

Abstract
Thyroid hormone action is involved in virtually all physiological processes. It is well known that the liver and thyroid are intimately linked, with thyroid hormone playing important roles in de novo lipogenesis, beta-oxidation (fatty acid oxidation), cholesterol metabolism, and carbohydrate metabolism. Clinical and mechanistic research studies have shown that thyroid hormone can be involved in chronic liver diseases, including alcohol-associated or NAFLD and HCC. Thyroid hormone action and synthetic thyroid hormone analogs can exert beneficial actions in terms of lowering lipids, preventing chronic liver disease and as liver anticancer agents. More recently, preclinical and clinical studies have indicated that some analogs of thyroid hormone could also play a role in the treatment of liver disease. These synthetic molecules, thyromimetics, can modulate lipid metabolism, particularly in NAFLD/NASH. In this review, we first summarize the thyroid hormone signaling axis in the context of liver biology, then we describe the changes in thyroid hormone signaling in liver disease and how liver diseases affect the thyroid hormone homeostasis, and finally we discuss the use of thyroid hormone-analog for the treatment of liver disease.
INTRODUCTION
Thyroid hormone action is involved in virtually all physiological processes and plays a critical role in development, growth, and metabolism.1
In normal physiology, the hypothalamus-pituitary-thyroid axis functions as a classical feedback system. From the hypothalamus, thyrotropin-releasing hormone (TRH) is released to the pituitary portal system promoting the secretion of thyroid-stimulating hormone (thyrotropin or TSH). TSH, in turn, drives the thyroid gland to secrete into circulation 2 hormones derived from the amino acid tyrosine, 5′,3,5,3′ tetraiodothyronine (thyroxine, T4) and 3,5,3′-triiodothyronine (T3), with T4 being the main product of the thyroid gland.2 Notably, while T3 is the active hormone, the majority of T3 derives from the peripheral conversion of T4 into T3.3 T4 conversion in peripheral tissues produces the active T3 and reverse T3 (rT3), which is thought to be metabolically inactive.4 Additionally, the secretion of leptin from the adipose tissue provides an important modulation of the hypothalamus-pituitary-thyroid axis by stimulating the release of TRH from the hypothalamus.5
T3 is involved in the regulation of several physiological activities, including cellular metabolic rate, cardiovascular and digestive functions, muscle development and activity, brain development, and bone turnover.6,7 Owing to the pleiotropy of thyroid hormones (TH) signaling, thyroid disease affects multiple systems including cardiovascular, hepatic function, carbohydrate, and lipid metabolism.1
Hepatic activity and thyroid signaling are closely intertwined. T3 and T4 are essential for regulation of hepatic function, while in turn the liver is involved in metabolism of TH.8 Moreover, TH action may play an important role in the pathogenesis of several hepatic disorders, such as alcohol-associated liver disease (ALD) and NASH, which may evolve into cirrhosis and HCC.9
On the other hand, there is evidence that TH action can exert beneficial actions in some liver diseases10,11 and several TH analogs have been developed to target liver disease.
The aims of this review are to provide a primer of TH signaling axis in the context of liver biology, to describe the changes in TH homeostasis during liver disease, to describe how liver diseases affect TH homeostasis, and to review the use of TH-analogu directed to the treatment of liver disease.
PRIMER ON THE THYROID HORMONE AXIS
In physiologic states, the thyroid gland produces ~100 mcg of T4 and only 8–10 mcg of T3.7
Extrathyroidal TH conversion is regulated by iodothyronine deiodinases-1, iodothyronine deiodinases-2, iodothyronine deiodinases-3 (D-1, D-2, and D-3, respectively),12 which are seleno-enzymes whose expression and activities vary among different tissues. Interestingly, intrathyroidal TH conversion is regulated by D-1 and D-2, accounting for a significant component of the T3 released by the thyroid gland.13,14
These enzymes regulate circulating and intracellular TH levels, thus allowing for a time- and tissue-specific modulation in TH concentration in circulation and in the tissues, ultimately regulating the availability of active hormones for thyroid hormone receptors (TRs) binding.15,16
The deiodinases are responsible for the activation of T4 to T3, inactivation of T4 to rT3, and for the conversion of rT3 and T3 to 3,5-diiodo-L-thyronine (T2), the latter are important steps in the recycling of iodine (Table 1).
TABLE 1.
Characteristics of the deiodinase enzymes activation and inactivation of TH, physiological tissue distribution, and changes in activity in relation to thyroid hormone status
| Type | D-1 | D-2 | D-3 |
|---|---|---|---|
| Action | T4⟶T3⟶T2; T4⟶rT3⟶T2; rT3⟶T2 |
T4⟶T3/rT3⟶T2 | T4⟶rT3; T3⟶T2; rT3⟶rT2 |
| Tissue | Liver, kidney, thyroid | Brain, pituitary gland, skeletal muscle, brown Fat | Brain, placenta, skin, fetal tissue, hypoxic tissues |
| Hypothyroidism | ↓ (kidney, liver) | ↑ | ↓ |
| Hyperthyroidism | ↑ (thyroid, liver) | ↓ (except in the thyroid) | ↑ |
Abbreviations: T2, 3,5-diiodo-L-thyronine; T3, 3,5,3′-triiodothyronine; T4, 5′,3,5,3′ tetraiodothyronine; rT3, reverse T3; TH, thyroid hormone.
D-1 catalyzes inner-ring and outer-ring deiodination, D-2 catalyzes exclusively outer-ring deiodination (activating pathway converting T4 into T3), and D-3 catalyzes only inner-ring mono-deiodination (inactivating pathway). The transcription of both D-1 and D-3 is positively regulated by T3, while thyroid hormone promotes the degradation of D-2.17
Hepatic D-1 has a prominent role in the peripheral metabolism of thyroid hormone by promoting the conversion of the prohormone T4 into its active metabolite T3. Moreover, D-1 is responsible for the disposal of rT3.
More than 99% of the circulating pool of TH is bound to plasma proteins. The large discrepancy between free and bound components as well as between T4 and T3 concentrations is reflected by the normal reference ranges for free (0.8–1.8 ng/dL) and total (4.9–10.5 mcg/dL) T4 and free (2.3–4.2 pg/mL) and total (76–181 ng/dL) T3.
But it can be rapidly exchanged to the “free” pool for entry into cells. The thyroid hormone–binding proteins comprise thyroid hormone–binding globulin (TBG, also known as SERPINA7), a glycoprotein synthesized in the liver with a half-life of 5 days in plasma.18 Hepatic nuclear factor-binding sites hepatocyte nuclear factor (-1, -3) homeobox (-α, -β) determine SERPINA7 transcription in hepatocytes.19 TBG has by far the highest affinity for T4, the result of which is that TBG binds 68% of serum T4 and 80% of serum T3.20
Another thyroid hormone-binding protein is transthyretin (TTR, or thyroxine-binding prealbumin), a 55-kDa homo-tetrameric protein comprising 127 amino acids with an extended β-sheet conformation21,22 synthesized in liver, with a half-life of 2 days in plasma. The average concentration of TTR in serum is 0.25 mg/mL and corresponds to a maximal binding capacity of approximately 300 µg T4/dL.23 TTR has high affinity but relatively low-binding capacity with respect to TBG so it binds to about 10% of T4 and T3.20
Human serum albumin (HSA) is also a thyroid hormone-binding protein synthesized by the liver. HSA is a 66.5 kD protein composed of 585 amino acids.24 After synthesis by hepatocytes, it is rapidly excreted into blood at the rate of about 10–15 g/d. HSA has low-affinity and high-binding capacity for TH and binds the remaining 10%–20% of serum T3 and T4.20
Mutations in TH binding proteins (mostly TBG and HSA) result in significant changes in total thyroid hormone concentrations with minimal changes in their free components.25,26
Thus, thyroid homeostasis is dependent on a close interaction between the thyroid axis and liver synthetic function.
GENOMIC PATHWAYS OF THYROID HORMONE ACTION
T3 and T4 enter cells from the extracellular fluid by diffusion and by means of transmembrane transporters; intracellular T3 binds to TR to exert their biological effects (Figure 1).27
FIGURE 1.
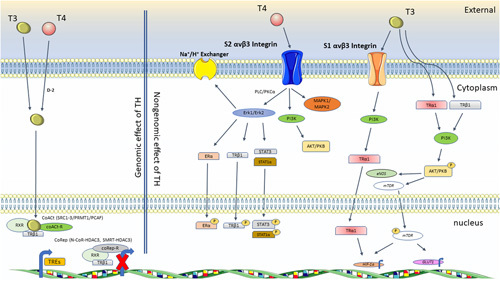
Genomic and nongenomic pathways of TH action. Genomic effects of TH (left side of figure). TR dimerizes with other nuclear receptors (in this case RXR) to recruit a set of coactivators forming a mediator-like complex that increase histone acetyltransferase activity (SRCs, PCAF) increasing the transcriptional activity of target genes. In the absence of thyroid hormone, TR/nuclear receptors dimers can recruit corepressors (NCoR, SMRT) with HDAC3 activity, reducing the transcriptional activity of target genes. SRCs and PCAF are histone acetyltransferases and PRMT1 is a histone methyltransferase. Nongenomic effect of TH (left side of figure). TH affect multiple physiological activities by means of interactions with S1 and S2 αvβ3 integrin. The interaction between T4 and S2 αvβ3 integrin, leading to regulation of PI3K, MAPK1/2, and ERK-1/2 by means of PLC and PKCα, promoting phosphorylation of nucleoproteins and modulation of intracellular protein trafficking. ERK-1/2 activity can activate the sodium proton exchanger (Na+/H+ ) in the plasma membrane. S1 αvβ3 integrin recognizes T3 and activates the PI3K pathway leading to direct trafficking of TRα1 from the cytoplasm to the nucleus and transcriptional activity of the target gene, HIF-1α. Furthermore, activation of the PI3K/Akt/PKB pathway can be rapidly stimulated by means of T3 interactions with TRβ1 or TRα1 and initiates downstream target gene transcription including HIF-1α and GLUT1. Abbreviations: ERK, extracellular signal-regulated kinase; HDAC3, histone deacetylase 3; HIF, hypoxia inducible factor; NCoR, nuclear receptor co-repressor; PCAF, P300/CBP-associated factor; PI3K, phosphatidylinositol-3-kinase; RXR, retinoid X receptor; SMRT, silencing mediator for retinoid or thyroid hormone receptors; SRCs, steroid receptor coactivator; TH, thyroid hormone; TR, thyroid hormone receptor.
TH action is exerted primarily through nuclear TR, which are members of the superfamily of hormone-responsive nuclear transcription factors.15
T3 binds nuclear TR located on thyroid hormone response elements within promoter regions of genes downstream of TH/nuclear TR28 and induces the transcription or repression of target genes.1
The 2 primary isoforms of TR, α and β, are differentially expressed during development and in adult tissue.15,29 Alternative splicing from TRα and TRβ genes generates TRα1, TRα2 isoforms and TRβ1, TRβ2, respectively.30
Human hepatocytes express an equal amount of TRα1 and TRβ1 isoform, conferring the relative dominance of TRβ1 when compared to other tissues (myocardium, bone, and adipose tissue) where the TRα1 is more prevalent. The transcriptional effect of TH is exerted by means of formation of homodimers or heterodimers with nuclear receptors, such as retinoid X receptor, other coactivator receptors, like vitamin D receptor, other retinoic acid receptor subtypes and peroxisome proliferator receptor. TR-cognate receptors heterodimers generally function as partner with several transcription factors to regulate target genes modulating the chromatin acetylation or methylation. Lack of ligand causes the binding of corepressors to the transcriptional complex, ultimately inhibiting the transcription of target genes (Figure 1).31,32
NONGENOMIC PATHWAYS OF THYROID HORMONE ACTION
TH can exert their function not only by genomic but also through nongenomic pathways such as integrin αvβ3, which has been identified as a membrane TH receptor that activates both the phosphatidyl-inositol-3-kinase (PI3K) and extracellular signal-regulated kinase (ERK)-1/2 pathways.33 TH binds integrin αvβ3 at 2 sites, S1 and S2, triggering several intracellular pathways and gene transcription (Figure 1).34–36
T4 preferentially binds the αvβ3 S2 site and activates ERK-1/2, which modulates intracellular protein trafficking from the cytoplasm to nucleus and can also induce the sodium proton exchanger (Na+/H+). The stimulation of the PI3K/Akt/protein kinase B pathway is another nongenomic TH-mediated action. T3 binds exclusively to αvβ3 S1 site, and the T3-αvβ3 S1 and the resulting PI3K activation promotes in turn trafficking of TRα1 from the cytoplasm to the nucleus. This generates an indirect increase in target gene expression, such as hypoxia inducible factor-1α. In the cytoplasm, nongenomic T3 signaling rapidly induces the PI3K pathway and initiates downstream gene transcription.
Table 2 describes the molecular pathways and function of genomic and nongenomic TH regulation as discussed by Chi et al.37 Of note, it is not clear whether nongenomic T3 signaling plays a clinically relevant role in humans.38
TABLE 2.
