidechains of substrates used would parallel the specificity for inhibitor sidechains. The information summarized in Table 4 supports this expectation. Thus, the TyrAc proteins of P. stutzeri and P. aeruginosa will accept either a pyruvyl (as with PPA) or an alanyl (as with AGN) sidechain in the alternative substrates used, and this is paralleled by recognition of either a pyruvyl (4-hydroxyphenylpyruvate) or an alanyl (TYR) sidechain in the competent inhibitor structures. In another case, the N. gonorrhoeae TyrAc exhibits an overwhelming substrate preference for PPA, and consistent with the foregoing, is subject to inhibition by 4-hydroxyphenylpyruvate but not by TYR. A variety of analog inhibitor structures were used by Xie et al. [1] to show that the minimal structure for binding at the substrate-binding site of P. stutzeri TyrAc is a six-membered ring with a 4-hydroxy substituent.
Table 4.
Cyclohexadienyl substrates and inhibitors of TyrA proteins possess identical sidechains
| Organism | Co-substrate | Substrate(s) | Inhibitor(s)a | Reference |
| Synechocystis sp. | NADP+ | AGN | TYR | [25] |
| Arabidopsis thaliana | NADP+ | AGN | TYR | [27, 28] |
| Nitrosomonas europaea | NADP+ | AGN | None | [21] |
| Corynebacterium glutamicum | NAD(P)+ | AGN | None | [42, 43] |
| Neisseria gonorrhoeae | NAD+ | PPAb | HPP | [21] |
| Pseudomonas stutzeri | NAD+ | PPA/AGN | HPP/TYR | [1] |
| Pseudomonas aeruginosa | NAD+ | PPA/AGN | HPP/TYR | [17] |
| Zymomonas mobilis | NAD+ | PPA/AGN | None | [18] |
aAbbreviation: HPP, 4-hydroxyphenylpyruvate. bThis TyrAc enzyme has an overwhelming preference for PPA, but will use AGN poorly.
In contrast to the TyrAc proteins just described, the Z. mobilis TyrAc is totally insensitive to inhibition by either 4-hydroxyphenylpyruvate or TYR. Since both of these compounds lack a 1-carboxy moiety, it is reasonable to assume that the 1-carboxy substituent present in the two substrates accepted may be required for binding at the catalytic center. Thus, although TyrAc from Z. mobilis will accept the same two substrates as does the TyrAc from P. stutzeri, the greatly different inhibition results suggest that Z. mobilis obeys more stringent rules for binding at the catalytic site (i.e., a ring carboxylate must be present).
Synechocystis sp. and Arabidopsis thaliana TyrAa proteins accept as a substrate only AGN, which has an alanyl sidechain. The ring-carboxylate moiety is evidently not absolutely required for binding since these TyrAa proteins can recognize TYR (alanyl sidechain) as a competitive inhibitor. In contrast, since N. europaea TyrAa is not inhibited by TYR, it resembles the Z. mobilis TyrAc in the putative requirement for a 1-carboxy substituent to secure successful binding at the catalytic site.
In summary, some TyrA proteins probably exercise greater discrimination in their requirement for a 1-carboxy moiety for binding at the catalytic site, and these are insensitive to competitive inhibition by the aromatic reaction products (which lack the 1-carboxy substituent). Other TyrA proteins that require the 1-carboxy moiety for the fundamental catalytic process, but presumably do not require it for binding, will recognize product inhibitors that have the same sidechain as any substrate recognized.
Specificity for the pyridine nucleotide co-substrate within the TyrA superfamily
NAD+ differs from NADP+ only in that NADP+ has a phosphate group esterified at the 2'-position of adenosine ribose. Therefore, the ability of a dehydrogenase to discriminate between those two lies in the particular enzyme region that contacts the ribose moiety. The glycine-rich region known to constitute the ADP-binding βαβ fold is well known to be this point of contact [26]. This Rossmann β α β fold is inevitably positioned at the extreme N terminus of TyrA proteins, and the typical GXGXXG motif is almost always observed, as illustrated in Fig. 4. This region is helpful for assessment of probable specificities for pyridine nucleotide. One can be fairly sure that TyrA proteins possessing D-32 (E. coli numbering, reference [26]) are NAD+-specific. A negatively charged residue (D or E) at position 32 is critical for hydrogen binding to the diol group of the ribose near the adenine moiety in NAD+-specific enzymes. NADP+-specific dehydrogenases cannot tolerate a negatively charged residue at position 32. TyrA proteins that possess an asparagine residue in the corresponding position appear to be broadly specific for both NAD+ and NADP+ as discussed above. No clearcut motif has been identified for NADP+-specific TyrA proteins, although at least one positively charged residue is expected in the region just beyond residue 32. By elimination, those sequences lacking D-32 or N-32 are strong candidates for NADP+ specificity. As with the cyclohexadienyl co-substrate, narrowed specificity for NAD+ (or NADP+) also seems to have occurred independently on many occasions (some examples given earlier).
The absolute specificity of TyrAp proteins for PPA tends to be accompanied by absolute specificity for NAD+, as illustrated by the large Bacillus/Staphylococcus/Listeria/Enterococcus clade at eight o'clock in Fig. 2. However, it is interesting that species of Streptococcus have retained the presumed ancestral breadth of specificity for the pyridine nucleotide substrate. The opposite relationship, whereby absolute specificity for AGN tends to be accompanied by absolute specificity for NADP+, is also observed. Here three of the four TyrAa lineages described earlier exhibit this pattern. Exceptions, though, are the aforementioned TyrAa proteins of Actinobacteridae_1 which accept either NAD+ or NADP+, as well as the TyrAa proteins of the sister Actinobacteridae_2 which are specialized for NAD+ [42,43].
The TyrAc proteins of most complete-genome organisms thus far have happened to be NAD+-specific, and this has been the property of the most rigorously characterized ones (from Z. mobilis, P. stutzeri, and P. aeruginosa). However, it is clear from extensive enzymological surveys [22] that TyrAc proteins having broad specificity for NAD+/NADP+ are common, examples including species of Ralstonia and Burkholderia. The spectrum of variation that can exist, even within a clade of organisms that are of fairly close relationship, is illustrated by one striking example. In the pseudomonad clade marked by a common tyrA•aroF fusion, the Acinetobacter sp. TyrAc is NADP+-specific [44], whereas the sister subclade Pseudomonas/Azotobacter exhibits NAD+ specificity (Fig. 2). Here the entire clade marked by a common ancestral fusion shares approximately the same profile of cyclohexadienyl substrate preference, but cofactor specificity has been narrowed in opposite directions.
We had previously suggested that there might be a general structural relationship of substrate pairing that tends to favor interaction between PPA and NAD+, on the one hand, and, on the other hand, between the greater positive charge of AGN and the greater negative charge of NADP+. These relationships may indeed be favored, but it increasingly appears that any combination can occur.
Beyond the catalytic core: allosteric domains
Various lineages have acquired an amino acid binding domain known as the ACT domain (pfam01842), which is known to bind a variety of amino acids, thus functioning as an allosteric domain for many proteins including phosphoglycerate dehydrogenase, aspartokinase, acetolactate synthase, phenylalanine hydroxylase, prephenate dehydratase and formyltetrahydrofolate deformylase. Recruitment of this domain by fusion with tyrAp appears to have occurred in a common ancestor of the large Bacillus/Staphylococcus/Listeria/Enterococcus/Streptococcus assemblage (Fig. 2). It is interesting that B. subtilis also possesses a gene encoding a free-standing ACT domain in its genome (incorrectly annotated as pheB). An additional fusion of genes encoding an ACT domain and tyrA (that arose independently, judging from the widely spaced tree positions) occurred in the common ancestor of Xanthomonas and Xylella. Actinobacteria usually possess a C-terminal extension that probably functions as an allosteric domain. The extension possessed by the Actinobacteridae_2 assemblage, which includes Streptomyces coelicolor and its relatives, appears to be an ACT domain. On the other hand, it is not all all clear that the C-terminal extension of the Actinobacteridae_2 assemblage is an ACT domain. This difference, in addition to the differing specificities for pyridine nucleotide substrate, may have contributed to the overall TyrAa divergence observed between the two Actinobacteridae groups. There is no correlation between presence of the ACT domain and specificity for cyclohexadienyl substrate since TyrAp from the Bacillus clade is PPA-specific, Xanthomonas/Xylella TyrAc is broadly specific, and Streptomyces TyrAa is AGN-specific.
B. subtilis, which belongs to the large clade having an ACT domain as a carboxy extension, has been extensively characterized [30]. 4-Hydroxyphenylpyruvate is an effective competitive inhibitor, as would be consistent with our proposed effects at the catalytic core for a PPA-specific enzyme. However, TYR, phenylalanine (PHE) and tryptophan were also inhibitors. The violation of the rule that the latter three amino acid inhibitors would not be expected to bind the catalytic core region (because they have alanyl sidechains even though the substrate-binding site only recognizes the pyruvyl sidechain of prephenate) and the finding that some of these were not competitive inhibitors can now be accounted for by the presence of the allosteric ACT domain. A carboxy extension shared by a number of Archaea (denoted 'REG' in Fig. 2) is presumably a regulatory domain as well. This is consistent with the recent result of Porat et al. [45] that not only 4-hydroxyphenylpyruvate, but also TYR, inhibited prephenate dehydrogenase activity of Methanococcus maripaludis.
The tyrA gene is a popular fusion partner
Fusion with aroQ
tyrA may be fused with a number of other catalytic domains, each of them relevant to aromatic biosynthesis (Fig. 2). aroQ (encoding chorismate mutase) is frequently fused with a number of other aromatic-pathway genes [46]. The lower-gamma Proteobacteria (enteric lineage) located at twelve o'clock in Fig. 2 possess an aroQ•tyrAc_Δ fusion. The fusion physically links chorismate mutase (which forms PPA) with TyrAc_Δ (which utilizes PPA). The two protein domains of AroQ•TyrAc_Δ may have co-evolved to produce cooperative protein-protein interactions since physical separation of the domains evoked relatively low activities of both activities in E. coli [32]. Substantial comparative work shows that the aroQ•tyrAc_Δ fusion has been stably maintained throughout the entire enteric lineage [47]. Exceptions in some genomes lacking this fusion altogether can be attributed to reductive evolutionary loss in pathogens (e.g., Haemophilus ducreyi) or endosymbionts (e.g., Buchnera aphidicola). An independent aroQ•tyrA fusion was generated in the common ancestor of Sulfolobus solfataricus and S. tokodaii (Fig. 2). Since the TyrA domain of Sulfolobus species lacks the indel structure of the TyrAc_Δ class, it would be interesting to see whether physical separation of the two domains would yield evidence of independent function, in contrast to the results mentioned just above for E. coli.
