

Abstract
We report that under ‘transcriptional stress' in budding yeast, when most pol II activity is acutely inhibited, rapid deposition of nucleosomes occurs within genes, particularly at 3′ positions. Whereas histone H3K4 trimethylation normally marks 5′ ends of highly transcribed genes, under ‘transcriptional stress' induced by 6-azauracil (6-AU) and inactivation of pol II, TFIIE or CTD kinases Kin28 and Ctk1, this mark shifted to the 3′ end of the TEF1 gene. H3K4Me3 at 3′ positions was dynamic and could be rapidly removed when transcription recovered. Set1 and Chd1 are required for H3K4 trimethylation at 3′ positions when transcription is inhibited by 6-AU. Furthermore, Δchd1 suppressed the growth defect of Δset1. We suggest that a ‘transcriptional stress' signal sensed through Set1, Chd1, and possibly other factors, causes H3K4 hypermethylation of newly deposited nucleosomes at downstream positions within a gene. This response identifies a new role for H3K4 trimethylation at the 3′ end of the gene, as a chromatin mark associated with impaired pol II transcription.
Keywords: Chd1, chromatin, histone H3K4 methylation, Set1, transcription
Introduction
How the pol II transcription complex interacts with its chromatin template is a key question in eukaryotic gene expression. Much attention has focused on remodeling of chromatin at promoters that modulates accessibility to the transcriptional apparatus (Almer et al, 1986; Hirschhorn et al, 1992; Boeger et al, 2003; Reinke and Horz, 2003; Sharma et al, 2003; Adkins et al, 2004). In contrast, less is known about how chromatin within a gene is affected when transcription rate changes. Transcription is accompanied by replication-independent turnover of specific histones, however (Ahmad and Henikoff, 2002; Thiriet and Hayes, 2005). Activation and repression of GAL genes in yeast correlate with altered micrococcal nuclease sensitivity (Cavalli and Thoma, 1993) and reversible loss of histone DNA contacts consistent with nucleosome eviction and replacement within the gene (Kristjuhan and Svejstrup, 2004; L Zhang and D Bentley, unpublished; Lee et al, 2004; Schwabisch and Struhl, 2004). A recent global analysis found a negative correlation between mRNA synthesis rate and histone occupancy at both intergenic and coding sequences (Lee et al, 2004). An important unresolved question is whether these chromatin changes are a cause or a consequence of altered pol II transcription.
Transcriptional activity is correlated with particular patterns of covalent modification of histone N-terminal tails (Strahl and Allis, 2000; Iizuka and Smith, 2003). Trimethylation of H3 tails on K4 (K4Me3) at the 5′ end of the transcribed region is a signature motif of highly expressed genes (Santos-Rosa et al, 2002; Ng et al, 2003; Sims et al, 2003). H3K4 trimethylation has been proposed to enhance transcriptional elongation (Gerber and Shilatifard, 2003) and to act as a memory signal (Ng et al, 2003). Methylation of H3K4 in yeast is carried out by the Set1 subunit of the COMPASS complex (Miller et al, 2001; Roguev et al, 2001; Nagy et al, 2002). The Paf complex, proteasome subunits and factors that ubiquitlylate H2B K123 all stimulate H3K4 methylation (Dover et al, 2002; Sun and Allis, 2002; Krogan et al, 2003; Ezhkova and Tansey, 2004). Set1 trimethylation activity is positively regulated by its RNA binding domain (Schlichter and Cairns, 2005; V Geli, personal communication), although whether or not binding to the nascent RNA transcript affects Set1 activity during transcription is unknown. It was recently found that dimethyl H3K4 tails are bound by one of the chromodomains of the chromatin assembly factor Chd1 (Tran et al, 2000; Robinson and Schultz, 2003; Pray-Grant et al, 2005) but the functional relationship between Chd1 and Set1 has not been explored.
Phosphorylation of heptad repeats (YS2PTS5PS) in the C-terminal domain of the pol II large subunit, Rpb1, modulates transcription elongation (Kobor and Greenblatt, 2002) and chromatin modification by H3K36 methylation (Xiao et al, 2003). In yeast, Kin28 is the major kinase that phosphorylates Ser5 residues at the 5′ end, and Ctk1 is the major kinase that phosphorylates Ser2 residues further downstream (Kobor and Greenblatt, 2002). Both Kin28 and Ctk1 appear to enhance transcriptional elongation (Schroeder et al, 2000; Jona et al, 2001). It has been suggested that Set1 binding to Ser5 phosphorylated pol II restricts H3K4 trimethylation to the 5′ end of genes (Ng et al, 2003). The effect of inactivating Kin28 on H3K4 methylation has not been determined.
How acute inhibition of pol II activity influences chromatin structure within genes is poorly understood. Budding yeast is well suited to investigation of this problem because pol II transcription can be specifically disrupted by mutations of the transcription apparatus or treatment with 6-azauracil (6-AU) as well as by physiological stress stimuli such as heat shock and glucose deprivation (Jona et al, 2000). 6-AU reduces GTP and UTP levels (Exinger and Lacroute, 1992) thereby inhibiting elongation rate and processivity (Lennon et al, 1998; Mason and Struhl, 2005). Growth of many mutants implicated in regulation of elongation is sensitive to 6-AU, whereas growth of some mutants including Δchd1 is particularly resistant to this drug (Woodage et al, 1997). The short-term effects of 6-AU on chromatin structure have not been investigated.
We used ChIP against histones to measure how transcription inhibition affects histone occupancy and methylation of H3K4 in budding yeast genes. ‘Transcriptional stress' induced by 6-AU or mutations in pol II and general transcription factors rapidly increased histone occupancy particularly near the 3′ end of the gene and in some cases also decreased occupancy at the 5′ end. These results suggest a dynamic balance between nucleosome displacement and replacement that is responsive to pol II activity (Kaplan et al, 2003; Lee et al, 2004). Transcriptional inhibition by 6-AU caused a remarkable increase in H3K4 trimethylation at the 3′ end, which is dependent on Chd1. We suggest that H3K4 trimethylation is not only a chromatin mark for the 5′ end of highly expressed genes, but also a mark of chromatin at the 3′ end of a gene when transcription is impaired.
Results
6-AU alters the distribution of pol II and histones within transcription units
To investigate how pol II transcription through a gene might affect its chromatin structure, we modulated pol II function as directly as possible in vivo and assayed the consequences for histone occupancy and modification. We initially chose to repress transcription by reducing cellular NTP concentrations with 6-AU. Pol II and histone occupancy were monitored in 6-AU-treated cells by ChIP at three positions along the highly transcribed TEF1 gene. TEF1 was among 136 mRNAs reduced over 2.5-fold after 1 h in 6-AU in an rpo21-18 mutant as determined by microarray analysis (G Geiger and D Bentley, unpublished). Note that TEF1 and TEF2 are almost identical within their coding regions so that they cannot be distinguished. Telomere VIR (TEL) and mitochondrial COXIII served as negative controls for pol II and histone ChIP. Quantitative PCR verified that signals from different PCR products are linear and multiplexing ensures that the signals are internally controlled relative to one another (Supplementary Figure 1A). A globin plasmid spiked into ChIP samples before reversing the crosslinks served as a control for DNA recovery and gel loading. 6-AU treatment for 1 h modestly reduced pol II crosslinking at the 3′ end of TEF1 relative to the middle and 5′ end (Figure 1A, lanes 3 and 4 and graph) consistent with impaired processivity. Ser5 phosphorylation of the pol II CTD remained high at the 5′ end of TEF1 and low at the 3′ end in 6-AU (data not shown).
Figure 1.
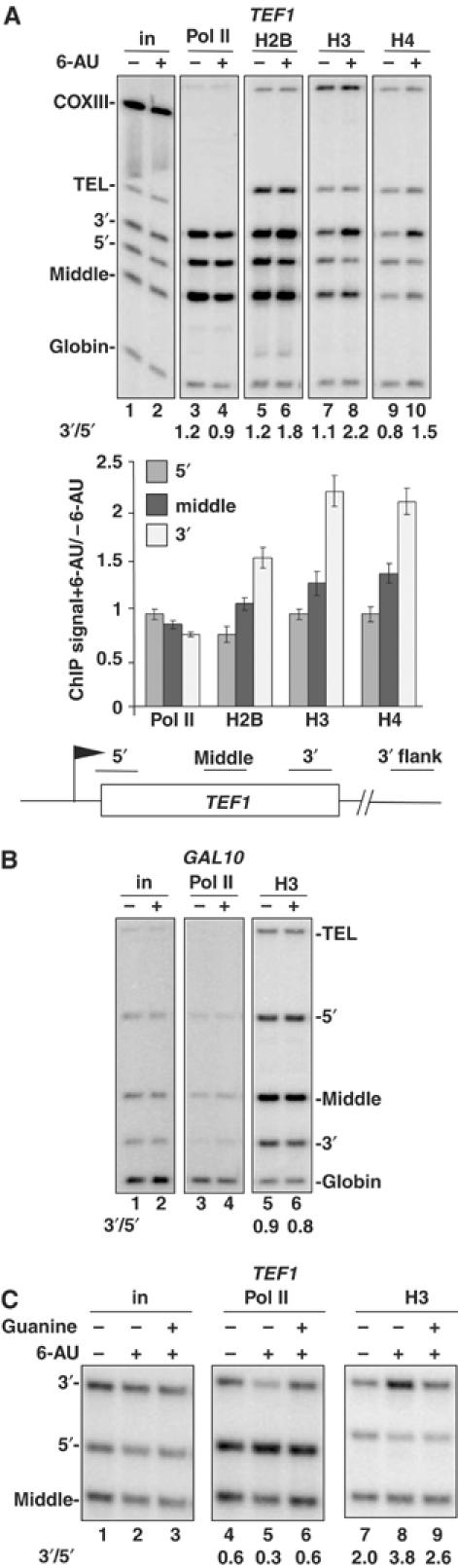
Inhibition of transcription by 6-AU alters the 5′-3′ distribution of histones within a gene. (A) Treatment with 6-AU results in an increase in histone occupancy at the 3′ end and a modest reduction at the 5′ end of TEF1. WT cells (BY4741) were treated without (−) or with (+) 6-AU. ChIP analysis was carried out using anti-pol II, -H2B, -H3 and -H4, and 32P-labeled PCR products of 5′, middle and 3′ region on TEF1 gene are shown. Mitochondrial COXIII, telomere VIR (TEL) and globin are controls. Note that the COXIII background signal is low relative to input chromatin (in). Ratios of ChIP signal +6-AU/−6-AU normalized to input are graphed in the bottom panel with mean and standard deviations from three different WT strains: BY4741, DY103 and HTB1. (B) 6-AU specifically affects histone occupancy on transcribed sequences. 6-AU had little effect on H3 occupancy in the nontranscribed gene GAL10 ORF (lanes 5 and 6) or telomere. 3′/5′ ratios of normalized ChIP signals are shown. (C) The 5′-3′ redistribution of pol II and histones caused by 6-AU is partially reversed by guanine. DY108 (rpb2-10Δdst1) was treated with 6-AU or 6-AU+guanine. Note that guanine restored pol II density (lanes 5 and 6) and reversed the increase in H3 occupancy at the TEF1 3′ end (lanes 8 and 9).