Summary of the genes/signals regulated by genomic or nongenomic action of TH/THR signal axis. Black arrows represent genes/signals that are upregulated (upward arrow) or downregulated (downward arrow) by TH
| Molecular function | Gene/signal name (↑increase, ↓decrease) | |
|---|---|---|
| Genomic regulation by TH/TR | Transcriptional coregulator of THR | ↑ SP1, p53, Oct-1, GHF-1 ↓ CTCF, LCOR |
| Autophagy regulator | ↑ DAPK2, Betatrophin | |
| Cell cycle regulator | ↓ UHRF1, STMN1, Mir-214 BC200 | |
| Apoptosis regulator | ↑ TRAIL | |
| Metastatic regulator | ↑ BSSP4, LCN2, mir-21 ↓ mir-130b |
|
| Nongenomic regulation by TH/TR | Membrane receptor of TH | ↑ integrin αvβ3 |
| Signal transductor | ↑ Src kinase, PI3K/Akt, p-ERK1/2, mTOR/p70S6K, eNOS | |
| Transcriptional factor | ↑ Estrogen receptor, STAT3, HIF1-α, β-catenin | |
| Metabolic regulator | ↑ GLUT1, PFKP, MCT4 | |
| Na-K-ATPase | ↓ KCNH2 | |
| Apoptosis regulator | ↓ FOXO1, BCL2L11 |
Abbreviations: FOXO1, forkhead box protein O1; GLUT, glucose transporter; HIF, hypoxia inducible factor; MCT, monocarboxylate transporter; PI3K, phosphatidylinositol-3-kinase; STAT3, signal transducer and activator of transcription 3; TH, thyroid hormone; TR, thyroid hormone receptor; TRH, thyrotropin-releasing hormone.
Adapted from Davis et al.47
THYROID HORMONE ACTION ON LIVER FUNCTION
In the liver, TRβ is the predominant TR isoform, and mutations can cause a liver-specific phenotype.39–41 Thus, the liver is an ideal target for therapeutic intervention with isoform-specific TH agonists (see the Thyromimetics section).42–44
TH plays a role in hepatic lipid homeostasis and they have direct effects on both cholesterol and fatty acid synthesis and metabolism. It has long been appreciated that the liver is the major metabolic site for cholesterol and triglycerides; TH promotes low‐density lipoprotein receptor (LDLR) expression and activity of lipid‐lowering liver enzymes, resulting in a reduction in LDL levels.45,46
The expression of apolipoprotein A1, a major component of HDL, is also increased by TH.47
A deeper, mechanistic understanding of the role of TH in hepatic metabolism was made possible thanks to the generation of TR knock-in mouse models. Using a dominant negative mutation in TRβ (Thrβ PV/PV) mice, Araki and colleagues established the role of THRβ in hepatic lipid metabolism. The authors showed that lack of TRβ causes hepatic steatosis and hepatomegaly within a few months from birth. In this Thrβ PV/PV mouse model, the decrease in fatty acid oxidation drives the lipid accumulation in the liver. On the other hand, mice expressing the same mutation in TRα (ThrαPV/PV) and TR α-null showed decreased lipogenesis, weight loss, and reduced hepatic accumulation of lipids.39 In the next section, we will discuss in more detail how TH is involved in hepatic lipid metabolism and in cholesterol metabolism.
LIPID METABOLISM AND HEPATIC THYROID HORMONE ACTION
TH action modulates the lipolysis of fat stores from white adipose tissue, which generates circulating free fatty acids (FFAs), the major source of lipids for the liver. FFAs can also derive from ″de novo″ lipogenesis (DNL) and from hydrolysis of dietary triglycerides (Figure 2).48
FIGURE 2.

Hepatic FFAs metabolism and TH effect in hepatocytes. Thyroid hormone stimulates lipolysis from fat stores in white adipose tissue and from dietary fat sources to generate FFAs that enter the hepatic cells by means of protein transporters such as fatty acid–binding protein (FABP), calcium-independent phospholipase A2 Beta (iPLA2-β), caveolin-1, and fatty acid transporter (FAT/CD36). FFAs are typically esterified to triacylglycerol and subsequently packaged into VLDL for export or stored as intracellular lipid droplets. Triacylglycerol stored as lipid droplets can also be hydrolyzed back to FFAs by means of classic lipases and lipophagy by regulating transcription factors (SIRT1, FOXO1), various coactivators or nuclear receptors such as (PPARα, FGF21, and PGC1α) and target the transcription of genes such as Cpt1a, Acadm, Pdk4, and Ucp2. Thyroid hormone induces DNL by means of the transcription of several key lipogenic genes such as Acaca, Fas, Me, and Thrsp. In addition, thyroid hormone indirectly controls the transcriptional regulation of hepatic DNL by regulating the expression and activities of other transcription factors such as SREBP1C, LXRs, and ChREBP. Abbreviations: CHREBP, carbohydrate-responsive element-binding protein; DNL, de novo lipogenesis; FABP, fatty acid-binding protein; FAT/CD36, fatty acid translocase; FFA, free fatty acid; FOXO1, forkhead box protein O1; LXR, liver X receptor; PGC1α, PPARγ co-activator 1α; PPAR, peroxisome proliferator-activated receptor; SREBP, sterol regulatory element-binding protein; SIRT1, NAD-dependent protein deacetylase sirtuin 1; TH, thyroid hormone.
Most of hepatic lipid homeostasis and some enzymatic carrier proteins, transporter and cell-carrier protein are under control of TH.49,50
TH increases liver uptake of FFAs through a tetrameric plasma membrane protein complex consisting of plasma membrane fatty acid–binding protein, caveolin-1, calcium-independent membrane phospholipase A2, and fatty acid translocase.51
T3 can induce hepatic lipolysis of triacylglycerol stored as lipid droplets which can subsequently be hydrolyzed into FFAs by classical lipase and lipophagy,52–55 which involves the engulfment of triacylglycerol stored in the fat droplets by autophagosomes, followed by autophagosomal-lysosomal fusion that delivers the triacylglycerols to lysosomes for degradation and hydrolysis into FFAs.54,55
FFAs are then shuttled into mitochondria where they undergo β-oxidation. The control action exerted by T3 takes place through positive regulation of NAD-dependent protein deacetylase sirtuin 1, forkhead box protein O1,50 and by increasing the activity of hormones like FGF21,56 various coactivators or nuclear receptors (such as peroxisome proliferator-activated receptor (PPAR)-α45 and PPARγ coactivator 1α (PGC1α) and target the transcription of genes such as carnitine palmitoyltransferase 1A, acyl-CoA dehydrogenase medium chain, pyruvate dehydrogenase kinase (PDK), isoenzyme 4, and uncoupling protein 2.57–61
TH action increases the number of lipid-laden autophagosomes and lysosomes in a TR-dependent manner both in mice and human hepatocytes.62
De novo lipogenesis involves a number of proteins and enzymes, including sterol regulatory element-binding protein (SREBP) 1C, thyroid hormone-responsive spot14 homolog, FA synthase, acetyl-CoA carboxylase 1, liver X receptor, carbohydrate-responsive element-binding protein (CHREBP), and malic enzyme, which are upregulated directly and indirectly by T3.63–65 After their synthesis, FFAs are typically esterified to triacylglycerol, which can be packaged into VLDL, stored as fat droplets or used to make and/or repair cellular constituents.
TH promotes hepatic cholesterol synthesis (Figure 3) by increasing the expression of 3-hydroxy-3-methylglutaryl-CoA reductase and farnesyl diphosphate synthase.66
FIGURE 3.

TH effect on cholesterol metabolism. TH increases the expression of HMGCR and FDPS to promote hepatic cholesterol synthesis. TH is involved also in the cholesterol uptake from peripheral tissue inducing the gene and protein expression of SRBP1, SREBP2, Apo A1, LDLR, CETP, LDLR, LPL, and HL. TH is also involved in the reverse cholesterol transport pathway increasing expression of CYP7A1. Furthermore, TH increases the efflux of bile by stimulating Abcg5/Abcg8 gene transcription. Additionally, TH can negatively modulate cholesterol ester formation by means of CDX2 and SOAT2. Abbreviations: Abcg, ATP-binding cassette subfamily G member; APOA1, apolipoprotein A1; CDX2, caudal-type homeobox 2; CETP, cholesteryl ester transfer protein; CYP7A1, cholesterol 7α-hydroxylase; FDPS, farnesyl diphosphate synthase; HL, hepatic lipase; HMGCR, 3-hydroxy-3-methylglutaryl-CoA reductase; LDLR, LDL receptor; LPL, lipoprotein lipase; ; SOAT2, sterol O-acyltransferase 2; SRB1, scavenger receptor class B member 1; SREBP, sterol regulatory element-binding protein; TH, thyroid hormone.
Furthermore, TH is involved in the downregulation of gene expression of caudal-type homeobox 2 and sterol O-acyltransferase 2 by means of miR181d, resulting in inhibition of cholesterol ester formation by human hepatocytes.67
Conversely, TH also induces protein and gene expression of apolipoprotein A1, scavenger receptor class B member 1 (SRB1) and sterol regulatory element-binding protein 2 (SREBP2), which then increase LDLR levels to increase cholesterol efflux from peripheral tissues to HDL through the reverse cholesterol transport pathway.68,69
TH increases HDL metabolism by stimulating cholesteryl ester transfer protein activity to accelerate serum cholesterol clearance.70
Cholesterol synthesis, mostly as VLDL, can be stimulated by TH starting with its precursor and acetyl-CoA.71
In addition, the cholesterol uptake from peripheral tissue is indirectly mediated by TH. In fact, TH stimulates the lipoprotein lipase (LPL), which catabolizes the triglycerides-rich lipoproteins, and the hepatic lipase to decrease HDL particle size, resulting in higher relative levels of lipid-poor apolipoprotein A1 in HDL that facilitates the cholesterol uptake.72,73 The cholesterol 7α-hydroxylase expression, which converts cholesterol into bile acids in the reverse cholesterol transport pathway, is increased by TH.46,74
In addition, TH increases the efflux of bile acids in both the liver and intestines, which are the last steps of the reverse cholesterol transport pathway, by stimulating ATP-binding cassette subfamily G member 5 and 8 gene transcription.75
THYROID HORMONE ACTION ON HEPATIC CARBOHYDRATE METABOLISM
The liver plays a unique role in carbohydrate metabolism owing to its ability to both store and release glucose to minimize changes in serum glucose concentration between fed and fasted states. In healthy individuals, the intake of a mixed meal results in modest hyperglycemia, accompanied by substantial storage of glycogen in the liver. In response to ingestion of glucose or a mixed meal and the resulting hyperinsulinemia and hyperglycemia, the fasting liver shifts from net output to net uptake of glucose.76
Glucose enters the hepatocyte by means of the insulin-independent glucose transporter-2 and is phosphorylated by hexokinase isoenzymes to glucose 6-phosphate, which may follow several metabolic pathways; during the post-prandial period, most glucose 6-phosphate is used to synthesize glycogen by means of the formation of glucose 1-phosphate and uracil-diphosphate (UDP)–glucose. Minor amounts of UDP–glucose are used to form UDP–glucuronate and UDP–galactose, which are donors of monosaccharide units used in glycosylation. A second pathway of glucose 6-phosphate metabolism is the formation of fructose 6-phosphate, which may either start the hexosamine pathway to produce UDP-N-acetylglucosamine or follow the glycolytic pathway to generate pyruvate and then acetyl-CoA. Acetyl-CoA may enter the tricarboxylic acid cycle to be oxidized or may be exported to the cytosol to synthesize fatty acids when excess glucose is present within the hepatocyte. Finally, glucose 6-phosphate may produce NADPH and ribose 5-phosphate through the pentose phosphate pathway. In addition to metabolizing carbohydrates, the liver produces glucose to be released in circulation from glycogen breakdown, or by means of de novo synthesis using primarily lactate and alanine (gluconeogenesis), including the hexosamine pathway, the pentose phosphate pathway, and oxidative routes. Excess glucose is used to synthesize fatty acids in the liver. This unique ability of the human liver to store and release glucose is crucial to endure periods of fasting.77
Centrally, TH action increases hepatic glucose production and reduces insulin sensitivity through a sympathetic pathway connecting the paraventricular hypothalamus to the liver and through glucagon hormone signaling.78
In the liver, T3 positively modulates genes involved in glycogenolysis and gluconeogenesis.79 Increased deacetylation and activation of the master gluconeogenic transcription factor forkhead box protein O1 by NAD-dependent protein deacetylase sirtuin 1 is also stimulated by T3 leading to increase in gluconeogenesis.50 Indeed, 2 key enzymes involved in gluconeogenesis, such as phosphoenolpyruvate carboxikinase 1 and glucose-6-phosphatase are modulated by T3 in the liver.80,81 TH positively regulates PDK4 expression82 promoting gluconeogenesis. Moreover, TRβ and CCAAT enhancer binding protein regulate the phosphoenolpyruvate carboxykinase, which is an enzyme critical for glucose homeostasis.80
Apart from regulating gluconeogenic gene transcription, increased alanine transport and inhibition of insulin signaling may also contribute to TH-induced hepatic glucose production. Ultimately, TH action results in an increase in hepatic gluconeogenesis and hepatic insulin resistance.
ROLE OF LIVER IN THE NONTHYROIDAL ILLNESS SYNDROME
Nonthyroidal illness syndrome (NTIS) also referred as “euthyroid sick syndrome” can be defined as any acute or chronic inflammatory condition not originating in the thyroid causing a perturbation in the serum concentrations of TH and TSH. NTIS is characterized by a low serum concentration of T3, normal or in some case reduced T4 serum concentration, and an increased serum concentration of rT383 with normal or low TSH concentration. The pathophysiology of NTIS involves several organs, including the liver,84 where a variety of changes in deiodinases, TH transporters, and TR action have been observed.