Fusion with aroF
Secondly, tyrAc has been fused with aroF on at least two separate occasions in Bacteria. (The aroF gene encodes enolpyruvylshikimate-3-P synthase, the sixth enzyme in the common pathway of aromatic biosynthesis; see [5,6] for nomenclature used.) One clade includes members of the upper-gamma Proteobacteria: P. aeruginosa, P. syringae, P. putida, P. stutzeri, P. fluorescens and Azotobacter vinelandii. It is interesting that P. syringae has experienced a deletion of about 200 residues at the N-terminal region of the AroF domain. This has been coupled with the acquisition of a stand-alone aroF gene that is absent in other members of the clade. Interestingly, the latter AroF shows high identity only with AroF from Agrobacterium tumefaciens, an alpha proteobacterium. The A. tumefaciens aroF, in turn, is unique compared to its α-subdivision relatives, both in having divergent sequence and in being unlinked to cmk and rpsA. Thus, it seems likely that the incongruence of AroF belonging to both P. syringae and A. tumefaciens reflects acquisition via LGT from some as yet unknown source. The disruption of the fused aroF domain in P. syringae is an unusual instance where the catalytic function of one fusion domain has become discarded while the function of the second domain has been retained. It is interesting to consider the possibility that the truncated remnant of the aroF fusion domain might be exploitable for use as an innovative source of a new regulatory domain. An additional fusion of tyrA with aroF has occurred independently within the beta Proteobacteria in the common ancestor of Burkholderia pseudomallei and B. mallei. This has been very recent since the closely related B. fungorum and B. cepacia organisms lack the fusion.
It has been suggested that presence of a given fusion may be useful for sorting out clades that diverged from a common ancestor, independent of other methods [48]. Different fusions offer the power of discriminating clades at various hierarchical levels, i.e., nested clades discriminated by nested gene fusions. The tyrA•aroF fusion occurred in the common ancestor of the clade that includes the upper-gamma Proteobacteria shown in Fig. 2. One can reasonably assume that relatively close upper-gamma organisms lacking the tyrA•aroF fusion diverged from the common ancestor of the fusion clade prior to the fusion event. Such would appear to be the case, for example, with Acidithiobacillus ferrooxidans, an outlying member of the upper-gamma Proteobacteria that lacks the fusion. It is reasonable to conclude that the fusion event must have pre-dated the differential specialization for the pyridine nucleotide cosubstrate that distinguishes Acinetobacter sp. (NADP+-specific) from the large grouping of pseudomonads that are NAD+-specific.
Fusion with hisHb
Thirdly, a single organism, Rhodobacter sphaeroides, possesses a hisHb•tyrA fusion that must have occurred very recently. hisHb encodes an aromatic aminotransferase that is closely related to (or sometimes even synonymous with) imidazole acetol phosphate aminotransferase [49]. The hisHb/tyrA/aroF linkage group is part of a supraoperon in some gram-negative bacteria in which a relatively conserved, yet frequently shuffled gene order is observed [5,6]. Hence, it is reasonable to assume that at the time just prior to fusion, hisHb, tyrA and aroF were adjacent. Note that among the fusions currently known, hisHb and aroF are fused to the N-terminal and C-terminal ends of tyrA, respectively. It would be interesting to know the substrate specificity of the R. sphaeroides TyrA domain. If it is AGN-specific the significance of hisHb presumably would be to transaminate PPA to form AGN, the substrate used by TyrAa (see Fig. 1). On the other hand, if the dehydrogenase is PPA-specific, the significance of the HisHb domain would be to transaminate the product of the TyrAp reaction. If the enzyme is a TyrAc enzyme (as is probable), then HisHb likely is competent to catalyze either of the foregoing reactions.
Fusion with ACT
The widespread ACT regulatory domain appears to have been acquired by independent fusions at least three separate times judging from the widely separated lineages that possess a TyrA•ACT fusion (Fig. 2). Xie et al. [5] initially noted homologous domains positioned at the N terminus of mammalian phenylalanine hydroxylase and at the C terminus of most microbial prephenate dehydratases. This domain is responsible for phenylalanine-mediated activation and phenylalanine-mediated inhibition of the hydroxylase and dehydratase enzymes, respectively. This domain was later named the ACT domain [50] and shown to be a widely distributed domain family that shares a conserved overall fold. Members of the ACT-domain family possess a wide variety of different ligand-binding capabilities. For example, the ACT domain of 3-phosphoglycerate dehydrogenase binds L-serine as a allosteric inhibitor.
Fusion with REG
Another putative regulatory domain fused to tyrA (denoted tyrA•REG) is thus far restricted to some of the Archaea. This domain is a predicted regulatory domain, as described in COG4937.
A novel 4-domain fusion
Archaeoglobus fulgidus exhibits a striking four-domain fusion consisting of three catalytic domains and a regulatory ACT domain (TyrA•AroQ•PheA•ACT). The TyrA domain is predicted to belong to the TyrAc_Δ class when used as a query input into the AroPath Specificity Predictor Tool [40]. We speculated earlier that the •AroQ fusion domain of Archaeoglobus may exercise cooperative interactions with TyrAc_Δ, as appears to occur between the AroQ•TyrAc_Δ domains of E. coli and its relatives.
tyrA in its syntenic context
Although the genes of prokaryotes have clearly been subject to frequent scrambling, some gene-gene associations persist more tenaciously than others. Xie et al. [5,6] asserted that one such ancestral gene string that has resisted scrambling forces is hisHb > tyrA > aroF. As suggested above, contemporary gene fusions can serve as frozen-in-time indicators of ancient gene organizations that were later obscured by gene-scrambling events. Another gene string that is often within the syntenic region of hisHb, tyrA, and aroF is cmk > rpsA. Gene synteny in prokaryotes has not been easily recognized because substantial manual scrutiny in combination with a sufficient density of genomic representation on a given portion of the phylogenetic tree is necessary to detect patterns of synteny that are camouflaged by frequent scrambling events (inversion, deletion and transposition).
The domain Bacteria is now represented by a collection of sequenced genomes that is progressively approaching the genomic densities needed for meaningful analysis. Figure 5 provides a visual sense of the frequency with which tyrA is closely positioned with other genes of aromatic biosynthesis, as well as the underlying patterns of overall synteny. These patterns are unstable, and yet persistent traces of synteny can be seen where genomic representation is sufficiently dense. The four genes of particular emphasis in this paper are color coded. Other genes that are engaged in aromatic biosynthesis are colored grey, and any other genes are white. At a very deep level of phylogenetic branching, Thermotoga exhibits a tyrA gene flanked by seven genes encoding all of the common steps of aromatic biosynthesis (two of them being fused). Since closely related genomes are not yet available here, we cannot judge whether these genes came together recently or whether an ancient pattern of synteny has been retained. Although tyrA is not linked to any functionally relevant genes in Aquifex, representing another point of deep phylogenetic branching, this does not necessarily mean that tyrA was not already generally associated with other aromatic-pathway genes at an early time. For reasons that are totally mysterious, certain scattered lineages exhibit a total lack of operon organization for aromatic-pathway genes (and indeed for most other biosynthetic pathways, such as that for histidine biosynthesis). These lineages (Fig. 5) include, besides Aquifex, those of Deinococcus, the actinomycetes, the cyanobacteria, and Chlorobium. Except for the actinomycetes, this phenomenon of total gene dispersal also applies to genes of tryptophan biosynthesis [7,8].
Figure 5.
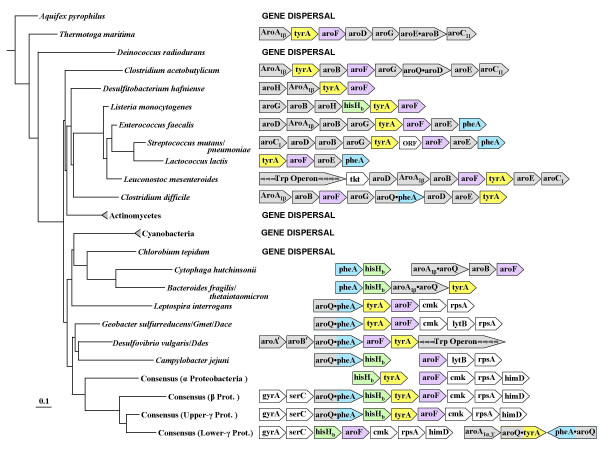
Context of gene organization for tyrA, profiled against the 16S rRNA tree of the domain Bacteria. pheA, hisHb, tyrA, and aroF are color coded. Lineages typified by complete dispersal of aromatic-pathway genes are indicated by "GENE DISPERSAL". Gmet refers to Geobacter metallireducens; Dace refers to Desulfuromonas acetoxidans; and Ddes refers to Desulfovibrio desulfuricans. Consensus gene organizations are shown for the alpha and beta divisions of the Proteobacteria. The gamma division is subdivided to yield consensus gene organizations for the upper- and lower-gamma (enteric lineage) organisms. Genes that are adjacent and share a common transcriptional direction appear to reside in operons (or supraoperons). Any white spacing indicates substantial separation of the gene clusters shown in the genome. Genes of special interest are color coded, other genes of aromatic biosynthesis are shown in gray and all other genes are shown in white.
When the various examples of hisHb > tyrA > aroF linkage are mapped on a 16S rRNA tree, they first appear in gram-positive bacteria. In Bacillus and related organisms (such as Listeria), the hisHb > tyrA > aroF unit is associated with a large ancestral operon consisting of aroG > aroB > aroH > hisHb > tyrAp> aroF. Bacillus additionally possesses the cmk > rpsA unit, albeit in a separate location. Interestingly, in one narrow subclade (B. subtilis, B. halodurans and B. stearothermophilus) the trp operon has been inserted between aroH and hisHb to yield a supraoperon that has been fully characterized as a complex functional unit [51]. See Xie et al. [7] for a full presentation of evolutionary interpretation relevant to the latter. Though highly scrambled, a pattern of association of pheA with hisHb > tyrA >aroF is suggested by linkage patterns seen at the hierarchical level of Cytophaga and Bacteroides (Fig. 5). aroQ became associated with pheA through gene fusion as early as the divergence of the Spirochaetes to yield an aroQ•pheA>tyrA>aroF>cmk>rpsA linkage unit (Leptospira interrogans in Fig. 5). The aroQ•pheA gene associated with tyrA and aroF in Clostridium difficile appears to have arisen from a distinctly different fusion event than that present in delta, epsilon, beta and upper-gamma Proteobacteria or from that present in lower-gamma Proteobacteria (based upon analysis of inter-domain linker regions; unpublished data).