Acute 6-AU treatment caused rearrangement of chromatin on the TEF1 ORF marked by increased crosslinking of H2B, H3 and H4 at the 3′ end relative to untreated controls (Figure 1A, graph). 6-AU also reduced crosslinking of H2B, H3 and H4 at the 5′ end of TEF1 (Figure 1A, compare lanes 5, 7 and 9 with 6, 8 and 10). Whether or not related mechanisms are involved in histone displacement at the 5′ end and histone deposition at the 3′ end in response to 6-AU is not known. As a convenient way of quantifying this response, we calculated the ratio of ChIP signals (normalized to input) at the 3′ end relative to the 5′ end before and after 6-AU treatment (Figure 1). The net effect is a significant increase in the 3′/5′ ratio of histone crosslinking in response to 6-AU. The fact that histone crosslinking can change in opposite directions in different parts of the same ORF indicates that nucleosome loading is not necessarily equal along a gene. This effect of 6-AU on histone crosslinking is specific to transcribed sequences with little or no effect in the 3′ flanking sequence of TEF1, telomere VIR or the repressed GAL10 gene (Figures 1B and 3A). The results of H2B, H3 and H4 crosslinking to the TEF1 5′ and 3′ ends + and −6-AU in five wild-type (WT) strains are summarized in Table I. H3 and H4 crosslinking at the 5′ end was reduced to 74 and 82% of control values on average after 1 h in 6-AU (Table I). Reduction of H2B crosslinking at the 5′ end in response to 6-AU was observed in some (Figure 1A) but not all strains. 6-AU elicited a marked increase in H2B, H3 and H4 crosslinking at the 3′ end of TEF1 by about 180% on average (Table I). Similar effects of 6-AU on histone occupancy were observed in 13 additional mutants affecting chromatin and transcription (isw1, isw2, ioc2, ioc3, ioc4, htz1, swr1, rtf1, bre1, snf2, set1, htb1k123R, and the rpb2-10, dst1 double mutant) (Supplementary Figures 1C, 4B, C and 5B, and data not shown). The coordinated increase in histone crosslinking at the 3′ end of TEF1 when transcription is inhibited by 6-AU is consistent with deposition of new nucleosomes. By comparison, the deposition of histones following repression of the GAL1 gene by addition of glucose resulted in a 67% increase in H3 crosslinking (Supplementary Figure 1B, lanes 1–4).
Figure 3.
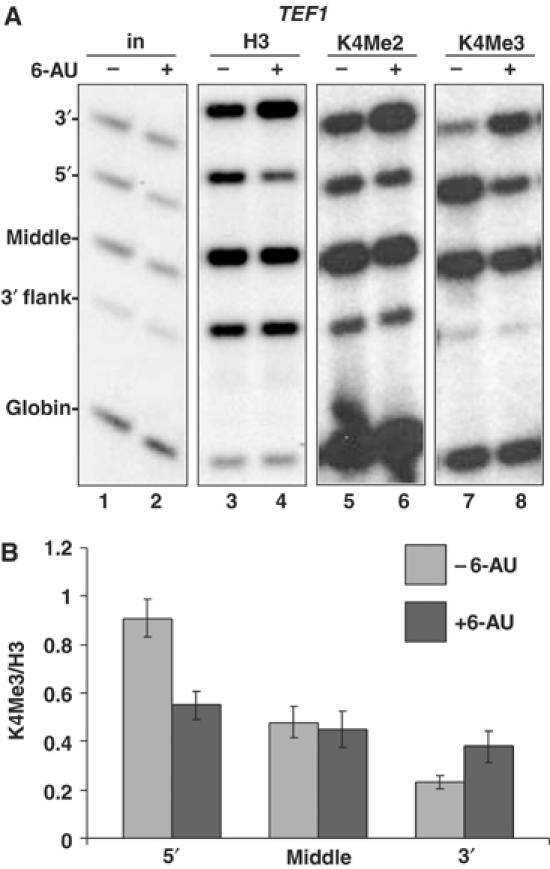
Inhibition of transcription by 6-AU increases H3K4Me3 at the 3′ end of TEF1 gene. (A) DY103 cells were treated without (−) and with (+) 6-AU for 1 h and ChIP analysis was carried out using anti-H3, -H3K4Me2 and -H3K4Me3. 32P-labeled PCR products of 5′, middle and 3′ region in TEF1 gene are shown. Note that H3K4Me3 crosslinking decreased at the 5′ end and increased at the 3′ end (lanes 7 and 8) whereas H3K4Me2 did not decrease at the 5′ end with 6-AU treatment (lanes 5 and 6). (B) 6-AU reduces K4 trimethylation of H3 at the 5′ end and increases it at the 3′ end of TEF1. ChIP signals for H3K4Me3 relative to total H3 were graphed from seven independent experiments: three in DY103 and one each in BY4741, W303mycSet1, W1588-4C and HTB1.
Table 1.
Histone occupancy changes within TEF1 in response to 6-AU
| H2B | H3 | H4 | |
|---|---|---|---|
| TEF1 5′+6-AU/−6-AU | 94±12% | 74±10% | 82±6% |
| TEF1 3′+6-AU/−6-AU | 171±26% | 182±36% | 187±25% |
| Inhibition of transcription by 6-AU increases histone occupancy at the 3′ end and decreases it at the 5′ end of TEF1. Histone ChIP signals normalized to input were determined before and after 1 h treatment with 6-AU. Histone occupancy changes at the 5′ and 3′ ends are expressed as ratios of the ChIP signals normalized to input +6-AU/−6-AU. H2B and H4 data are from 10 PCRs from eight experiments in four strains (BY4741, DY103, W1588-4C and HTB1); H3 data are from 12 PCRs from 10 experiments in five strains (BY4741, W303mycSet1, DY103, W1588-4C and HTB1). | |||
Restoring cellular GTP by adding guanine to 6-AU-containing medium reversed the loss of pol II at the 3′ end of TEF1 (Figure 1C, lanes 5 and 6). DY108 (rpb2-10 dst1) was chosen for this experiment because of the relatively large drop in pol II density at the 3′ end in this mutant in 6-AU (lanes 4 and 5). Restoration of a normal pol II distribution by guanine addition partially reversed the 5′-3′ shift in histone distribution along TEF1 that is normally caused by 6-AU (Figure 1C, lanes 8 and 9). This result strongly suggests that impairment of pol II transcription due to reduced NTP concentrations caused the redistribution of histones along the TEF1 ORF in 6-AU.
6-AU also increased crosslinking of H2B, H3 and H4 on the PMA1 and ASC1 genes (Figure 2). Unlike TEF1, 6-AU reduced pol II density at both 5′ and 3′ ends of these genes (Figure 2A and B, lanes 3 and 4) whereas histone occupancy increased by approximately 20–75% throughout the length of these genes although the greatest changes still occurred at promoter distal regions (Figure 2A and B, lanes 5–10).
Figure 2.
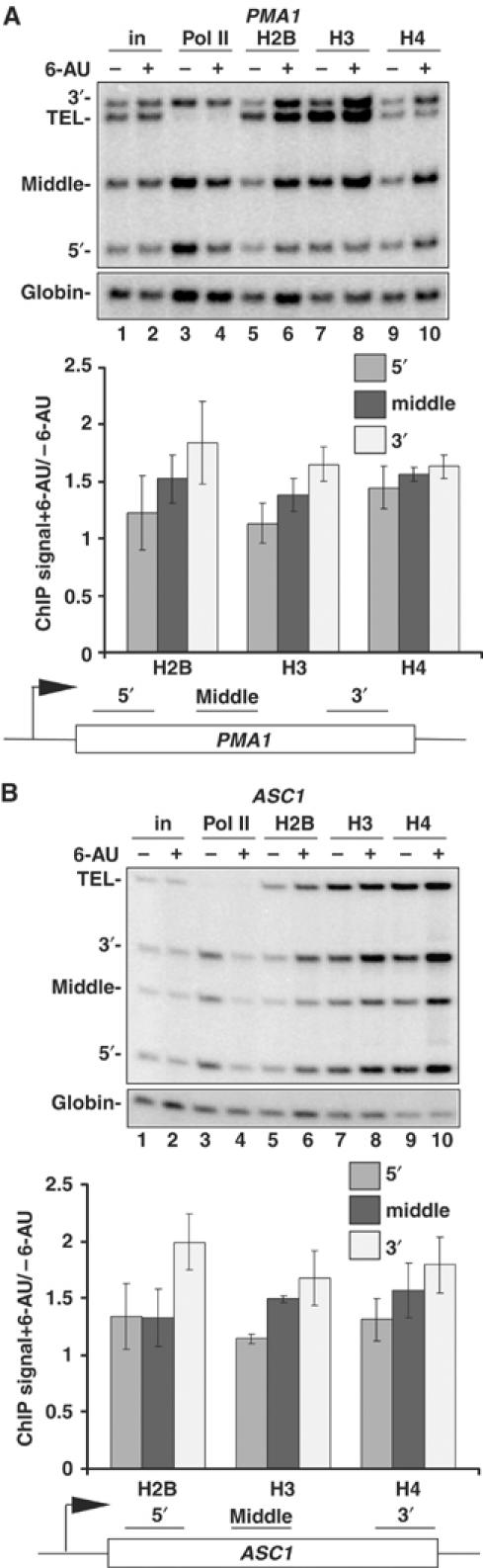
6-AU-induced transcription inhibition increases histone occupancy within the PMA1 and ASC1 genes. DY103 cells were treated without (−) or with (+) 6-AU. ChIP analysis was carried out using anti-pol II, -H2B, -H3 or -H4, and PCR products of 5′, middle and 3′ region on PMA1(A) or ASC1(B) are shown. TEL and globin are controls. Ratios of ChIP signals normalized to input and the globin control + and −6-AU were determined. Means and standard deviation from three PCR determinations are shown.
