

Abstract
Abstract
Preeclampsia (PE) is a hypertensive disorder during human pregnancy. Aryl hydrocarbon receptor (AhR) is a ligand‐activated transcription factor. Exogenous and endogenous AhR ligands can induce hypertension in male rats and mice. Herein, using rats as a model, we tested the hypothesis that over‐regulation of endogenous AhR ligands during pregnancy impairs vascular functions by disrupting the transcriptome in the placenta, contributing to the development of PE. Pregnant rats were injected daily with an endogenous AhR ligand, 2‐(1′H‐indole‐3′‐carbonyl)‐thiazole‐4‐carboxylic acid methyl ester (ITE), from gestational day (GD) 10 to 19. Maternal mean blood pressure was measured on GD16–20. Proteinuria and uteroplacental blood flow were monitored on GD20. Placentas collected on GD20 were used to determine changes in vascular density and transcriptome. Compared with the vehicle control, ITE elevated maternal mean blood pressure by 22% and 16% on GD16 and 17, respectively. ITE increased proteinuria by 50% and decreased uteroplacental blood flow by 26%. ITE reduced the placental vascular density by 18%. RNA sequencing analysis revealed that ITE induced 1316 and 2020 differentially expressed genes (DEGs) in female and male placentas, respectively. These DEGs were enriched in pathways relevant to heart diseases, vascular functions and inflammation. Bioinformatics analysis also predicted that ITE altered immune cell infiltration in placentas depending on fetal sex. These data suggest that over‐regulation of endogenous AhR ligands may lead to PE with impaired vascular functions and disrupted fetal sex‐specific transcriptomes and immune cell infiltration in placentas. These AhR ligand‐induced DEGs and pathways may represent promising therapeutic targets for PE‐induced cardiovascular dysfunctions.
Key points
An endogenous AhR ligand (ITE) elevated maternal blood pressure and proteinuria in pregnant rats, and decreased uteroplacental blood flow and fetal and placental growth, all of which are hallmarks of preeclampsia.
ITE reduced vascular density and altered immune cell distribution in rat placentas.
ITE dysregulated transcriptomes in rat placentas in a fetal sex‐specific manner. These ITE‐dysregulated genes and pathways are highly relevant to diseases of heart, vascular functions and inflammatory responses.
Keywords: AhR ligand, placentas, preeclampsia, sexual dimorphism, transcriptomics, vasculature
Abstract figure legend An endogenous AhR ligand (ITE) elevates maternal blood pressure and proteinuria in pregnant rats, and decreases uteroplacental blood flow and fetal and placental growth, all of which are hallmarks of preeclampsia. ITE also reduces vascular density, alters immune cell distribution and fetal sex‐specifically disturbs transcriptomes in rat placentas. These ITE‐dysregulated genes are highly relevant to diseases of heart, vascular functions and inflammatory responses.
Introduction
Preeclampsia (PE) is a hypertensive disorder which poses a major risk to both mothers and fetuses during human pregnancy (Rana et al., 2019; Roberts et al., 2021). Beside inducing vascular dysfunction in mothers, PE also impairs fetal vascular functions (Rana et al., 2019). Specifically, PE reduces fetal blood flow and increases vascular resistance and vascular permeability in the placenta (Khankin et al., 2010). We have reported that PE dysregulates the transcriptome and angiogenic responses in human umbilical vein endothelial cells (HUVECs) in a fetal sex‐specific manner (Zhou et al., 2023, 2019), supporting the significance of sexually dimorphic regulation of endothelial functions in PE. Currently, the aetiology of PE is unclear, but defects in placental vascular functions are considered a major factor contributing to the development of PE (Roberts et al., 2021). Thus, further deciphering of the mechanisms underlying PE is crucial for developing proper diagnostic tools and therapeutic strategies for this pregnancy complication.