With respect to the deiodinases, a healthy liver predominantly expresses D-1, with very low expression of D-3. The low serum concentration of T3 during NTIS has been ascribed to reduced activity of D-1, which is responsible for conversion of T4 to T3.85–87 Inflammatory states inhibit D-1 transcription causing a decrease in circulating T3 and accumulation of rT3, which are the hallmark of NTIS.6 Liver D-3 is differentially regulated during illness: acute inflammation in mice results in decreased D-3 levels85 but prolonged critical illness, and hypoxia both in patients and in rabbits, increase D-3.87,88
Furthermore, a report indicated that serum from patients with NTIS inhibits T4 uptake, mediated in part by increased bilirubin and nonesterified fatty acids, which also inhibit T4 to T3 conversion.89 In systemically ill patients, nonesterified fatty acids levels rise in parallel with the severity of the illness.83
During acute illness, the concentration of serum binding protein and the affinity of TH binding to transport proteins are altered. TBG levels may be increased while serum levels of TTR and albumin are decreased, especially during prolonged illness, resulting in a decrement of total T3 serum level.90
Reduced expression of TRH in the hypothalamus appears to play a key role in the prolonged phase of critical illness, although the processes that trigger this upstream disturbance are somewhat unclear; a paradoxical increase in hypothalamic D2 activity leads to the suppression of TSH secretion.91
Interestingly, TH transport in the human liver is differentially affected during illness: MCT 8 expression increases in prolonged critical illness compared to acute illness.4 This means that patients with the lowest T3 serum show the highest upregulation of MCT8 mRNA. The positive correlation in patients between MCT8 mRNA and low tissue T3 levels could suggest that the upregulated MCT8 represents a compensatory mechanism to increase T3 levels in tissues while the circulating T3 concentrations are low.92
Furthermore, hepatic TRα and TRβ mRNA expression is differentially affected during illness, depending on severity and duration. In rodents during acute illness but not in chronic illness, liver TRα and TRβ expression decrease.85,93 In contrast, biopsy studies performed in patients with chronic liver disease prior to liver transplantation indicated that TRα and TRβ mRNA expression are increased in association with low serum T3 and T4 levels.94 However, in patients with a variety of chronic liver diseases, hepatic TRα1, TRα2, and TRβ1 and T3-target gene expression were unchanged despite reduced serum TH levels.31
The differential regulation of TH transporters, deiodinases, and THR expression in acute and chronic states of disease supports the concept that there is altered hepatic TH metabolism during NTIS. The clinical importance of NTIS is still not clear because this phenomenon may protect against the catabolic state associated with the underlying condition or represent a maladaptive response leading to a decreased delivery of TH to target tissues.
ALD AND THYROID HORMONE HOMEOSTASIS
Alcohol is one of the world’s largest modifiable risk factors for disease and disability, affecting almost all organs and systems.95 Alcohol is metabolized by the liver, which is the primary site of damage, and its abuse is the primary cause of several diseases, such as fatty liver, alcohol-associated hepatitis, and cirrhosis.9,16 In hepatocytes, cellular levels of TH are closely associated with ALD.96
The hepatic dysfunction due to alcohol, in turn, affects thyroid function including hormone levels and gland size. In fact, a significant reduction in thyroid gland volume has been reported in patients with ALD.97–99 Alcohol causes a moderate suppression of serum T4 levels with more significant suppression of T3 levels in heavy drinkers. In addition, individuals with dependence to alcohol may present with features of NTIS,100,101 which correlates with the severity of liver dysfunction and mortality risk.102–104
Papineni and colleagues conducted a prospective cohort study for 1 year on a group of 70 males aged 30–80 years with alcohol use disorder who had a regular alcohol intake of more than 60–80 g/d for the past 10 years. The authors investigated whether TH levels could be used as markers of ALD and assessed the changes in thyroid hormone levels with treatment and concluded that TH levels, particularly free-T3 (fT3) and free-T4 (fT4), should be evaluated in patients with chronic ALD. Additionally, the authors suggest that assessing free TH levels during the withdrawal and abstinent periods is important because decreased hormone levels may be associated with increase withdrawal symptoms and craving for alcohol.105
Alcohol-induced liver steatosis is a highly prevalent liver pathology.106 The effects of TH on this condition have been partially studied in animal models, and preliminary observations suggested that a decrease in TH action, presumably by reducing cell metabolic rate and oxidative stress, may ameliorate necrosis and hypoxia in the liver.107,108
Clinically, propylthiouracil, a thiourea-derivate antithyroid medication was tested in clinical trials in alcohol-associated hepatitis and cirrhosis to mitigate the presumed negative effects of excess of TH in the liver.109 Of note, propylthiouracil use is associated with hepatotoxicity particularly in the pediatric population, and it carries a FDA “black box.”110
NAFLD AND THYROID HORMONE AXIS
Chronic liver disease is a major public health problem worldwide, estimated to affect up to one-third of the US population111 often in association with obesity and/or type 2 diabetes mellitus.112
NAFLD is initiated by lipid accumulation in hepatocytes,113,114 which leads to a spectrum of liver dysfunction ranging from excess lipid storage (hepato-steatosis) to progressive NASH, that in turn increases the risk for cirrhosis and HCC.115 NAFLD is considered the hepatic phenotype of the metabolic syndrome, associated with insulin resistance or established type 2 diabetes mellitus, increased visceral adiposity, overweight/obesity, dyslipidemia, and arterial hypertension116 and is associated with increased risk of cardiovascular, hepatic, and metabolic diseases. In the United States, NAFLD is recognized as one of the leading causes of chronic liver disease in adults117 and children,118 and has an estimated prevalence of 20%–40% in western countries.119
As discussed, TH exerts important effects on hepatic lipid metabolism. In hypothyroid states, centrilobular hepatocytes have increased steatosis.120,121 These studies support the role of the thyroid axis in the development and progression of NAFLD, which often occurs in association with endocrine disorders.122,123
Hypothyroidism has been proposed as a contributory mechanism in the pathophysiology of NAFLD,1,124 but to date, no human studies showed an association between circulating TH and histological features of NAFLD despite TH are crucial in the regulation of many hepatic metabolic processes125–128 and the transcription of several lipogenic genes, whose expression are altered in NAFLD.5,129
While TH action in hepatocytes promotes a decrease in cholesterol and triglyceride levels, patients with NAFLD show elevated cholesterol, LDL, and triglycerides levels.130,131
At the transcriptional level, many of the genes whose expression is altered in NAFLD132 such as genes regulating lipid synthesis (eg, acetyl-CoA carboxylase 1, fatty acid synthase, diacylglycerol o-acyltransferase) and mitochondrial β-oxidation (eg, long-chain acyl-CoA dehydrogenase, hydroxyacyl-CoA dehydrogenase trifunctional multienzyme complex subunit alpha, UCP2, acyl-CoA oxidase) are upregulated,133 potentially as a secondary compensatory mechanism to respond to the increased flow of substrate due to calorie overload. Of interest, these genes are positively regulated by TH as well.132 Furthermore, altered expression and activation of proinflammatory genes (eg IL-1 receptor family and genes regulating fibrotic responses (TGF beta-1 , FGFR2 receptor 2) that may contribute to the development of NASH are also under the control of TH.134,135
These observations have provided the rationale for therapeutic interventions with TH in preclinical models of NAFLD,136 as well a small clinical trial which showed that low-dose levothyroxine (synthetic T4) administration resulted in a significant reduction in hepatic fat content from baseline.137
Observational and meta-analysis data linked TH and NAFLD. Several studies have investigated the association between either subclinical (ie, elevated TSH with normal TH levels) or overt hypothyroidism (elevated TSH with subnormal TH levels) and NAFLD. Data are conflicting: some studies indicated that hypothyroidism, mainly subclinical, was common, occurring in ~25% of the individuals with imaging-defined or biopsy-proven NAFLD,9 other studies, however, failed to identify a significant association between hypothyroidism and NAFLD.138 In a large population-based cohort study conducted on 3144 euthyroid subjects, Chen et al139 found positive associations of fT3 levels and fT3/fT4 ratios with NAFLD in euthyroid women but not in euthyroid men.
In addition, a study conducted in children with obesity and NAFLD showed that adolescents with hepatic steatosis had elevated values for fT3/fT4 ratio. This finding is consistent with peripheral conversion of T4 to T3 due to increased deiodinase activity as a compensatory mechanism to increase energy expenditure to prevent excessive fat accumulation.140 Conversely, it is also possible that the shift to the right of the thyroid axis is driven by the adiposity resulting in elevated leptin levels, which in turn stimulate the hypothalamic release of TRH ultimately stimulating the thyroid axis.141 A significant positive relationship between TSH levels and hepatic steatosis, independent of age, sex, and stage of puberty was also observed in euthyroid severely obese children and adolescents. Higher TSH levels were associated with a greater degree of fatty infiltration, emphasizing the possible significance of subclinical hypothyroidism as a predictor of metabolic comorbidity.142 Collectively, the human data do not demonstrate whether the changes in circulating TH observed in NAFLD are causative or rather the consequence of obesity, which is associated per se with NAFLD.
The axiom that hypothyroidism is associated with fatty liver has been partly challenged by recent mouse model data. Experimental observations indicate that mice develop NAFLD when TH levels are mildly reduced but, counterintuitively, not when there is a severe reduction of TH levels. The disease process of hypothyroidism-induced NAFLD therefore appears to involve both intrahepatic and extrahepatic factors, with important differences between mild (subclinical) and severe hypothyroidism.143 Future mechanistic and clinical studies are needed clarify this emerging concept.
HEPATIC CANCER AND THYROID HORMONE SIGNALING
HCC is the fifth-most common form of cancer worldwide and one of the most prevalent causes of tumor-related mortality.144 HCC is linked to high mortality rates, and the majority (80%–90%) develop from cirrhotic livers.145
Two case-control studies suggested that hypothyroidism represents a risk factor for HCC.
In the first study, Hassan et al146 observed an increased association between hypothyroidism and HCC in women, independent of established HCC risk factors; in the second study, Reddy et al147 observed that hypothyroidism was more prevalent in patients with HCC of unknown etiology.
Furthermore, several studies indicate that disruption of TH signaling is involved in the development of HCC (Figure 4).146–148 Other studies have shown that mutated or truncated forms of TRα and TRβ are expressed at high frequencies in human HCC (Figure 4).149,150 In rat models of HCC, T3/TR was shown to suppress the carcinogenic process. Intriguingly, in vivo studies showed that a short treatment with T3 accelerated the regression of chemically induced hepatic preneoplastic lesions in rats subjected to the resistant-hepatocyte (R-H) model of hepatocarcinogenesis.151 The authors observed that only 50% of the rats exposed to repeated cycles of T3 developed HCC, with no sign of lung metastases.152 An anti-preneoplastic effect of T3 was also observed in the choline-devoid methionine-deficient nutritional model.153 Interestingly, T3 has also been recognized as a strong inducer of hepatocyte proliferation in rats and mice.154,155 The potent mitogenic effect of T3 was demonstrated not only for intact liver, but also during the regenerative response in rodents after 70% partial or 90% subtotal hepatectomy.156,157 Hepatic mitogenic effects of TH were shown also in the absence of activation of other transcription factors, such as activator protein 1, NF-κB, or signal transducer and activator of transcription 3, while c-fos, c-jun, or c-myc proto-oncogenes and cyclin D1 mRNA and protein levels were increased.155
FIGURE 4.

TH action promotes differentiation and inhibits neoplastic proliferation in hepatocytes, while hypothyroidism and somatic mutations in the TRα and TRβ genes are involved in HCC progression. TH action is also involved in HCC inhibition by means of Wnt/β-catenin/PDK1/PGC1α or Wnt/β-catenin/DKK-4 pathway. TH signaling indirectly activates PPARγ, that is also involved in HCC inhibition by means of cell cycle arrest and apoptosis. Abbreviations: DKK, Dickkopf; PDK, pyruvate dehydrogenase kinase; PGC1α, PPARγ co-activator 1α; PPAR, peroxisome proliferator-activated receptor; TH, thyroid hormone; TR, thyroid hormone receptor.
Collectively, these results suggest a possible protective effect of T3 against malignant transformation in conditions characterized by an impaired regenerative ability (such as aged livers) or when a rapid growth stimulation of the liver is required.158,159 On the other hand, Chen et al160 have shown that T3/TR signaling upregulates members of the urokinase plasminogen activator system, which increases hepatoma cell metastasis. Among them, urokinase plasminogen activator is highly expressed in multiple types of malignancies, including liver, lung, colon, stomach, breast, and ovarian cancers.161 It is important to note that low levels of PGC1α, whose transcription is positively regulated by TH signaling, were associated with poor prognosis in HCC (Figure 4). Moreover, PGC1α suppressed metastasis of HCC cells by inhibiting the Warburg effect by means of the Wnt/β-catenin/PDK1 axis, in a PPARγ-dependent manner (Figure 4). Collectively, the data indicate that TH-mediated PGC1α may act as a candidate therapeutic target for patients with HCC.162 Moreover, in vitro and in vivo models indicate that the activation of PPARγ can inhibit HCC cell proliferation and tumor growth by inducing cell cycle arrest and apoptosis by means of the regulation of a panel of downstream effector molecules, such as p18, p21, p27, p53, and caspase-3/7 and caspase-8, respectively (Figure 4).163 β-catenin expression in HCC is correlated with poor prognosis164,165 but TH can suppress the Wnt/β-catenin pathway by means of Dickkopf-4 induction, leading to inhibition of hepatoma cell proliferation and metastasizing (Figure 4).166 Conversely, the proteolytic enzyme cathepsin H is induced in TR-expressing hepatoma cells on T3 treatment and its upregulation results in increased cell migration and activation of matrix metalloproteinase or ERK activation.163
In aggregate, TH signaling in the liver has a mostly protective effect against neoplastic transformation and progression.