6-AU causes a 5′-3′ shift in the domain of K4 trimethylated H3
We investigated whether inhibition of transcription by 6-AU also affected the 5′-3′ distribution of H3K4 methylation. Consistent with previous observations (Ng et al, 2003), H3K4Me3 localized preferentially to the 5′ end and middle of the TEF1 ORF and not the 5′ or 3′ flanking sequences (Figure 3A, lane 7, and data not shown). Addition of 6-AU caused a remarkable shift in the distribution of H3K4Me3 along the TEF1 gene. Within 1 h, H3K4Me3 decreased at the 5′ end and increased at the 3′ end (Figure 3A, lanes 7 and 8). Similar results were obtained with homemade anti-K4Me3 antisera from two rabbits and a commercial antiserum (Supplementary Figure 1C). In five strains (BY4741, W303mycSet1, DY103, W1588-4C, HTB1), the average ratio of H3K4Me3/total H3 at 3′ end of TEF1 increased by a factor of 1.7 (0.22±0.026 versus 0.38±0.066, P=0.0005; Figure 3B) in 6-AU. Conversely, at the 5′ end, 6-AU reduced H3K4Me3 relative to total H3 by 1.7-fold (0.91±0.10 versus 0.55±0.06, P=0.001; Figure 3B). The chromatin response to transcriptional inhibition by 6-AU differs from that observed during normal repression of transcription of the GAL1 gene. Although histones are deposited at the GAL1 3′ end during glucose repression (Lee et al, 2004; Schwabisch and Struhl, 2004), they are not hypermethylated on H3K4 (Supplementary Figure 1B, lane 4 and graph).
Unlike H3K4Me3, the 5′-3′ distribution of H3K4Me2 is low at the 5′ end and high at the 3′ end of TEF1 consistent with previous observations (Ng et al, 2003). Like H3K4Me3, H3K4Me2 also increased at the 3′ end in 6-AU, but in contrast, little or no decrease in K4Me2 occurred at the 5′ end (Figure 3A, lanes 5 and 6). This difference between di- and trimethylated H3K4 (Figure 3A, lanes 5–8) suggests a functional distinction between these two covalent marks.
One possible explanation for the shift in H3K4Me3 caused by 6-AU is that nucleosomes bearing this modification are preferentially removed at the 5′ end and that new nucleosomes, deposited at the 3′ end, are modified by H3K4 trimethylation. To determine if de novo H3K4 methylation could occur at the 3′ end of TEF1, we localized N-terminally myc-tagged Set1 by ChIP before and after 6-AU treatment. Previously, Set1 was found to be restricted to the 5′ ends of several genes in yeast (Ng et al, 2003); however, we observed that Set1 was enriched throughout the TEF1 ORF relative to the 3′ flank or telomere (Figure 4A, lanes 3 and 4; see also Figure 5C). A similar distribution of Set1 at both the 5′ and 3′ ends has also been found on other yeast genes (V Geli, personal communication). In 6-AU, Set1 decreased slightly at the 5′ end of TEF1 and increased slightly at the 3′ end; however, this small change is not sufficient to account for the large shift in H3K4Me3 toward the 3′ end. Rather, this result suggests that trimethylation activity of Set1 is specifically enhanced at the 3′ end when transcription is inhibited by 6-AU.
Figure 4.
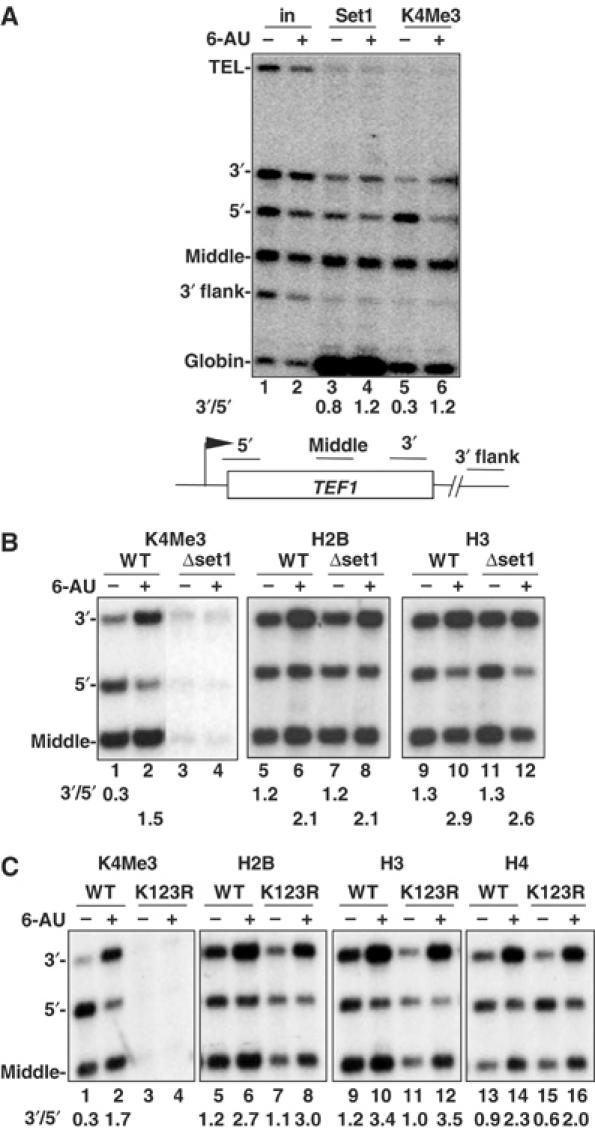
5′-3′ histone redistribution in 6-AU is independent of H3K4 methylation. (A) Set1 is localized throughout the TEF1 gene − and +6-AU. Set1myc W303 cells were treated without (−) or with (+) 6-AU and analyzed by ChIP using anti-myc and -H3K4Me3. A small enrichment of Set1 within TEF1 relative to the 3′ flank and telomere was observed. 3′/5′ ratios of normalized ChIP signals are shown. Note that stronger globin signals that appear in lanes 3 and 4 are due to larger amount of samples analyzed. (B) Set1 is not required for the 5′-3′ repositioning of histones on TEF1 in 6-AU. Isogenic WT (DY103) and Δset1 (DBY531) strains were treated without (−) or with (+) 6-AU, and analyzed by ChIP with the indicated antibodies. H2B was immunoprecipitated with antibody against N-terminal Flag epitope. Although there is no methylated H3K4 in Δset1 (lanes 3 and 4), 5′-3′ redistribution of H2B and H3 still occurred as shown by the increased 3′/5′ ratios of ChIP signals with 6-AU treatment (lanes 7, 8, 11 and 12). (C) H2B K123 ubiquitylation is not required for the 5′-3′ repositioning of histones on TEF1 with 6-AU treatment. Isogenic HTB1 and htb1K123R cells were treated without (−) or with (+) 6-AU and analyzed by ChIP with antibodies as indicated. 3′/5′ ratios of ChIP signals show that the 5′-3′ redistribution of histones H2B, H3 and H4 with 6-AU treatment is approximately equivalent in WT and htb1K123R although the mutation abolished methylation of H3K4 (lanes 3 and 4).
Figure 5.