Aryl hydrocarbon receptor (AhR) is a ligand‐activated transcription factor that is involved in metabolizing xenobiotics, for example, dioxin (2,3,7,8‐tetrachlorodibenzodioxin; TCDD) (Denison et al., 2011). Additionally, AhR participates in mediation of vascular functions (Li et al., 2020; Walisser et al., 2004) and immune responses (Rothhammer & Quintana, 2019; Stockinger et al., 2014). For instance, AhR‐mutant mice exhibit defects in the fetal vasculature (Zhang, 2011). Adult AhR null mice develop hypertension with elevated angiotensin II and endothelin 1 (Lund et al., 2008, 2006). In contrast, endothelial cell‐specific AhR‐null mice are hypotensive, and exhibit increases in endothelial nitric oxide (NO) synthase activity and NO production (Agbor et al., 2011). Moreover, treating male mice with an exogenous AhR ligand (3‐methylcholanthrene; 3MC) induces hypertension (Chang et al., 2017). Activation of AhR by an endogenous AhR ligand (6‐formylindolo[3,2‐b] carbazole; FICZ) causes pulmonary arterial hypertension in male rats (Masaki et al., 2021). Collectively, these data suggest a pivotal role of the AhR pathway in controlling blood pressure.
Besides being expressed in trophoblast cells (Jiang et al., 2010; Stejskalova et al., 2011), AhR is localized in endothelium of large blood vessels in villi and endothelium of human umbilical vessels as well as in human fetal tissues (e.g. lung, kidney, pancreas, liver, testicle) (Jiang et al., 2010). Previous studies have shown that AhR mediates function of human trophoblast cells and umbilical endothelial cells (Li et al., 2017; Stejskalova et al., 2011), indicating the importance of AhR during pregnancy.
Many of endogenous AhR ligands have been identified including tryptophan (Trp)‐derived 2‐(1′H‐indole‐3′‐carbonyl)‐thiazole‐4‐carboxylic acid methyl ester (ITE) (Nguyen & Bradfield, 2008; Platten et al., 2019). Recently, we have reported the presence of several Trp‐derived AhR ligands in maternal and fetal sera (Zhao et al., 2022). As a potent AhR ligand, ITE has been used for investigating actions of endogenous AhR ligands in various biological processes including vascular function, immune responses and tumour biology (Li et al., 2020). ITE can also easily cross the placental barrier and enter the mouse fetus, activating the AhR pathway (Henry et al., 2006; Wu et al., 2014). ITE inhibits endothelial growth without altering the phosphorylation (or activation) of extracellular signal‐regulated kinases 1 and 2, AKT1, c‐Jun N‐terminal kinase or p38 (Li et al., 2017; Pang et al., 2017), and suppresses the expansion and function of T helper cells and promotes differentiation of T‐regulatory cells (Gutiérrez‐Vázquez & Quintana, 2018; Rothhammer & Quintana, 2019). Given that AhR is expressed in human villous endothelium and endothelium of human umbilical cord arteries and veins (Jiang et al., 2010) and PE elevates the levels of indole‐3‐lactic acid (another Trp‐derived AhR ligand) in maternal and fetal serum (Zhao et al., 2022), these data suggest that Trp‐derived AhR ligands might contribute to the pathophysiology of PE by disrupting endothelial and immune cell functions.
In this study, using pregnant rats as a model, we tested the hypothesis that over‐regulation of endogenous AhR ligands induces hypertension during pregnancy and disrupts the transcriptome in the placenta, contributing to the development of PE. Pregnant rats were treated daily with ITE for 10 days after gestational day (GD) 10. The mean blood pressure was monitored on GD16–20. Uteroplacental blood flow, placental and fetal growth were assayed on GD20. Vascular growth and cell apoptosis in the placentas on GD20 were determined. RNA sequencing (RNA‐seq) analysis was conducted to profile transcriptomic changes in the placentas. Bioinformatics and gene ontology analyses were performed to identify genes and pathways dysregulated by ITE in female and male placentas.