THYROID HORMONE AND THYROID HORMONE ANALOGS IN NAFLD AND HCC
The development of TH analog compounds started in the 1980s when 2 compounds, L-94901 and CG-23425, were shown to reduce cholesterol levels in rats without unfavorable effects on the heart.167
In the late 1980s and early 1990s, crystallography studies provided new information on TR structure and function. The crystal structure of the ligand-binding pocket of the TRα and TRβ revealed that they varied only in 1 amino acid residue (Asn331, TRβ, Ser277, and TRα) and that the selectivity originated from this difference.167–169 These discoveries allowed to improve the design of TR-targeting drugs, in particular for TRβ1-selective thyromimetics, and a number of different ligands were subsequently developed: (3,5-dimethyl-4 (4′-hydroxy-3′- isopropylbenzyl)-phenoxy) acetic acid (GC-1) known as sobetirome, 3-[(3,5-dibromo-4-(4-hydroxy-3-(1-methylethyl)-phenoxy)-phenyl)-amino-3-oxopropanoic acid) known as eprotirome, KB2115, (3,5-dimethyl-4-(4′-hydroxy-3′-isopropylbenzyl)phenoxy] methylphosphonic acid known as MB07344/VK2809, and 2-[3,5-Dichloro-4-(5-isopropyl-6-oxo-1,6-dihydropyridazin-3-yloxy)phenyl]-3,5-dioxo-2,3,4,5-tetrahydro[1,2,4]triazine-6-carbonitrile, also known as MGL-3196 and resmetirom.
TH could be useful in the treatment of hyperlipidemia and NAFLD. However, supraphysiologic TH levels trigger significant end-organ side effects, such as sarcopenia, osteoporosis, cardiac arrhythmia, and heart failure.170 Conversely, some TH derivatives may exert beneficial actions without considerable side effects.10,11,170
As discussed, TH and thyromimetics regulate lipid parameters through numerous associated pathways, such as bile acid synthesis, reverse cholesterol transport, and cholesterol reabsorption by means of LDL receptors.167
THYROMIMETICS
Sobetirome. Sobetirome, developed in 1998, was the first synthetic TRβ agonist.171 It accumulates predominantly in the liver and binds all major TRβ isoforms with similar affinity to T3 binding preferentially to TRβ relative to TRα, and was initially studied as a lipid-lowering compound able to stimulate hepatic pathways with minimal side effects due to its hepatic tropism.172 Sobetirome binds selectively to TRβ1 with half maximal EC50 of 0.16 vs. 0.58 μM for TRα1,173 exhibiting gene-specific actions relative to the native TH form.6 Sobetirome (48 nmol/kg) reduces HDL cholesterol and VLDL triglyceride levels in euthyroid mice, reduces HDL cholesterol and triglyceride levels, increases hepatic HDL receptors and stimulates bile acid synthesis in hypercholesterolemic mice.174
In the choline-methionine deficient diet rats, a model of NAFLD, sobetirome prevented and reverted choline-methionine deficient-induced fat accumulation, ameliorated steatohepatitis and promoted regression to pre-existing fat accumulation without significant side effects on heart rate. Sobetirome ameliorated hepatic steatosis also in other animal experimental models, such as obese/obese mice and western diet-fed LDLR−/− mice.141
In rats and mice, sobetirome also replicates the T3 effects as a powerful inducer of hepatocyte proliferation,175,176 thus sobetirome might be beneficial in the area of hepatic regenerative medicine. Indeed, preclinical studies demonstrated that mice pretreated with sobetirome showed a significant increase in hepatocyte proliferation following partial hepatectomy.177 Sobetirome exerts therapeutic effects on HCC. T3 or sobetirome administration in rats treated with diethylnitrosamine combined with a choline-deficient diet dramatically reduced the preneoplastic lesions caused by diethylnitrosamine.178–180
Eprotirome. Eprotirome is another TRβ liver-selective agonist. Eprotirome prevented hepatic steatosis development and significantly lowered serum total and LDL cholesterol.141,181,182 Rats treated with eprotirome had increased hepatocyte proliferation with no evidence of significant liver toxicity.183 Eprotirome appeared to be more liver-specific than sobetirome and did not cause myocardial off-target effects.181,182 At present, no data regarding the effect of eprotirome on HCC development/progression are available.
MB07344/VK2809. The active compound MB07344/VK2809, derived from hepatic enzymatic cleavage of MB07811/VK2809, has been characterized as a liver-selective TRβ agonist. MB07344 displays around 12-fold selectivity for TRβ over TRα and approximately half of the affinity of T3.171,174 MB07811/VK2809 treatment for 2 weeks in high-fat diet exposed or diabetic obese animals significantly decreased cholesterol and both serum and hepatic triglycerides without effects on body weight, glycemia, and the thyroid hormone axis, while showing an acceptable side effect profile.130
MB07811/VK2809 is a promising drug for the treatment of metabolic disorders, including NASH. In a rodent model of diet-induced NASH, VK2809 caused a significant reduction in plasma and liver lipids, as well as improvements in liver fibrosis. In animal models of hypercholesterolemia, VK2809 demonstrated a promising reduction in plasma cholesterol with minimal effect on the thyroid hormone.184
MB07811 markedly reduced hepatic steatosis, plasma FFAs, and triglycerides in several experimental models of NAFLD, such as Zucker diabetic fatty rats, obese/obese mice, and diet-induced obese mice.184 Importantly, after treatment with MB07811, no sign of liver fibrosis or other histological liver damage was observed.
Resmetirom. Resmetirom is a highly selective TRβ agonist with an EC50 value of 0.21 μM and it is 28-fold selective for TRβ (EC50 = 0.21 μM) over TRα (EC50 = 3.74 μM).171 Resmetirom showed a very positive myocardial safety profile in rats and was efficacious in a preclinical model at doses that showed no impact on the central thyroid axis. In studies conducted in healthy volunteers, resmetirom administered orally once daily at 50 mg or higher dose for 2 weeks exhibited an excellent safety profile and decreased LDL cholesterol and triglycerides.171
In diet-induced obese mice treated at human-equivalent doses, resmetirom reversed and prevented progression of lipid accumulation, inflammatory, and fibrotic markers of NASH without the adverse effects of T3. In humans, 60% reduction in triglycerides was observed for doses ranging from 50 to 200 mg, in the absence of drug-related adverse events.185
CLINICAL USE OF THYROMIMETICS
Eprotirome phase 3 clinical trial was undertaken to compare eprotirome (at the daily doses of 50 and 100 μg)versus placebo in 236 patients with familial hypercholesterolemia. The trial was interrupted because of a significant increase in liver enzymes in 4 patients and the contemporary report of cartilage side effects in animals.186 Conversely, after achieving excellent results in experimental models, clinical trials have been started with resmetirom and MB07811/VK2809, and the initial reports are extremely encouraging.187,188
NAFLD/NASH. Thyromimetic compounds (Table 3) actions are mediated in the liver by TRβ and have been proven to be beneficial in dyslipidemia and NAFLD.141,190,191
TABLE 3.
Thyromimetic compounds in clinical trials
| Compound | NCT number | Sponsor | Condition | Status | Phase |
|---|---|---|---|---|---|
| Sobetirome | QuatRX | Hypercholesterolemia | Complete | 1 | |
| Eprotirome | NCT00677248 | Karo Bio AB | Hypercholesterolemia | Complete | 2 |
| NCT00776321 | Karo Bio AB | Primary hypercholesterolemia | Complete | 2 | |
| NCT00593047 | Karo Bio AB | Dyslipidemia | Complete | 2 | |
| NCT01410383 | Karo Bio AB | Heterozygous familiar hypercholesterolemia | Complete | 3 | |
| VK2809 | NTC04173065 | Viking Therapeutics, Inc. | NASH | Active | 2 |
| NCT02927184 | Viking Therapeutics, Inc. | Hyperlipidemia/NAFLD | Complete | 2 | |
| Resmetirom | NCT04197479 | Madrigal Pharmaceuticals, Inc. | NAFLD | Active | 3 |
| NCT02912260 | Madrigal Pharmaceuticals, Inc. | NASH | Active | 2 | |
| NCT03900429 | Madrigal Pharmaceuticals, Inc. | NASH | Activea | 3 | |
| NCT04951219 | Madrigal Pharmaceuticals, Inc. | NAFLD | Active | 3 | |
| NCT04643795 | Madrigal Pharmaceuticals, Inc. | Hepatic impairment/NASH/cirrhosis | Active | 1 |
Results reported in abstract form.189
To date, 4 different compounds have been employed in clinical trials, originally studied for their ability to reduce LDL cholesterol, in particular sobetirome and eprotirome, however, in recent years, TRβ agonists have been considered for NAFLD/NASH treatment in particular, MB07344/VK2809, and resmetirom.
Sobetirome completed phase 1 clinical trials in 2008 and demonstrated lipid-lowering effects with both single and multiple dosing192 with no phase II trials being planned due to the reported elevation of liver enzymes.
Eprotirome progressed to phase 3 clinical trial but was terminated due to unexpected side effects in animal studies.193 Four clinical trials were conducted between 2008 and 2012 (identifier: NCT00677248, NTC00776321, NCT00593047, and NCT01410383).
In 2021, Younossi and colleagues studied 125 patients with NASH, of whom 84 were treated with 80 mg/d with resmetirom and 41 with placebo for 36. Compared to placebo, resmetirom-treated patients showed significant improvement of health-related quality of life.194
In a randomized, double-blind, placebo control in phase 2 clinical trial (NCT02912260), resmetirom was studied in 116 patients with NASH (fibrosis stages 1–3). Seventy-eight patients received 80 mg of resmetirom 38 patients were treated with placebo for 36 weeks.187 Patients treated with resmetirom (n = 78) showed a relative reduction of hepatic fat at week 12 and week 36. Biopsy performed at 36 weeks showed that histological markers of inflammation were reduced. Adverse events were mostly mild or moderate and were balanced between groups, except for a higher incidence of transient mild diarrhea and nausea in the resmetirom-treated group.
MAESTRO‐NAFLD‐1 (NCT04197479), a 52‐week phase 3 “real‐life NASH study” that enrolls patients based on NASH diagnosis using noninvasive assessments is currently ongoing.
An explorative 36-week active treatment open-label extension study was conducted in 31 consenting patients with NASH (14 treated with placebo and 17 treated with 80 or 100 mg resmetirom orally per day).195 At the end of the open-label extension study after 36 weeks, patients treated with resmetirom had improvements in proton density fat fraction, reduction of lipoprotein (LDL cholesterol, ApoB, triglycerides, and ApoC3), reduction of liver enzymes, and increased sex hormone–binding globulin from baseline. However, given the small number of patients treated with 100 mg compared with 80 mg, no statistical comparison was possible between the 2 doses. The ratio N-terminal type III collagen pro-peptide and matrix metalloproteinase-degraded C3 indicated an overall decrease in fibrosis. Resmetirom was well tolerated, without serious adverse events.
Based on these results, a resmetirom phase 3 clinical trial “MAESTRO‐NASH” (NCT03900429) was designed. Patients with NASH and stage F2 or F3 fibrosis are currently recruited for a 52-week double-blind, 3-arm placebo-controlled trial with resmetirom 80 mg, 100 mg daily, or placebo. Endpoints of the study are resolution of NASH, or significant reduction of fibrosis on liver biopsy. The results of this study, presented very recently in abstract form, indicate that resmetirom treatment 80 mg, and 100 mg compared to placebo resulted in highly significant increase in NASH resolution 26%, 30% vs. 10%, and reduction in NAFLD activity score 24%, 26% versus 14%, respectively. Moreover, resmetirom treatment 80 mg, and 100 mg compared to placebo resulted in significant decrease in LDL cholesterol −12%, −16% versus 1%. Resmetirom was well tolerated with only significant increase in diarrhea and nausea.189
Currently, (Table 4), 5 resmetirom clinical trials are ongoing, all in active recruitment state (ClinicalTrials.gov identifier: NCT02912260, NCT04197479, NCT03900429, NCT04951219, and NCT04643795). Long-term and postapproval surveillance studies will be necessary to confirm safety and assure absence of off-target effects.
TABLE 4.
Thyromimetic compounds in clinical trials (↑ increase, ↓ decrease)
| Results | No. patients (treated vs. placebo) | Type of treatment | |
|---|---|---|---|
| NCT02912260 | ↓ Hepatic fat ↓ Inflammation markers |
116 (78 vs. 38) | 80 mg daily OL |
| ↑ PDFF ↓ Lipoprotein (LDL-C, ApoB, ApoCIII, tryglicerides) ↓ Liver enzyme (ALT, AST, GGT) ↑ SHBG ↑ Fibrosis (↑ collagen markers ↑ Pro-C3 and ↓ C3M) |
31 (17 vs. 14) | 80 or 100 mg daily OL | |
| NCT04197479 | Current enrolling data not available | ||
| NCT03900429 | Current enrolling data not available | ||
| NCT04951219 | Current enrolling data not available | ||
| NCT04643795 | Current enrolling data not available | ||
Abbreviations: C3M, matrix metalloproteinase-degraded C3; PDFF, proton density fat fraction; PRO-C3, N-terminal type III collagen pro-peptide; SHBG, sex hormone–binding globulin.
VK2809 was tested in a multicenter, randomized, double-blind, placebo-controlled, phase 2 trial for safety, tolerability, and efficacy (NCT02927184). In this study, 337 patients with NAFLD and liver fat content ≥8% were assigned to 3 different doses of VK2809 or placebo for 12 weeks. Safety was also assessed at 16 weeks (4 wk following completion of the treatment with active compound). Following 12 weeks of therapy, patients treated with VK2809 experienced statistically significant reductions in liver fat content relative to placebo. These effects were maintained at the week-16 assessment. VK2809 was well tolerated in this study and no serious adverse events were reported in any cohort, and overall excellent side effect profile. These data indicate that a 12-week treatment with low-dose (1.0, 2.5, or 5.0 mg) VK2809 produced significant and robust improvements in liver fat content in patients with NAFLD that were similar to the high dose (10 mg) every other day.196,197
Currently, VK2809 has progressed to phase 2 clinical trial in patients with primary hypercholesterolemia and NAFLD (ClinicalTrials.gov identifier: NCT04173065).