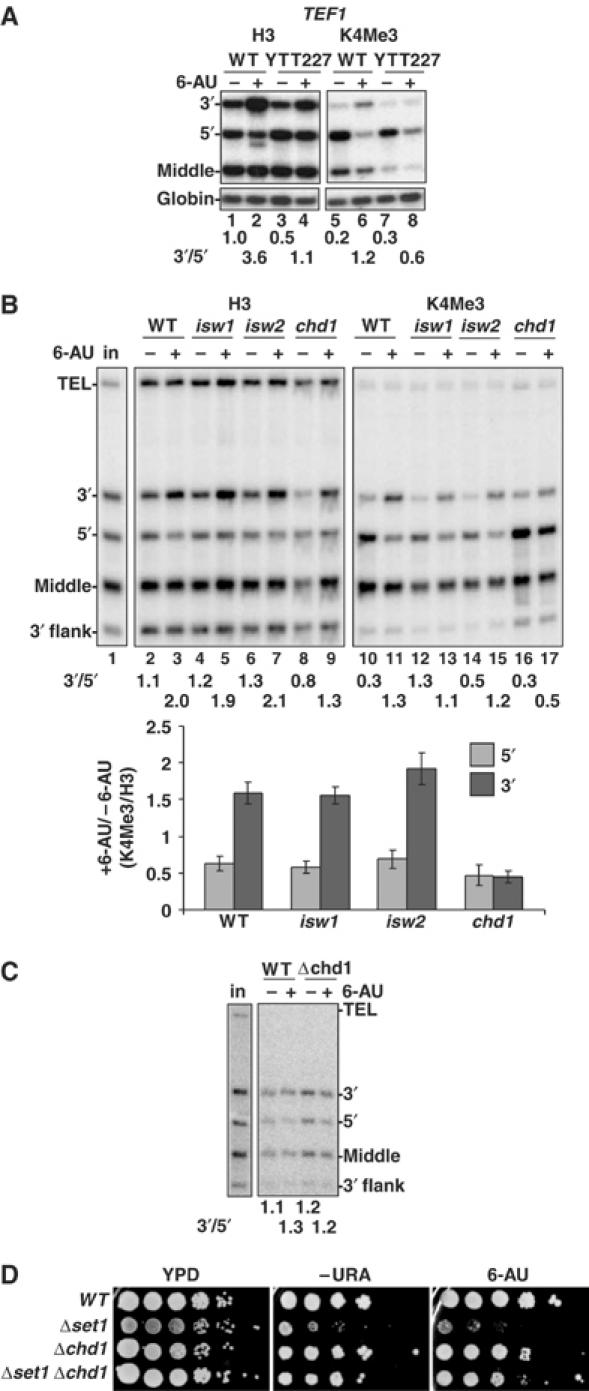
Chd1 is required for 6-AU-induced H3K4 trimethylation at the 3′ end of TEF1 gene. (A) Increased H3K4 trimethylation at the TEF1 3′ end with 6-AU treatment is abolished in Δisw1Δisw2Δchd1 triple mutant (YTT227). WT (W1588-4C) and mutant (YTT227) cells were treated without (−) or with (+) 6-AU and ChIP analysis was carried out with anti-H3 and -H3K4Me3. 3′/5′ ratio of normalized ChIP signals is shown. The loss of H3 at the 5′ end and the increase of H3K4Me3 at the 3′ end in 6-AU are both inhibited in the mutant (lanes 3, 4, 7 and 8). (B) Δchd1 but not Δisw1 or Δisw2 disrupts the chromatin response to 6-AU. ChIP analysis was carried out as in panel A with cells with single deletion of ISW1, ISW2 or CHD1. The enhanced H3K4 trimethylation normalized to total H3 at the 3′ end in 6-AU is abolished only in Δchd1 (lanes 16 and 17 and graph). Results from three determinations are summarized in the graph. (C) Set1 localization is independent of Chd1. Set1 localization in BY4741 (WT) and Δchd1 cells was determined by ChIP analysis with monoclonal anti-Set1. 3′/5′ ratios of normalized ChIP signals are shown. (D) Δchd1 suppresses the growth defect of Δset1. Dilution series of isogenic strains WT (BY4741), Δchd1, Δset1 (DBY804) and Δset1Δchd1 (DBY805) were plated on YPD, −Ura, and −Ura+100 μg/ml 6-AU at 25°C.
The 5′-3′ redistribution of histones in 6-AU is not dependent on H3K4 methylation
We asked whether H3K4 methylation is required for the 5′-3′ redistribution of histones in 6-AU. Trimethylation of H3K4 at the 3′ end of TEF1 in 6-AU was abolished in a Δset1 mutant, confirming that Set1 is required for K4 trimethylation at both ends of the gene − and +6-AU (Figure 4B, lanes 1–4). The net recruitment of histones H2B and H3 to the 3′ end of TEF1 in 6-AU was unaffected by SET1 deletion (Figure 4B, lanes 5–12).
We determined whether ubiquitylation of H2B was required for 5′-3′ histone redistribution in 6-AU using a K123R mutant that cannot be ubiquitylated (Robzyk et al, 2000). No H3K4Me3 was detected in the K123R mutant + or −6-AU (Figure 4C, lanes 3 and 4). H3, H4 and H2B crosslinking increased in the middle and 3′ end of TEF1 in the H2B K123R mutant treated with 6-AU as in WT (Figure 4C). bre1 and rtf1 mutants, which lack H3K4Me3, also repositioned histones toward the 3′ end of TEF1 gene in 6-AU (data not shown). In summary, deposition of histones in promoter distal regions of TEF1 when transcription was inhibited by 6-AU does not require either H2BK123 ubiquitylation or H3K4 methylation.
The chromatin response to 6-AU was also examined in strains bearing deletions of the chromatin remodeling factors Snf2, Ioc2, Ioc3 and Ioc4, the H2A variant Htz1, and its assembly factor Swr1, but no differences from WT were observed (Supplementary Figure 1D and E, and data not shown). Nor did we observe skewing of H3K4Me3 or pol II toward the 3′ end of TEF1 or PMA1 in isw1 or ioc2 mutants prior to 6-AU treatment in contrast to previous observations on MET16 (Morillon et al, 2003) (Supplementary Figure 1E and F; W Luo and D Bentley, unpublished).
Chd1 is required for H3K4 trimethylation at the 3′ end in 6-AU
Unlike other mutants we examined, the isw1, isw2, chd1 triple deletion YTT227 (Tsukiyama et al, 1999) differed from WT in both the 5′-3′ distribution of total H3 and H3K4Me3 in response to 6-AU. The 3′:5′ ratio of total H3 on TEF1 was reduced in the triple mutant relative to WT − and +6-AU (Figure 5A, lanes 1–4). When treated with 6-AU, the shift in H3K4Me3 to the 3′ end observed in the WT (Figure 5A, lanes 5 and 6) was almost abolished in the triple mutant (Figure 5A, lanes 7 and 8). We asked whether a single mutation could explain this failure to add Me3 to H3K4 at the 3′ end in 6-AU. No substantial defect in the distribution of total H3 or H3K4Me3 occurred when ISW1 or ISW2 was deleted (Figure 5B) but remarkably, deletion of CHD1 prevented the shift of H3K4Me3 to the 3′ end of TEF1 in 6-AU. In contrast to WT (Figure 5B, lanes 10 and 11), H3K4Me3 remained mostly at the 5′ end in chd1 after 6-AU addition (Figure 5B, lanes 16 and 17) as it did in the chd1, isw1, isw2 triple mutant (Figure 5A, lanes 7 and 8). We conclude that Chd1 is required for accumulation of H3K4Me3 at the 3′ end of TEF1 when transcription was inhibited by 6-AU.
Δchd1 also reduced the 3′:5′ ratio of H3 crosslinking across TEF1 relative to WT (Figure 5B, compare lanes 2 and 3 with 8 and 9) similar to the chd1, isw1, isw2 triple mutant (Figure 5A, lanes 1–4) implying that Chd1 may promote nucleosome assembly at the 3′ end of this gene. Although Chd1 stimulates H3 acetylation by the SLIK complex (Pray-Grant et al, 2005), there was no reduction of H3 K9/14 acetylation on the TEF1 ORF in Δchd1 − or +6-AU (data not shown).
Consistent with the 6-AU resistance of chd1 mutants (Woodage et al, 1997), TEF1 mRNA was not diminished by 6-AU treatment (1 h) in this strain, whereas it fell by 2.2-fold on average (n=3) relative to a control mRNA in the isogenic WT strain (BY4741) as determined by multiplex RT–PCR (data not shown). The fact that chd1 mutants maintain the normal distribution of high H3K4Me3 at the 5′ end and low at the 3′ end in 6-AU, therefore, correlates with maintenance of TEF1 mRNA levels and 6-AU-resistant growth.
We asked whether failure to trimethylate H3K4 at the 3′ end of TEF1 in Δchd1 cells treated with 6-AU correlated with loss of Set1 from this region of the gene. Set1 ChIP with a monoclonal antibody in WT and Δchd1 showed that it was relatively evenly distributed over the TEF1 coding region in both strains + and −6-AU (Figure 5C) as we observed previously (Figure 4A). We suggest that although Set1 is present at the 3′ end of TEF1 in WT cells, it is apparently not active until transcription is inhibited. CHD1 deletion appears to prevent activation of the Set1 K4 trimethylation activity in 6-AU.
The functional relationship between Chd1 and Set1 was further investigated by determining if they interact genetically. We compared growth of isogenic single and double deletions of SET1 and CHD1. Consistent with previous results, Δset1 inhibited growth especially on minimal medium (Briggs et al, 2001) and in 6-AU whereas Δchd1 had little effect. Remarkably, the double mutant showed strong suppression of the Δset1 growth defect by Δchd1 both with and without 6-AU (Figure 5D). We conclude that there is a significant genetic interaction between CHD1 and SET1 consistent with the fact that H3K4 trimethylation in 6-AU depends on Chd1 and with binding of Chd1 to H3K4Me2 (Pray-Grant et al, 2005).
Physiological stress and inactivation of pol II or TFIIE cause rapid chromatin reconfiguration on transcribed sequences
The chromatin response to 6-AU suggests that histone deposition is activated in response to reduced pol II activity. To investigate this idea further, we asked whether histone occupancy was affected when global pol II transcription was inhibited by glucose starvation or heat shock (Jona et al, 2000). Transferring cells to medium lacking glucose for 30 min reduced pol II on TEF1 by about 50% (Figure 6A, lanes 3 and 4) and increased H2B and H3 by 50–100% on the ORF but not the 3′ flanking region (Figure 6A, lanes 5–8). Unlike 6-AU, the extent of H3K4 trimethylation relative to total H3 did not increase significantly in glucose-starved cells (Figure 6A, lanes 7–10 and graph).
Figure 6.
