

Abstract
ATP-sensitive K+ (KATP) channels, comprised of pore-forming Kir6.2 and regulatory SUR1 subunits, play a critical role in regulating insulin secretion. Binding of ATP to Kir6.2 inhibits, whereas interaction of MgATP with SUR1 activates, KATP channels. We tested the functional effects of two Kir6.2 mutations (Y330C, F333I) that cause permanent neonatal diabetes mellitus, by heterologous expression in Xenopus oocytes. Both mutations reduced ATP inhibition and increased whole-cell currents, which in pancreatic β-cells is expected to reduce insulin secretion and precipitate diabetes. The Y330C mutation reduced ATP inhibition both directly, by impairing ATP binding (and/or transduction), and indirectly, by stabilizing the intrinsic open state of the channel. The F333I mutation altered ATP binding/transduction directly. Both mutations also altered Kir6.2/SUR1 interactions, enhancing the stimulatory effect of MgATP (which is mediated via SUR1). This effect was particularly dramatic for the Kir6.2-F333I mutation, and was abolished by SUR1 mutations that prevent MgATP binding/hydrolysis. Further analysis of F333I heterozygous channels indicated that at least three SUR1 must bind/hydrolyse MgATP to open the mutant KATP channel.
Keywords: diabetes, KATP channel, Kir6.2, SUR1
Introduction
ATP-sensitive potassium (KATP) channels couple metabolism to electrical activity in many tissues by regulating K+ fluxes across the cell membrane. Increased metabolism leads to channel closure, membrane depolarization and electrical activity, and, conversely, metabolic inhibition opens KATP channels and suppresses electrical activity. KATP channels play many functional roles in the organism, but are of particular importance in regulating insulin secretion from pancreatic β-cells (Seino and Miki, 2003; Ashcroft and Rorsman, 2004). At substimulatory glucose concentrations, K+ efflux through open KATP channels keeps the β-cell membrane hyperpolarized, thus preventing opening of voltage-gated Ca2+ channels, Ca2+ influx and insulin secretion (Ashcroft and Rorsman, 1989). When β-cell metabolism increases in response to elevated plasma glucose, KATP channels close, leading to membrane depolarization, Ca2+ channel opening, Ca2+ influx and thereby exocytosis of insulin granules. Sulphonylurea drugs, which are used to treat type II diabetes, bypass metabolism and stimulate insulin secretion by binding to, and closing, KATP channels directly (Gribble and Reimann, 2003).
KATP channels are octameric complexes of two different proteins (Shyng and Nichols, 1997; Ashcroft and Gribble, 1998). Four inwardly rectifying K+ channel subunits form a tetrameric pore: in almost all tissues except vascular smooth muscle, Kir6.2 serves this role (Inagaki et al, 1995; Sakura et al, 1995). Each Kir subunit is associated with a regulatory sulphonylurea receptor (SUR) subunit, the SUR1 isoform being found in pancreatic β-cells (Aguilar-Bryan et al, 1995). Metabolic regulation of KATP channel activity is mediated by changes in the intracellular concentrations of adenine nucleotides, which interact with both Kir6.2 and SUR1 subunits. Binding of ATP or ADP to Kir6.2 produces channel inhibition (Tucker et al, 1997; Tanabe et al, 1999), whereas interaction of Mg-nucleotides with the nucleotide-binding domains (NBDs) of SUR stimulates channel activity (Nichols et al, 1996; Tucker et al, 1997). Thus in the presence of MgATP, channel activity is determined by the balance between these inhibitory and stimulatory effects.
Recently, mutations in Kir6.2 have been found to be a common cause of permanent neonatal diabetes mellitus (PNDM) (Gloyn et al, 2004; Sagen et al, 2004; Vaxillaire et al, 2004). Some mutations were associated with a more severe phenotype in which neonatal diabetes was accompanied by developmental delay, epilepsy or muscle weakness (we refer to this condition as DEND syndrome or severe disease). All patients show an impaired insulin secretory response to glucose, and those tested respond to sulphonylureas such as tolbutamide and glibenclamide with insulin release (Gloyn et al, 2004; Sagen et al, 2004). This suggests that their diabetes is caused by gain-of-function mutations that result in a larger β-cell KATP current at a given glucose concentration thereby reducing or abolishing glucose-dependent depolarization and Ca2+ influx. Consistent with this idea, the mutation R201H (which causes PNDM alone) produces a reduction in the ATP sensitivity of the KATP channel, and a larger resting KATP current when expressed in heterologous systems (Gloyn et al, 2004). Mutations that cause neonatal diabetes with neurological symptoms (e.g. I296L) cause a greater shift in ATP sensitivity and a larger increase in the resting KATP current, suggesting that non-pancreatic tissues are affected only when the reduction in ATP inhibition is substantial (Proks et al, 2004, 2005). More than 20 other PNDM mutations have been identified to date, which lie scattered throughout the cytosolic regions of the channel, but their effects on KATP channel function are unknown.
In this paper, we analyse the functional effects of the Y330C and F333I mutations, identified in Caucasian families from Norway, France and the USA (Sagen et al, 2004; Vaxillaire et al, 2004). As found for other PNDM mutations, all patients were heterozygotes. Patients with the Y330C mutation presented with blood glucose levels of 22–70 mM within 5 weeks of birth. In addition, one patient showed motor and mental retardation when young (Sagen et al, 2004) and another had hemiplegia (Vaxillaire et al, 2004). The patient with the F333I mutation presented with neonatal diabetes within 10 weeks of birth but had no additional symptoms (Sagen et al, 2004). She was successfully treated with sulphonylureas, and insulin therapy was discontinued after 6 months.
We show here that the Y330C and F333I mutations both impair the ability of ATP to block the KATP channel (at Kir6.2) and enhance channel activation by MgATP (via SUR1). These effects combine to produce a large reduction in the overall inhibition of the channel by MgATP. In the presence of 1 mM MgATP, the KATP current is larger for heterozygous Y330C channels than for heterozygous F333I channels, and much greater than for wild-type channels, consistent with the difference in clinical phenotypes. We analyse the molecular basis of the reduction in ATP inhibition and demonstrate that it is primarily due to impaired ATP binding/transduction (F333I) and/or secondary to a change in channel gating (Y330C). Both mutations also alter by modulation by SUR1, suggesting that this region of Kir6.2 may lie in close proximity to SUR1.
Results
Effects on whole-cell currents
We analysed the effects of the Y330C and F333I mutations on the metabolic regulation of the KATP channel by the two-electrode voltage-clamp method. In oocytes, wild-type KATP channels are normally closed by resting levels of intracellular ATP ([ATP]i) but they can be opened by the metabolic inhibitor azide, which lowers [ATP]i. Mutations that impair ATP sensitivity normally show an increased whole-cell current before azide addition, which reflects the fact that they are less blocked by resting levels of [ATP]i (Gloyn et al, 2004). Figure 1A shows that significant resting whole-cell KATP currents were present in oocytes expressing homomeric (hom) Y330C channels. These currents were further increased by azide, suggesting that the channel is not fully open at resting [ATP]i, and they were partially blocked by the sulphonylurea tolbutamide.
Figure 1.

(A) Whole-cell currents recorded from Xenopus oocytes coexpressing SUR1 and either Kir6.2, Kir6.2-F333I or Kir6.2-Y330C, as indicated, in response to voltage steps of ±20 mV from a holding potential of −10 mV. The bars indicate azide and tolbutamide (tolb) application. (B) Mean steady-state whole-cell currents evoked by a voltage step from −10 to −30 mV before (control, white bars) and after application of 3 mM azide (pale grey bars) and in the presence of 3 mM azide plus 0.5 mM tolbutamide (dark grey bars). The number of oocytes is indicated below the bars.