A therapeutic strategy based on thyromimetic use, either as single agents or in combination with other drugs, may be effective for the treatment of liver diseases, as NAFLD and NASH are currently devoid of any effective treatment. Future data from thyromimetic clinical trials will provide more clarity on their safety and their possible clinical indications.
Liver is one of the major target tissues of TH action and TH levels are closely correlated with liver-associated diseases with a spectrum ranging from hepatic steatosis to HCC. There is a complex interplay between the thyroid axis and the liver, and the role of TH in the pathogenesis of liver diseases has been established. Conversely, liver disorders may affect TH levels, and at the present time, it is not clear whether they are adaptive or maladaptive. In this review, we have discussed the growing complexity of TH signaling in the liver and highlighted the potential benefits associated with TH-based therapeutic strategies for liver-related diseases as well as recent encouraging results from clinical trials on the use of thyromimetics for the treatment of liver disease, especially NASH.
Footnotes
Abbreviations: Abcg, ATP-binding cassette subfamily G member; ALD, alcohol-associated liver disease; APOA1, apolipoprotein A1; C3M, matrix metalloproteinase-degraded C3; CDX2, caudal-type homeobox 2; CETP, cholesteryl ester transfer protein; CHREBP, carbohydrate-responsive element-binding protein; CYP7A1, cholesterol 7α-hydroxylase; DKK, Dickkopf; DNL, de novo lipogenesis; ERK, extracellular signal-regulated kinase; FABP, fatty acid–binding protein; FAT/CD36, fatty acid translocase; FDPS, farnesyl diphosphate synthase; FFA, free fatty acid; FOXO1, forkhead box protein O1; fT3, free-T3; fT4, free-T4; GLUT, glucose transporter; HDAC3, histone deacetylase 3; HIF, hypoxia inducible factor; HL, hepatic lipase; HMGCR, 3-hydroxy-3-methylglutaryl-CoA reductase; iPLA2β, calcium-independent membrane phospholipase A2; LDLR, LDL receptor; LPL, lipoprotein lipase; LXR, liver X receptor; MCT, monocarboxylate transporter; NCoR, nuclear receptor co-repressor; NTIS, nonthyroidal illness syndrome; PCAF, P300/CBP-associated factor; PDFF, proton density fat fraction; PDK, pyruvate dehydrogenase kinase; PGC1α, PPARγ coactivator 1α; PI3K, phosphatidylinositol-3-kinase; PPAR, peroxisome proliferator-activated receptor; PRO-C3, N-terminal type III collagen pro-peptide; rT3, reverse T3; RXR, retinoid X receptor; SHBG, sex hormone–binding globulin; SIRT1, NAD-dependent protein deacetylase sirtuin 1; SMRT, silencing mediator for retinoid or thyroid hormone receptors; SOAT2, sterol O-acyltransferase 2; SRB1, scavenger receptor class B member 1; SRC, steroid receptor coactivator; SREBP, sterol regulatory element-binding protein; STAT3, signal transducer and activator of transcription 3; T2, 3,5-diiodo-L-thyronine; T3, 3,5,3′-triiodothyronine; T4, 5′,3,5,3′ tetraiodothyronine; TBG, thyroid hormone binding globulin; TH, thyroid hormone; TR, thyroid hormone receptor; TRH, thyrotropin-releasing hormone; TSH, thyroid stimulating hormone; TTR, transthyretin; UDP, uracil-diphosphate.
Contributor Information
Luigi Marino, Email: lmarino@uchc.edu.
Adam Kim, Email: adkim@uchc.edu.
Bin Ni, Email: bni@alliancepharmaco.com.
Francesco S. Celi, Email: celi@uchc.edu.
CONFLICTS OF INTEREST
The authors have no conflicts to report.
REFERENCES
- 1.Sinha RA, Singh BK, Yen PM. Thyroid hormone regulation of hepatic lipid and carbohydrate metabolism. Trends Endocrinol Metab. 2014;25:538–45. [DOI] [PubMed] [Google Scholar]
- 2.Dayan CM, Panicker V. Novel insights into thyroid hormones from the study of common genetic variation. Nat Rev Endocrinol. 2009;5:211–8. [DOI] [PubMed] [Google Scholar]
- 3.Hassi J, Sikkila K, Ruokonen A, Leppaluoto J. The pituitary-thyroid axis in healthy men living under subarctic climatological conditions. J Endocrinol. 2001;169:195–203. [DOI] [PubMed] [Google Scholar]
- 4.Mebis L, van den Berghe G. The hypothalamus-pituitary-thyroid axis in critical illness. Neth J Med. 2009;67:332–40. [PubMed] [Google Scholar]
- 5.Ghamari-Langroudi M, Vella KR, Srisai D, Sugrue ML, Hollenberg AN, Cone RD. Regulation of thyrotropin-releasing hormone-expressing neurons in paraventricular nucleus of the hypothalamus by signals of adiposity. Mol Endocrinol. 2010;24:2366–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huang YH, Tsai MM, Lin KH. Thyroid hormone dependent regulation of target genes and their physiological significance. Chang Gung Med J. 2008;31:325–34. [PubMed] [Google Scholar]
- 7.Pilo A, Iervasi G, Vitek F, Ferdeghini M, Cazzuola F, Bianchi R. Thyroidal and peripheral production of 3,5,3’-triiodothyronine in humans by multicompartmental analysis. Am J Physiol. 1990;258:E715–26. [DOI] [PubMed] [Google Scholar]
- 8.Khemichian S, Fong TL. Hepatic dysfunction in hyperthyroidism. Gastroenterol Hepatol (NY). 2011;7:337–9. [PMC free article] [PubMed] [Google Scholar]
- 9.Eshraghian A, Hamidian Jahromi A. Non-alcoholic fatty liver disease and thyroid dysfunction: A systematic review. World J Gastroenterol. 2014;20:8102–09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baxter JD, Webb P. Thyroid hormone mimetics: potential applications in atherosclerosis, obesity and type 2 diabetes. Nat Rev Drug Discov. 2009;8:308–20. [DOI] [PubMed] [Google Scholar]
- 11.Brenta G, Danzi S, Klein I. Potential therapeutic applications of thyroid hormone analogs. Nat Clin Pract Endocrinol Metab. 2007;3:632–40. [DOI] [PubMed] [Google Scholar]
- 12.Schweizer U, Schlicker C, Braun D, Kohrle J, Steegborn C. Crystal structure of mammalian selenocysteine-dependent iodothyronine deiodinase suggests a peroxiredoxin-like catalytic mechanism. Proc Natl Acad Sci USA. 2014;111:10526–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Celi FS, Coppotelli G, Chidakel A, Kelly M, Brillante BA, Shawker T, et al. The role of type 1 and type 2 5’-deiodinase in the pathophysiology of the 3,5,3’-triiodothyronine toxicosis of McCune-Albright syndrome. J Clin Endocrinol Metab. 2008;93:2383–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brtko J, Bobalova J, Podoba J, Schmutzler C, Kohrle J. Thyroid hormone receptors and type I iodothyronine 5’-deiodinase activity of human thyroid toxic adenomas and benign cold nodules. Exp Clin Endocrinol Diabetes. 2002;110:166–70. [DOI] [PubMed] [Google Scholar]
- 15.Brent GA. Mechanisms of thyroid hormone action. J Clin Invest. 2012;122:3035–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chi HC, Chen CY, Tsai MM, Tsai CY, Lin KH. Molecular functions of thyroid hormones and their clinical significance in liver-related diseases. Biomed Res Int. 2013;2013:601361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sabatino L, Vassalle C, Del Seppia C, Iervasi G. Deiodinases and the three types of thyroid hormone deiodination reactions. Endocrinol Metab (Seoul). 2021;36:952–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Refetoff S, Murata Y, Mori Y, Janssen OE, Takeda K, Hayashi Y. Thyroxine-binding globulin: Organization of the gene and variants. Horm Res. 1996;45:128–38. [DOI] [PubMed] [Google Scholar]
- 19.Hayashi Y, Mori Y, Janssen OE, Sunthornthepvarakul T, Weiss RE, Takeda K, et al. Human thyroxine-binding globulin gene: complete sequence and transcriptional regulation. Mol Endocrinol. 1993;7:1049–60. [DOI] [PubMed] [Google Scholar]
- 20.Schweizer U, Kohrle J. Function of thyroid hormone transporters in the central nervous system. Biochim Biophys Acta. 2013;1830:3965–73. [DOI] [PubMed] [Google Scholar]
- 21.Blake CC, Geisow MJ, Swan ID, Rerat C, Rerat B. Strjcture of human plasma prealbumin at 2-5 A resolution. A preliminary report on the polypeptide chain conformation, quaternary structure and thyroxine binding. J Mol Biol. 1974;88:1–12. [DOI] [PubMed] [Google Scholar]
- 22.Blake CC, Geisow MJ, Oatley SJ, Rerat B, Rerat C. Structure of prealbumin: secondary, tertiary and quaternary interactions determined by Fourier refinement at 1.8 A. J Mol Biol. 1978;121:339–56. [DOI] [PubMed] [Google Scholar]
- 23.Soprano DR, Herbert J, Soprano KJ, Schon EA, Goodman DS. Demonstration of transthyretin mRNA in the brain and other extrahepatic tissues in the rat. J Biol Chem. 1985;260:11793–798. [PubMed] [Google Scholar]
- 24.Peters T, Jr. Serum albumin. Adv Protein Chem. 1985;37:161–245. [DOI] [PubMed] [Google Scholar]
- 25.Bartalena L, Robbins J. Variations in thyroid hormone transport proteins and their clinical implications. Thyroid. 1992;2:237–45. [DOI] [PubMed] [Google Scholar]
- 26.Yeo PP, Yabu Y, Etzkorn JR, Rajatanavin R, Braverman LE, Ingbar SH. A four generation study of familial dysalbuminemic hyperthyroxinemia: diagnosis in the presence of an acquired excess of thyroxine-binding globulin. J Endocrinol Invest. 1987;10:33–38. [DOI] [PubMed] [Google Scholar]
- 27.Evans RM. The steroid and thyroid hormone receptor superfamily. Science. 1988;240:889–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cheng SY, Leonard JL, Davis PJ. Molecular aspects of thyroid hormone actions. Endocr Rev. 2010;31:139–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Margolis RN. The Nuclear Receptor Signaling Atlas: Catalyzing understanding of thyroid hormone signaling and metabolic control. Thyroid. 2008;18:113–22. [DOI] [PubMed] [Google Scholar]
- 30.Lazar MA. Thyroid hormone receptors: Multiple forms, multiple possibilities. Endocr Rev. 1993;14:184–93. [DOI] [PubMed] [Google Scholar]
- 31.Chamba A, Neuberger J, Strain A, Hopkins J, Sheppard MC, Franklyn JA. Expression and function of thyroid hormone receptor variants in normal and chronically diseased human liver. J Clin Endocrinol Metab. 1996;81:360–7. [DOI] [PubMed] [Google Scholar]
- 32.Shi YB. Dual functions of thyroid hormone receptors in vertebrate development: the roles of histone-modifying cofactor complexes. Thyroid. 2009;19:987–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Song Y, Shan S, Zhang Y, Liu W, Ding W, Ren W, et al. Ligand-dependent corepressor acts as a novel corepressor of thyroid hormone receptor and represses hepatic lipogenesis in mice. J Hepatol. 2012;56:248–54. [DOI] [PubMed] [Google Scholar]
- 34.Bergh JJ, Lin HY, Lansing L, Mohamed SN, Davis FB, Mousa S, et al. Integrin alphaVbeta3 contains a cell surface receptor site for thyroid hormone that is linked to activation of mitogen-activated protein kinase and induction of angiogenesis. Endocrinology. 2005;146:2864–71. [DOI] [PubMed] [Google Scholar]
- 35.Hones GS, Rakov H, Logan J, Liao XH, Werbenko E, Pollard AS, et al. Noncanonical thyroid hormone signaling mediates cardiometabolic effects in vivo. Proc Natl Acad Sci USA. 2017;114:E11323–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Flamant F, Cheng SY, Hollenberg AN, Moeller LC, Samarut J, Wondisford FE, et al. Thyroid hormone signaling pathways: time for a more precise nomenclature. Endocrinology. 2017;158:2052–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chi HC, Tsai CY, Tsai MM, Yeh CT, Lin KH. Molecular functions and clinical impact of thyroid hormone-triggered autophagy in liver-related diseases. J Biomed Sci. 2019;26:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Araki O, Ying H, Zhu XG, Willingham MC, Cheng SY. Distinct dysregulation of lipid metabolism by unliganded thyroid hormone receptor isoforms. Mol Endocrinol. 2009;23:308–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chaves C, Bruinstroop E, Refetoff S, Yen PM, Anselmo J. Increased hepatic fat content in patients with resistance to thyroid hormone beta. Thyroid. 2021;31:1127–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moran C, McEniery CM, Schoenmakers N, Mitchell C, Sleigh A, Watson L, et al. Dyslipidemia, insulin resistance, ectopic lipid accumulation, and vascular function in resistance to thyroid hormone beta. J Clin Endocrinol Metab. 2021;106:e2005–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang XY, Kaneshige M, Kamiya Y, Kaneshige K, McPhie P, Cheng SY. Differential expression of thyroid hormone receptor isoforms dictates the dominant negative activity of mutant Beta receptor. Mol Endocrinol. 2002;16:2077–92. [DOI] [PubMed] [Google Scholar]