Methods
Ethical approval
All animal care and use procedures followed the National Institutes of Health guidelines (Guide for the Care and Use of Laboratory Animals, NIH Publication No. 85‐23, revised 1996) with approval from the Institutional Animal Care and Use Committee at the University of Wisconsin‐Madison (Protocol ID V005847).
Animals
Timed pregnant Sprague‒Dawley rats (6–8 weeks old, n = 20) were purchased from Envigo Bioproducts (Madison, WI, USA). All animals were housed in designated animal rooms under controlled conditions (lights on from 07.00 to 19.00 h at 22‒23°C). Standard rodent chow and water were available ad libitum throughout the experimental protocols. After acclimatization on GD10, the rats were randomly divided into ITE treatment and vehicle control groups.
A stock solution (100 mM) of ITE (cat. no. A163287, AmBeed, Arlington Heights, IL, USA) was prepared in dimethyl sulfoxide (DMSO, cat. no. 2650, Sigma‐Aldrich, St Louis, MO, USA), stored at room temperature and used within 1 week. Rats were injected daily via the tail vein with ITE (5 mg/kg body weight (BW) per day, n = 10) or an equivalent amount of DMSO (vehicle control, n = 10) from GD10 to GD19. The volume (ml) of ITE or DMSO injected daily into each rat was calculated using the formula 0.172 × BW (kg), and was ∼40–60 μl of ITE or DMSO per day depending on the rat weight. This dose of ITE likely represents a pharmacological level since ITE levels are low in tissues (Song et al., 2002). However, this dose is comparable to those that regulate placental expression of angiogenic factors (Wang et al., 2013; Wu et al., 2014) and immune cell differentiation (Gutiérrez‐Vázquez & Quintana, 2018; Rothhammer & Quintana, 2019). The dam weights were recorded daily from GD10 to GD20, the period of dramatic vascular adaptations during rat pregnancy (Moll, 2003; Osikoya et al., 2019). The mean blood pressure was monitored in the first set of rats (n = 6/group) from GD16 to GD20, using the tail‐cuff method with a computerized CODA system (Kent Scientific Torrington, CT, USA) as previously described (Blesson et al., 2015; Mishra et al., 2019). Uteroplacental blood flow was measured in the second set of rats on GD20 (n = 4/group) under isoflurane anaesthesia and on a heated platform using a 30 MHz transducer and Vevo 2100 ultrasound system (Visual Sonics, Toronto, ON, Canada) as described (Gopalakrishnan et al., 2016). In this set of animals, one rat in each group did not become pregnant and was removed from the study. Urine samples were collected on GD20 before blood flow measurement and stored at −80°C as described (Kurien et al., 2004). Due to technical limitations, we collected urine samples only from six rats (n = 3/group). The urine samples were centrifuged at 600 g for 3 min at 4°C. Urinary albumin levels were determined using a rat albumin ELISA kit (cat. no. NR002, Exocell, Philadelphia, PA, USA).
Tissue preparation
The dams were sacrificed on GD20 by CO2 inhalation. Following euthanasia, the maternal heart, liver, lung, kidney, and placenta and fetus were immediately collected and weighed. Fetal viability and sex were not determined to avoid RNA degradation. Fetal body weights less than the 10th percentile of the mean fetal weight in the DMSO group were considered to indicate fetal growth restriction (FGR) (Zhang et al., 2010). Placental tissue samples were fixed in 4% buffered formaldehyde or snap frozen in liquid nitrogen and then stored at −80°C for subsequent experiments.
RNA isolation
Total RNA was isolated from rat placentas using the RNeasy Mini Kit (Qiagen, Germantown, MD, USA) (Zhou et al., 2019, 2017). The concentration and quality of each RNA sample were assessed using a NanoDrop ND‐2000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA) and an Agilent 2100 bioanalyser (Agilent Technologies, Santa Clara, CA, USA). RNA samples with a high RNA integrity number (>7.0) were used.