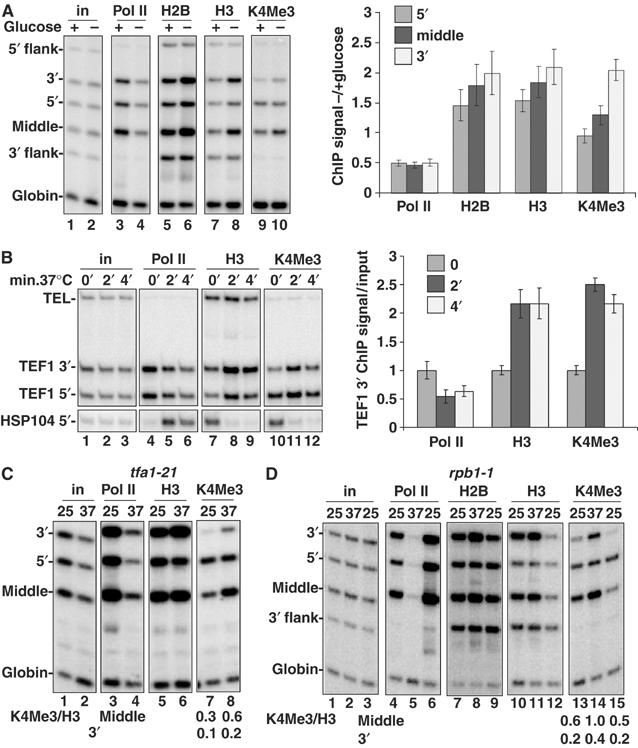
‘Transcriptional stress' causes chromatin reconfiguration of transcribed sequences. (A) Glucose starvation reduced pol II and increased histone occupancy in the TEF1 ORF but not in the nontranscribed 5′ or 3′ flank regions. DY103 cells were shifted from YPD (2% glucose (+)) to YP lacking glucose (−) for 30 min, and analyzed by ChIP. Mean ratios of normalized ChIP signals from − and +glucose were quantified and are indicated in the graph (n=3). (B) Heat shock rapidly stimulates histone deposition on TEF1 but eviction from HSP104. WT cells (DBY175) in YPD were shifted from 25 to 37°C for 2 and 4 min and then analyzed by ChIP. Mean ChIP signals for the TEF1 3′ end relative to values at 25°C are indicated in the graph (n=3). (C) TFIIE inactivation stimulates H3 deposition and H3K4 trimethylation on TEF1. tfa1-21 cells in YPD were shifted from 25 to 37°C for 1 h and analyzed by ChIP. 3′/5′ ratios of normalized ChIP signals for H3 before and after Tfa1 inactivation are 1.4 and 2.9 (lanes 5 and 6) indicating a 5′:3′ shift in histone occupancy. The ratios of H3K4Me3/H3 ChIP signals for the middle and 3′ end of TEF1 gene are shown. (D) Histone deposition and H3K4 trimethylation in response to inactivation of Rpb1 are reversible. rpb1-1 (DBY120) cells were shifted from 25 to 37°C for 1 h and then recovered at 25°C for 1 h. The ratios of H3K4Me3/H3 ChIP signals for the middle and 3′ end of TEF1 gene are shown. 3′/5′ ratios of normalized H3 ChIP signals for lanes 10–12 are 1.5, 2.4 and 1.0.
The chromatin response to transcriptional inactivation by heat shock was extremely rapid. An increase in total H3 and an equivalent increase in H3K4M3 throughout the TEF1 gene were detected within 2 min at 37°C (Figure 6B, lanes 7–12 and graph). This rapid increase in histone deposition was restricted to the transcribed region and not the 3′ flank (data not shown). As a control, we examined the HSP104 gene whose transcription is induced by heat shock (Figure 6B, lanes 4–6). A high level of H3K4Me3 was detected at the 5′ end of HSP104 prior to transcriptional activation (Figure 6B, lane 10). In contrast to TEF1, H3 and H3K4Me3 were eliminated from the HSP104 5′ end within 2 min at 37°C, consistent with eviction of nucleosomes bearing the K4Me3 mark (Figure 6B, lanes 7–12).
To determine if a chromatin rearrangement also occurs within a gene when a promoter-bound general transcription factor is inactivated, we analyzed the ts mutant in the large subunit of TFIIE, tfa1-21 (Sakurai et al, 1997). Inactivation of Tfa1 for 1 h depleted pol II throughout TEF1 (Figure 6C, lanes 3 and 4) and triggered net deposition of H3 at the 3′ end (Figure 6C, lanes 5 and 6). Inactivation of Tfa1 caused hypermethylation of H3K4 as shown by the two-fold increase in H3K4Me3 ChIP signals relative to total H3 in the middle and 3′ end of the gene (Figure 6C, lanes 7 and 8).
We also examined the effect on chromatin when pol II function was directly disrupted by the rpb1-1 ts mutation (Nonet et al, 1987). Within 1 h at 37°C, pol II was eliminated from the TEF1 gene (Figure 6D, lanes 4 and 5) and histone H2B and H3 occupancy increased specifically in the middle and 3′ end of the ORF but not in the 3′ flank (Figure 6D, lanes 7–12). Histone occupancy on TEF1 returned to a lower level within 1 h when transcription was restored by shifting rpb1-1 cells back to 25°C (Figure 6D, lanes 6, 9 and 12). Remarkably, inactivation and reactivation of Rpb1 were accompanied by rapid reversible changes in H3K4 trimethylation. Rpb1 inactivation caused a roughly two-fold increase in H3K4Me3 relative to total H3 in the middle and 3′ end of the gene (Figure 6D, lanes 10, 11, 13 and 14). Within 1 h of reactivating transcription by shifting the cells back to 25°C, the H3K4Me3 mark had been partly erased, as demonstrated by the approximately two-fold reduction in H3K4Me3 relative to total H3 (Figure 6D, lanes 14 and 15). We conclude that H3K4 trimethylation under these conditions is surprisingly dynamic.
Inactivation of CTD kinases increases H3K4 trimethylation at the 3′ end of TEF1
Both major CTD kinases in yeast, Kin28 and Ctk1, have been suggested to enhance elongation. We analyzed whether inactivation of these kinases affected chromatin structure within a gene. Δctk1 abolished Ser2 phosphorylation of the CTD (Figure 7A, lanes 5–8) as expected, although it did not markedly affect the 5′:3′ distribution of pol II on TEF1 + or −6-AU (Figure 7A, lanes 1–4). Loss of Ctk1 enhanced H3K4 trimethylation relative to total H3 at the 3′ end of TEF1 by over four-fold relative to WT (Figure 7A, lanes 9, 11, 13 and 15). The 3′ shifted distribution of H3K4 trimethylation on TEF1 in Δctk1 cells resembled the pattern in WT cells treated with 6-AU (Figure 7A, lanes 14 and 15). This observation is consistent with the idea that CTK1 deletion causes chronic ‘transcription stress', which alters the 5′-3′ distribution of H3K4 trimethylation.
Figure 7.
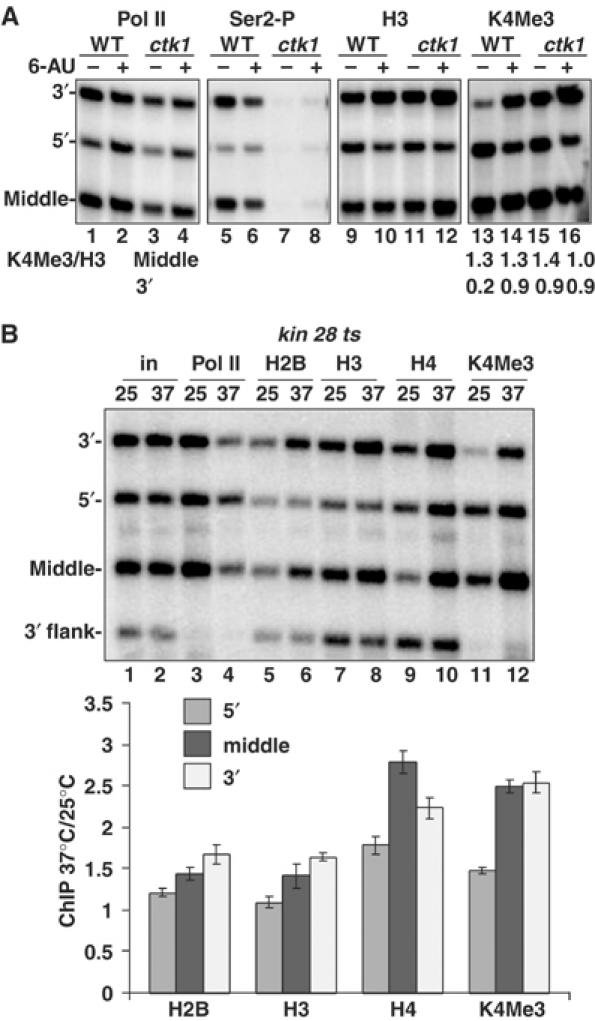
Inactivation of CTD kinases causes chromatin reconfiguration of transcribed sequences. (A) Ctk1 deletion causes a shift of H3K4Me3 toward the 3′ end of TEF1. Isogenic WT (BY4741) and Δctk1 cells were treated with 6-AU and analyzed by ChIP. Ser-2P is an antibody against Ser2 phosphorylated CTD. The ratios of H3K4Me3/H3 ChIP signals for the middle and 3′ end are shown. Note the elevated H3K4Me3 level at the 3′ end of TEF1 in untreated Δctk1 cells (lane 15) mimicking the effect of 6-AU on WT cells (lane 14). (B) Histone deposition and H3K4 trimethylation on TEF1 are elevated with Kin28 inactivation. kin28 ts3 cells in YPD were shifted from 25 to 37°C for 1 h. Loss of pol II (lanes 3 and 4) was associated with increased histone occupancy (lanes 5–10) particularly in the middle and 3′ regions of the TEF1 ORF relative to the 3′ flank. Note the elevated H3K4Me3 relative to total H3, particularly at the middle and 3′ end at 37°C (lanes 7, 8, 11 and 12 and graph). The graph shows mean ratios of normalized ChIP signals at 37°C relative to 25°C (n=3).