- 42.Ritter MJ, Amano I, Hollenberg AN. Thyroid hormone signaling and the liver. Hepatology. 2020;72:742–752. [DOI] [PubMed] [Google Scholar]
- 43.Li L, Song Y, Shi Y, Sun L. Thyroid hormone receptor-beta agonists in NAFLD therapy: possibilities and challenges. J Clin Endocrinol Metab. 2023;108:1602–13. [DOI] [PubMed] [Google Scholar]
- 44.Ness GC, Lopez D, Chambers CM, Newsome WP, Cornelius P, Long CA, et al. Effects of L‐triiodothyronine and the thyromimetic L‐94901 on serum lipoprotein levels and hepatic low‐density lipoprotein receptor, 3‐hydroxy‐3‐methylglutaryl coenzyme A reductase, and apo A‐I gene expression. Biochem Pharmacol. 1998;56:121–9. [DOI] [PubMed] [Google Scholar]
- 45.Ness GC, Lopez D. Transcriptional regulation of rat hepatic low-density lipoprotein receptor and cholesterol 7 alpha hydroxylase by thyroid hormone. Arch Biochem Biophys. 1995;323:404–8. [DOI] [PubMed] [Google Scholar]
- 46.Taylor AH, Stephan ZF, Steele RE, Wong NC. Beneficial effects of a novel thyromimetic on lipoprotein metabolism. Mol Pharmacol. 1997;52:542–7. [DOI] [PubMed] [Google Scholar]
- 47.Davis PJ, Davis FB, Mousa SA, Luidens MK, Lin HY. Membrane receptor for thyroid hormone: physiologic and pharmacologic implications. Annu Rev Pharmacol Toxicol. 2011;51:99–115. [DOI] [PubMed] [Google Scholar]
- 48.Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, Parks EJ. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest. 2005;115:1343–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Singh BK, Sinha RA, Zhou J, Tripathi M, Ohba K, Wang ME, et al. Hepatic FOXO1 target genes are co-regulated by thyroid hormone via RICTOR protein deacetylation and MTORC2-AKT protein inhibition. J Biol Chem. 2016;291:198–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Singh BK, Sinha RA, Zhou J, Xie SY, You SH, Gauthier K, et al. FoxO1 deacetylation regulates thyroid hormone-induced transcription of key hepatic gluconeogenic genes. J Biol Chem. 2013;288:30365–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stremmel W, Staffer S, Wannhoff A, Pathil A, Chamulitrat W. Plasma membrane phospholipase A2 controls hepatocellular fatty acid uptake and is responsive to pharmacological modulation: Implications for nonalcoholic steatohepatitis. FASEB J. 2014;28:3159–70. [DOI] [PubMed] [Google Scholar]
- 52.Quiroga AD, Lehner R. Liver triacylglycerol lipases. Biochim Biophys Acta. 2012;1821:762–9. [DOI] [PubMed] [Google Scholar]
- 53.Kihara S, Wolle J, Ehnholm C, Chan L, Oka K. Regulation of hepatic triglyceride lipase by thyroid hormone in HepG2 cells. J Lipid Res. 1993;34:961–70. [PubMed] [Google Scholar]
- 54.Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, et al. Autophagy regulates lipid metabolism. Nature. 2009;458:1131–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cingolani F, Czaja MJ. Regulation and functions of autophagic lipolysis. Trends Endocrinol Metab. 2016;27:696–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Adams AC, Astapova I, Fisher FM, Badman MK, Kurgansky KE, Flier JS, et al. Thyroid hormone regulates hepatic expression of fibroblast growth factor 21 in a PPARalpha-dependent manner. J Biol Chem. 2010;285:14078–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Thakran S, Sharma P, Attia RR, Hori RT, Deng X, Elam MB, et al. Role of sirtuin 1 in the regulation of hepatic gene expression by thyroid hormone. J Biol Chem. 2013;288:807–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mullur R, Liu YY, Brent GA. Thyroid hormone regulation of metabolism. Physiol Rev. 2014;94:355–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Djouadi F, Riveau B, Merlet-Benichou C, Bastin J. Tissue-specific regulation of medium-chain acyl-CoA dehydrogenase gene by thyroid hormones in the developing rat. Biochem J. 1997;324(Pt 1):289–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Holness MJ, Bulmer K, Smith ND, Sugden MC. Investigation of potential mechanisms regulating protein expression of hepatic pyruvate dehydrogenase kinase isoforms 2 and 4 by fatty acids and thyroid hormone. Biochem J. 2003;369:687–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jekabsons MB, Gregoire FM, Schonfeld-Warden NA, Warden CH, Horwitz BA. T(3) stimulates resting metabolism and UCP-2 and UCP-3 mRNA but not nonphosphorylating mitochondrial respiration in mice. Am J Physiol. 1999;277:E380–9. [DOI] [PubMed] [Google Scholar]
- 62.Sinha RA, You SH, Zhou J, Siddique MM, Bay BH, Zhu X, et al. Thyroid hormone stimulates hepatic lipid catabolism via activation of autophagy. J Clin Invest. 2012;122:2428–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang Y, Viscarra J, Kim SJ, Sul HS. Transcriptional regulation of hepatic lipogenesis. Nat Rev Mol Cell Biol. 2015;16:678–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hashimoto K, Matsumoto S, Yamada M, Satoh T, Mori M. Liver X receptor-alpha gene expression is positively regulated by thyroid hormone. Endocrinology. 2007;148:4667–75. [DOI] [PubMed] [Google Scholar]
- 65.Hashimoto K, Ishida E, Matsumoto S, Okada S, Yamada M, Satoh T, et al. Carbohydrate response element binding protein gene expression is positively regulated by thyroid hormone. Endocrinology. 2009;150:3417–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ness GC, Pendleton LC, Li YC, Chiang JY. Effect of thyroid hormone on hepatic cholesterol 7 alpha hydroxylase, LDL receptor, HMG-CoA reductase, farnesyl pyrophosphate synthetase and apolipoprotein A-I mRNA levels in hypophysectomized rats. Biochem Biophys Res Commun. 1990;172:1150–6. [DOI] [PubMed] [Google Scholar]
- 67.Yap CS, Sinha RA, Ota S, Katsuki M, Yen PM. Thyroid hormone negatively regulates CDX2 and SOAT2 mRNA expression via induction of miRNA-181d in hepatic cells. Biochem Biophys Res Commun. 2013;440:635–9. [DOI] [PubMed] [Google Scholar]
- 68.Mooradian AD, Wong NC, Shah GN. Age-related changes in the responsiveness of apolipoprotein A1 to thyroid hormone. Am J Physiol. 1996;271:R1602–7. [DOI] [PubMed] [Google Scholar]
- 69.Lopez D, Abisambra Socarras JF, Bedi M, Ness GC. Activation of the hepatic LDL receptor promoter by thyroid hormone. Biochim Biophys Acta. 2007;1771:1216–25. [DOI] [PubMed] [Google Scholar]
- 70.Lagrost L. Regulation of cholesteryl ester transfer protein (CETP) activity: Review of in vitro and in vivo studies. Biochim Biophys Acta. 1994;1215:209–36. [DOI] [PubMed] [Google Scholar]
- 71.Lesmana R, Sinha RA, Singh BK, Zhou J, Ohba K, Wu Y, et al. Thyroid hormone stimulation of autophagy is essential for mitochondrial biogenesis and activity in skeletal muscle. Endocrinology. 2016;157:23–38. [DOI] [PubMed] [Google Scholar]
- 72.Santamarina-Fojo S, Gonzalez-Navarro H, Freeman L, Wagner E, Nong Z. Hepatic lipase, lipoprotein metabolism, and atherogenesis. Arterioscler Thromb Vasc Biol. 2004;24:1750–4. [DOI] [PubMed] [Google Scholar]
- 73.Liu YY, Brent GA. Thyroid hormone crosstalk with nuclear receptor signaling in metabolic regulation. Trends Endocrinol Metab. 2010;21:166–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Goldberg IJ, Huang LS, Huggins LA, Yu S, Nagareddy PR, Scanlan TS, et al. Thyroid hormone reduces cholesterol via a non-LDL receptor-mediated pathway. Endocrinology. 2012;153:5143–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bonde Y, Plosch T, Kuipers F, Angelin B, Rudling M. Stimulation of murine biliary cholesterol secretion by thyroid hormone is dependent on a functional ABCG5/G8 complex. Hepatology. 2012;56:1828–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Moore MC, Coate KC, Winnick JJ, An Z, Cherrington AD. Regulation of hepatic glucose uptake and storage in vivo. Adv Nutr. 2012;3:286–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Adeva-Andany MM, Perez-Felpete N, Fernandez-Fernandez C, Donapetry-Garcia C, Pazos-Garcia C. Liver glucose metabolism in humans. Biosci Rep. 2016;36:e00416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Klieverik LP, Sauerwein HP, Ackermans MT, Boelen A, Kalsbeek A, Fliers E. Effects of thyrotoxicosis and selective hepatic autonomic denervation on hepatic glucose metabolism in rats. Am J Physiol Endocrinol Metab. 2008;294:E513–20. [DOI] [PubMed] [Google Scholar]
- 79.Lopez M, Alvarez CV, Nogueiras R, Dieguez C. Energy balance regulation by thyroid hormones at central level. Trends Mol Med. 2013;19:418–27. [DOI] [PubMed] [Google Scholar]
- 80.Feng X, Jiang Y, Meltzer P, Yen PM. Thyroid hormone regulation of hepatic genes in vivo detected by complementary DNA microarray. Mol Endocrinol. 2000;14:947–955. [DOI] [PubMed] [Google Scholar]
- 81.Park EA, Song S, Vinson C, Roesler WJ. Role of CCAAT enhancer-binding protein beta in the thyroid hormone and cAMP induction of phosphoenolpyruvate carboxykinase gene transcription. J Biol Chem. 1999;274:211–17. [DOI] [PubMed] [Google Scholar]
- 82.Attia RR, Sharma P, Janssen RC, Friedman JE, Deng X, Lee JS, et al. Regulation of pyruvate dehydrogenase kinase 4 (PDK4) by CCAAT/enhancer-binding protein beta (C/EBPbeta). J Biol Chem. 2011;286:23799–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Stockigt JR, Lim CF. Medications that distort in vitro tests of thyroid function, with particular reference to estimates of serum free thyroxine. Best Pract Res Clin Endocrinol Metab. 2009;23:753–67. [DOI] [PubMed] [Google Scholar]
- 84.Boelen A, Kwakkel J, Fliers E. Beyond low plasma T3: Local thyroid hormone metabolism during inflammation and infection. Endocr Rev. 2011;32:670–93. [DOI] [PubMed] [Google Scholar]
- 85.Boelen A, Kwakkel J, Thijssen-Timmer DC, Alkemade A, Fliers E, Wiersinga WM. Simultaneous changes in central and peripheral components of the hypothalamus-pituitary-thyroid axis in lipopolysaccharide-induced acute illness in mice. J Endocrinol. 2004;182:315–23. [DOI] [PubMed] [Google Scholar]
- 86.Peeters RP, Kester MH, Wouters PJ, Kaptein E, van Toor H, Visser TJ, et al. Increased thyroxine sulfate levels in critically ill patients as a result of a decreased hepatic type I deiodinase activity. J Clin Endocrinol Metab. 2005;90:6460–5. [DOI] [PubMed] [Google Scholar]
- 87.Debaveye Y, Ellger B, Mebis L, Darras VM, Van, den Berghe G. Regulation of tissue iodothyronine deiodinase activity in a model of prolonged critical illness. Thyroid. 2008;18:551–60. [DOI] [PubMed] [Google Scholar]
- 88.Peeters RP, Wouters PJ, Kaptein E, van Toor H, Visser TJ, Van, et al. Reduced activation and increased inactivation of thyroid hormone in tissues of critically ill patients. J Clin Endocrinol Metab. 2003;88:3202–11. [DOI] [PubMed] [Google Scholar]
- 89.Hennemann G, Docter R, Friesema EC, de Jong M, Krenning EP, Visser TJ. Plasma membrane transport of thyroid hormones and its role in thyroid hormone metabolism and bioavailability. Endocr Rev. 2001;22:451–76. [DOI] [PubMed] [Google Scholar]
- 90.Farwell AP. Nonthyroidal illness syndrome. Curr Opin Endocrinol Diabetes Obes. 2013;20:478–84. [DOI] [PubMed] [Google Scholar]
- 91.Fekete C, Gereben B, Doleschall M, Harney JW, Dora JM, Bianco AC, et al. Lipopolysaccharide induces type 2 iodothyronine deiodinase in the mediobasal hypothalamus: implications for the nonthyroidal illness syndrome. Endocrinology. 2004;145:1649–55. [DOI] [PubMed] [Google Scholar]
- 92.Mebis L, Paletta D, Debaveye Y, Ellger B, Langouche L, D’Hoore A, et al. Expression of thyroid hormone transporters during critical illness. Eur J Endocrinol. 2009;161:243–50. [DOI] [PubMed] [Google Scholar]
- 93.Beigneux AP, Moser AH, Shigenaga JK, Grunfeld C, Feingold KR. Sick euthyroid syndrome is associated with decreased TR expression and DNA binding in mouse liver. Am J Physiol Endocrinol Metab. 2003;284:E228–36. [DOI] [PubMed] [Google Scholar]
- 94.Williams GR, Franklyn JA, Neuberger JM, Sheppard MC. Thyroid hormone receptor expression in the “sick euthyroid” syndrome. Lancet. 1989;2:1477–81. [DOI] [PubMed] [Google Scholar]