Consensus ancestral gene organizations for the most densely represented divisions of Proteobacteria have been deduced as shown at the bottom of Fig. 5. Detailed information that supports a deduced consensus for ancestral gene organizations with respect to beta Proteobacteria, upper-gamma Proteobacteria, and lower-gamma Proteobacteria are shown later (Figs. 6, 7). We suggest that the last common ancestor of all Proteobacteria possessed the gene organization aroQ•pheA>hisHb>tyrA>aroF>cmk>rpsA. This is similar to the synteny that has been retained in general by the beta Proteobacteria and the upper-gamma Proteobacteria. The aroQ•pheA>hisHb>tyrA portion likely specified all the catalytic requirements for conversion of chorismate to PHE and conversion of chorismate to TYR. Chorismate mutase activity specified by the aroQ domain could supply PPA for both PHE and TYR biosynthesis. Likewise, HisHb, widely utilized as an aromatic aminotransferase [49], could also function for both PHE and TYR biosynthesis. Though currently available members of delta and epsilon Proteobacteria exhibit substantial gene scrambling, the various fragmentary linkage patterns seen provide support for the ancestor proposed. Geobacter (and other Delta_1 members) has the aroQ•pheA > tyrA > aroF > cmk > rpsA linkage group (with lytB inserted between cmk and rpsA). Desulfovibrio vulgaris, another delta Proteobacterium (Delta_2) that is highly divergent from Geobacter, has a very interesting pattern of conservation and scrambling. aroQ•pheA > aroF > tyrA has been attached to a complete 7-gene trp operon. hisHb > cmk (not shown in Fig. 5) is completely separated from rpsA. The supraoperonic gene organization shown for D. vulgaris begins with two recently discovered genes, herein denoted aroA' and aroB', that encode enzymes specifying an alternative biochemical route to dehydroquinate [52]. The epsilon Proteobacteria all display significant gene scrambling, but piecemeal evidence for the unscrambled ancestor proposed is present. For example, Campylobacter jejuni possesses an aroQ•pheA > hisHb unit, as well as aroF > lytB > rpsA (Fig. 5). Wollinella succinogenes and Helicobacter hepaticus both possesses an aroF > lytB > rpsA unit.
Figure 6.
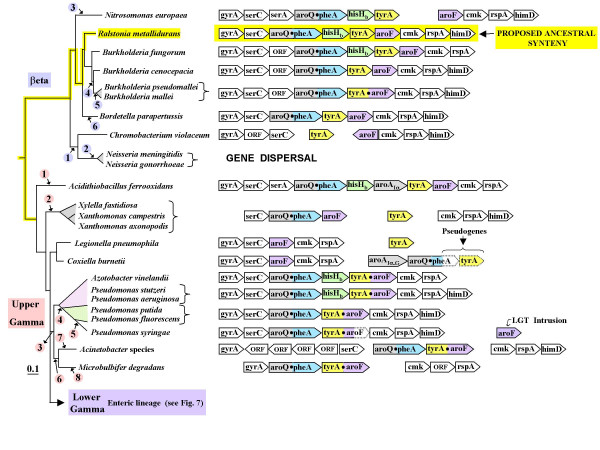
Zoom-in from Fig. 5 showing tyrA synteny for the beta Proteobacteria and the upper-gamma Proteobacteria. The tree shown, based upon 16S rRNA sequences of the indicated organisms, indicates correct branching orders, but (to facilitate presentation) is not strictly correct in proportion. Circled numbers (in violet) indicate deduced evolutionary events for the beta Proteobacteria (see top of Table 5), whereas circled numbers (in pink; see bottom of Table 5) correspond to deduced evolutionary events for the upper-gamma Proteobacteria. Gene organizations of organisms indicated are shown on the right. The dotted outlining of some gene boxes in Coxiella burnetii and in P. syringae indicates pseudogene status.
Figure 7.
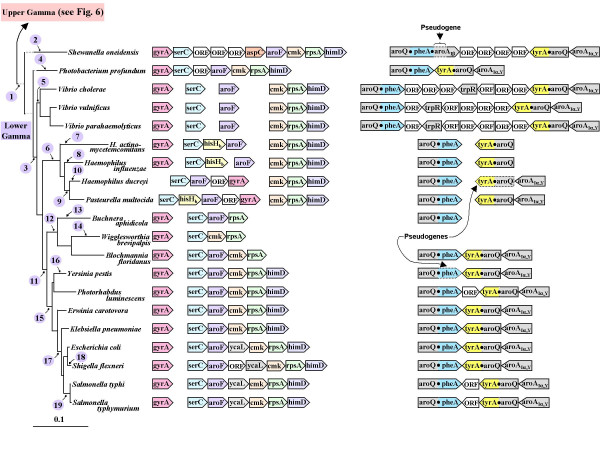
Zoom-in from Fig. 5 showing tyrA synteny for the lower-gamma Proteobacteria (enteric lineage). Deduced phylogenetic events numbered on the left are described in Table 6. The branching position for Buchnera is as suggested in ref. [7]. Dotted horizontal lines near the top of the tree indicate branch lengths that were shortened for convenience of presentation. Dotted outlining of boxes around some genes indicates their pseudogene status. It is unknown if the various open reading frame (ORF) insertions are functional.
The ancestor of alpha Proteobacteria has lost the aroQ•pheA fusion, and a stand-alone pheA is consistently observed. Members of this group are quite uniform in the stable possession of hisHb > tyrA and aroF > cmk > rpsA as two separated linkage groups. The beta Proteobacteria are represented by members that have the gene organization: serC > aroQ•pheA > hisHb > tyrA > aroF > cmk > rpsA. This is also seen in the members of the upper-gamma Proteobacteria.
Figure 5 includes organisms that illustrate the traces of synteny that can be detected in Bacteria where overall genome representation is just barely adequate. The following two figures illustrate how syntenic patterns of more resolution and refinement become evident with denser genome representation.
Zooming in on syntenic contexts of proteobacteria
Beta proteobacteria and upper-gamma proteobacteria
The beta Proteobacteria exhibit a dynamic but still interpretable pattern of altered synteny (Fig. 6 and Table 5). Species of Ralstonia have retained the proposed ancestral synteny that is marked with yellow highlighting in Fig. 6. This syntenic organization is such that the aromatic-gene unit aroQ•pheA > hisHb > tyrA > aroF is nested between gyrA > serC at the leftward flank and cmk > rpsA > himD at the rightward flank. Species of Burkholderia (the next closest lineage) are almost identical, but exhibit individual evolutionary events (marked by circled numbers on the left, which correspond to a description of the proposed evolutionary events given in companion Table 5). These events include gene insertion, loss of hisHb, translocation of genes away from the ancestral supraoperon, and fusion of tyrA and aroF (in the common ancestor of B. mallei and B. pseudomallei). At a deeper level in the beta Proteobacteria section of the tree, Nitrosomonas europaea exhibits a separation of the ancestral supraoperon between tyrA and aroF. Either a very large insertion was made between tyrA and aroF, or one of the two gene clusters shown was transposed as part of a sufficiently large segment to include all of the conserved flanking genes. In Chromobacterium violaceum tyrA has become completely isolated from other gene members of the ancestral supraoperon, and aroF has assumed an inverted orientation with respect to cmk. Species of Neisseria exhibit no remnants of supraoperon synteny at all, and wholesale dispersal of all the supraoperon genes has occurred. (It is interesting that among the beta Proteobacteria, Neisseria species are also unique in that all of the trp-pathway genes are dispersed [7]).
Table 5.
Key to evolutionary events asserted in Figure 6
| Group | Evolutionary event(s) proposed | |
| Beta | 1 | Dispersal of aroQ•pheA > hisHb > tyrA away from one another and away from gyrA > serC and from cmk > rspA > himD; inversion of aroF with respect to cmk. |
| 2 | Complete dispersal of all nine genes originally in the gyrA/himD linkage group. | |
| 3 | Insertion of serA after serCa; separation of tyrA and aroF to yield the separated 6-gene unit and 4-gene unit shown. | |
| 4 | Expulsion of hisHbfrom the genome; insertion of 'ORF' after serC. | |
| 5 | Fusion of tyrA with aroF. | |
| 6 | Loss of hisHb from genome. | |
| Upper-Gamma | 1 | Insertion of serA after serCa; insertion of aroAIα after hisHb. |
| 2 | Translocation of hisHb and tyrA to other regions, leaving two separated 3-gene units. | |
| 3 | Fusion of tyrA with aroF. | |
| 4 | Loss of hisHb. | |
| 5 | N-terminal deletion of •aroF domain, and acquisition of new aroF gene (probable LGT). | |
| 6 | Separation of cmk > rpsA > himD from aroQ•pheA > tyrA•aroF. | |
| 7 | Insertion of 4 unknown genes between gyrA and serC in opposite orientation and separation of gyrA > ORF > ORF > ORF > serC from aroQ•pheA > tyrA•aroF. | |
| 8 | Loss of himD; translocation of serC away from gyrA and aroQ•pheA. | |
aSince both Nitrosomonas (beta Proteobacteria) and Acidothiobacillus (upper-gamma Proteobacteria) emerge at deep positions in the tree of Fig. 5, an almost equally parsimonius possibility is that the ancestral serA was retained in this syntenic position in these two genera, but was transposed elsewhere shortly after early divergence.
The gamma Proteobacteria have separated into two distinctly different synteny patterns. The lower-gamma Proteobacteria have undergone marked syntenic change (see below). The assemblage portrayed between Acidithiobacillus and Microbulbifer in the lower part of Fig. 6 (termed the upper-gamma Proteobacteria) exhibit a strong overall syntenic resemblance of supraoperon genes to that of the beta Proteobacteria. Acidithiobacillus possesses a near-intact ancestral supraoperon, differing only in having two insertions: one gene encoding 3-deoxy-D-arabino-heptulosonate 7-phosphate (DAHP) synthase between hisHb and tyrA, and the other being the insertion of serA between serC and aroQ•pheA. Pseudomonas aeruginosa and P. stutzeri have also retained nearly intact ancestral supraoperons, differing only in the fusion of tyrA and aroF. The serC > aroQ•pheA > hisHb > tyrA•aroF > cmk > rpsA supraoperon has been studied in P. stutzeri [5,6]. The tyrA•aroF fusion occurred in the common ancestor of the clade shown between Azotobacter and Microbulbifer in Fig. 6. The supraoperons of P. syringae, P. fluorescens and P. putida lack hisHb. P. syringae exhibits a recent C-terminal truncation of the aroF domain, coupled with acquisition elsewhere in the genome of a free-standing •aroF that is not phylogenetically congruent (probably of LGT origin). Acinetobacter sp. and Microbulbifer degradans possess an aroQ•pheA > tyrA•aroF unit that has become dissociated from serC at one end and from cmk on the other end. In Xylella and Xanthomonas, hisHb has been deleted from the genome and tyrA has been transposed away from serC > aroQ•pheA > aroF. The latter unit has been transposed away from gyrA, the ancestral flanking gene. On the other hand, cmk > rpsA has remained next to himD, the gene usually flanking rpsA.