Inactivation of Kin28 in a ts mutant diminished overall pol II crosslinking, and increased the 5′:3′ ratio of polymerase density on TEF1, consistent with previous results (Figure 7B, lanes 3 and 4, compare 5′ versus 3′ signals) (Schroeder et al, 2000). Kin28 inactivation also elevated H2B, H3 and H4 occupancy on TEF1 particularly in the middle and 3′ end but not 3′ flank region (Figure 7B, lanes 5–10 and graph). This result is consistent with a recent report of greater histone occupancy on GAL genes after Kin28 inactivation (Schwabisch and Struhl, 2004). Unexpectedly, inactivation of Kin28, like Ctk1, also enhanced H3K4 trimethylation by about two-fold in the middle and 3′ end of the gene (Figure 7B, lanes 7, 8, 11 and 12 and graph). Whereas K4Me3 at the 5′ end was previously linked with CTD phosphorylation by Kin28 (Ng et al, 2003), our results showed that robust H3K4 trimethylation can occur at the 3′ end of the gene independently of Kin28 activity.
Discussion
Histone eviction and deposition in response to ‘transcriptional stress'
We investigated how acute inhibition of pol II transcription affects histone occupancy and modification within transcribed sequences. Chromatin structure was reconfigured, in some cases within a few minutes, in response to ‘transcriptional stress' caused by 6-AU, glucose starvation, heat shock or inactivation of Rpb1, Tfa1 or Kin28. All of these ‘stress' stimuli caused a rapid increase in crosslinking of H2B, H3 and H4 particularly in the promoter distal regions of the transcription unit but not in the 5′ or 3′ flanking regions. In the case of heat shock, increased histone deposition on TEF1 was detectable within 2 min (Figure 6B). It is unlikely that the altered histone ChIP signals we observed under ‘transcriptional stress' are caused by changes in epitope accessibility because very similar results were obtained with antibodies against natural epitopes and tags on three different histones H2B, H3 and H4 (Figure 4C, and data not shown).
Increased histone occupancy at the 3′ end of TEF1 in response to 6-AU was observed in 19 different strains. Analysis of five WT strains in Table I shows approximately equal increases in crosslinking of H2B, H3 and H4, suggesting that the effect involves deposition of whole nucleosomes rather than exchange of H2A/H2B dimers. Histone occupancy at the 3′ end of TEF1 increased by approximately 180% in 6-AU (Table I), suggesting that nucleosome loading almost doubles on this portion of the gene. The factors responsible for depositing nucleosomes within the gene under ‘transcriptional stress' remain unknown. The fact that this response can occur within a few minutes may indicate that the histones deposited are already localized close to the gene. One possible function of nucleosome deposition in response to ‘transcriptional stress' is to prevent spurious transcriptional initiation events within genes by repackaging DNA that has been exposed by transcription into nucleosomes (Kaplan et al, 2003; Mason and Struhl, 2003). 6-AU also consistently decreased H3/H4 occupancy at the 5′ end of TEF1 by about 20% (Table I). This effect could be related to polymerase stalling at the promoter or in the 5′ end of the transcribed sequence.
Impaired pol II transcription and Chd1-dependent H3K4 trimethylation by Set1
H3K4 trimethylation has been viewed as a signature mark of highly transcribed genes (Santos-Rosa et al, 2002), which is placed exclusively in the 5′ region downstream of the promoter (Ng et al, 2003). Contrary to this view, we show that the H3K4Me3 mark is appended to nucleosomes in the 3′ region of TEF1 when pol II function is impaired by 6-AU, or inactivation of Rpb1, Tfa1, Kin28 and Ctk1 (Figures 3 and 5, 6 and 7). In five strains (BY4741, W303mycSet1, DY103, W1588-4C, HTB1), the average ratio of H3K4Me3/total H3 at the 3′ end of TEF1 increased by a factor of 1.7 (Figure 3B) after 1 h in 6-AU. 6-AU-induced H3K4 trimethylation at the TEF1 3′ end was dependent on Set1, Bre1, Rtf1 and ubiquitylation of H2B K123 (Figure 4, and data not shown). 6-AU also caused loss of H3K4Me3 at the 5′ end of the transcribed region (Figures 3, 5 and 7). The net result of these changes is to make H3K4 trimethylation higher at the 3′ end than at the 5′ end under ‘transcriptional stress' thereby reversing the normal 5′:3′ distribution of this modification. H3K4 trimethylation is therefore not simply restricted to the 5′ ends of genes that are being highly transcribed but is also rapidly added at 3′ ends under conditions of impaired transcription.
Although lysine methylation is usually thought of as a stable modification, we observed relatively rapid (within 1 h) removal of H3K4Me3 from the 5′ end of TEF1 in 6-AU (Figures 3, 5A and B) and from 3′ sequences when the rpb1-1 ts mutant was returned from 37 to 25°C (Figure 6D). K4Me3-modified H3 was also rapidly lost from the 5′ end of HSP104 when it was activated by heat shock (Figure 6B). Loss of K4 trimethylation is probably achieved by selective eviction of H3, H3/H4 complexes or whole nucleosomes bearing this modification. We cannot exclude the possibility however that an unknown demethylase is responsible for selective removal of methyl groups from trimethylated H3K4 in 6-AU-treated cells. Unlike H3K4Me3, we did not observe preferential loss of K4Me2 from the 5′ end of TEF1 in 6-AU (Figure 3A, lanes 5 and 6), suggesting a functional distinction between these two modifications. Nucleosomes marked by H3K4Me3 at the 5′ end could be selectively removed either by sliding or displacement in trans by chromatin-remodeling factors.
How is H3K4Me3 specifically enhanced at the 3′ end of the gene? Although 6-AU enhanced K4 trimethylation at promoter distal positions within the gene, it did not cause a major shift in the position of Set1, which was fairly evenly distributed over the TEF1 coding region (Figures 4A and 5C). We suggest that Set1 trimethylation activity is stimulated at 3′ positions in response to stimuli that impair pol II transcription through the gene. Set family proteins bind to RNA and single-stranded DNA (Krajewski et al, 2005) and the RNA binding domain of Set1 is important for its trimethylation activity (Schlichter and Cairns, 2005; V Geli, personal communication). Impaired transcriptional elongation may induce formation of abnormal nascent mRNPs (Jensen et al, 2004) that could interact with Set1 and activate trimethylation. The function of H3K4 trimethylation induced by ‘transcriptional stress' is unknown, but it may serve to promote rapid recovery of transcription when the cells return to permissive conditions.
The chromodomain protein Chd1 is specifically required for the increase in H3K4Me3 trimethylation at the 3′ end of TEF1 in 6-AU (Figures 5 and 8). Chd1 binds directly to the Paf complex subunit Rtf1 (Simic et al, 2003), which is necessary for H3K4 methylation (Krogan et al, 2003). Moreover, a Chd1 chromodomain binds to K4 dimethylated H3 N-terminal tails (Pray-Grant et al, 2005). CHD1 deletion also inhibits transcription termination and confers resistance to high concentrations of 6-AU (Woodage et al, 1997; Alen et al, 2002). Our results are consistent with the idea that when transcription is inhibited by 6-AU, Chd1 recognition of dimethylated K4 may promote conversion to the trimethylated form by Set1. A functional interaction between Chd1 and Set1 is strongly suggested by our observation (Figure 5D) that Δchd1 almost totally suppressed the growth defect of Δset1. One model consistent with these results is that methylation by Set1 normally prevents Chd1 from carrying out inappropriate chromatin remodeling or assembly that inhibits growth.
Figure 8.
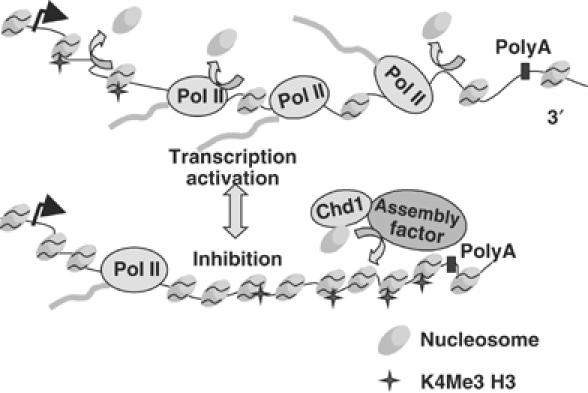
Model for chromatin reconfiguration in response to ‘transcriptional stress'. Impaired pol II transcription causes preferential recruitment of nucleosomes to promoter distal regions of the transcription unit, which can be accompanied by increased H3K4 trimethylation indicated with stars. This chromatin reconfiguration could be mediated by Chd1 and yet unidentified assembly factor(s). Inhibition of transcription by 6-AU also caused net loss of H3 and H3K4 trimethylation at the 5′ end.
Pol II as an effector of chromatin structure within genes
Similar changes in histone occupancy and H3K4 methylation were elicited by inhibitory stimuli that retain polymerases on the gene but probably hamper their movement (6-AU, kin28 and ctk1; Figures 1, 2, 3, 4 and 7) and by those that eliminate most polymerases from the gene (rpb1-1 and tfa1; Figure 6). These observations suggest that pol II stalling as well as outright release from the template may provoke local histone mobilization and H3K4 trimethylation. In this context, it is interesting to note that pol II itself is required for reconfiguration of the RNR3 promoter when it is activated by DNA damage (Sharma et al, 2003). Together, these results support the idea that on highly transcribed genes at least, there is a rapid dynamic balance between nucleosome displacement and replacement (Kaplan et al, 2003; Kristjuhan and Svejstrup, 2004; Lee et al, 2004; Schwabisch and Struhl, 2004) (Figure 8). We propose that inhibition of pol II activity under conditions of ‘transcriptional stress' rapidly resets the balance in favor of nucleosome replacement manifested by increased histone crosslinking and H3K4 trimethylation, especially at more 3′ positions within a gene. Our results suggest a new role for H3K4 trimethylation as a mark of chromatin through which pol II transcription has been impaired. In summary, our results show that in addition to the well-established control of pol II transcription by modulation of chromatin structure at promoters, nucleosome occupancy and histone modification within genes can be modulated as a consequence of altered pol II activity.