Sex typing
RT‐qPCR was used to detect the Y chromosome‐specific sex‐determining region Y (SRY) gene in rat placentas. RNA samples from both F and M placentas were prepared for RNA‐seq.
RNA‐seq analysis and RT‐qPCR verification
RNA‐seq was conducted at the UW‐Madison Biotechnology Centre (Zhou et al., 2019). RNA libraries were generated using the Illumina TruSeq Stranded Total RNA Sample Preparation Kit (Illumina, San Diego, CA, USA). Briefly, 300 ng of total RNA from each sample was fragmented using divalent cations at an elevated temperature and purified. Double‐stranded complementary DNAs (cDNAs) were synthesized, ligated to unique dual‐index adapters, and cleaned with two rounds of AMPure XP beads (0.8x). Adapter‐ligated DNA was amplified by PCR and cleaned with AMPure XP beads (0.8x).
The quality and quantity of the libraries were assessed using an Agilent DNA1000 series chip assay and an Invitrogen Qubit HS cDNA Kit (Thermo Fisher Scientific Waltham, MA, USA), respectively. Libraries were standardized to 2 nM. Paired‐end 150 bp sequencing was performed using standard SBS chemistry on an Illumina NovaSeq 6000 sequencer. Images were analysed using bcl2fastq v2.17 software.
To validate the RNA‐seq data, RT‐qPCR analysis was conducted using TaqMan Gene Expression Assays (Supporting information, Table S1), TaqMan Fast Advanced Master Mix, and a StepOnePlus qPCR system (Thermo Fisher Scientific). For rat placentas, 15 genes with different expression patterns were selected. The data were normalized to the levels of internal housekeeping genes (GAPDH, YWHAZ and RPLP0). The normalized data were analysed using the method.
Functional genomic analysis
We obtained 67 million reads per sample from rat placentas from the University of Wisconsin‐Madison Biotechnology Sequencing Centre. The reads were trimmed of the TruSeq adapters using Skewer software with the default parameters. The quality of the individual trimmed sequence reads was evaluated by the University of Wisconsin‐Madison Bioinformatics Resource Centre. The average Phred quality score from the per‐base sequence quality analysis was approximately 37, which is a conventional threshold denoting high‐quality base calls. Trimmed reads were aligned to the GRCH38 genome using the STAR aligner, which is a splice junction‐aware RNA‐seq alignment algorithm. Consistent with the sample‐level RNA QC steps using a Bioanalyser 2100 (Agilent Technologies, Santa Clara, CA, USA), gene body coverage indicated no evidence of RNA degradation. The mapped reads in each sample were counted via RNA‐seq with Expectation Maximization (RSEM) software. Empirical Analysis of Digital Gene Expression Data in R software was used to determine the differentially expressed genes (DEGs; fold change (FC) >|2| and Benjamini‒Hochberg false discovery rate (FDR)‐adjusted P‐value <0.05). Circos plots showing the locations of the DEGs at the chromosome sites were generated using Circa software (Circa Information Technologies, Downey, CA, USA). Gene Ontology analysis was performed to determine the enriched biological functions, disease/canonical pathways, and gene networks using Ingenuity Pathway Analysis (IPA; www.qiagenbioinformatics.com).
CIBERSORTx analysis
The ITE‐induced DEGs were subjected to the CYBERSORTx algorithm to predict ITE‐altered immune cell infiltration in placentas. The signature matrix LM22 was used to define 22 subtypes of immune cells. The reads of ITE‐induced DEGs were uploaded to the CIBERSORTx web portal (https://cibersortx.stanford.edu/). The algorithm was set in B‐mode with batch correction at 1000 permutations and run following the instructions of CIBERSORTx.