To simulate the heterozygous state, we coinjected a 1:1 mixture of wild-type and mutant Kir6.2, together with SUR1. The population of channels that result will contain a variable number of mutant subunits (between zero and four) in the Kir6.2 tetramer. We refer to this mixed channel population as heterozygous (het) channels. Resting hetY330C currents were greater than wild-type but much less than for homY330C channels (Table IA and Figure 1A). Like homY330C channels, however, they were further activated by azide, to approximately the same amplitude as wild-type channels. Tolbutamide (500 μM) substantially blocked the azide-activated current.
Table 1.
Mean data for wild-type and mutant Kir6.2/SUR1 and Kir6.2ΔC channels
| (A) Wild-type and mutant Kir6.2/SUR1 channels | ||||||
|---|---|---|---|---|---|---|
| Mutation | Irest (μA) | IC50 (μM), Mg-free | h, Mg-free | IC50 (μM), 2 mM Mg | h, 2 mM Mg | %I unblocked at 1 mM MgATP |
| Kir6.2 (WT) | 0.08±0.01 (n=8) | 10.6±1.8 (n=10) | 1.39±0.07 (n=10) | 16.7±2.6 (n=6) | 0.98±0.05 (n=7) | 3.7±0.8 (n=7) |
| Kir6.2-F333I (hetero) | 0.22±0.02 (n=17)*** | 22.8±4.6 (n=5)** | 1.01±0.04 (n=5)** | 39±4 (n=6)*** | 0.74±0.03 (n=6)*** | 10±0.06 (n=6)*** |
| Kir6.2-F333I (homo) | 2.66±0.29 (n=17)*** | 211±11 (n=6)*** | 0.98±0.05 (n=5)*** | NM | NM | NM |
| Kir6.2-Y330C (hetero) | 0.38±0.06 (n=12)*** | 19.7±2.0 (n=6)** | 1.08±0.05 (n=6) | 116±19 (n=6)*** | 0.67±0.02 (n=6)*** | 20±1 (n=6)*** |
| Kir6.2-Y330C (homo) | 1.46±0.25 (n=9)*** | 168±17 (n=11)*** | 1.45±0.11 (n=11) | 2.9±3.3 (n=7)*** | 0.80±0.04 (n=7)** | 71±3 (n=6)*** |
| (B) Wild-type and mutant Kir6.2ΔC channels | ||||||
|---|---|---|---|---|---|---|
| Mutation | IC50 (μM), Mg-free | h, Mg-free | ||||
| Kir6.2ΔC (WT) | 124±12 (n=7) | 1.13±0.03 (n=7) | ||||
| Kir6.2ΔC-F333I | 6450±523 (n=5)*** | 1.22±0.08 (n=8) | ||||
| Kir6.2ΔC-Y330C | 480±40 (n=6)*** | 0.95±0.06 (n=6)* | ||||
| Values represent mean values of resting whole-cell KATP current (Irest), ATP concentration producing half-maximal of the channel (IC50) and Hill coefficient (h). WT, wild type. NM, not measurable (as very ATP insensitive). Statistical significance against wild type is indicated by *P<0.05, **P< 0.01 and ***P<0.001. | ||||||
The results of similar experiments with hetF333I and homF333I channels are also shown in Figure 1. Resting currents for homF333I channels were substantially larger, and those for hetF333I channels slightly larger, than wild-type channels (Table IA). Consistent with the less severe phenotype, hetF333I resting currents were smaller than those of hetY330C channels.
Effects on ATP sensitivity in the absence of Mg2+
The ability of ATP to block wild-type and mutant channels was assessed by measuring concentration–inhibition curves in inside-out membrane patches. We first carried out experiments in the absence of Mg2+ to avoid the stimulatory effects of MgATP, which are mediated via SUR1 (Figures 2A and B). Both homF333I and homY330C channels were much less sensitive to ATP, being half maximally blocked (IC50) by 168 and 211 μM ATP, respectively, compared with 11 μM for wild-type channels (Table IA). Heterozygous channels were also significantly less sensitive to ATP, with IC50 equal to 20 μM (hetY330C) and 23 μM (hetF333I) (Figures 2A and B and Table IA).
Figure 2.

(A) Currents recorded in inside-out patches excised from Xenopus oocytes coexpressing SUR1 and either wild type Kir6.2, Kir6.2-F333I or Kir6.2-Y330C, as indicated, in response to 3 s voltage ramps from −110 to +100 mV. ATP (100 μM) was applied as indicated by the horizontal bars. (B) Mean relationship between [ATP] and KATP conductance (G), expressed relative to the conductance in the absence of nucleotide (Gc) for wild-type and mutant KATP channels. Experiments were carried out in the absence of Mg2+. The smooth curves are the best fit to the Hill equation (equation (4)), using the values given in Table IA. (a) Kir6.2/SUR1 (open symbols, n=10), heterozygous (semifilled symbols, n=6) and homomeric (filled symbols, n=11) Kir6.2-Y330C/SUR1 channels. (b) Kir6.2/SUR1 (open symbols, n=10), heterozygous (semifilled symbols, n=5), and homomeric (filled symbols, n=6) Kir6.2-F333I/SUR1 channels.
It is well established that even in the absence of Mg2+, SUR1 enhances the ATP sensitivity of the KATP channel (Tucker et al, 1997; Gribble et al, 1998). To determine if the change in ATP sensitivity of mutant channels is intrinsic to Kir6.2 or is conferred by SUR1, we measured the ATP sensitivity of Kir6.2ΔC, a C-terminally truncated Kir6.2 that expresses in the absence of SUR1 (Tucker et al, 1997). As Figure 3 shows, the ATP sensitivity of Kir6.2ΔC, expressed in the absence of SUR1, was also impaired by the F333I and Y330C mutations, the IC50 being 124, 6450 and 480 μM for wild-type, homF333I and homY330C channels, respectively (Table IB). This suggests that the altered ATP sensitivity of F333I and Y330C channels is, at least in part, intrinsic to Kir6.2 rather than due to impaired regulation by SUR1.
Figure 3.

Mean relationship between [ATP] and KATP conductance (G), expressed relative to the conductance in the absence of nucleotide (Gc) for Kir6.2ΔC (circles, n=7), Kir6.2ΔC-F333I (squares, n=5) and Kir6.2ΔC-Y330C (triangles, n=6) channels. SUR1 was not coinjected in these experiments. The smooth curves are the best fit of equation (4), using the values given in Table IB.
Molecular mechanism for reduced ATP sensitivity
The molecular mechanism by which PNDM mutations reduce KATP channel ATP sensitivity might involve impaired ATP binding, a failure of ATP binding to induce a change in channel gating (transduction) or an indirect effect that is secondary to changes in the intrinsic (unliganded) gating of the channel. As previously reported, mutations that stabilize the intrinsic open state of the channel result in reduced sensitivity to ATP (Trapp et al, 1998; Enkvetchakul et al, 2001; Proks et al, 2004).
We therefore examined the effect of the Y330C and F333I mutations on single-channel currents in the absence of ATP, where intrinsic gating can be assessed. Neither mutation had any effect on the single-channel current amplitude (Table II). In the case of F333I channels, the open probability in the absence of ligand (Po(0)) was also unaffected (Figure 4 and Table IIA). However, the Y330C mutation was associated with a marked increase in intrinsic Po(0) (Figure 4 and Table IIA). This suggests that whereas the F333I mutation impairs ATP binding/transduction, the Y330C mutation exerts its effect on ATP sensitivity both directly, via changes in ATP binding/transduction, and indirectly, via an increase in Po(0).