Materials and methods
Yeast strains
Strains used are listed in Table II. DBY531 was made by one-step disruption of SET1 in DY103. To make DBY804 (Δset1) and DBY805 (Δset1Δchd1), a SET1 URA3 plasmid (Briggs et al, 2001) was transformed into BY4741 and Δchd1. The chromosomal copy of SET1 was then disrupted by one-step integration of set1Δ::HIS3 and the SET1 plasmid was shuffled out by FOA selection.
Table 2.
Yeast strains used in this study
| Strain | Genotype | Reference |
|---|---|---|
| BY4741 | MATa his3Δ leu2Δ met15Δ ura3Δ | Euroscarf |
| isw1 | MATa his3Δ leu2Δ met15Δ ura3Δ isw1Δ∷kan | Euroscarf |
| isw2 | MATa his3Δ leu2Δ met15Δ ura3Δ isw2Δ∷kan | Euroscarf |
| ioc2 | MATa his3Δ leu2Δ met15Δ ura3Δ ioc2Δ∷kan | Euroscarf |
| ioc3 | MATa his3Δ leu2Δ met15Δ ura3Δ ioc3Δ∷kan | Euroscarf |
| ioc4 | MATa his3Δ leu2Δ met15Δ ura3Δ ioc4Δ∷kan | Euroscarf |
| chd1 | MATa his3Δ leu2Δ met15Δ ura3Δ chd1Δ∷kan | Euroscarf |
| Htz1 | MATa his3Δ leu2Δ met15Δ ura3Δ htz1Δ∷kan | Euroscarf |
| swr1 | MATa his3Δ leu2Δ met15Δ ura3Δ swr1Δ∷kan | Euroscarf |
| Rtf1 | MATa his3Δ leu2Δ met15Δ ura3Δ rtf1Δ∷kan | Euroscarf |
| Bre1 | MATa his3Δ leu2Δ met15Δ ura3Δ bre1Δ∷kan | Euroscarf |
| Snf2 | MATa his3Δ leu2Δ met15Δ ura3Δ snf2Δ∷kan | Euroscarf |
| Ctk1 | MATa his3Δ leu2Δ met15Δ ura3Δctk1Δ∷kan | Euroscarf |
| DBY120 | MATa ura3-52 rpb1-1 trp1∷HisG | McNeil et al (1998) |
| W303mycSet1 | MAT a, ura3, trp1, leu2, his3, SET1∷myc9TRP1 | V Geli |
| DBY531 | MATα ura3-52 leu2-3,112 his3-200delta set1∷kanMX4 rpb2-d297∷HIS3 [pRP214 RPB2 pRS316] isogenic with DY103 | This work |
| DY103 | MATα ura3-52 leu2-3,112 his3-200delta rpb2-d297∷HIS3 [pRP214 RPB2 pRS316] | Lennon et al (1998) |
| DY108 | MATα ura3-52 leu2-3,112 his3-200delta rpb2-d297∷HIS3 dst1∷hisG [pRP2 10L rpb2-10 pRS316] | Lennon et al (1998) |
| W1588-4C | MATa ade2-1 ura3-1 his3-11,15 trp1-1 leu2-3,112 , can1-100RAD5 | Tsukiyama et al (1999) |
| YTT227 | MATa ade2-1 ura3-1 his3-11,15 trp1-1 leu2-3,112 , can1-100RAD5 isw1∷ADE2 isw2∷LEU2 chd1∷TRP1 | Tsukiyama et al (1999) |
| HTB1 | MATa hta1-htb1Δ∷LEU2 hta2-htb2Δ pRS413HTA1-FlagHTB1 | Robzyk et al (2000) |
| htb1K123R | MATa hta1-htb1Δ∷LEU2 hta2-htb2Δ pRS413HTA1-Flaghtb1K123R | Robzyk et al (2000) |
| tfa1-21 | MATα tfa1∷ADE2 ura3-1 ade2-1 leu2-3,112 trp1-1 can1-100 [YCpADH1-HA-tfa1-21]] | Sakurai et al (1997) |
| kin28 | MAT a trp1 leu2 ura3 kin28ts3 his3 | Valay et al (1995) |
| DBY 175 | MAT a trp1 leu2 ura3 his3 RPB3∷RPB3(HA)3Kanr isogenic with kin28 | Schroeder et al (2000) |
| DBY804 | MAT a trp1 leu2 ura3 his3 set1Δ∷HIS3 isogenic with BY4741 | This study |
| DBY805 | MAT a trp1 leu2 ura3 his3 set1Δ∷HIS3, chd1Δ∷kan isogenic with DBY804 | This study |
ChIP assay
Saccharomyces cerevisiae grown at 30°C in SC-Ura 2% glucose to OD 0.8–1.0 were pelleted, resuspended in fresh SC-Ura plus 6-AU (100 μg/ml) and then grown for 1 h at 30°C. Strains were made URA+ with pRS316. Guanine was added to 100 μg/ml from a 10 mg/ml solution in 1 M NaOH. Controls had an equal volume of NaOH added. Crosslinking was in 1% formaldehyde for 15 min at room temperature (Schroeder et al, 2000). In some experiments, a human β-globin plasmid (20 pg) was added after washing and before elution of the immune complexes from the beads to serve as a recovery and gel loading control. Multiplex PCR reactions (20 cycles) were labeled with [32P]dCTP for the last two cycles. Products resolved on 6% denaturing acrylamide gels were quantified by Phosphorimager. PCR signals from immunoprecipitated samples were normalized to input and in some cases to the globin control. TEF1 primer pairs amplified the following fragments relative to the ATG: −435 to −667 (5′ flank); +1 to +175 (5′); +570 to +734 (middle); +1176 to +1374 (3′); +2400 to +2550 (3′ flank). The 5′, middle and 3′ primer pairs also amplify TEF2. GAL10 5′ +2–200, GAL10 middle +698–858; GAL10 3′ +1866–2006; ASC1 5′ +29–189; ASC1 middle +511–690; ASC1 3′ 1013–1226; PMA1 5′ +168–376; PMA1 middle +1101–1334; PMA1 3′ +2018–2290; HSP104 5′ +1 to +145.
Antibodies
Rabbit antisera were raised against the C-terminal peptides of yeast H2B (CKHAVSEGTRAVTKYSSSTQA), and human H3 (CIQLARRIRGERA), the acetylated N-terminus of yeast H4 (SGRGK(Ac)GGK(Ac)GLGK(Ac)GGAK(Ac)RHRKC), and yeast H3 TriMeK4 peptide (ARTK(Me3)QTARGGC). Peptides were coupled to KLH using the Imject kit (Pierce). Similar results were obtained with homemade and commercial anti-H3 and -H3K4Me3 (Abcam #ab1791 and #8580). The H4 antiserum was sequentially affinity purified on sulfo-link columns coupled to the acetylated peptide and then unacetylated peptide in order to purify the subpopulation that crossreacts with both forms. Rabbit anti-pol II pan CTD has been described (Schroeder et al, 2000). Anti-dimethyl H3K4 was from Upstate (#07-369), anti-myc 9E10 beads were from Santa Cruz, anti-Flag M2 was from Sigma and monoclonal anti-Set1 was a gift of M Cleary.
Supplementary Material
Supplementary Information 1
Supplementary Information 2
Supplementary Information 3
Acknowledgments
We thank H Sakurai, T Tsukiyama, R Young, M Osley, G Faye, V Geli, M Cleary and D Reines for yeast strains, plasmids and antibodies, K Struhl and V Geli for communicating results prior to publication and J Tyler, P Megee, T Blumenthal, J Jaehning and members of the Bentley lab for valuable discussions. We also thank B Soriano and the UCHSC microarray facility. Supported by NIH grant GM063873.