The enteric lineage
The lower-gamma Proteobacteria differ sharply from upper-gamma Proteobacteria in their possession of the tyrAc_Δ class of tyrA and its fusion with aroQ. In Fig. 2 this clade of AroQ•TyrAc_Δ fusions was presented as one exhibiting absolute specificity for NAD+, combined with an overwhelming but not complete specificity for PPA. In Fig. 7 the gene synteny associated with tyrAc_Δ is profiled against the 16S rRNA phylogenetic trees of the lower-gamma Proteobacteria possessing these genes, and the proposed evolutionary events are summarized in the companion Table 6. Figure 5 has indicated a synteny consensus for the common ancestor at this hierarchical level whereby gyrA > serC > hisHb > aroF > cmk > rpsA parallels the ancestral synteny of β-Proteobacteria, but without aroQ•pheA or tyrA in the middle of the linkage group. Many dynamic evolutionary events of altered aromatic biosynthesis have occurred within the lower-gamma Proteobacteria since their divergence from the upper-gamma Proteobacteria. This includes the emergence of three allosterically distinct DAHP synthases, one of which now comprises the two-gene, three-domain tyr operon (aroAIα_Y > aroQ•tyrAc_Δ). The upper-gamma Proteobacteria characteristically possess the aroAIα paralogs encoding AroAIα_H (TRP-inhibited DAHP synthase) and AroAIα_Y (TYR-inhibited DAHP synthase). It has been asserted that AroAIα_F (PHE-inhibited DAHP synthase) was the most recent paralog, acquired just after divergence of the lower-gamma Proteobacteria [53]. It is bizarre that Shewanella oneidensis possesses a pseudogene of aroAIβ fused to the C terminus of aroQ•pheA. The aroAIβ subclass of Family-I DAHP synthases is not usually observed in gram-negative bacteria [54].
Table 6.
Key to evolutionary events asserted in Figure 7
| Number | Evolutionary events proposed |
| 1 | Escape of aroQ•pheA and tyrA from the ancestral gyrA > serC > aroQ•pheA > hisHb> tyrA > aroF > cmk > rpsA > himD supraoperon. Origin of an aroQ•tyrA fusion. Origin of the aroAIα_Y > aroQ•tyrA operon. Addition of tyrR. Addition of third aroAIα species: aroAIα_F. |
| 2 | Fusion of aroQ•pheA with aroAIβ pseudogene of unknown origin. Replacement of hisHb by aspC duplicate linked with three ORFs. |
| 3 | Dissociation of gyrA and serC. |
| 4 | Removal of all genes intervening between aroQ•pheA and aroQ•tyrA. |
| 5 | Dissociation of aroF from both serC and cmk > rpsA > himD. Insertion of trpR within the intervening region between aroQ•pheA and aroQ•tyrA. |
| 6 | Dissociation of serC > hisHb > aroF from cmk > rpsA > himD. |
| 7 | Loss of aroAIα_Y from tyr operon. |
| 8 | aroF becomes dissociated from hisHb, and aroAIα_Y is removed from the tyrA operon. |
| 9 | ORF > gyrA is inserted after aroF. |
| 10 | aroQ•tyrA becomes a pseudogene. |
| 11 | hisHb is lost. |
| 12 | himD is lost. |
| 13 | cmk, himD and aroAIα_Y > aroQ•tyrA are lost. |
| 14 | aroF, himD, aroQ•pheA, and aroAIα_Y > aroQ•tyrA are lost. |
| 15 | All intervening genes between aroQ•pheA and aroQ•tyrA are eliminated. |
| 16 | pheA domain of aroQ•pheA becomes a pseudogene. |
| 17 | Insertion of ycaL between aroF and cmk. |
| 18 | Insertion of ORF between aroF and ycaL. |
| 19 | Insertion of ORF between aroQ•pheA and aroQ•tyrA. |
The dissociation of tyrAc_Δ from the serC/rpsA linkage group correlates with the fusion of aroQ with tyrAc_Δ. The aroQ•pheA fusion has also escaped from the serC/rpsA linkage grouping and has become linked with the newly emerged tyr operon. Some sort of duplication and recombinational event between aroQ•pheA and tyrAc_Δ may have led to the creation of aroQ•tyrAc_Δ since the AroQ•PheA proteins of lower-gamma Proteobacteria are distinct from AroQ•PheA proteins of other Proteobacteria with respect to the inter-domain linker length and the indel content (data not shown).
Although it usually is absent from the lower-gamma Proteobacteria, HisHb has persisted as the broad-specificity aromatic aminotransferase in the Pasteurella/Haemophilus grouping where two hisH paralogs are generally present, one of narrow specificity (denoted hisHn) being within the histidine operon. The aspC gene next to aroF in Shewanella is a paralog that probably functions as an aromatic aminotransferase, suggestive of the situation in the E. coli grouping where tyrB is a close paralog relative of aspC, tyrB having become specialized for aromatic biosynthesis [49]. Gene reduction associated with both endosymbiotic and pathogenic lifestyles are evident. Thus, Buchnera lacks tyrA, cmk, hisH, tyrB, and possesses only a single aroAIα species (aroAIα_H). Haemophilus ducreyi also lacks tyrA, as well as aroAIα_H and the entire trp operon [5].
TyrA in its context of regulation
TyrR regulon
Knowledge of the gene regulation impacting TyrA in prokaryotes is sparse, being limited to the lower-gamma Proteobacteria. Here, extensive information gathered from E. coli has revealed that aroQ•tyrAc_Δ belongs to a large regulon controlled by the TyrR repressor. The limited phylogenetic distribution of TyrR, being present only in the lower-gamma Proteobacteria (Fig. 8), indicates that it is a recent evolutionary acquisition. In E. coli the regulon members that are under the control of tyrR are the aroAIα_Y > tyrA operon, the aroLM operon, tyrP, tyrB, aroP, mtr, aroAIα_F and tyrR itself [55]. Thus, tyrR not only regulates the tyrosine branch of the pathway, but heavily impacts the common pathway and the transport of all three aromatic amino acids as well.
Figure 8.
Distribution of modules of aromatic catabolism mapped on a 16S rRNA tree. In this figure, only presence or absence (not gene order) is indicated. The Phh module (orange) consists of phenylalanine hydroxylase (PhhA), carbinolamine dehydratase (PhhB), and tyrosine aminotransferase (not shown, see Text), and accomplishes the overall conversion of PHE to 4-hydroxyphenylpyruvate. The Hpd module (yellow) is 4-hydroxyphenylpyruvate dioxygenase, which converts 4-hydroxyphenylpyruvate to homogentisate. The Hmg module (blue) catalyzes the 3-step conversion of homogentisate to acetoacetate and fumarate. The distribution of PhhR and TyrR is shown in boxes. In some cases the HmgC member is shaded light blue to indicate that the gene encoding this isomerase could not be found and is probably encoded by an as yet unknown analog. Some long branches are drawn with gaps that represent 25% of the length of the scale bar.
Although outside the scope of this study, a logical expansion of it would be to examine the individual evolutionary histories of all the members of the contemporary E. coli TyrR regulon, i.e., asking when and in what order did these genes come under the influence of tyrR? Clearly, the recruitment of structural genes by tyrR has been recent, quite dynamic and even now, exhibits evidence of further ongoing change. For example, tyrosine phenol-lyase (a catabolic enzyme that is only sparsely present in gamma Proteobacteria) has been recruited to the TyrR regulons of Erwinia herbicola [56] and Citrobacter freundii [57]. In these cases, not only does TyrR perform as a transcriptional activator, but it requires cyclic AMP receptor protein and integration host factor to do so.
As exemplified by E. coli, TyrR is generally a repressor. However, the transcriptional expression of mtr is activated by TyrR in the presence of TYR, and tyrP is activated in the presence of PHE (although it is repressed in the presence of TYR). The N-terminal domain of TyrR has been associated with the ability of TyrR to activate transcription in the case of mtr and tyrP [55]. Members of the Haemophilus/Pasteurella lineage have all lost the N-terminal domain and presumably all lack the ability to accomplish transcriptional activation, as has been demonstrated experimentally with H. influenzae TyrR [58].
In view of the interesting complexity that two operons (mtr and aroLM) in E. coli are regulated by both tyrR and trpR [55], it may be more than coincidental that tyrR and trpR seem to have emerged at about the same evolutionary time, i.e., coincident with the divergence of the upper-gamma Proteobacteria from the lower-gamma Proteobacteria (Fig. 7). A possible interaction between the TyrR and TrpR proteins has been noted [55].
PhhR in relationship to aromatic catabolism
Arias-Barrau et al. [59] have recently characterized a central catabolic pathway (Hmg) that degrades homogentisate in three steps to fumarate and acetoacetate as a source of carbon and energy. One of several peripheral pathways feeding into the central pathway begins with PHE and produces homogentisate via the reactions of phenylalanine hydroxylase (Phh), aromatic aminotransferase, and 4-hydroxyphenylpyruvate dioxygenase (Hpd). In the absence of Phh, a shorter version of the peripheral pathway is one that can use TYR, but not PHE, as a source of carbon and energy. In Fig. 8 the presence of Phh, Hpd, and Hmg segments of catabolism are mapped on a 16S rRNA tree. (The aromatic aminotransferase distribution is not shown since a multiplicity of aromatic aminotransferases having overlapping substrate specificities makes it particularly challenging to identify the functional role [49].) The cyanobacteria are unique among Bacteria in the use of Hpd for a completely different metabolic role unrelated to aromatic catabolism, i.e., the synthesis of vitamin E derivatives [60].
PhhR is a homolog of TyrR that has been shown in P. aeruginosa to be a divergently transcribed activator of a 3-gene operon needed for PHE and TYR catabolism [61]. The structural genes encode phenylalanine hydroxylase (phhA), carbinolamine dehydratase (phhB) and 4-hydroxyphenylpyruvate aminotransferase (phhC), and are powered by a σ54 promoter [61,62]. PhhR evolved relatively recently since it is only present in some gamma Proteobacteria (Fig. 8). The ancestral regulatory gene for the Phh peripheral pathway may have been a member of the leucine-responsive regulatory protein/asparagine synthase C (Lrp/AsnC) family judging from the adjacent and divergently oriented position of asnC genes to phhA in organisms such as Xanthomonas axonopodis and Mesorhizobium loti. A recent overview of the many different regulator families involved in the control of aromatic catabolism conveys an emerging sense of the variety and dynamic evolutionary processes that underlie aromatic catabolism [63]. Occasional distant homologs of phhR that appear in erratic fashion (see Fig. 9) may have some other regulatory function. For example, Clostridium tetani may use its PhhR homolog as a transcriptional activator of the gene encoding tyrosine phenol-lyase, as occurs in species of Erwinia [56] and Citrobacter [57].