Table 2.
Mean single-channel data for wild-type and mutant Kir6.2/SUR1 and Kir6.2ΔC channels
| Mutation | Po(0) | i (pA) |
|---|---|---|
| (A) Wild-type and mutant Kir6.2/SUR1 channels | ||
| Kir6.2 (WT) | 0.27±0.04 (n=4) | 4.0±0.1 (n=4) |
| Kir6.2-F333I | 0.31±0.05 (n=6) | 3.9±0.2 (n=6) |
| Kir6.2-Y330C | 0.75±0.04 (n=8)*** | 4.0±0.1 (n=8) |
| (B) Wild-type and mutant Kir6.2ΔC channels | ||
| Kir6.2ΔC (WT) | 0.023±0.01 (n=8) | 3.7±0.1 (n=8) |
| Kir6.2ΔC-Y330C | 0.019±0.01 (n=8) | 3.7±0.2 (n=8) |
| Mean values of intrinsic open probability (Po(0)) and single-channel current amplitude (i), measured at −60 mV. WT, wild type. Statistical significance against wild type is indicated by *P<0.05, **P<0.01 and ***P<0.001. | ||
Figure 4.
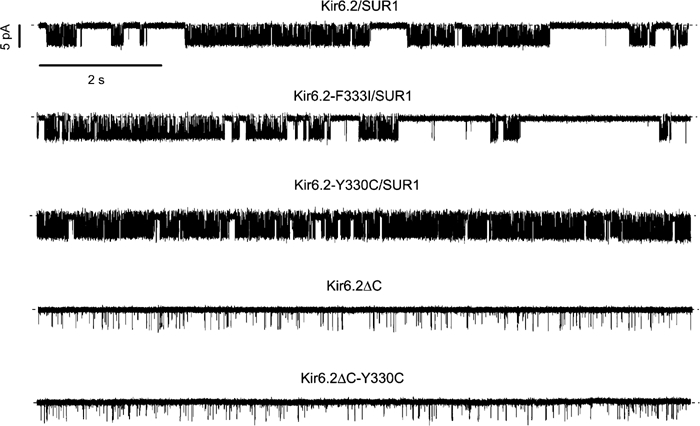
Single-channel currents recorded at −60 mV from inside-out membrane patches excised from oocytes expressing Kir6.2/SUR1, Kir6.2-F333I/SUR1, Kir6.2-Y330C/SUR1, Kir6.2ΔC and Kir6.2ΔC-Y330C. The dashed line indicates the zero current level.
To explore this question further, we used a simple kinetic model of the hetY330C channel (Proks et al, 2004). We assumed that the energy of the open state is simply the sum of the contributions of the four individual subunits in the channel (G0,j):
![]()
The IC50 of channels containing i mutant and 4−i wild-type subunits can be estimated as (for further details, see Proks et al, 2004)
![]()
where IC50,wt is the IC50 measured for wild-type channels, IC50,i is the IC50 for channels formed from i (0<i<4) mutant subunits and λ (equal to exp(G0,M−G0,wt)) is a factor reflecting the change in the Gibbs free energy of the open state of a single Kir6.2 subunit induced by the Y330C mutation. Given independent mixing of wild-type and mutant subunits (Shyng and Nichols, 1997; Markworth et al, 2000), the relative numbers of Kir6.2 subunits in the heteromeric channel will follow the binomial distribution. The value of IC50 for the heterozygous mixture (IC50,H) is then obtained from
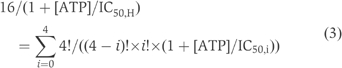
The value of IC50,H observed experimentally (∼20 μM) was smaller than that predicted from equation (3) (43 μM) (Figure 5).
Figure 5.

Simulated ATP dose-inhibition curves (dashed lines) for heteromeric Kir6.2-Y330C/SUR1 (left) and Kir6.2-F333I/SUR1 (right) channels. The predicted IC50 values are 43 μM (Kir6.2-Y330C/SUR1) and 25 μM (Kir6.2-F333I/SUR1). The data are the same as those in Figure 2.
Binomial analysis also predicts that mutations that impair ATP binding alone produce an ∼2-fold shift in the ATP sensitivity of the heterozygous channels. This is because ATP binding to a single subunit is sufficient to close the channel (Markworth et al, 2000), so that channels will exhibit a markedly reduced ATP sensitivity only when all four subunits are mutant (heteromeric channels will show a slightly reduced sensitivity, which depends on the number of mutant subunits they contain). According to this model, the predicted IC50,H for F333I is 25 μM, which is very close to the experimentally determined value of 23 μM (Figure 5).
We next examined the single-channel kinetics of Kir6.2ΔC-Y330C, expressed in the absence of SUR1. As Figure 4 shows, the intrinsic open probability of these channels was not different from Kir6.2ΔC (Table IIB). This suggests that the effect of the mutation on channel gating results from an altered interaction with SUR1 and is not intrinsic to Kir6.2.
Effects on ATP sensitivity in the presence of Mg2+
In order to isolate the effects of ATP on Kir6.2, it is essential to use Mg2+-free solutions, as described above. However, in β-cells, KATP channel activity is governed by the balance between ATP inhibition via Kir6.2 and Mg2+-nucleotide stimulation mediated by SUR. We therefore next explored the ATP sensitivity of wild-type and mutant channels in the presence of 2 mM Mg2+. Under these conditions, the differences in ATP sensitivity were more marked (Figures 6A and B and Table IA). Homomeric mutant channels displayed a very low sensitivity to MgATP, the IC50 for inhibition of homY330C channels being 2.8 mM. In striking contrast, MgATP caused a dramatic stimulation of homF333I channels (Figure 6A). However, both heterozygous Y330C and F333I channels were blocked by MgATP: the IC50 for the hetY330C channel (116 μM) was about seven-fold greater than wild type (16 μM), and that for the hetF333I channel (39 μM) was about 2.5-fold greater.
Figure 6.

(A) Currents recorded in inside-out patches excised from Xenopus oocytes coexpressing SUR1 and either wild type, Kir6.2-F333I or Kir6.2-Y330C as indicated, in response to voltage ramps from −110 to +100 mV. MgATP (1 mM) was applied as indicated by the horizontal bars. (B) Mean relationship between [MgATP] and KATP conductance (G), expressed relative to the conductance in the absence of nucleotide (Gc) for wild-type and mutant KATP channels. The smooth curves are the best fit to equation (4), using the values given in Table IA. (a) Kir6.2/SUR1 (open symbols, n=6), heterozygous (semifilled symbols, n=6) and homomeric (filled symbols, n=7) Kir6.2-Y330C/SUR1 channels. (b) Kir6.2/SUR1 (open symbols, n=6) and heterozygous Kir6.2-F333I/SUR1 (semifilled symbols, n=6) channels. The shaded bar indicates the predicted range of physiological ATP concentrations found in β-cells.
It is notable that in the physiological range of [MgATP]i (1–5 mM), there is a significant current for both heterozygous and homomeric mutant channels, but not for wild-type channels. At 1 mM ATP, this amounted to 4, 10 and 20% of the maximal current for wild-type, hetF333I and hetY330C channels, respectively (Table IA). This current accounts for the observed differences in the resting whole-cell currents. In β-cells, a similar current would be expected to hyperpolarize the membrane and impair insulin secretion, even in the heterozygous state, thus accounting for the diabetic phenotype.