- 95.Balhara Y, Mathur S. Alcohol: A major public health problem—South Asian perspective. Addict Disord Their Treat. 2012;11:101–20. [Google Scholar]
- 96.Ozsoy S, Esel E, Izgi HB, Sofuoglu S. Thyroid function in early and late alcohol withdrawal: relationship with aggression, family history, and onset age of alcoholism. Alcohol Alcohol. 2006;41:515–21. [DOI] [PubMed] [Google Scholar]
- 97.Knudsen N, Bulow I, Laurberg P, Perrild H, Ovesen L, Jorgensen T. Alcohol consumption is associated with reduced prevalence of goitre and solitary thyroid nodules. Clin Endocrinol (Oxf). 2001;55:41–6. [DOI] [PubMed] [Google Scholar]
- 98.Liappas I, Piperi C, Malitas PN, Tzavellas EO, Zisaki A, Liappas AI, et al. Interrelationship of hepatic function, thyroid activity and mood status in alcohol-dependent individuals. In Vivo. 2006;20:293–300. [PubMed] [Google Scholar]
- 99.Pienaar WP, Roberts MC, Emsley RA, Aalbers C, Taljaard FJ. The thyrotropin releasing hormone stimulation test in alcoholism. Alcohol Alcohol. 1995;30:661–7. [PubMed] [Google Scholar]
- 100.Loosen PT, Dew BW, Prange AJ. Long-term predictors of outcome in abstinent alcoholic men. Am J Psychiatry. 1990;147:1662–6. [DOI] [PubMed] [Google Scholar]
- 101.Sudha S, Balasubramanian K, Arunakaran J, Govindarajulu P. Preliminary study of androgen, thyroid & adrenal status in alcoholic men during deaddiction. Indian J Med Res. 1995;101:268–72. [PubMed] [Google Scholar]
- 102.Hegedus L. Decreased thyroid gland volume in alcoholic cirrhosis of the liver. J Clin Endocrinol Metab. 1984;58:930–3. [DOI] [PubMed] [Google Scholar]
- 103.Walfish PG, Orrego H, Israel Y, Blake J, Kalant H. Serum triiodothyronine and other clinical and laboratory indices of alcoholic liver disease. Ann Intern Med. 1979;91:13–6. [DOI] [PubMed] [Google Scholar]
- 104.Burra P, Franklyn JA, Ramsden DB, Elias E, Sheppard MC. Severity of alcoholic liver disease and markers of thyroid and steroid status. Postgrad Med J. 1992;68:804–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Papineni JK, Pinnelli VBK, Davanum R. Thyroid hormone levels in chronic alcoholic liver disease patients before and after treatment. J Clin Diagn Res. 2017;11:BC13–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kondili LA, Taliani G, Cerga G, Tosti ME, Babameto A, Resuli B. Correlation of alcohol consumption with liver histological features in non-cirrhotic patients. Eur J Gastroenterol Hepatol. 2005;17:155–9. [DOI] [PubMed] [Google Scholar]
- 107.Hernandez A. Thyroid hormone and alcoholic fatty liver: the developmental input. Alcohol Clin Exp Res. 2019;43:1834–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Israel Y, Orrego H. Hypermetabolic state and hypoxic liver damage. Recent Dev Alcohol. 1984;2:119–33. [DOI] [PubMed] [Google Scholar]
- 109.Orrego H, Blake JE, Blendis LM, Compton KV, Israel Y. Long-term treatment of alcoholic liver disease with propylthiouracil. N Engl J Med. 1987;317:1421–7. [DOI] [PubMed] [Google Scholar]
- 110.Glinoer D, Cooper DS. The propylthiouracil dilemma. Curr Opin Endocrinol Diabetes Obes. 2012;19:402–7. [DOI] [PubMed] [Google Scholar]
- 111.Younossi ZM, Stepanova M, Afendy M, Fang Y, Younossi Y, Mir H, et al. Changes in the prevalence of the most common causes of chronic liver diseases in the United States from 1988 to 2008. Clin Gastroenterol Hepatol. 2011;9:524–30. [DOI] [PubMed] [Google Scholar]
- 112.Bellentani S, Scaglioni F, Marino M, Bedogni G. Epidemiology of non-alcoholic fatty liver disease. Dig Dis. 2010;28:155–61. [DOI] [PubMed] [Google Scholar]
- 113.Farrell GC, Wong VW, Chitturi S. NAFLD in Asia--as common and important as in the West. Nat Rev Gastroenterol Hepatol. 2013;10:307–18. [DOI] [PubMed] [Google Scholar]
- 114.Federico A, Zulli C, de Sio I, Del Prete A, Dallio M, Masarone M, et al. Focus on emerging drugs for the treatment of patients with non-alcoholic fatty liver disease. World J Gastroenterol. 2014;20:16841–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Sinha RA, Yen PM. Thyroid hormone-mediated autophagy and mitochondrial turnover in NAFLD. Cell Biosci. 2016;6:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Krawczyk M, Bonfrate L, Portincasa P. Nonalcoholic fatty liver disease. Best Pract Res Clin Gastroenterol. 2010;24:695–708. [DOI] [PubMed] [Google Scholar]
- 117.Friedman SL, Neuschwander-Tetri BA, Rinella M, Sanyal AJ. Mechanisms of NAFLD development and therapeutic strategies. Nat Med. 2018;24:908–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Love-Osborne KA, Nadeau KJ, Sheeder J, Fenton LZ, Zeitler P. Presence of the metabolic syndrome in obese adolescents predicts impaired glucose tolerance and nonalcoholic fatty liver disease. J Adolesc Health. 2008;42:543–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Chalasani N, Younossi Z, Lavine JE, Diehl AM, Brunt EM, Cusi K, et al. The diagnosis and management of non-alcoholic fatty liver disease: practice Guideline by the American Association for the Study of Liver Diseases, American College of Gastroenterology, and the American Gastroenterological Association. Hepatology. 2012;55:2005–23. [DOI] [PubMed] [Google Scholar]
- 120.Das K, Chainy GB. Modulation of rat liver mitochondrial antioxidant defence system by thyroid hormone. Biochim Biophys Acta. 2001;1537:1–13. [DOI] [PubMed] [Google Scholar]
- 121.Messarah M, Boumendjel A, Chouabia A, Klibet F, Abdennour C, Boulakoud MS, et al. Influence of thyroid dysfunction on liver lipid peroxidation and antioxidant status in experimental rats. Exp Toxicol Pathol. 2010;62:301–10. [DOI] [PubMed] [Google Scholar]
- 122.Singeap AM, Stanciu C, Huiban L, Muzica CM, Cuciureanu T, Girleanu I, et al. Association between nonalcoholic fatty liver disease and endocrinopathies: clinical implications. Can J Gastroenterol Hepatol. 2021;2021:6678142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Marino L, Jornayvaz FR. Endocrine causes of nonalcoholic fatty liver disease. World J Gastroenterol. 2015;21:11053–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Loria P, Carulli L, Bertolotti M, Lonardo A. Endocrine and liver interaction: the role of endocrine pathways in NASH. Nat Rev Gastroenterol Hepatol. 2009;6:236–47. [DOI] [PubMed] [Google Scholar]
- 125.Sinha RA, Singh BK, Yen PM. Direct effects of thyroid hormones on hepatic lipid metabolism. Nat Rev Endocrinol. 2018;14:259–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Guo Z, Li M, Han B, Qi X. Association of non-alcoholic fatty liver disease with thyroid function: a systematic review and meta-analysis. Dig Liver Dis. 2018;50:1153–62. [DOI] [PubMed] [Google Scholar]
- 127.Lonardo A, Mantovani A, Lugari S, Targher G. NAFLD in some common endocrine diseases: Prevalence, pathophysiology, and principles of diagnosis and management. Int J Mol Sci. 2019;20:2841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Lonardo A, Ballestri S, Mantovani A, Nascimbeni F, Lugari S, Targher G. Pathogenesis of hypothyroidism-induced NAFLD: Evidence for a distinct disease entity? Dig Liver Dis. 2019;51:462–70. [DOI] [PubMed] [Google Scholar]
- 129.Bohinc BN, Michelotti G, Xie G, Pang H, Suzuki A, Guy CD, et al. Repair-related activation of hedgehog signaling in stromal cells promotes intrahepatic hypothyroidism. Endocrinology. 2014;155:4591–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Erion MD, Cable EE, Ito BR, Jiang H, Fujitaki JM, Finn PD, et al. Targeting thyroid hormone receptor-beta agonists to the liver reduces cholesterol and triglycerides and improves the therapeutic index. Proc Natl Acad Sci USA. 2007;104:15490–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Singh BK, Sinha RA, Yen PM. Novel transcriptional mechanisms for regulating metabolism by thyroid hormone. Int J Mol Sci. 2018;19:3284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Pihlajamaki J, Boes T, Kim EY, Dearie F, Kim BW, Schroeder J, et al. Thyroid hormone-related regulation of gene expression in human fatty liver. J Clin Endocrinol Metab. 2009;94:3521–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Kohjima M, Enjoji M, Higuchi N, Kato M, Kotoh K, Yoshimoto T, et al. Re-evaluation of fatty acid metabolism-related gene expression in nonalcoholic fatty liver disease. Int J Mol Med. 2007;20:351–8. [PubMed] [Google Scholar]
- 134.Chiappini F, Barrier A, Saffroy R, Domart MC, Dagues N, Azoulay D, et al. Exploration of global gene expression in human liver steatosis by high-density oligonucleotide microarray. Lab Invest. 2006;86:154–65. [DOI] [PubMed] [Google Scholar]
- 135.Younossi ZM, Gorreta F, Ong JP, Schlauch K, Del Giacco L, Elariny H, et al. Hepatic gene expression in patients with obesity-related non-alcoholic steatohepatitis. Liver Int. 2005;25:760–71. [DOI] [PubMed] [Google Scholar]
- 136.Zhou J, Tripathi M, Ho JP, Widjaja AA, Shekeran SG, Camat MD, et al. Thyroid hormone decreases hepatic steatosis, inflammation, and fibrosis in a dietary mouse model of nonalcoholic steatohepatitis. Thyroid. 2022;32:725–38. [DOI] [PubMed] [Google Scholar]
- 137.Bruinstroop E, Dalan R, Cao Y, Bee YM, Chandran K, Cho LW, et al. Low-dose levothyroxine reduces intrahepatic lipid content in patients with type 2 diabetes mellitus and NAFLD. J Clin Endocrinol Metab. 2018;103:2698–706. [DOI] [PubMed] [Google Scholar]
- 138.Lugari S, Mantovani A, Nascimbeni F, Lonardo A. Hypothyroidism and nonalcoholic fatty liver disease—a chance association? Horm Mol Biol Clin Investig. 2018;41. doi: 10.1515/hmbci-2018-0047 [DOI] [PubMed] [Google Scholar]
- 139.Chen P, Hou X, Wei L, Feng L, Zhong L, Jiao L, et al. Free triiodothyronine is associated with the occurrence and remission of nonalcoholic fatty liver disease in euthyroid women. Eur J Clin Invest. 2019;49:e13070. [DOI] [PubMed] [Google Scholar]
- 140.Bilgin H, Pirgon O. Thyroid function in obese children with non-alcoholic fatty liver disease. J Clin Res Pediatr Endocrinol. 2014;6:152–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Martagon AJ, Lin JZ, Cimini SL, Webb P, Phillips KJ. The amelioration of hepatic steatosis by thyroid hormone receptor agonists is insufficient to restore insulin sensitivity in ob/ob mice. PLoS One. 2015;10:e0122987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Kaltenbach TE, Graeter T, Oeztuerk S, Holzner D, Kratzer W, Wabitsch M, et al. Thyroid dysfunction and hepatic steatosis in overweight children and adolescents. Pediatr Obes. 2017;12:67–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Ferrandino G, Kaspari RR, Spadaro O, Reyna-Neyra A, Perry RJ, Cardone R, et al. Pathogenesis of hypothyroidism-induced NAFLD is driven by intra- and extrahepatic mechanisms. Proc Natl Acad Sci USA. 2017;114:E9172–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. [DOI] [PubMed] [Google Scholar]
- 145.Fattovich G, Stroffolini T, Zagni I, Donato F. Hepatocellular carcinoma in cirrhosis: incidence and risk factors. Gastroenterology. 2004;127:S35–50. [DOI] [PubMed] [Google Scholar]
- 146.Hassan MM, Kaseb A, Li D, Patt YZ, Vauthey JN, Thomas MB, et al. Association between hypothyroidism and hepatocellular carcinoma: a case-control study in the United States. Hepatology. 2009;49:1563–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Reddy A, Dash C, Leerapun A, Mettler TA, Stadheim LM, Lazaridis KN, et al. Hypothyroidism: A possible risk factor for liver cancer in patients with no known underlying cause of liver disease. Clin Gastroenterol Hepatol. 2007;5:118–23. [DOI] [PubMed] [Google Scholar]
- 148.Perra A, Plateroti M, Columbano A. T3/TRs axis in hepatocellular carcinoma: new concepts for an old pair. Endocr Relat Cancer. 2016;23:R353–69. [DOI] [PubMed] [Google Scholar]
- 149.Lin KH, Zhu XG, Shieh HY, Hsu HC, Chen ST, McPhie P, et al. Identification of naturally occurring dominant negative mutants of thyroid hormone alpha 1 and beta 1 receptors in a human hepatocellular carcinoma cell line. Endocrinology. 1996;137:4073–81. [DOI] [PubMed] [Google Scholar]