Figure 9.
Protein tree of TyrR homologs. Nodes supported by bootstrap values of 998 or more are marked with solid circles, and the bootstrap values for nodes internal to these are shown. Generic names relevant to the organism abbreviations can be viewed in Fig. 8. A conserved region containing the σ54 contact motif GAFTGA is highlighted as an orange band. "Imperfect" residues in this region are shown in lower-case fonts. Residue numbers are shown at the right. TyrR and PhhR are regulators of σ70 and σ54 promoters, respectively. Four σ54 proteins of unknown function have very long branches, and to facilitate the visual presentation, the gaps in branch continuity shown represent a scale-bar distance of 0.1. Clades possessing σ54 regulators are indicated with blue stripes, and σ70 regulators are indicated with green stripes.
Relationship of TyrR and PhhR
What might be of origin of TyrR? TyrR is an anomalous member of the large prokaryote family of σ54 enhancer-binding proteins that activate promoters dependent upon σ54. TyrR is unique within its homology grouping in that it targets σ70 promoters for regulation, usually (but not always) being a repressor. Its closest homolog relative is PhhR, a canonical member of σ54 enhancer-binding proteins. σ54-dependent enhancer proteins possess a highly conserved σ54-contact motif, GAFTGA, that is intimately involved in formation of the ternary complex of enhancer and σ54-RNA polymerase holoenzyme [64]. This is perfectly or nearly perfectly retained in the upper clades shown in Fig. 9, but is disrupted or completely absent in the clades between Shewanella oneidensis and Pasteurella multocida. The deeper phylogenetic distribution of PhhR (Fig. 8) suggests that TyrR evolved as a variant of PhhR. If correct, a regulatory gene that is oriented to catabolism (phhR), and itself of relatively recent origin, was conscripted even more recently for a completely new role in the regulation of primary biosynthesis (tyrR).
Consistent with the latter supposition, the gain of TyrR generally correlates with the loss of competence for aromatic catabolism (Fig. 8). In contrast to the Citrobacter/Salmonella/Escherichia/Shigella and the Pasteurella/Haemophilus clades (whose TyrR homologs completely lack the GAFTGA motif), the remaining enteric clades have retained some residues in this region. These residues appear to be more than random remnants. It would be interesting to know if these residues have any functional significance. Indeed, the Photobacterium/Vibrio clade has retained the ancestral catabolic capabilities (Fig. 8) that would appear to demand retention of regulation via PhhR; yet the parallelism of the overall features of biosynthesis that are shared with other lower-gamma Proteobacteria would seem, on the other hand, to demand TyrR-mediated regulation. Perhaps this "TyrR" species participates in the regulation of both catabolic and biosynthetic genes. In this connection, it is interesting that Chaney et al. [64] found that a change in the GAFTGA motif of NifA could be partially "suppressed" by mutational changes in the N-terminal region of σ54.
Even more striking as a possible evolutionary intermediate is the most outlying member of the lower- gamma Proteobacteria, Shewanella oneidensis. The position of its TyrR on the protein tree parallels expectations based on the 16S rRNA tree. This, plus the conservation of the TyrR regulon features and the overall gene synteny suggest E. coli-like function as TyrR, i.e. acting as a general repressor of regulon-member σ70 promoters engaged in aromatic biosynthesis. However, the location of "tyrR" in S. oneidensis between phhA and phhB on one side, and hmgB and hmgC on the other side, strongly implies some kind of regulatory relationship with the catabolic genes. It would be quite interesting to determine experimentally whether "TyrR" in S. oneidensis (and maybe Vibrio, as well) can function as a repressor of the usual suite of σ70 promoters, as well as an activator of σ54 promoters for phhA/phhB and/or hmgB/hmgC.
We suggest that TyrR evolved as a modified version of PhhR as follows. In view of the distribution of genes encoding PhhR and TyrR, as well as the aforementioned catabolic enzymes, the most parsimonious evolutionary scenario may be that central and peripheral catabolic pathways depicted in Fig. 8 are quite ancient, but acquisition of PhhR as a σ54-dependent activator of phenylalanine hydroxylase was quite recent, originating about the time of divergence of gamma Proteobacteria. The clade defined by Shewanella/Vibrio/Photobacterium retained the catabolic pathway, whereas the other enteric lineages discarded the catabolic pathway, but retained PhhR, which was then recruited as a σ70-dependent regulator of aromatic biosynthesis (TyrR).
Regulation by attenuation
A widespread mechanism of regulation is via an attenuation mechanism whereby transcripts initiated at given promoters can be terminated prior to reaching the structural genes of an operon. Whether termination occurs usually depends on the balance (dictated by a variety of mechanisms) between mutually exclusive terminator and anti-terminator structures [65].
Merino has developed a website [66] to provide a database of putative attenuators ahead of various operons in Bacteria. We screened this database for likely attenuators relevant to the regulation of tyrA. Table 7 shows intriguing results that point to significant experimental work that would be desirable. tyrA is frequently a member of apparent supraoperons, as alluded to elsewhere in this paper, and some of these appear to be large gene clusters controlled by attenuation. Substantial work is needed to establish the depth of clades possessing a given attenuator. For example, the hisHb > tyrA operon is reliably present throughout all alpha Proteobacteria. Since Agrobacterium tumefaciens has been found to possess an attenuator ahead of the hisHb > tyrA operon, one might reasonably expect that most of the alpha Proteobacteria would possess the attenuator as well. If not, this attenuator would have been a very recent evolutionary innovation. Likewise, since the aroAIα_Y > tyrA operon is widely present throughout the lower-gamma Proteobacteria, it would be interesting to confirm whether only the several species of Vibrio identified on the Merino website have an attenuator ahead of this operon (or whether other attenuators present are too weak to exceed the threshold imposed for preliminary detection).
Table 7.
Putative attenuatorsa associated with tyrA
| Organism | Gene organizationb | Figc |
| Agrobacterium tumefaciens | ¬hisHb > tyrA | 4d |
| Bacillus anthracis | ¬aroG > hisHb > tyrA > aroF | (11) |
| Bacillus cereus | ¬aroG > hisHb > tyrA > aroF | (11) |
| Bacillus halodurans | ¬tyrA > aroF | (11) |
| Bacteroides thetaiotaomicron | ¬pheA > hisHb > aroAIβ•aroQ > tyrA | 4 |
| Bordetella parapertussis | ¬gyrA > serC > aroQ•pheA > tyrA > aroF > cm k> rpsA > himD | 5 |
| Desulfovibrio vulgaris | ¬aroA' > aroB' > aroQ•pheA > aroF > tyrA > [trp operon] | 4 |
| Enterococcus faecalis | ¬aroD > aroAIβ > aroB > aroG > tyrA > aroF > aroE > pheA | 4 |
| Lactococcus lactis | ¬ysaA > blrG > kinG > tyrA > aroF > aroE > pheA | 4 |
| Lactobacillus plantarum | ¬ORF > aroG > ORF > aroF > tyrA > aroE | |
| Listeria innocua | ¬aroG > aroB > aroH > hisH b > tyrA > aroF | 4,(11) |
| Streptococcus pneumoniae | ¬ORF>aroCI>aroD>aroB>aroG>tyrA > ¬ORF>aroF>aroE>pheA | 4 |
| Thermoanaerobacter tencongensis | ¬pheA > aroAIβ > tyrA > aroF > ORF > ORF | |
| Thermus thermophilus | ¬aroAIβ > tyrA | |
| Vibrio parahaemolyticus | ¬aroAIα_Y > tyrA | 6 |
| Vibrio vulnificus | ¬aroAIα_Y > tyrA | 6 |
aAttenuators were extracted from the website of Merino [66]. Links are provided for viewing the complete data, including a visualization of the putative attenuator structures.
bThe symbol ¬ is used for attenuators. Genes encoding the alternative biochemical steps that were recently reported for formation of dehydroquinate from aspartate semialdehyde and ketohexose 1-phosphate [52] are designated aroA' and aroB'.
cRefers to figures within this manuscript or, if enclosed within parentheses, to the figure in ref. [7].
dSee the consensus gene organization for α Proteobacteria.
Some of the supraoperons that appear to be controlled by attenuation are interesting in that they contain the majority of genes needed for both PHE and TYR biosynthesis, e.g., the supraoperons in Enterococcus faecalis and Streptococcus pneumoniae. The latter organism displays two attenuator units. The supraoperon of Desulfovibrio vulgaris is novel in that it begins with two relatively rare genes encoding alternative enzyme steps for aromatic biosynthesis [52], denoted here as aroA' and aroB'. The leading five genes are adjacent to the seven-gene trp operon.
Conclusion
Protein divergence within a vertical genealogy is not necessarily smooth and progressive. Qualitative biochemical innovations can result in a barrage of new selective pressures that result in evolutionary jumps. The consequent incongruence might easily be mistaken for LGT. The basis for evolutionary jumps will usually only be recognized by detailed and comprehensive analyses of any given subsystem. Examples in this study are as follows. (i) The tyrAc_Δ gene of the lower-gamma Proteobacteria has diverged markedly from tyrAc of the upper-gamma Proteobacteria. Here the milestone event was fusion of aroQ to a putative tyrAc in the ancestor of lower-gamma Proteobacteria to produce aroQ•tyrAc_Δ. Indels within the •tyrAc_Δ domain presumably reflect a multiplicity of selections for functional interactions known to exist between the two fused domains as discussed earlier. (ii) Members of the subclass taxon Actinobacteridae possess TyrAa proteins that separate into two distinct groupings. The presumed ancestral NAD(p)TyrAa that is still present in the Actinobacteridae_1 clade very likely spawned the divergent NAD+-specific variety of TyrAa to yield the contemporary Actinobacteridae_2 clade.
The previous evolutionary analysis of trp-pathway genes [7,8] can be viewed as a model for comparable studies with other gene systems. Expansion to the greater aromatic pathway is a logical extension. The dynamics of evolutionary change for tyrA can be matched to the dynamics exhibited by the trp system. For example, the lower-gamma Proteobacteria separate as a distinct phylogenetic unit from beta Proteobacteria and upper-gamma Proteobacteria on criteria defined by milestone evolutionary events that altered many character states of both tryptophan and tyrosine biosynthesis in the lower-gamma Proteobacteria. In the future one can anticipate that comprehensive and systematic phylogenetic analysis of each protein member of the TYR, PHE and TRP branches, the common aromatic-pathway trunk, and minor vitamin-like branches (such as the 4-aminobenzoate/folate branch) will accommodate a progressively integrated picture of the entire aromatic network, including catabolic pathways and many other specialized pathways.