It is obvious that the ATP sensitivity of homF333I channels is dramatically less in the presence of Mg2+ than in the absence of the cation (compare Figures 2 and 6; see also Supplementary Figure 1). Indeed, no inhibition was observed even with as much as 10 mM MgATP. The ATP sensitivity of homY330C channels is also reduced by Mg2+ (the IC50 increased ∼17-fold; Supplementary Figure 2B), although to a lesser extent than homF333I channels. However, this is still greater than the 1.5-fold shift in IC50 observed for wild-type channels when Mg2+ is increased (Supplementary Figure 2A). Interestingly, in the heterozygous condition, the shift in the IC50 for ATP inhibition produced by Mg2+ is greater for Y330C channels (∼6-fold) than for F333I channels (∼1.5-fold; Supplementary Figures 2C and D). No difference in ATP sensitivity in the absence and presence of 2 mM Mg2+ was observed for Kir6.2ΔC channels (Gribble et al, 1998) or Kir6.2ΔC-F333I channels expressed in the absence of SUR1. ATP (10 mM) blocked Kir6.2ΔC-F333I currents by 70.2±2% in the absence, and 71.1±1% in the presence, of 2 mM Mg2+ (n=4). Thus the effect of Mg2+ is not intrinsic to either wild-type or mutant Kir6.2 and must be conferred by SUR1.
There are two explanations for the shift in ATP sensitivity produced by Mg2+. First, it could result from MgATP-dependent generation of phosphoinositol bisphosphate (PIP2), which stimulates channel activity (Baukrowitz et al, 1998; Shyng and Nichols, 1998; Fan and Makielski, 1999). Alternatively, it might reflect KATP channel activation due to MgATP binding and hydrolysis at the NBDs of SUR1 (Gribble et al, 1998). The latter appears to account for the shift in ATP sensitivity of wild-type channels (Gribble et al, 1998). The mechanism is unknown, but it is thought that the presence of the hydrolytic product, MgADP, at NBD2 of SUR1 leads to a conformational change in SUR1 that is transduced to Kir6.2 and results in opening of the pore (Nichols et al, 1996; Gribble et al, 1997b; Zingman et al, 2001). Whichever explanation is correct, the F333I mutation must markedly enhance the stimulatory effect (of PIP2 and/or SUR1) on the channel and the Y330C mutation must do so to a lesser extent. We have focused our analysis on the F333I mutation, as it has a much greater effect.
Activatory effect of F333I mutation in the presence of MgATP
To determine if the stimulatory influence of SUR1 on Kir6.2 is modified by the F333I mutation, we coexpressed Kir6.2-F333I with a mutant form of SUR1 that does not support channel activation (Gribble et al, 1997b, 1998). In this construct (SUR1-KA/KM), a consensus lysine in the Walker A motif of NBD1 is replaced by alanine, and the equivalent lysine in NBD2 is mutated to methionine. Figure 7A compares the effect of 1 mM MgATP on F333I channels containing either wild-type SUR1 (Figure 7Aa) or SUR1-KA/KM (Figure 7Ab). The nucleotide produced a gradual increase in Kir6.2-F333I/SUR1 currents, of approximately five-fold. In contrast, MgATP produced an immediate and substantial block of Kir6.2-F333I/SUR1-KA/KM currents, which was followed by a small time-dependent increase in current. Mean data are given in Figure 7B. The results are consistent with the idea that channel activation by MgATP is primarily the result of interaction with SUR1 and not secondary to generation of PIP2. Abolition of the stimulatory effect of MgATP (mediated via SUR1) unmasks the inhibitory effect of the nucleotide on Kir6.2.
Figure 7.

(A) Currents recorded in inside-out patches excised from Xenopus oocytes coexpressing Kir6.2-F333I and either SUR1 (a) or SUR1-KA/KM (b) in response to voltage ramps from −110 to +100 mV. MgATP (1 mM) was applied as indicated by the bars. (B) Mean slope conductance (G), expressed relative to the conductance in the absence of nucleotide (Gc), plotted against time, for Kir6.2-F333I/SUR1 (open circles, n=5) and Kir6.2-F333I/SUR1-KA/KM (filled circles, n=7). MgATP (1 mM) was added as indicated by the bar. (C) Mean slope conductance (G), expressed relative to the conductance in the absence of nucleotide (Gc), plotted against time for Kir6.2-F333I/SUR1-KA/KM. The bar indicates application of 1 mM MgATP (open circles, n=10) or of 1 mM MgATP plus 10 μM LY294002 (filled squares, n=5). (D) Currents recorded in inside-out patches excised from Xenopus oocytes coexpressing Kir6.2-F333I and SUR1 in response to voltage ramps from −110 to +100 mV. MgATP and MgADP (1 mM) were applied as indicated by the bars.
The small time-dependent increase in Kir6.2-F333I/SUR1-KA/KM currents observed in the maintained presence of MgATP was prevented by 10 μM LY294002 (Figure 7C). The drug (10 μM) had no effect Kir6.2/SUR1 or Kir6.2-F333I/SUR1-KA/KM currents when applied in the absence of ATP (n=3) (Supplementary Figure 3). At a concentration of 10 μM, LY294002 blocks PI-4 kinase and hence generation of PIP3 (Rosado and Sage, 2000). This suggests that the very small activation of Kir6.2-F333I/SUR1-KA/KM channels produced by MgATP results from generation of PIP3. Like PIP2, PIP3 activates Kir6.2/SUR1 channels when applied directly to the patch (Shyng and Nichols, 1998; Song and Ashcroft, 2001).
The time course of activation of Kir6.2-F333I/SUR1 currents in the presence of MgATP is quite slow (Figure 7B). One possibility is that this reflects the rate of hydrolysis to MgADP. We therefore compared the rate of activation by MgATP and MgADP (Figure 7D). In both cases, the rate of current activation could be fit with a single exponential with time constants of 107±38 s for 1 mM MgATP and 5.1±0.1 s for 1 mM MgADP, measured in the same patch (n=4). The decrease in current on nucleotide removal, however, was quite similar. In most patches, a fast transient increase in current was also seen on MgATP removal (Figure 7D).
Another explanation for the greater stimulation of Kir6.2-F333I/SUR1 currents by MgATP is that the interaction between SUR1 and mutant Kir6.2-F333I subunits results in a conformational change in the MgATP-binding site on SUR1 that now allows MgATP to act directly as a positive channel modulator (rather than via hydrolysis to MgADP). To test this idea, we examined the effects of two nonhydrolysable ATP analogues AMP-PCP and AMP-PNP. Neither compound supported channel activation: Kir6.2-F333I/SUR1 currents were blocked by 45±5% (n=5) and 62±2% (n=6) with 1 mM AMP-PCP or AMP-PNP, respectively (Supplementary Figure 4). This is consistent with the idea that ATP hydrolysis is required to stimulate both mutant and wild-type channels.
It is evident that the F333I mutation produces a marked increase in the ability of MgATP to activate the homomeric mutant channel. This effect is not due to a reduced ability of ATP to block Kir6.2 when Mg2+ is present, as MgATP and ATP blocked Kir6.2ΔC-F333I channels with similar potency. Instead, enhanced activation of homF333I channels involves MgATP binding/hydrolysis at the NBDs of SUR1, as it was abolished when Kir6.2-F333I was coexpressed with SUR1-KA/KM. The inability of AMP-PNP and AMP-PCP to stimulate channel activity also supports this idea. MgADP activated homF333I channels, but the rate of activation was about 20-fold faster than for MgATP: it is possible that this reflects the fact that MgATP has to be hydrolysed to MgADP (by NBD2 of SUR1) in order to stimulate channel activity.