Morphological and immunohistochemical analyses
Fixed placentas were embedded in paraffin. CD31, an endothelial marker, was immunolocalized in placentas as described (Li et al., 2015). Two adjacent tissue sections (5 μm thick) per sample were deparaffinized, dehydrated and boiled in a 10 mM citrate buffer solution (pH 6.5) in a microwave for 10 min for antigen retrieval. Endogenous peroxidase activity was quenched by immersing the tissue sections in 3% H2O2 for 10 min. After blocking non‐specific binding sites, one tissue section per slide was probed with a CD31 goat polyclonal antibody (4 μg/ml, cat. no. AF3628, Novus Biologicals, Littleton, CO, USA) for 1 h. The second tissue section on the same slide was probed with preimmune goat IgG (4 μg/ml; cat. no. 55926, MP Biomedicals, Irvine, CA, USA) as a negative control. After washing, the tissue sections were incubated with a biotinylated horse anti‐goat antibody for 30 min. The specific immunoreactivity was visualized by 3‐amino‐9‐ethylcarbazole (SK‐4200, Vector Laboratories, Burlingame, CA, USA).
The whole tissue sections were scanned at ×20 using the Aperio Digital Pathology Slide Scanner system (Aperio‐AT2, Leica Biosystems, Buffalo Grove, IL, USA). The thickness of the labyrinth (L) and basal (B) zones at the middle of the cross‐section of each tissue as well as the areas of L (marked by yellow dotted lines) and B (marked by red dotted lines) were measured using NIH ImageJ software. Vascular area density (areas occupied by vessel wells) and CD31 staining intensity were analysed using InForm 2.4 imaging software, a machine learning software (PerkinElmer, Waltham, MA, USA). For each tissue section, 10 areas were randomly photographed with a ×20 objective, representing 21% of the total L area. Whole‐tissue scanning and CD31 image acquisition and imaging analysis were performed at Translational Research Initiatives in the Pathology Laboratory, University of Wisconsin‐Madison.
TUNEL assay
Apoptosis in placental tissues was assayed using the DeadEnd colorimetric terminal deoxynucleotidyl transferase dUTP nick end labelling (TUNEL) system (cat. no. G7130, Promega, Madison, WI, USA). The deparaffinized tissue sections were fixed in 10% buffered formalin, followed by antigen retrieval (100 μg/ml proteinase K solution for 30 min). The tissue sections were incubated with recombinant terminal deoxynucleotidyl transferase reaction mixture for 60 min at 37°C. The reaction was stopped with a 2× saline sodium citrate buffer. Endogenous horseradish peroxidase (HRP) was blocked with 1% H2O2. HRP–streptavidin was added, and the sections were incubated for 30 min. The tissue sections were incubated with diaminobenzidine until brownish staining developed (5‒10 min).
Western blotting
Rat placental samples were lysed in lysis buffer (cat. no.: 39000, Thermo Fisher Scientific). Lysate protein samples (20 or 25 μg/sample) were separated on 10 or 15% SDS‐PAGE gels and electrically transferred to polyvinylidene difluoride membranes. The membranes were probed with primary antibodies against the target proteins (Supporting information, Table S2). Proteins were visualized by enhanced chemiluminescence (ECL) or ECL plus (Thermo Fisher Scientific) using Amersham ImageQuant 800 (Cytiva, Marlborough, MA, USA). The absorbance was measured using ImageJ. The data were normalized to the level of glyceraldehyde 3‐phosphate dehydrogenase (GAPDH). When appropriate, HUVECs pooled from five individual cell preparations from normotensive pregnant patients (Li, Wang et al., 2015; Li et al., 2017) were used as a positive control.