Methods
TyrA sequences
Most TyrA sequences were obtained from the National Center for Biotechnology Information (NCBI) [16]. TyrA sequences from incomplete genomes were retrieved from the PEDANT database [67]. Several sequences in our curated TyrA collection have been corrected for incorrect translation start sites. Various curated TyrA sequence files can be downloaded from our website. These files include complete sequences, trimmed catalytic-core domains, and amino-acid sequence segments that are relevant to specificity for pyridine nucleotide or to specificity for the cyclohexadienyl substrate. The sequence files are summarized in Table 3.
Congruency groupings
TyrA proteins that cluster together on the TyrA protein tree in congruence with the 16S rRNA tree are called congruency groups. Exact correspondence of branching orders is not necessarily observed. So far, congruency groupings have been assembled for tryptophan-protein concatenates [8] and for TyrA proteins. Completion of equivalent work with the remaining aromatic-pathway segments will identify the repertoire of bacterial organisms in possession of a "pure" vertical genealogy with respect to aromatic biosynthesis. Congruency groups for TyrA can be accessed at our AroPath website [9], where a listing of the membership of congruency groups is maintained and updated. Any members of congruency-group clusters, whose position there is incongruent with 16S rRNA expectations, probably (but not necessarily) originated by LGT. The donor lineage may not be obvious, but as more genomes come on line, many cases where donor identities are currently unknown may become revealed. A listing of "orphan" TyrA proteins that belong to no current congruency group is given. Such orphans reflect the lack of sufficient genome representation in particular phylogenetic regions and undoubtedly will become the nucleus for additional congruency groups in due course.
Alignments
Multiple alignments were obtained by use of the ClustalW or ClustalX programs (Version 1.83) [68]. Manual adjustments were needed in the region of the GxGxxG motif for binding of pyridine nucleotide cofactor in the N-terminal region of TyrA proteins. Guidance for alignment was assisted by maximizing conformation with the Wierenga fingerprint, making allowance for a variable loop of 2–5 residues [26]. This was done with the assistance of the BioEdit multiple alignment tool of Hall (5.0.9 Edition) [69]. The refined multiple alignment was used as input for generation of a phylogenetic tree using the phylogeny inference package (Version 3.2), PHYLIP [70]. The neighbor-joining program was used to obtain a distance-based tree. The distance matrix was obtained by use of Protdist with a Dayhoff Pam matrix. The Seqboot and Consense programs were then applied to assess the statistical support of the tree using bootstrap resampling (1,000 replications). We also used the ANCESCON package [71], which produced similar results as shown in Fig. 2 (albeit with even wider separation of many groups). The presence of regulatory domains (ACT and REG) was accepted when indicated by the Domain Architecture Retrieval Tool (DART) on the BLAST menu at NCBI [16].
Profile HMMs
Profile hidden Markov models for each of the four TyrA subfamilies, TyrAa, TyrAc, TyrAp and tyrAc_Δ, were built using Sean Eddy's HMMER package [72]. The HMMs were generated from our file of curated cyclohexadienyl-substrate core segments (see Table 3). The seed sequences for each subfamily were first aligned using ClustalW [68]. The resulting multiple sequence alignments were then manually edited to produce more accurate alignment of the seed sequences. Finally, the edited multiple sequence alignments were used to generate the profile HMMs for each TyrA subfamily.
Appraisal of gene fusions as one-time or multiple events
Whether any given contemporary gene fusions tracked back to a fusion event in a common ancestor or whether they occurred independently was evaluated by phylogenetic analysis of the individual protein domains and by inspection of the inter-domain linker region. Linker regions were determined by multiple alignments of fusion sequences with corresponding free-standing domains present in the closest relatives to organisms that lack the gene fusions.
Authors' contributions
JS and MW integrated this specific effort with the broader and ongoing objective of implementing a dynamic and progressively updateable website (AroPath). JS also made substantial contributions to the bioinformatic work. CB did all of the art work and a majority of the bioinformatic analyses. RJ provided initial guiding concepts, a general organizational overview, and assembled the initial manuscript draft. CB, RJ, and JS contributed to the formulation of conclusions made, and all of the authors read and approved the final version of the manuscript.
Supplementary Material
Table S1, entitled "Key to organism acronyms and sequence identifiers", is provided as supplementary material in an html document. This table contains the full collection of sequence data and annotations contained in this paper, and gene identification (gi) numbers are included and hyperlinked to facilitate access to the corresponding GenBank records. For future reference to a progressively updated table, refer to the AroPath website [73].
Table 2.
Key to organism acronyms
| Organism |
Abbreviation in Paper |
Abbreviation on websiteb |
| Acidithiobacillus ferrooxidans ATCC 23270 | Aferr | |
| Acinetobacter sp. ADP1 | ACIN | |
| Actinobacillus actinomycetemcomitans HK1651 | Aact | |
| Actinomyces naeslundii MG1 | Anae | |
| Actinoplanes teichomyceticus | Atei | |
| Agrobacterium tumefaciens strain C58 | Atum | |
| Amycolatopsis balhimycina | Abal | |
| Amycolatopsis orientalis | Aori | |
| Anabaena sp. PCC 7120 | ANAB | |
| Arabidopsis thaliana | Atha | |
| Archaeoglobus fulgidus DSM 4304 | Aful | Aful_1 |
| Azotobacter vinelandii | Avin | Avin_1 |
| Bacillis anthracis str. A2012 | Bant | |
| Bacillus cereus ATCC 14579 | Bcer | |
| Bacillus halodurans C-125 | Bhal | Bhal_2 |
| Bacillus stearothermophilus | Bste | |
| Bacillis subtilis | Bsub | |
| Bacillus thuringiensis israelensis | Bthu | |
| Bifidobacterium longum NCC2705 | Blon | Blon_1 |
| Blochmannia floridanus | Bflo | |
| Bordetella bronchisepticus | Bbro | |
| Burkholderia cepacia J2315 | Bcep | |
| Burkholderia fungorum LB400 | Bfun | |
| Burkholderia mallei ATCC 23344 | Bmal | |
| Burkholderia pseudomallei K96243 | Bpse | Bpse_6 |
| Campylobacter jejuni | Cjej | |
| Chromobacterium violaceum ATCC 12472 | Cvio | |
| Corynebacterium diphtheriae NCTC 13129 | Cdip | |
| Corynebacterium efficiens YS-314 | Ceff | |
| Corynebacterium glutamicum ATCC 13032 | Cglu | Cglu_1 |
| Desulfovibrio desulfuricans G20 | Ddes | |
| Desulfovibrio vulgaris subsp. vulgaris strain Hildenborough | Dvul | |
| Desulfuromonas acetoxidans | Dace | Dace_5 |
| Enterococcus faecalis V583 | Efae_2 | |
| Enterococcus faecium | Efae_1 | Efae_1 |
| Erwinia carotovoa subsp.atroseptica SCRI1043 | Ecar | |
| Escherichia coli K12 | Ecol | |
| Geobacter metallireducens GS-15 | Gmet | |
| Geobacter sulfurreducens PCA | Gsul | |
| Gloeobacter violaceus PCC 7421 | Gvio | |
| Haemophilus influenzae Rd KW20 | Hinf | |
| Helicobacter hepaticus ATCC 51449 | Hhep | |
| Helicobacter pylori 26695 | Hpyl | |
| Klebsiella pneumoniae subsp. pneumoniae MGH 78578 | Kpne | |
| Leifsonia xyli subsp. Xyli strain CTCB07 | Lxyl | |
| Listeria innocua Clip 11262 | Linn | |
| Listeria monocytogenes EGD-e | Lmon | |
| Lotus corniculatus var. japonicus | Lcor | Lcor_3 |
| Lycopersicon esculentum | Lesc | |
| Methanococcus jannaschii | Mjan | |
| Methanopyrus kandleri AV19 | Mkan | Mkan_1 |
| Methanosarcina barkeri strain Fusaro | Mbar | |
| Methanothermobacter thermoautotrophicus strain Delta H | Mthe | Mthe_7 |
| Microbulbifer degradans 2–40 | Mdeg | |
| Mycobacterium avium subsp. paratuberculosis strain k10 | Mavi | |
| Mycobacterium bovis TrEMBL | Mbov | Mbov_2 |
| Mycobacterium leprae TN | Mlep | |
| Mycobacterium tuberculosis CDC1551 | Mtub | |
| Myxococcus xanthus DK 1622 | Mxan | |
| Neisseria gonorrhoeae FA 1090 | Ngon | |
| Nitrosomonas europaea ATCC 19718 | Neur | |
| Nocardia farcinica IFM 10152 | Nfar | |
| Nonomuraea sp. | NONO | |
| Nostoc punctiforme PCC73102 | Npun | Npun_1 |
| Novosphingomonas aromaticivorans DSM 12444 | Naro | |
| Oceanobacillus iheyensis THE831 | Oihe | |
| Oryza sativa ssp. japonica | Osat | |
| Pantoea agglomerans | Pagg | |
| Pasteurella multocida subsp. multocida strain Pm70 | Pmul | |
| Photorhabdus luminescens subsp. laumondii TT01 | Plum | |
| Prochlorococcus marinus subsp. pastoris strain CCMP1378 | Pmar_1 | Pmar_3 |
| Prochlorococcus marinus MIT9313 | Pmar_2 | Pmar_10 |
| Propionibacterium acnes KPA171202 | Pacn | |
| Pseudomonas aeruginosa PAO1 | Paer | Paer_1 |
| Pseudomonas fluorescens PfO-1 | Pflu | |
| Pseudomonas putida KT2440 | Pput | |
| Pseudomonas stutzeri | Pstu | |
| Ralstonia eutropha JMP134 | Reut | |
| Ralstonia solanacearum GMI1000 | Rsol | |
| Rhodobacter capsulatus | Rcap | |
| Rhodobacter sphaeroides 2.4.1 | Rsph | |
| Rhodopseudomonas palustris CGA009 | Rpal | |
| Rhodospirillum rubrum | Rrub | Rrub_1 |
| Rubrobacter xylanophilus DSM 9941 | Rxyl | |
| Saccharomyces cerevisiae | Scer | |
| Salmonella typhimurium LT2 | Styp | Styp_1 |
| Schizosaccharomyces pombe | Spom | |
| Shewanella oneidensis MR-1 | Sone | |
| Shewanella putrifacians | Spu | |
| Staphylococcus aureus subsp. Aureus MW2 | Saur | Saur_2 |
| Streptococcus gordonii str. Challis | Sgor | |
| Streptococcus pneumoniae R6 | Spne | |
| Streptomyces avermitilis MA-4680 | Save | |
| Streptomyces caeruleus | Scae | Scae_2 |
| Streptomyces coelicolor A3(2) | Scoe | Scoe_1 |
| Streptomyces lavendulae | Slav | |
| Streptomyces pristinaespiralis | Spri | |
| Streptomyces roseochromogenes subsp. Oscitans | Sros | Sros_1 |
| Streptomyces toyocaensis strain 7 | Stoy | |
| Sulfolobus solfataricus P2 | Ssol | |
| Sulfolobus tokodaii strain 7 | Stok | |
| Synechococcus sp. WH8102 | SYNE_1 | SYNE_1 |
| Synechococcus sp. PCC7002 | SYNE_2 | |
| Synechocystis sp. PCC6803 | SYNE_3 | SYNE_3 |
| Thermobifida fusca | Tfus | |
| Thermosynechococcus elongates BP-1 | Telo | |
| Trichodesmium erythraeum IMS101 | Tery | |
| Tropheryma whipplei TW08/27 | Twhi | |
| Vibrio cholerae O1 biovar eltor strain N16961 | Vcho | |
| Vibrio parahaemolyticus RIMD 2210633 | Vpar | |
| Wolinella succinogenes DSM 1740 | Wsu | |
| Xanthomonas campestris pv. campestris strain ATCC 33913 | Xcam | |
| Xylella fastidiosa 9a5c | Xfas | |
| Yersinia enterocolitica (type 0:8) | Yent | |
| Zymomonas mobilis subsp. mobilis ZM4 | Zmob |
aThe system of acronym usage is: the first letter (capital) is the first letter of the genus followed by the first three letters (lower-case) of the species. If there is no species designation the first four letters of the genus are used (all in capitals). Redundant 4-letter acronyms are distinguished by unique following numbers. See [74] for a comprehensive listing with hyperlinks to the Taxonomy database records and the GenBank records at NCBI.