The fact that hetF333I channels were blocked, rather than strongly activated, by MgATP suggests that several (probably all four) Kir6.2 subunits must be mutated in order for MgATP to stimulate channel activity dramatically, and that heteromeric channels behave like wild type with regard to MgATP activation. Because MgATP activation is conferred by SUR1, which must interact with Kir6.2 to open the pore, these data argue that the F333I mutation influences the interaction of SUR1 with Kir6.2. In the case of hetY330C, however, the IC50 for MgATP inhibition is intermediate between that of homY330C and wild type (Table IA): thus, it is likely that in this case fewer Kir6.2 subunits must be mutated in order for MgATP to stimulate channel activity.
Activation of three or four SUR1 subunits is needed to open hetF333I KATP channels
The F333I mutation is the first Kir6.2 mutation reported to influence channel activation by Mg-nucleotides without affecting intrinsic gating. As such, it offers an opportunity to determine the number of SUR1 subunits that must bind/hydrolyse ATP in order to open the channel. Figure 8A shows the mixed population of channel types expected when wild-type and F333I Kir6.2 subunits are coexpressed with wild-type SUR1, assuming they distribute randomly. If activation of all four SUR1 subunits was required to open the channel, then only about 1/16 of channels in the heterozygous state would exhibit marked activation on exposure to MgATP (i.e. those containing four F333I subunits). On the other hand, if only one SUR must be activated to open the channel, then 15/16 channels will show the enhanced channel activation associated with the F333I subunit. The fact that hetF333I channels are blocked rather than activated by MgATP (Figure 6) favours the former hypothesis.
Figure 8.

(A) Schematic of the different channel types expected when wild-type and mutant Kir6.2 are coexpressed with SUR1 (as in the heterozygous state). The box indicates channel types expected to have altered MgATP activation if four activated SUR1 are needed to open the channel. (B) Schematic of the different channel types expected when wild-type and mutant SUR1 are coexpressed with Kir6.2-F333I (as in the heterozygous state). The box indicates channel types expected to have altered MgATP activation if four activated SUR1 are needed to open the channel. (C) Mean slope conductance (G), expressed relative to the conductance in the absence of nucleotide (Gc), plotted against time, for the heterozygous mixture of channel types that occur when Kir6.2-F333I is coexpressed with both SUR1 and SUR1-KA/KM subunits (semifilled circles, n=9). The blue line indicates the Kir6.2-F333I/SUR1 data and the pink line the Kir6.2-F333I/SUR1-KAKM data (same data as in Figure 7B). MgATP (1 mM) was added as indicated by the bar.
To test this idea further, we coexpressed Kir6.2-F333I with an equal amount of wild-type SUR1 and SUR1-KA/KM. The latter is not activated by MgATP: thus, if all four SUR1 must be activated to open the pore, only about 1/16 of channels will be opened by the nucleotide, whereas many more channels will open if one or two SUR1 must be activated (Figure 8B). As Figure 8C shows, 1 mM MgATP substantially blocked Kir6.2-F333I/hetSUR1-KA/KM channels. This excludes the possibility that activation of a single SUR1 subunit is sufficient to open the channel. Assuming a binomial distribution of wild-type and mutant SUR1 subunits, the extent of activation we observe is somewhat greater than that expected if all four subunits must be activated but slightly less than that expected if three SUR1 subunits are required.
Previous studies on membrane trafficking have indicated that each Kir6.2 couples to one SUR1 (Shyng and Nichols, 1997; Clement et al, 1997; Zerangue et al, 1999). Our data indicate that (at least for channels containing Kir6.2-F333I mutant subunits) in order for MgATP to open the Kir6.2 pore, three or four SUR1 subunits probably must be activated (i.e. bind/hydrolyse MgATP) and transduce a conformational change to each Kir6.2 subunit in the tetramer. This seems reasonable, given that binding of ATP to a single Kir6.2 subunit induces a global conformational change that closes the pore (Markworth et al, 2000): conversely, one might expect that opening of the pore would also require an inverse global conformational change affecting all four Kir6.2 subunits.
It is interesting that F333I markedly enhances MgADP activation but does not affect gating, whereas Y330C produces a smaller effect on MgADP activation and also alters the ability of SUR1 to influence channel gating. This suggests that these effects may be conferred by separate interactions of SUR1 with Kir6.2, but that the interaction domains lie close together.
Discussion
Both F333I and Y330C mutations result in a marked decrease in the sensitivity of the KATP channel to inhibition by ATP. However, they appear to do so by somewhat different molecular mechanisms, with F333I causing impaired ATP binding/transduction, whereas Y330C both impairs ATP binding/transduction and also influences ATP inhibition indirectly, via an increase in intrinsic Po. In addition, both mutations enhance MgATP activation by SUR1, with F333I having a greater effect in the homomeric state and Y330C in the heterozygous state. The effects of Kir6.2 mutations on MgATP inhibition (at Kir6.2) and activation (at SUR1) combine to produce a significant reduction in the overall block of heterozygous channels by MgATP, and a significant increase in the KATP current amplitude at physiological [ATP]i. As Table I shows, in the absence of Mg2+, both hetY330C and F333I channels are blocked to a similar extent by ATP, whereas in the presence of Mg2+, ATP blocks hetF333I channels more strongly. This suggests that activation by MgATP determines the difference in overall sensitivity to ATP between the two kinds of mutant channels.
Structural considerations
In a homology model of Kir6.2 (Antcliff et al, 2005), residues Y330 and F333 both lie within the outer mouth of the ATP-binding pocket (Figure 9). In particular, F333 sits within 3.0 Å of the alpha phosphate of ATP (Trapp et al, 2003; Antcliff et al, 2005). Previous studies have shown that mutation of F333 to leucine also strongly reduced KATP channel inhibition (Antcliff et al, 2005). Likewise, mutation of the adjacent residue, G334, to aspartate markedly decreases ATP sensitivity (Drain et al, 1998). Residues R201, L181, V202 and P254 also lie within 3.0 Å of F333. It is noteworthy that mutation of R201 markedly impairs ATP inhibition (John et al, 2003; Gloyn et al, 2004; Antcliff et al, 2005), and that naturally occurring mutations at this residue are a common cause of PNDM (Gloyn et al, 2004; Sagen et al, 2004; Vaxillaire et al, 2004). Homology modelling thus supports the idea that the F333I mutation interferes with ATP binding.
Figure 9.

(A) Homology model of Kir6.2 (Antcliff et al, 2005). For clarity, only two subunits are shown, and the intracellular and transmembrane domains are from separate subunits (each subunit is shown in a different colour). ATP (yellow) is shown in its binding site. The red circle illustrates the ATP-binding site. (B) Close-up of the ATP-binding sites shown in A. ATP is shown in yellow and residues lining the ATP-binding site are labelled. Residues Y330 and F333 are shown in red.
In our homology model, residue 330 lies within 3.0 Å of several residues, including F333 and L181 in the C-terminal domain (C-domain) of the same subunit and H46 in the N-terminal domain (N-domain) of the adjacent subunit. Mutation of H46 to alanine had no effect on ATP sensitivity (Cukras et al, 2002). Mutations at L181 have not been reported, but mutation of T180 to alanine causes an increase in Po(0) of Kir6.2ΔC channels and thereby reduces KATP channel ATP sensitivity (Light et al, 2000), and mutation of I182 is associated with a decrease in ATP sensitivity but no change in the Po(0) of either Kir6.2ΔC or Kir6.2/SUR1 (Li et al, 2000). Furthermore, the I182V mutation causes PNDM (Gloyn et al, 2005). Residue 182 forms part of the hydrophobic pocket that accommodates the adenine ring of ATP (Antcliff et al, 2005). The ability of the Y330C mutation to affect the ATP sensitivity of Kir6.2ΔC, without altering gating, is consistent with its predicted proximity to I182. Interestingly, mutation of Y330 to leucine was without effect on the ATP sensitivity of Kir6.2ΔC (Antcliff et al, 2005). This suggests that a large nonpolar residue (like Y or L) is required at this position and that a small polar residue (like C) is unable to substitute.