References
- Adkins MW, Howar SR, Tyler JK (2004) Chromatin disassembly mediated by the histone chaperone Asf1 is essential for transcriptional activation of the yeast PHO5 and PHO8 genes. Mol Cell 14: 657–666 [DOI] [PubMed] [Google Scholar]
- Ahmad K, Henikoff S (2002) The histone variant H3.3 marks active chromatin by replication-independent nucleosome assembly. Mol Cell 9: 1191–1200 [DOI] [PubMed] [Google Scholar]
- Alen C, Kent NA, Jones HS, O'Sullivan J, Aranda A, Proudfoot NJ (2002) A role for chromatin remodeling in transcriptional termination by RNA polymerase II. Mol Cell 10: 1441–1452 [DOI] [PubMed] [Google Scholar]
- Almer A, Rudolph H, Hinnen A, Horz W (1986) Removal of positioned nucleosomes from the yeast PHO5 promoter upon PHO5 induction releases additional upstream activating DNA elements. EMBO J 5: 2689–2696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeger H, Griesenbeck J, Strattan JS, Kornberg RD (2003) Nucleosomes unfold completely at a transcriptionally active promoter. Mol Cell 11: 1587–1598 [DOI] [PubMed] [Google Scholar]
- Briggs SD, Bryk M, Strahl BD, Cheung WL, Davie JK, Dent SY, Winston F, Allis CD (2001) Histone H3 lysine 4 methylation is mediated by Set1 and required for cell growth and rDNA silencing in Saccharomyces cerevisiae. Genes Dev 15: 3286–3295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavalli G, Thoma F (1993) Chromatin transitions during activation and repression of galactose-regulated genes in yeast. EMBO J 12: 4603–4613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dover J, Schneider J, Tawiah-Boateng MA, Wood A, Dean K, Johnston M, Shilatifard A (2002) Methylation of histone H3 by COMPASS requires ubiquitination of histone H2B by Rad6. J Biol Chem 277: 28368–28371 [DOI] [PubMed] [Google Scholar]
- Exinger F, Lacroute F (1992) 6-Azauracil inhibition of GTP biosynthesis in Saccharomyces cerevisiae. Current Genetics 22: 9–11 [DOI] [PubMed] [Google Scholar]
- Ezhkova E, Tansey WP (2004) Proteasomal ATPases link ubiquitylation of histone H2B to methylation of histone H3. Mol Cell 13: 435–442 [DOI] [PubMed] [Google Scholar]
- Gerber M, Shilatifard A (2003) Transcriptional elongation by RNA polymerase II and histone methylation. J Biol Chem 278: 26303–26306 [DOI] [PubMed] [Google Scholar]
- Hirschhorn JN, Brown SA, Clark CD, Winston F (1992) Evidence that SNF2/SWI2 and SNF5 activate transcription in yeast by altering chromatin structure. Genes Dev 6: 2288–2298 [DOI] [PubMed] [Google Scholar]
- Iizuka M, Smith MM (2003) Functional consequences of histone modifications. Curr Opin Genet Dev 13: 154–160 [DOI] [PubMed] [Google Scholar]
- Jensen TH, Boulay J, Olesen JR, Colin J, Weyler M, Libri D (2004) Modulation of transcription affects mRNP quality. Mol Cell 16: 235–244 [DOI] [PubMed] [Google Scholar]
- Jona G, Choder M, Gileadi O (2000) Glucose starvation induces a drastic reduction in the rates of both transcription and degradation of mRNA in yeast. Biochim Biophys Acta 1491: 37–48 [DOI] [PubMed] [Google Scholar]
- Jona G, Wittschieben BO, Svejstrup JQ, Gileadi O (2001) Involvement of yeast carboxy-terminal domain kinase I (CTDK-I) in transcription elongation in vivo. Gene 267: 31–36 [DOI] [PubMed] [Google Scholar]
- Kaplan CD, Laprade L, Winston F (2003) Transcription elongation factors repress transcription initiation from cryptic sites. Science 301: 1096–1099 [DOI] [PubMed] [Google Scholar]
- Kobor M, Greenblatt J (2002) Regulation of transcription elongation by phosphorylation. Biochim Biophys Acta 1577: 261. [DOI] [PubMed] [Google Scholar]
- Krajewski WA, Nakamura T, Mazo A, Canaani E (2005) A motif within SET-domain proteins binds single-stranded nucleic acids and transcribed and supercoiled DNAs and can interfere with assembly of nucleosomes. Mol Cell Biol 25: 1891–1899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristjuhan A, Svejstrup JQ (2004) Evidence for distinct mechanisms facilitating transcript elongation through chromatin in vivo. EMBO J 23: 4243–4252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogan NJ, Dover J, Wood A, Schneider J, Heidt J, Boateng MA, Dean K, Ryan OW, Golshani A, Johnston M, Greenblatt JF, Shilatifard A (2003) The Paf1 complex is required for histone H3 methylation by COMPASS and Dot1p: linking transcriptional elongation to histone methylation. Mol Cell 11: 721–729 [DOI] [PubMed] [Google Scholar]
- Lee CK, Shibata Y, Rao B, Strahl BD, Lieb JD (2004) Evidence for nucleosome depletion at active regulatory regions genome-wide. Nat Genet 36: 900–905 [DOI] [PubMed] [Google Scholar]
- Lennon JC III, Wind M, Saunders L, Hock MB, Reines D (1998) Mutations in RNA polymerase II and elongation factor SII severely reduce mRNA levels in Saccharomyces cerevisiae. Mol Cell Biol 18: 5771–5779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason PB, Struhl K (2003) The FACT complex travels with elongating RNA polymerase II and is important for the fidelity of transcriptional initiation in vivo. Mol Cell Biol 23: 8323–8333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason PB, Struhl K (2005) Distinction and relationship between elongation rate and processivity of RNA polymerase II in vivo. Mol Cell 17: 831–840 [DOI] [PubMed] [Google Scholar]
- McNeil JB, Agah H, Bentley D (1998) Activated transcription independent of the RNA polymerase II holoenzyme in budding yeast. Genes Dev 12: 2510–2521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller T, Krogan NJ, Dover J, Erdjument-Bromage H, Tempst P, Johnston M, Greenblatt JF, Shilatifard A (2001) COMPASS: a complex of proteins associated with a trithorax-related SET domain protein. Proc Natl Acad Sci USA 98: 12902–12907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morillon A, Karabetsou N, O'Sullivan J, Kent N, Proudfoot N, Mellor J (2003) Isw1 chromatin remodeling ATPase coordinates transcription elongation and termination by RNA polymerase II. Cell 115: 425–435 [DOI] [PubMed] [Google Scholar]
- Nagy PL, Griesenbeck J, Kornberg RD, Cleary ML (2002) A trithorax-group complex purified from Saccharomyces cerevisiae is required for methylation of histone H3. Proc Natl Acad Sci USA 99: 90–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng HH, Robert F, Young RA, Struhl K (2003) Targeted recruitment of Set1 histone methylase by elongating Pol II provides a localized mark and memory of recent transcriptional activity. Mol Cell 11: 709–719 [DOI] [PubMed] [Google Scholar]
- Nonet M, Scafe C, Sexton J, Young R (1987) Eukaryotic RNA polymerase conditional mutant that rapidly ceases mRNA synthesis. Mol Cell Biol 7: 1602–1611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pray-Grant MG, Daniel JA, Schieltz D, Yates JR III, Grant PA (2005) Chd1 chromodomain links histone H3 methylation with SAGA- and SLIK-dependent acetylation. Nature 433: 434–438 [DOI] [PubMed] [Google Scholar]
- Reinke H, Horz W (2003) Histones are first hyperacetylated and then lose contact with the activated PHO5 promoter. Mol Cell 11: 1599–1607 [DOI] [PubMed] [Google Scholar]
- Robinson KM, Schultz MC (2003) Replication-independent assembly of nucleosome arrays in a novel yeast chromatin reconstitution system involves antisilencing factor Asf1p and chromodomain protein Chd1p. Mol Cell Biol 23: 7937–7946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robzyk K, Recht J, Osley MA (2000) Rad6-dependent ubiquitination of histone H2B in yeast. Science 287: 501–504 [DOI] [PubMed] [Google Scholar]
- Roguev A, Schaft D, Shevchenko A, Pijnappel WW, Wilm M, Aasland R, Stewart AF (2001) The Saccharomyces cerevisiae Set1 complex includes an Ash2 homologue and methylates histone 3 lysine 4. EMBO J 20: 7137–7148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurai H, Ohishi T, Fukasawa T (1997) Promoter structure-dependent functioning of the general transcription factor iie in Saccharomyces-cerevisiae. J Biol Chem 272: 15936–15942 [DOI] [PubMed] [Google Scholar]
- Santos-Rosa H, Schneider R, Bannister AJ, Sherriff J, Bernstein BE, Emre NC, Schreiber SL, Mellor J, Kouzarides T (2002) Active genes are tri-methylated at K4 of histone H3. Nature 419: 407–411 [DOI] [PubMed] [Google Scholar]
- Schlichter A, Cairns BR (2005) Histone trimethylation by Set1 is coordinated by the RRM, autoinhibitory, and catalytic domains. EMBO J 24: 1222–1231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder S, Schwer B, Shuman S, Bentley D (2000) Dynamic association of capping enzymes with transcribing RNA polymerase II. Genes Dev 14: 2435–2440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwabisch M, Struhl K (2004) Evidence for eviction and rapid deposition of histones upon transcriptional elongation by RNA polymerase II. Mol Cell Biol 24: 10111–10117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma VM, Li B, Reese JC (2003) SWI/SNF-dependent chromatin remodeling of RNR3 requires TAF(II)s and the general transcription machinery. Genes Dev 17: 502–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simic R, Lindstrom DL, Tran HG, Roinick KL, Costa PJ, Johnson AD, Hartzog GA, Arndt KM (2003) Chromatin remodeling protein Chd1 interacts with transcription elongation factors and localizes to transcribed genes. EMBO J 22: 1846–1856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims RJ III, Nishioka K, Reinberg D (2003) Histone lysine methylation: a signature for chromatin function. Trends Genet 19: 629–639 [DOI] [PubMed] [Google Scholar]
- Strahl BD, Allis CD (2000) The language of covalent histone modifications. Nature 403: 41–45 [DOI] [PubMed] [Google Scholar]
- Sun ZW, Allis CD (2002) Ubiquitination of histone H2B regulates H3 methylation and gene silencing in yeast. Nature 418: 104–108 [DOI] [PubMed] [Google Scholar]
- Thiriet C, Hayes JJ (2005) Replication-independent core histone dynamics at transcriptionally active loci in vivo. Genes Dev 19: 677–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran HG, Steger DJ, Iyer VR, Johnson AD (2000) The chromo domain protein chd1p from budding yeast is an ATP-dependent chromatin-modifying factor. EMBO J 19: 2323–2331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukiyama T, Palmer J, Landel CC, Shiloach J, Wu C (1999) Characterization of the imitation switch subfamily of ATP-dependent chromatin-remodeling factors in Saccharomyces cerevisiae. Genes Dev 13: 686–697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valay J-G, Simon M, Dubois M-F, Bensaude O, Facca C, Faye G (1995) The KIN28 gene is required for RNA polymerase II mediated transcription and phosphorylation of the Rpb1p CTD. J Mol Biol 249: 535–544 [DOI] [PubMed] [Google Scholar]
- Woodage T, Basrai MA, Baxevanis AD, Hieter P, Collins FS (1997) Characterization of the CHD family of proteins. Proc Natl Acad Sci USA 94: 11472–11477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao T, Hall H, Kizer KO, Shibata Y, Hall MC, Borchers CH, Strahl BD (2003) Phosphorylation of RNA polymerase II CTD regulates H3 methylation in yeast. Genes Dev 17: 654–663 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information 1
Supplementary Information 2
Supplementary Information 3