Acknowledgments
Acknowledgements
R. Jensen thanks the National Library of Medicine (Grant G13 LM008297) for partial support. This research is partially supported by the U. S. Army Research Institute of Infectious Diseases (USAMRIID). This analysis would not have been possible were it not for the yeoman efforts in comparative enzymology carried out over a period of more than 25 years by many students and postdoctoral fellows, most notably Graham S. Byng, Robert Whitaker, Alan X. Berry and Suhail Ahmad. This has produced an invaluable resource of comprehensive data, some of it unpublished. This paper is dedicated to our colleague and collaborator, John E. Gander, on the occasion of his 80th birthday.
Contributor Information
Jian Song, Email: jian@lanl.gov.
Carol A Bonner, Email: cbonner@ufl.edu.
Murray Wolinsky, Email: murray@lanl.gov.
Roy A Jensen, Email: rjensen@ufl.edu.
References
- Xie G, Bonner CA, Jensen RA. Cyclohexadienyl dehydrogenase from Pseudomonas stutzeri exemplifies a widespread type of tyrosine-pathway dehydrogenase in the TyrA protein family. Comp Biochem Physiol C Toxicol Pharmacol. 2000;125:65–83. doi: 10.1016/s0742-8413(99)00090-0. [DOI] [PubMed] [Google Scholar]
- Jensen RA. Tyrosine and phenylalanine biosynthesis: relationship between alternative pathways, regulation and subcellular location. Rec Adv Phytochem. 1986;20:57–82. [Google Scholar]
- Todd AE, Orengo CA, Thornton JM. Evolution of function in protein superfamilies, from a structural perspective. J Mol Biol. 2001;307:1113–1143. doi: 10.1006/jmbi.2001.4513. [DOI] [PubMed] [Google Scholar]
- Teichmann SA, Rison SCG, Thornton JM, Riley M, Gough J, Clothia C. The evolution and structural anatomy of the small molecule metabolic pathways in Escherichia coli. J Mol Biol. 2001;311:693–708. doi: 10.1006/jmbi.2001.4912. [DOI] [PubMed] [Google Scholar]
- Xie G, Brettin T, Bonner CA, Jensen RA. Mixed-function supraoperons that exhibit overall conservation, albeit shuffled gene organization, across wide intergenomic distances within eubacteria. Microb Comp Genomics. 1999;4:5–28. doi: 10.1089/omi.1.1999.4.5. [DOI] [PubMed] [Google Scholar]
- Xie G, Bonner CA, Jensen RA. A probable mixed-function supraoperon in Pseudomonas exhibits gene organization features of both intergenomic conservation and gene shuffling. J Mol Evol. 1999;49:108–121. doi: 10.1007/pl00006523. [DOI] [PubMed] [Google Scholar]
- Xie G, Keyhani N, Bonner CA, Jensen RA. Ancient origin of the tryptophan operon and the dynamics of evolutionary change. Microbiol Mol Biol Rev. 2003;67:303–342. doi: 10.1128/MMBR.67.3.303-342.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie G, Bonner CA, Song J, Keyhani NO, Jensen RA. Inter-genomic displacement via lateral gene transfer of bacterial trp operons in an overall context of vertical genealogy. BMC Biology. 2004;2:15. doi: 10.1186/1741-7007-2-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- AroPath http://AroPath.lanl.gov/Phylogeny/CG/tyrCG.html
- Gil R, Silva FJ, Zientz E, Delmotte F, Gonzalez-Candelas F, Latorre A, Rausell C, Kamerbeek J, Gadau J, Holldobler B, et al. The genome sequence of Blochmannia floridanus : comparative analysis of reduced genomes. Proc Natl Acad Sci USA. 2003;100:9388–9393. doi: 10.1073/pnas.1533499100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gevers D, Vandepoole K, Simillion C, Van de Pere Y. Gene duplication and biased functional retention of paralogs in bacterial genomes. Trends Microbiol. 2004;12:148–154. doi: 10.1016/j.tim.2004.02.007. [DOI] [PubMed] [Google Scholar]
- AroPath http://AroPath.lanl.gov/Phylogeny/CG/index.html
- Blanc V, Gil P, Bamasjacques N, Lorenzon S, Zagorec M, Schleuniger J. Identification and analysis of genes from Streptomyces pristinaespiralis encoding enzymes involved in the biosynthesis of the 4-dimethylamino-L-phenylalanine precursor of pristinamycin I. Mol Microbiol. 1997;23:191–202. doi: 10.1046/j.1365-2958.1997.2031574.x. [DOI] [PubMed] [Google Scholar]
- Lingens F, Göbel W, Üsseler H. Regulation der biosynthesis der aromatischen aminosauren in Saccharomyces cerevisiae, I. Hemmung der Enzymaktivitaten (Feedback-Wirkung) Biochem Z. 1966;346:357–67. [Google Scholar]
- Zamir LO, Jung E, Jensen RA. Co-accumulation of prephenate L-arogenate and spiro-arogenate in a mutant of Neurospora. 1983;258:6492–6496. [PubMed] [Google Scholar]
- National Center for Biotechnology Information http://www.ncbi.nlm.nih.gov
- Xia T, Jensen RA. A single cyclohexadienyl dehydrogenase specifies the prephenate dehydrogenase and arogenate dehydrogenase components of the dual pathways to L-tyrosine in Pseudomonas aeruginosa. J Biol Chem. 1990;265:20033–20036. [PubMed] [Google Scholar]
- Zhao G, Xia T, Ingram L, Jensen RA. An allosterically insensitive class of cyclohexadienyl dehydrogenase from Zymomonas mobilis. Eur J Biochem. 1993;212:157–165. doi: 10.1111/j.1432-1033.1993.tb17646.x. [DOI] [PubMed] [Google Scholar]
- Jensen RA. Enzyme recruitment in evolution of new function. Annu Rev Microbiol. 1976;30:409–425. doi: 10.1146/annurev.mi.30.100176.002205. [DOI] [PubMed] [Google Scholar]
- Hall GC, Flick MB, Gherna RL, Jensen RA. Biochemical diversity for biosynthesis of aromatic amino acids among the cyanobacteria. J Bacteriol. 1982;149:65–78. doi: 10.1128/jb.149.1.65-78.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramaniam P, Bhatnagar R, Hooper A, Jensen RA. The dynamic progression of evolved character states for aromatic amino acid biosynthesis in gram-negative bacteria. Microbiology. 1994;140:3431–3440. doi: 10.1099/13500872-140-12-3431. [DOI] [PubMed] [Google Scholar]
- Byng GS, Whitaker RJ, Gherna RL, Jensen RA. Variable enzymological patterning in tyrosine biosynthesis as a means of determining natural relatedness among the Pseudomonadaceae. J Bacteriol. 1980;144:247–257. doi: 10.1128/jb.144.1.247-257.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller B, Keller E, Gorisch H, Lingens F. Biosynthesis of phenylalanine and tyrosine in Streptomycetes. Hoppe Seylers Z Physiol Chem. 1983;364:455–459. [PubMed] [Google Scholar]
- Keller B, Keller E, Lingens F. Arogenate dehydrogenase from Streptomyces phaeochromogenes. Purification and properties. Biol Chem Hoppe Seyler. 1985;366:1063–1066. doi: 10.1515/bchm3.1985.366.2.1063. [DOI] [PubMed] [Google Scholar]
- Bonner CA, Jensen RA, Gander JE, Kehani NO. A core catalytic domain of the TyrA protein family: arogenate dehydrogenase from Synechocystis. Biochem J. 2004;382:279–291. doi: 10.1042/BJ20031809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wierenga RK, Terpstra P, Hol WGJ. Prediction of the occurrence of the ADP-binding β α β-fold in proteins, using an amino-acid sequence fingerprint. J Mol Biol. 1986;187:101–107. doi: 10.1016/0022-2836(86)90409-2. [DOI] [PubMed] [Google Scholar]
- Rippert P, Matringe M. Molecular and biochemical characterization of an Arabidopsis thaliana arogenate dehydrogenase with two highly similar and active protein domains. Plant Mol Biol. 2002;48:361–368. doi: 10.1023/A:1014018926676. [DOI] [PubMed] [Google Scholar]
- Rippert P, Matringe M. Purification and kinetic analysis of the two recombinant arogenate dehydrogenase isoforms of Arabidopsis thaliana. Eur J Biochem. 2002;269:4753–4761. doi: 10.1046/j.1432-1033.2002.03172.x. [DOI] [PubMed] [Google Scholar]
- Xie G, Forst C, Bonner CA, Jensen RA. Significance of two distinct types of tryptophan synthase beta chain in Bacteria, Archaea and higher plants. Genome Biol. 2002;3:Research0004.1–0004.13. doi: 10.1186/gb-2001-3-1-research0004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Champney WS, Jensen RA. The enzymology of prephenate dehydrogenase in Bacillus subtilis. J Biol Chem. 1970;245:3763–3770. [PubMed] [Google Scholar]
- Xie G, Bonner CA, Brettin T, Gottardo R, Keyhani NO, Jensen RA. Lateral gene transfer and ancient paralogy of operons containing redundant copies of tryptophan-pathway genes in Xylella species and heterocystous cyanobacteria. Genome Biol. 2003;4:R14. doi: 10.1186/gb-2003-4-2-r14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Vincent S, Wilson DB, Ganem B. Mapping of chorismate mutase and prephenate dehydrogenase domains in the Escherichia coli T-protein. Eur J Biochem. 2003;270:757–763. doi: 10.1046/j.1432-1033.2003.03438.x. [DOI] [PubMed] [Google Scholar]
- Mavrodi DV, Ksenzenko VM, Bonsall RF, Cook RJ, Boronin AM, Thomashow LS. A seven-gene locus for synthesis of phenazine-1-carboxylic acid by Pseudomonas fluorescens 2–79. J Bacteriol. 1998;180:2541–2548. doi: 10.1128/jb.180.9.2541-2548.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierson LS, Gaffney T, Lamb S, Gong F. Molecular analysis of genes encoding phenazine biosynthesis in the biological control bacterium. Pseudomonas aureofaciens 30–84. FEMS Lett. 1995;134:299–307. doi: 10.1016/0378-1097(95)00423-X. [DOI] [PubMed] [Google Scholar]