How the Y330C mutation influences gating when SUR1 is present remains unclear. However, the region of the C-domain in which Y330 resides lies very close to the N-domain of the adjacent Kir6.2 subunit. There is evidence that the N-domain is involved in interactions between Kir6.2 and SUR1, and that Po(0) is increased when these are disturbed (Babenko et al, 1999; Reimann et al, 1999; Babenko and Bryan, 2002). Thus, the mutation may distort subunit–subunit interactions, and thereby channel gating. The fact that the Y330C mutation only affects gating when SUR1 is present suggests that Y330 might lie close to SUR1 in the three-dimensional structure of the octameric KATP channel complex, and is consistent with its predicted location on the outer surface of Kir6.2 (Figure 9A).
Molecular basis of reduced ATP sensitivity (in the absence of Mg2+)
The Po(0) of homomeric channels containing the F333I mutation was not significantly different from wild type. Coupled with its position within the predicted ATP-binding pocket, this suggests that this mutation reduces ATP binding/transduction per se. Because the KATP channel pore is composed of four Kir6.2 subunits (Shyng and Nichols, 1997), in the heterozygous state most channels will contain a mixture of wild-type and mutant subunits (Figure 8A). Since ATP binding to a single subunit is sufficient to close the channel and the number of mutant subunits in the tetramer follows the binomial distribution (Shyng and Nichols, 1997; Markworth et al, 2000), F333I channels will exhibit a markedly reduced ATP sensitivity only when all four subunits are mutant (i.e. in about 1/16 of channels in the heterozygous state). Binomial analysis using the IC50 for wild-type and F333I subunits predicts an IC50 of 25 μM for hetF333I channels, close to that observed experimentally (23 μM). This is consistent with the F333I mutation (in the absence of Mg2+) principally affecting ATP binding. The reduced ATP sensitivity observed for Kir6.2ΔC-F333I channels is also harmonious with this idea.
Mutation of Y330 to cysteine impaired ATP binding/transduction, but the mechanism appears to differ from that of F333I. In Kir6.2/SUR1 channels, this mutation enhances the free energy of the open state and thereby decreases ATP sensitivity indirectly. Simulating an ensemble of Kir6.2 tetramers containing a binomial mixture of wild-type and mutant subunits exhibiting free energy additivity in their effects of gating produced an IC50 for hetY330C channels of 43 μM, which is more than twice that observed experimentally (20 μM). One possible explanation for this discrepancy is that the subunits do not assemble according to a perfect binomial distribution, and that there is a reduced number of Y330C subunits in the heterozygous population. An alternative idea is that the mutation potentiates the mechanism by which SUR1 enhances the ATP sensitivity of Kir6.2 (Tucker et al, 1997). However, the reduced ATP sensitivity of Kir6.2ΔC-Y330C channels argues that there may also be a direct effect on ATP binding/transduction at Kir6.2.
It is noteworthy that the decrease in ATP sensitivity observed for Y330C (compared to the wild type) is less for Kir6.2ΔC (four-fold) than for full-length Kir6.2 coexpressed with SUR1 (20-fold). This suggests that the mutation may have an additional effect that is only manifest in the presence of SUR1. Consistent with this idea, Kir6.2ΔC-Y330C channels had normal gating, whereas the Po(0) of Kir6.2-Y330C/SUR1 channels was increased. The four-fold shift in the ATP sensitivity of Kir6.2ΔC can be attributed solely to changes in ATP binding/transduction; however, it is not correct to attribute all of the remaining shift in the ATP sensitivity of Kir6.2-Y330C/SUR1 to the observed changes in gating, as additional effects might also be involved.
Physiological implications
Both F333I and Y330C mutations cause an increase in the KATP current observed at 2 mM MgATP, a concentration close to that found in oocytes following metabolic poisoning (Gribble et al, 1997a, 2000). This accounts for the larger whole-cell KATP currents observed for both heterozygous and homomeric mutant channels under these conditions. In pancreatic β-cells, where ATP levels are around 1 mM in the absence of glucose and 2–5 mM in the presence of glucose (Detimary et al, 1995; for review, see Tarasov et al, 2004), an equivalent increase in the KATP current will hyperpolarize the plasma membrane and suppress electrical activity. This will lead to a reduction in insulin secretion and thereby diabetes (Ashcroft and Rorsman, 2004).
Our results suggest that disease severity is correlated with the extent of KATP channel block by ATP and with the magnitude of the whole-cell KATP current under resting conditions. It is noteworthy that in the presence of 1 mM MgATP, hetY330C currents are approximately twice as large as hetF333I currents, and the whole-cell currents are also correspondingly larger. This may explain why two out of the three patients with the Y330C mutation exhibited neurological problems. Further support for the idea that magnitude of the current amplitude at high [MgATP]i is correlated with clinical phenotype is provided by studies of other naturally occurring Kir6.2 mutations. Thus, the I296L mutation associated with DEND syndrome (Proks et al, 2005) causes a larger resting current than mutations that produce neonatal diabetes alone (R201H; Gloyn et al, 2005) or cause transient neonatal diabetes (G35S/R, I182V; Gloyn et al, 2005). At 3 mM ATP, which lies with the range found in pancreatic β-cells in the presence of glucose (Detimary et al, 1995), the KATP current amplitude was 32, 8, 4, 6 and 5% of that in the absence of nucleotide for I296L, R201H, G35S, G35R and I182V, respectively (Gloyn et al, 2005; Proks et al, 2005). Values for Y330C and F333I were 10 and 3.5%, respectively.
Kir6.2 is expressed not only in the β-cell but also in other endocrine cells, skeletal muscle, cardiac muscle and neurones throughout the brain. In many of these tissues, KATP channels are normally silent and open only under metabolic stress. Furthermore, the different complement of other ion channels that contribute to electrical activity in these tissues means that the KATP current may not have as large an effect on excitability as in the β-cell. Thus, a greater reduction in ATP sensitivity is likely to be required to increase the resting KATP current sufficiently to influence electrical activity. Studies of other mutations that cause neonatal diabetes support this view. Thus, the I296L mutation associated with DEND syndrome causes a larger resting current than mutations that produce neonatal diabetes alone or cause transient neonatal diabetes (Gloyn et al, 2005; Proks et al, 2005).
Conclusion
Analysis of two PNDM mutations, F333I and Y330C, demonstrates that the severity of the disease phenotype is correlated with a reduced sensitivity to MgATP. This leads to an enhanced KATP current, which inhibits insulin secretion, and in the case of the Y330C mutation may also reduce electrical activity in extrapancreatic tissues. The functional effects of the mutations support a homology model of Kir6.2 (Antcliff et al, 2005), and provide fresh insights into the relationship between KATP channel structure and function. In particular, they demonstrate that this part of the C-domain of Kir6.2 is involved in ATP binding and in transducing the effects of SUR1 on intrinsic gating and MgADP activation.