- 150.Lin KH, Wu YH, Chen SL. Impaired interaction of mutant thyroid hormone receptors associated with human hepatocellular carcinoma with transcriptional coregulators. Endocrinology. 2001;142:653–62. [DOI] [PubMed] [Google Scholar]
- 151.Solt D, Farber E. New principle for analysis of liver carcinogenesis. Am J Pathol. 1976;88:595–618. [PMC free article] [PubMed] [Google Scholar]
- 152.Ledda-Columbano GM, Perra A, Loi R, Shinozuka H, Columbano A. Cell proliferation induced by triiodothyronine in rat liver is associated with nodule regression and reduction of hepatocellular carcinomas. Cancer Res. 2000;60:603–9. [PubMed] [Google Scholar]
- 153.Perra A, Kowalik MA, Pibiri M, Ledda-Columbano GM, Columbano A. Thyroid hormone receptor ligands induce regression of rat preneoplastic liver lesions causing their reversion to a differentiated phenotype. Hepatology. 2009;49:1287–96. [DOI] [PubMed] [Google Scholar]
- 154.Francavilla A, Carr BI, Azzarone A, Polimeno L, Wang Z, Van Thiel DH, et al. Hepatocyte proliferation and gene expression induced by triiodothyronine in vivo and in vitro. Hepatology. 1994;20:1237–41. [PubMed] [Google Scholar]
- 155.Pibiri M, Ledda-Columbano GM, Cossu C, Simbula G, Menegazzi M, Shinozuka H, et al. Cyclin D1 is an early target in hepatocyte proliferation induced by thyroid hormone (T3). FASEB J. 2001;15:1006–13. [DOI] [PubMed] [Google Scholar]
- 156.Higgins GM, Anderson RM. Experimental pathology of the liver. Arch Pathol. 1931;12:186–202. [Google Scholar]
- 157.Malik R, Habib M, Tootle R, Hodgson H. Exogenous thyroid hormone induces liver enlargement, whilst maintaining regenerative potential—a study relevant to donor preconditioning. Am J Transplant. 2005;5:1801–7. [DOI] [PubMed] [Google Scholar]
- 158.Bockhorn M, Frilling A, Benko T, Best J, Sheu SY, Trippler M, et al. Tri-iodothyronine as a stimulator of liver regeneration after partial and subtotal hepatectomy. Eur Surg Res. 2007;39:58–63. [DOI] [PubMed] [Google Scholar]
- 159.Alisi A, Demori I, Spagnuolo S, Pierantozzi E, Fugassa E, Leoni S. Thyroid status affects rat liver regeneration after partial hepatectomy by regulating cell cycle and apoptosis. Cell Physiol Biochem. 2005;15:69–76. [DOI] [PubMed] [Google Scholar]
- 160.Chen CY, Chi LM, Chi HC, Tsai MM, Tsai CY, Tseng YH, et al. Stable isotope labeling with amino acids in cell culture (SILAC)-based quantitative proteomics study of a thyroid hormone-regulated secretome in human hepatoma cells. Mol Cell Proteomics. 2012;11:M111 011270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Paschos KA, Canovas D, Bird NC. Enzymatic function of multiple origins regulates the progression of colorectal cancer and the development of metastases. Hippokratia. 2009;13:23–31. [PMC free article] [PubMed] [Google Scholar]
- 162.Zuo Q, He J, Zhang S, Wang H, Jin G, Jin H, et al. PPARgamma coactivator-1alpha suppresses metastasis of hepatocellular carcinoma by inhibiting Warburg effect by PPARgamma-dependent WNT/beta-catenin/pyruvate dehydrogenase kinase isozyme 1 axis. Hepatology. 2021;73:644–660. [DOI] [PubMed] [Google Scholar]
- 163.Wu CW, Farrel GC, Yu J. Functional role of peroxisome-proliferator-activated receptor γ in hepatocellular carcinoma. J Gastroenterol Hepatol. 2012;27:1665–9. [DOI] [PubMed] [Google Scholar]
- 164.Suzuki T, Yano H, Nakashima Y, Nakashima O, Kojiro M. Beta-catenin expression in hepatocellular carcinoma: a possible participation of beta-catenin in the dedifferentiation process. J Gastroenterol Hepatol. 2002;17:994–1000. [DOI] [PubMed] [Google Scholar]
- 165.Inagawa S, Itabashi M, Adachi S, Kawamoto T, Hori M, Shimazaki J, et al. Expression and prognostic roles of beta-catenin in hepatocellular carcinoma: correlation with tumor progression and postoperative survival. Clin Cancer Res. 2002;8:450–6. [PubMed] [Google Scholar]
- 166.Liao CH, Yeh CT, Huang YH, Wu SM, Chi HC, Tsai MM, et al. Dickkopf 4 positively regulated by the thyroid hormone receptor suppresses cell invasion in human hepatoma cells. Hepatology. 2012;55:910–20. [DOI] [PubMed] [Google Scholar]
- 167.Jakobsson T, Vedin LL, Parini P. Potential rle of thyroid receptor beta agonists in the treatment of hyperlipidemia. Drugs. 2017;77:1613–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Wagner RL, Apriletti JW, McGrath ME, West BL, Baxter JD, Fletterick RJ. A structural role for hormone in the thyroid hormone receptor. Nature. 1995;378:690–7. [DOI] [PubMed] [Google Scholar]
- 169.Ribeiro RC, Apriletti JW, Wagner RL, Feng W, Kushner PJ, Nilsson S, et al. X-ray crystallographic and functional studies of thyroid hormone receptor. J Steroid Biochem Mol Biol. 1998;65:133–41. [DOI] [PubMed] [Google Scholar]
- 170.Baxter JD, Dillmann WH, West BL, Huber R, Furlow JD, Fletterick RJ, et al. Selective modulation of thyroid hormone receptor action. J Steroid Biochem Mol Biol. 2001;76:31–42. [DOI] [PubMed] [Google Scholar]
- 171.Kelly MJ, Pietranico-Cole S, Larigan JD, Haynes NE, Reynolds CH, Scott N, et al. Discovery of 2-[3,5-dichloro-4-(5-isopropyl-6-oxo-1,6-dihydropyridazin-3-yloxy)phenyl]-3,5-dio xo-2,3,4,5-tetrahydro[1,2,4]triazine-6-carbonitrile (MGL-3196), a highly selective thyroid hormone receptor beta agonist in clinical trials for the treatment of dyslipidemia. J Med Chem. 2014;57:3912–23. [DOI] [PubMed] [Google Scholar]
- 172.Wagner RL, Huber BR, Shiau AK, Kelly A, Cunha Lima ST, Scanlan TS, et al. Hormone selectivity in thyroid hormone receptors. Mol Endocrinol. 2001;15:398–410. [DOI] [PubMed] [Google Scholar]
- 173.Gierach I, Li J, Wu WY, Grover GJ, Wood DW. Bacterial biosensors for screening isoform-selective ligands for human thyroid receptors alpha-1 and beta-1. FEBS Open Bio. 2012;2:247–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Johansson L, Rudling M, Scanlan TS, Lundasen T, Webb P, Baxter J, et al. Selective thyroid receptor modulation by GC-1 reduces serum lipids and stimulates steps of reverse cholesterol transport in euthyroid mice. Proc Natl Acad Sci USA. 2005;102:10297–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Columbano A, Pibiri M, Deidda M, Cossu C, Scanlan TS, Chiellini G, et al. The thyroid hormone receptor-beta agonist GC-1 induces cell proliferation in rat liver and pancreas. Endocrinology. 2006;147:3211–8. [DOI] [PubMed] [Google Scholar]
- 176.Kowalik MA, Perra A, Pibiri M, Cocco MT, Samarut J, Plateroti M, et al. TRbeta is the critical thyroid hormone receptor isoform in T3-induced proliferation of hepatocytes and pancreatic acinar cells. J Hepatol. 2010;53:686–92. [DOI] [PubMed] [Google Scholar]
- 177.Alvarado TF, Puliga E, Preziosi M, Poddar M, Singh S, Columbano A, et al. Thyroid hormone receptor beta agonist induces beta-catenin-dependent hepatocyte proliferation in mice: implications in hepatic regeneration. Gene Expr. 2016;17:19–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Chi HC, Chen SL, Tsai CY, Chuang WY, Huang YH, Tsai MM, et al. Thyroid hormone suppresses hepatocarcinogenesis via DAPK2 and SQSTM1-dependent selective autophagy. Autophagy. 2016;12:2271–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Kowalik MA, Columbano A, Perra A. Thyroid hormones, thyromimetics and their metabolites in the treatment of liver disease. Front Endocrinol (Lausanne). 2018;9:382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Ledda-Columbano GM, Perra A, Piga R, Pibiri M, Loi R, Shinozuka H, et al. Cell proliferation induced by 3,3’,5-triiodo-L-thyronine is associated with a reduction in the number of preneoplastic hepatic lesions. Carcinogenesis. 1999;20:2299–304. [DOI] [PubMed] [Google Scholar]
- 181.Berkenstam A, Kristensen J, Mellstrom K, Carlsson B, Malm J, Rehnmark S, et al. The thyroid hormone mimetic compound KB2115 lowers plasma LDL cholesterol and stimulates bile acid synthesis without cardiac effects in humans. Proc Natl Acad Sci USA. 2008;105:663–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Ladenson PW, Kristensen JD, Ridgway EC, Olsson AG, Carlsson B, Klein I, et al. Use of the thyroid hormone analogue eprotirome in statin-treated dyslipidemia. N Engl J Med. 2010;362:906–16. [DOI] [PubMed] [Google Scholar]
- 183.Szydlowska M, Pibiri M, Perra A, Puliga E, Mattu S, Ledda-Columbano GM, et al. The thyromimetic KB2115 (eprotirome) induces rat hepatocyte proliferation. Gene Expr. 2017;17:207–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Cable EE, Finn PD, Stebbins JW, Hou J, Ito BR, van Poelje PD, et al. Reduction of hepatic steatosis in rats and mice after treatment with a liver-targeted thyroid hormone receptor agonist. Hepatology. 2009;49:407–17. [DOI] [PubMed] [Google Scholar]
- 185.Taub R, Chiang E, Chabot-Blanchet M, Kelly MJ, Reeves RA, Guertin MC, et al. Lipid lowering in healthy volunteers treated with multiple doses of MGL-3196, a liver-targeted thyroid hormone receptor-beta agonist. Atherosclerosis. 2013;230:373–80. [DOI] [PubMed] [Google Scholar]
- 186.Sjouke B, Langslet G, Ceska R, Nicholls SJ, Nissen SE, Ohlander M, et al. Eprotirome in patients with familial hypercholesterolaemia (the AKKA trial): A randomised, double-blind, placebo-controlled phase 3 study. Lancet Diabetes Endocrinol. 2014;2:455–63. [DOI] [PubMed] [Google Scholar]
- 187.Harrison SA, Bashir MR, Guy CD, Zhou R, Moylan CA, Frias JP, et al. Resmetirom (MGL-3196) for the treatment of non-alcoholic steatohepatitis: A multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet. 2019;394:2012–24. [DOI] [PubMed] [Google Scholar]
- 188.Loomba R, Neutel J, Bernard D, Severance R, Mohseni R, Dao M, et al. VK2809, a novel liver-directed thyroid receptor beta agonist, significantly reduces liver fat in patients with nonalcoholic fatty liver disease: A phase 2 randomized placebo-controlled trial [abstract]. Hepatology. 2018;68:1447A. [Google Scholar]
- 189.Harrison S, Bedossa P, Guy C, Schattenberg J, Loomba R, Taub R, et al. Primary results from MAESTRO-NASH a pivotal phase 3 52-week serial liver biopsy study in 966 patients with NASH and fibrosis. EASL 2023 Journal of Hepatology. 2023;78(S1):GS–001. [Google Scholar]
- 190.Elbers LP, Kastelein JJ, Sjouke B. Thyroid hormone mimetics: The past, current status and future challenges. Curr Atheroscler Rep. 2016;18:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Coppola M, Glinni D, Moreno M, Cioffi F, Silvestri E, Goglia F. Thyroid hormone analogues and derivatives: Actions in fatty liver. World J Hepatol. 2014;6:114–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Chiellini G, Apriletti JW, Yoshihara HA, Baxter JD, Ribeiro RC, Scanlan TS. A high-affinity subtype-selective agonist ligand for the thyroid hormone receptor. Chem Biol. 1998;5:299–306. [DOI] [PubMed] [Google Scholar]
- 193.Boyer SH, Jiang H, Jacintho JD, Reddy MV, Li H, Li W, et al. Synthesis and biological evaluation of a series of liver-selective phosphonic acid thyroid hormone receptor agonists and their prodrugs. J Med Chem. 2008;51:7075–93. [DOI] [PubMed] [Google Scholar]
- 194.Younossi ZM, Stepanova M, Taub RA, Barbone JM, Harrison SA. Hepatic fat reduction due to resmetirom in patients with nonalcoholic steatohepatitis is associated with improvement of quality of life. Clin Gastroenterol Hepatol. 2022;20:1354–61. [DOI] [PubMed] [Google Scholar]
- 195.Harrison SA, Bashir M, Moussa SE, McCarty K, Pablo Frias J, Taub R, et al. Effects of resmetirom on noninvasive endpoints in a 36-week phase 2 active treatment extension study in patients with NASH. Hepatol Commun. 2021;5:573–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Inc., V.T. (2023). Viking Therapeutics Announces Positive Top-Line Results from Phase 2b VOYAGE Study of VK2809 in Patients with Biopsy-Confirmed Non-Alcoholic Steatohepatitis (NASH). Accessed June 8, 2023. https://www.prnewswire.com/news-releases/viking-therapeutics-announces-positive-top-line-results-from-phase-2b-voyage-study-of-vk2809-in-patients-with-biopsy-confirmed-non-alcoholic-steatohepatitis-nash-301825395.html
- 197.Manalac T. Viking Therapeutics Clears Mid-Stage NASH Trial. 2023. Accessed June 8, 2023. https://www.biospace.com/article/viking-therapeutics-clears-mid-stage-nash-trial/