- AroPath http://AroPath.lanl.gov/Annotation/CuratedAASeqForDownload.html
- Pfam http://www.sanger.ac.uk/Software/Pfam/
- Interpro http://www.ebi.ac.uk/interpro/
- Eddy SR. Profile-hidden Markov models. Bioinformatics. 1998;14:755–763. doi: 10.1093/bioinformatics/14.9.755. [DOI] [PubMed] [Google Scholar]
- Park J, Kaplus K, Barrett C, Hughey R, Haussler D, Hubbard T, Chothia C. Sequence comparisons using multiple sequences detect three times as many remote homologues as pairwise methods. J Mol Biol. 1998;284:1201–1210. doi: 10.1006/jmbi.1998.2221. [DOI] [PubMed] [Google Scholar]
- AroPath http://AroPath.lanl.gov/Biosynthesis/TyrPath/hmmPfamTyrA.html
- AroPath http://AroPath.lanl.gov/Biosynthesis/TyrPath/index.html
- Fazel A, Jensen R. Obligatory biosynthesis of L-tyrosine via the pretyrosine branchlet in coryneform bacteria. J Bacteriol. 1979;138:805–815. doi: 10.1128/jb.138.3.805-815.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fazel AM, Bowen JR, Jensen RA. Arogenate (pretyrosine) is an obligatory intermediate of L-tyrosine biosynthesis: confirmation in a microbial mutant. Proc Natl Acad Sci USA. 1980;77:1270–1273. doi: 10.1073/pnas.77.3.1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byng GS, Berry A, Jensen RA. Evolutionary implications of features of aromatic amino acid biosynthesis in the genus Acinetobacter. Arch Microbiol. 1985;143:122–129. doi: 10.1007/BF00411034. [DOI] [PubMed] [Google Scholar]
- Porat I, Waters BW, Teng Q, Whitman WB. Two biosynthetic pathways for aromatic amino acids in the archaeon Methanococcus maripaludis. J Bacteriol. 2004;186:4940–4950. doi: 10.1128/JB.186.15.4940-4950.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calhoun DH, Bonner CA, Gu W, Xie G, Jensen RA. The emerging periplasm-localized subclass of AroQ chorismate mutases, exemplified by those from Salmonella typhimurium and Pseudomonas aeruginosa. Genome Biol. 2001. pp. 2research0030.1–0030.16. [DOI] [PMC free article] [PubMed]
- Ahmad S, Jensen RA. The stable evolutionary fixation of a bifunctional tyrosine-pathway protein in enteric bacteria. Microbiol Lett. 1988;52:109–116. doi: 10.1016/0378-1097(88)90309-6. [DOI] [Google Scholar]
- Jensen RA, Ahmad S. Nested gene fusions as markers of phylogenetic branchpoints in prokaryotes. Trends Ecol Evol. 1990;5:219–224. doi: 10.1016/0169-5347(90)90135-Z. [DOI] [PubMed] [Google Scholar]
- Jensen RA, Gu W. Evolutionary recruitment of biochemically specialized subdivisions of Family I within the protein superfamily of aminotransferases. J Bacteriol. 1996;178:2161–2171. doi: 10.1128/jb.178.8.2161-2171.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aravind L, Koonin EV. Gleaning non-trivial structural, functional and evolutionary information about proteins by iterative database searches. J Mol Biol. 1999;287:1023–1040. doi: 10.1006/jmbi.1999.2653. [DOI] [PubMed] [Google Scholar]
- Henner D, Yanofsky C. Bacillus subtilis and other gram-positive bacteria. In: Sonenshein AL, Hoch J, Losick R, editor. Biochemistry, physiology, and molecular genetics. Washington, DC: ASM Press; 1993. [Google Scholar]
- White RH. L-Aspartate semialdehyde and a 6-deoxy-5-ketohexose 1-phosphate are the precursors to the aromatic amino acids in Methanocaldococcus jannashii. Biochemistry. 2004;43:7618–7627. doi: 10.1021/bi0495127. [DOI] [PubMed] [Google Scholar]
- Ahmad S, Johnson JL, Jensen RA. The recent evolutionary origin of the phenylalanine-sensitive isozyme of 3-deoxy-D-arabino-heptulosonate 7-phosphate synthase in the enteric lineage of bacteria. J Mol Evol. 1987;25:159–167. doi: 10.1007/BF02101758. [DOI] [PubMed] [Google Scholar]
- Jensen RA, Xie G, Calhoun DH, Bonner CA. The correct phylogenetic relationship of KdsA (3-deoxy-D-manno-octulosonate 8-phosphate synthase) with one of two independently evolved classes of AroA (3-deoxy-D-arabino-heptulosonate 7-phosphate synthase) J Mol Evol. 2002;54:416–423. doi: 10.1007/s00239-001-0031-z. [DOI] [PubMed] [Google Scholar]
- Pittard AJ, Camakaris H, Yang J. The TyrR regulon. Mol Microbiol. 2005;55:16–26. doi: 10.1111/j.1365-2958.2004.04385.x. [DOI] [PubMed] [Google Scholar]
- Katayama T, Suzuki H, Koyanagi T, Kumagai H. Cloning and random mutagenesis of the Erwinia herbicola tyrR gene for high-level expression of tyrosine phenol-lyase. Appl Envir Microbiol. 2000;66:4764–4771. doi: 10.1128/AEM.66.11.4764-4771.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai Q, Somerville R. Integration host factor and cyclic AMP receptor proein are required for TyrR-mediated activation of tpl in Citrobacter freundii. J Bacteriol. 1998;180:6173–6186. doi: 10.1128/jb.180.23.6173-6186.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S, Somerville RL. Isolated operator binding and ligand response domains of the TyrR protein of Haemophilus influenzae associate to reconstitute functional repressor. J Biol Chem. 1999;274:1842–1847. doi: 10.1074/jbc.274.3.1842. [DOI] [PubMed] [Google Scholar]
- Arias-Barrau E, Olivera E, Luengo J, Fernandez C, Galan B, Garcia J, Diaz E, Miñambres B. The homogentisate pathway: a central catabolic pathway involved in the degradation of L-phenylalanine, L-tyrosine, and 3-hydroxyphenylacetate in Pseudomonas putida. J Bacteriol. 2004;186:5062–5077. doi: 10.1128/JB.186.15.5062-5077.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dähnhardt D, Falk J, Appel J, van der Kooij A, Schulz-Friedrich R, Krupinska K. The hydroxyphenylpyruvate dioxygenase from Synechocystis sp. PCC 6803 is not required for plastoquinone biosynthesis. FEBS Lett. 2002;523:177–181. doi: 10.1016/S0014-5793(02)02978-2. [DOI] [PubMed] [Google Scholar]
- Song J, Jensen RA. PhhR, a divergently transcribed activator of the phenylalanine hydroxylase gene cluster of Pseudomonas aeruginosa. Mol Microbiol. 1996;22:497–507. doi: 10.1046/j.1365-2958.1996.00131.x. [DOI] [PubMed] [Google Scholar]
- Zhao G, Xia T, Song J, Jensen R. Pseudomonas aeruginosa possesses homologues of mammalian phenylalanine hydroxylase and 4a-carbinolamine dehydratase/DCoH as part of a three-component gene cluster. Proc Natl Acad Sci USA. 1994;91:1366–1370. doi: 10.1073/pnas.91.4.1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tropel D, van der Meer J. Bacterial transcriptional regulators for degradation pathways of aromatic compounds. Microbiol Mol Biol Rev. 2004;68:474–500. doi: 10.1128/MMBR.68.3.474-500.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaney M, Grande R, Wigneshweraraj S, Cannon W, Casaz P, Gallegos M-T. Binding of transcriptional activators to sigma 54 in the presence of the transition state analog ADP-aluminum fluoride: insights into activator mechanochemical action. Genes Dev. 2001;15:2282–2294. doi: 10.1101/gad.205501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanofsky C. The different roles of tryptophan transfer RNA in regulating trp operon expression in E. coli versus B. subtilis. Trends Genet. 2004;20:367–374. doi: 10.1016/j.tig.2004.06.007. [DOI] [PubMed] [Google Scholar]
- Predicted attenuators in bacteria http://cmgm.stanford.edu/~merino
- Riley ML, Schmidt T, Wagner c, Mewes H-W, Frishman D. The PEDANT genome database in 2005. Nuc Ac Res. 2005;33:D308–D310. doi: 10.1093/nar/gki019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chenna R, Sugawara H, Koike T, Lopez R, Gibson T, Higgins D, Thompson J. Multiple sequence alignment with the Clustal series of programs. Nucl Ac Res. 2003;31:3497–3500. doi: 10.1093/nar/gkg500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BioEdit http://www.mbio.ncsu.edu/BioEdit/bioedit.html
- Felsenstein J. PHYLIP-Phylogeny Inference Package (version 3.2) Cladistics. 1989;5:164–166. [Google Scholar]
- Cai W, Pei J, Grishin NV. Reconstruction of ancestral protein sequences and its applications. BMC Evol Biol. 2004;4:33. doi: 10.1186/1471-2148-4-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddy S. HMMER package. 1995. http://hmmer.wustl.edu
- AroPath http://AroPath.lanl.gov/Annotation/TyrA/TyrA_substrateSpc.html
- AroPath http://AroPath.lanl.gov/Organisms/Species_sorted_by_acronym.html
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1, entitled "Key to organism acronyms and sequence identifiers", is provided as supplementary material in an html document. This table contains the full collection of sequence data and annotations contained in this paper, and gene identification (gi) numbers are included and hyperlinked to facilitate access to the corresponding GenBank records. For future reference to a progressively updated table, refer to the AroPath website [73].