Materials and methods
Human Kir6.2 (GenBank NM000525 with E23 and I377) and rat SUR1 (GenBank L40624: Ref 29) were used in this study. Site-directed mutagenesis of Kir6.2 was performed using the QuickChange™XL system (Stratagene, La Jolla, CA, USA). Wild-type and mutant cDNAs were cloned in the pBF vector, and capped mRNA prepared using the mMESSAGE mMACHINE large-scale in vitro transcription kit (Ambion, Austin, TX, USA), as described previously (Gribble et al, 1997a).
Currents were recorded from Xenopus laevis oocytes 1–3 days after injection with 0.8 ng of wild-type or mutant Kir6.2 mRNA and ∼4 ng of SUR1 mRNA (giving a 1:5 ratio). For each batch of oocytes, all mutations were injected, to enable direct comparison of their effects. To simulate the heterozygous state, SUR1 was coexpressed with a 1:1 mixture of wild-type and mutant Kir6.2. Whole-cell currents were recorded from intact oocytes using the two-electrode voltage-clamp method, filtered at 1 kHz and digitized at 4 kHz. Oocytes were constantly perfused at 20–22°C with a solution containing (in mM) 90 KCl, 1 MgCl2, 1.8 CaCl2 and 5 HEPES (pH 7.4 with KOH). Metabolic inhibition was produced by 3 mM Na-azide. Whole-cell currents were monitored in response to voltage steps of ±20 mV from a holding potential of −10 mV.
Macroscopic currents were recorded from giant excised inside-out patches using the patch-clamp technique in response to 3 s voltage ramps from −110 to +100 mV (holding potential, 0 mV) and 20–22°C. Currents were filtered at 0.15 kHz and digitized at 0.5 kHz. The pipette solution contained (in mM) 140 KCl, 1.2 MgCl2, 2.6 CaCl2 and 10 HEPES (pH 7.4 with KOH). The Mg-free internal (bath) solution contained (in mM) 107 KCl, 1 K2SO4, 10 EGTA, 10 HEPES (pH 7.2 with KOH) and nucleotides as indicated. The Mg-containing internal was the same as the Mg-free solution except that 2 mM MgCl2 was added and MgATP (instead of ATP) was added as indicated. Rapid exchange of internal solutions was achieved by using a local perfusion system consisting of eight tubes of ∼200 μm diameter in which the tip of the patch pipette was inserted.
The macroscopic slope conductance was measured between −100 and +10 mV. ATP concentration–response curves were fit with the Hill equation
![]()
where [ATP] is the ATP concentration, IC50 is the ATP concentration at which inhibition is half-maximal and h is the slope factor (Hill coefficient). To account for possible rundown, Gc was taken as the mean of the conductance in control solution before and after ATP application. Single-channel currents were recorded at −60 mV from small inside-out patches, low-pass filtered at 5 kHz and sampled at 20–50 kHz (Trapp et al, 1998). Open probability was determined from single-channel patches as the fraction of time spent in the open state.
Data were analysed with custom software written in the Igor Pro (Wavemetrics, Lake Oswego, OR, USA) environment and with Origin 6.02 (Microcal Software, Northampton, MA, USA), and are given as mean±s.e.m. Statistical significance was evaluated using an unpaired two-tailed Student's t-test. A probability value of P<0.05 was taken as the criteria for a significant difference.
Supplementary Material
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Acknowledgments
We thank the Wellcome Trust, the Royal Society, Diabetes UK, the University of Bergen and the Haukeland University Hospital for support. We thank Dr Nathan Absalom for help with Figure 9.
References
- Aguilar-Bryan L, Nichols CG, Wechsler SW, Clement IV JP, Boyd AE III, Gonzalez G, Herrera-Sosa H, Nguy K, Bryan J, Nelson DA (1995) Cloning of the β-cell high-affinity sulfonylurea receptor: a regulator of insulin secretion. Science 268: 423–426 [DOI] [PubMed] [Google Scholar]
- Antcliff JF, Haider S, Proks P, Sansom MSP, Ashcroft FM (2005) Functional analysis of a structural model of the ATP-binding site of the KATP channel Kir6.2 subunit. EMBO J 24: 229–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashcroft FM, Gribble FM (1998) Correlating structure and function in ATP-sensitive K+ channels. Trends Neurosci 21: 288–294 [DOI] [PubMed] [Google Scholar]
- Ashcroft FM, Rorsman P (1989) Electrophysiology of the pancreatic β-cell. Prog Biophys Mol Biol 54: 87–143 [DOI] [PubMed] [Google Scholar]
- Ashcroft FM, Rorsman P (2004) Type-2 diabetes mellitus: not quite exciting enough? Hum Mol Genet 13: R21–R31 [DOI] [PubMed] [Google Scholar]
- Babenko AP, Bryan J (2002) SUR-dependent modulation of KATP channels by an N-terminal KIR6.2 peptide. Defining intersubunit gating interactions. J Biol Chem 277: 43997–44004 [DOI] [PubMed] [Google Scholar]
- Babenko AP, Gonzalez G, Bryan J (1999) The N-terminus of KIR6.2 limits spontaneous bursting and modulates the ATP-inhibition of KATP channels. Biochem Biophys Res Commun 255: 231–238 [DOI] [PubMed] [Google Scholar]
- Baukrowitz T, Schulte U, Oliver D, Herlitze S, Krauter T, Tucker SJ, Ruppersberg JP, Fakler B (1998) PIP2 and PIP as determinants for ATP inhibition of KATP channels. Science 282: 1141–1144 [DOI] [PubMed] [Google Scholar]
- Clement JP IV, Kunjilwar K, Gonzalez G, Schwanstecher M, Panten U, Aguilar-Bryan L, Bryan J (1997) Association and stoichiometry of K(ATP) channel subunits. Neuron 18: 827–838 [DOI] [PubMed] [Google Scholar]
- Cukras CA, Jeliazkova I, Nichols CG (2002) The role of NH2-terminal positive charges in the activity of inward rectifier KATP channels. J Gen Physiol 120: 437–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Detimary P, Jonas JC, Henquin JC (1995) Possible links between glucose-induced changes in the energy state of pancreatic B cells and insulin release: unmasking by decreasing a stable pool of adenine nucleotides in mouse islets. J Clin Invest 96: 1738–1745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drain P, Li L, Wang J (1998) KATP channel inhibition by ATP requires distinct functional domains of the cytoplasmic C terminus of the pore-forming subunit. Proc Natl Acad Sci USA 95: 13953–13958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enkvetchakul D, Loussouarn G, Makhina E, Nichols CG (2001) ATP interaction with the open state of the KATP channel. Biophys J 80: 719–728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Z, Makielski JC (1999) Phosphoinositides decrease ATP sensitivity of the cardiac ATP-sensitive K+ channel. A molecular probe for the mechanism of ATP-sensitive inhibition. J Gen Physiol 114: 251–269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gloyn AL, Pearson ER, Antcliff JF, Proks P, Bruining GJ, Slingerland AS, Howard N, Srinivasan S, Silva JM, Molnes J, Edghill EL, Frayling TM, Temple IK, Mackay D, Shield JPH, Sumnik Z, van Rhijn A, Wales JKH, Clark P, Gorman S, Aisenberg J, Ellard S, Njolstad PR, Ashcroft FM, Hattersley AT (2004) Activating mutations in the gene encoding the ATP-sensitive potassium channel subunit Kir6.2 gene and permanent neonatal diabetes. N Engl J Med 350: 1838–1849 [DOI] [PubMed] [Google Scholar]
- Gloyn AL, Reimann F, Girard C, Edghill EL, Proks P, Pearson ER, Temple IK, Mackay DJG, Shield JPH, Freedenberg D, Noyes K, Ellard S, Ashcroft FM, Gribble FM, Hattersley AT (2005) Moderately activating mutations in KCNJ11 may result in relapsing diabetes. Hum Mol Genet 14: 925–934 [DOI] [PubMed] [Google Scholar]
- Gribble FM, Ashfield R, Ämmälä C, Ashcroft FM (1997a) Properties of cloned ATP-sensitive K-currents expressed in Xenopus oocytes. J Physiol 4981: 87–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gribble FM, Loussouarn G, Tucker SJ, Zhao C, Nichols CG, Ashcroft FM (2000) A novel method for measurement of sub-membrane ATP concentration. J Biol Chem 275: 30046–30049 [DOI] [PubMed] [Google Scholar]
- Gribble FM, Reimann F (2003) Sulphonylurea action revisited: the post-cloning era. Diabetologia 46: 875–891 [DOI] [PubMed] [Google Scholar]
- Gribble FM, Tucker SJ, Ashcroft FM (1997b) The essential role of the Walker A motifs of SUR1 in KATP channel activation by MgADP and diazoxide. EMBO J 16: 1145–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gribble FM, Tucker SJ, Haug T, Ashcroft FM (1998) MgATP activates the β-cell KATP channel by interaction with its SUR1 subunit. Proc Natl Acad Sci USA 95: 7185–7190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inagaki N, Gonoi T, Clement IV JP, Namba N, Inazawa J, Gonzalez G, Aguilar-Bryan L, Seino S, Bryan J (1995) Reconstitution of IKATP: an inward rectifier subunit plus the sulfonylurea receptor. Science 270: 1166–1169 [DOI] [PubMed] [Google Scholar]
- John SA, Weiss JN, Xie LH, Ribalet B (2003) Molecular mechanism for ATP-dependent closure of the K+ channel Kir6.2. J Physiol 552: 23–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Wang J, Drain P (2000) The I182 region of K(ir)6.2 is closely associated with ligand binding in KATP channel inhibition by ATP. Biophys J 79: 841–852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Light PE, Bladen C, Winkfein RJ, Walsh MP, French RJ (2000) Molecular basis of protein kinase C-induced activation of ATP-sensitive potassium channels. Proc Natl Acad Sci USA 97: 9058–9063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markworth E, Schwanstecher C, Schwanstecher M (2000) ATP4− mediates closure of pancreatic beta-cell ATP-sensitive potassium channels by interaction with 1 of 4 identical sites. Diabetes 49: 1413–1418 [DOI] [PubMed] [Google Scholar]
- Nichols CG, Shyng SL, Nestorowicz A, Glaser B, Clement JP IV, Gonzalez G, Aguilar-Bryan L, Permutt MA, Bryan J (1996) Adenosine diphosphate as an intracellular regulator of insulin secretion. Science 272: 1785–1787 [DOI] [PubMed] [Google Scholar]
- Proks P, Antcliff JF, Lippiat J, Gloyn A, Hattersley AT, Ashcroft FM (2004) Molecular basis of Kir6.2 mutations associated with neonatal diabetes or neonatal diabetes plus neurological features. Proc Natl Acad Sci USA 101: 17539–17544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proks P, Girard C, Haider S, Gloyn AL, Hattersley AT, Sansom MSP, Ashcroft FM (2005) A novel gating mutation at the internal mouth of the Kir6.2 pore is associated with DEND syndrome. EMBO Rep 6: 470–475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimann F, Tucker SJ, Proks P, Ashcroft FM (1999) Involvement of the N-terminus of Kir6.2 in coupling to the sulphonylurea receptor. J Physiol 518: 325–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosado JA, Sage SO (2000) Phosphoinositides are required for store-mediated calcium entry in human platelets. J Biol Chem 275: 9110–9113 [DOI] [PubMed] [Google Scholar]
- Sagen JV, Raeder H, Hathout E, Shehadeh N, Gudmundsson K, Baevre H, Abuelo D, Phornphutkul C, Molnes J, Bell GI, Gloyn AL, Hattersley AT, Molven A, Sovik O, Njolstad PR (2004) Permanent neonatal diabetes due to mutations in KCNJ11 encoding Kir6.2: patient characteristics and initial response to sulfonylurea therapy. Diabetes 53: 2713–2718 [DOI] [PubMed] [Google Scholar]
- Sakura H, Ammala C, Smith PA, Gribble FM, Ashcroft FM (1995) Cloning and functional expression of the cDNA encoding a novel ATP-sensitive potassium channel subunit expressed in pancreatic β-cells, brain, heart and skeletal muscle. FEBS Lett 77: 338–344 [DOI] [PubMed] [Google Scholar]
- Seino S, Miki T (2003) Physiological and pathophysiological roles of ATP-sensitive K+ channels. Prog Biophys Mol Biol 81: 133–176 [DOI] [PubMed] [Google Scholar]
- Shyng S, Nichols CG (1997) Octameric stoichiometry of the KATP channel complex. J Gen Physiol 110: 655–664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyng S, Nichols CG (1998) Membrane phospholipid control of nucleotide sensitivity of KATP channels. Science 282: 1138–1141 [DOI] [PubMed] [Google Scholar]
- Song D-K, Ashcroft FM (2001) ATP-modulation of KATP channel ATP-sensitivity varies with the type of SUR subunit. J Biol Chem 276: 7143–7149 [DOI] [PubMed] [Google Scholar]
- Tanabe K, Tucker SJ, Matsuo M, Proks P, Ashcroft FM, Seino S, Amachi T, Ueda K (1999) Direct photoaffinity labeling of the Kir6.2 subunit of the ATP-sensitive K+ channel by 8-azido-ATP. J Biol Chem 274: 3931–7149 [DOI] [PubMed] [Google Scholar]
- Tarasov A, Dusonchet J, Ashcroft FM (2004) Metabolic regulation of the pancreatic β-cell KATP channel: a Pas de Deux. Diabetes 53: S123–S127 [DOI] [PubMed] [Google Scholar]
- Trapp S, Haider S, Jones P, Sansom MSP, Ashcroft FMA (2003) Identification of residues contributing to the ATP binding site of Kir6.2. EMBO J 22: 2903–2912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapp S, Proks P, Tucker SJ, Ashcroft FM (1998) Molecular analysis of KATP channel gating and implications for channel inhibition by ATP. J Gen Physiol 112: 333–349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker SJ, Gribble FM, Zhao C, Trapp S, Ashcroft FM (1997) Truncation of Kir6.2 produces ATP-sensitive K-channels in the absence of the sulphonylurea receptor. Nature 387: 179–183 [DOI] [PubMed] [Google Scholar]
- Vaxillaire M, Populaire C, Busiah K, Cave H, Gloyn AL, Hattersley AT, Czernichow P, Froguel P, Polak M (2004) Kir6.2 mutations are a common cause of permanent neonatal diabetes in a large cohort of French patients. Diabetes 53: 2719–2722 [DOI] [PubMed] [Google Scholar]
- Zerangue N, Schwappach B, Jan YN, Jan LY (1999) A new ER trafficking signal regulates the subunit stoichiometry of plasma membrane K(ATP) channels. Neuron 22: 537–548 [DOI] [PubMed] [Google Scholar]
- Zingman LV, Alekseev AE, Bienengraeber M, Hodgson D, Karger AB, Dzeja PP, Terzic A (2001) Signaling in channel/enzyme multimers: ATPase transitions in SUR module gate ATP-sensitive K+ conductance. Neuron 31: 233–245 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4