Statistical analyses
All the data except the RNA‐seq and phosphoproteomic data were analysed using SigmaPlot software (Systat Software, San Jose, CA, USA). For comparing two groups, data analyses were performed using the Mann‒Whitney rank‐sum test or Student's t test. For comparing multiple groups, the data were analysed using one‐way or two‐way ANOVA. When an F test was significant, the data were analysed using Dunnett's method (multiple comparisons versus control group) or the Holm‒Šidák or Dunn's test (pairwise comparisons) when appropriate. The Benjamini‒Hochberg FDR‐adjusted P‐value was used to determine the significance of the RT‐qPCR and phosphoproteomic data. Correlations between RNA‐seq and RT‐qPCR data as well as phosphoproteomic and verification western blotting data were analysed using Pearson's product moment correlation analysis. The data are presented as means ± SD. Differences and correlations were considered significant when P < 0.05 unless stated otherwise.
Results
ITE induces PE‐like phenotypes in rats
No animal death occurred throughout the study. Compared to DMSO, ITE did not alter the body weights of dams from GD10 to GD20 (Fig. 1A ). On GD20, ITE increased maternal lung, kidney and liver weights but not heart weights (Fig. 1B ).
Figure 1. ITE dysregulates tissue growth and cardiovascular functions in pregnant rats.
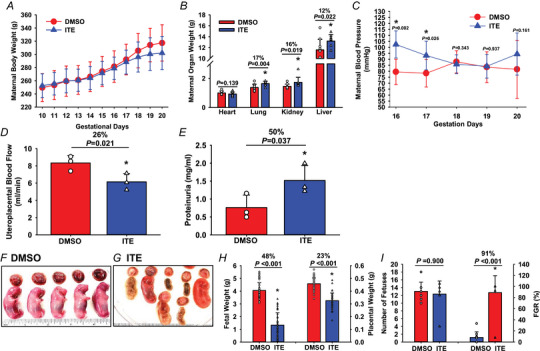
Pregnant rats were treated with ITE (5 mg/kg body weight/day) or DMSO from GD10 to GD19. A and B, maternal body (A) and organ (B) weights were recorded. Each maternal organ weight was adjusted to the body weight of corresponding dam in B. n = 9 and 10 for the ITE and DMSO groups, respectively. C and D, maternal blood pressure (C; n = 6/group for the DMSO and ITE groups, respectively) and uteroplacental blood flow (D; n = 3/group) were measured using the tail‐cuff method and Vevo 2100 ultrasound system, respectively. E, maternal proteinuria was measured (n = 3/group). F‒I, placentas and fetuses were collected (n = 9 and 10 dams for the ITE and DMSO groups, respectively) and weighed. The rulers are in inches. The Mann‒Whitney rank‐sum test or Student's t test was performed to compare differences between the ITE and DMSO groups. FGR: fetal growth restriction. *P < 0.05 versus DMSO (B, D, E, H, I) or DMSO at each corresponding time point (C). [Colour figure can be viewed at wileyonlinelibrary.com]
The mean maternal blood pressure values in the DMSO group (Fig. 1C ) were consistent with those previously reported (Blesson et al., 2015; Mishra et al., 2019). ITE elevated maternal blood pressure by 22% and 16% compared with DMSO on GD16 and GD17, respectively (Fig. 1C ). ITE decreased uteroplacental blood flow by 26% but elevated proteinuria by 50% (Fig. 1D and E ) on GD20, indicating that ITE induced PE‐like phenotypes in pregnant rats.
Compared to DMSO, ITE caused morphological changes in the placentas and fetuses (Fig. 1F and G ). Many placentas and fetuses in the ITE group were smaller in size (Fig. 1F‒I ). Many fetuses degenerated or started to degenerate, as indicated by their smaller size and darker appearance (Fig. 1F and G ). The number of fetuses and placentas was similar between the ITE and DMSO groups (Fig. 1H ). However, ITE reduced fetal and placental weights by 48% and 23%, respectively, on GD20 (Fig. 1H ), which might have contributed to slight decreases in dam body weights on GD18–20 (Fig. 1A ). As compared with DMSO, ITE increased the FGR rate by 91% (Fig. 1I ).
ITE decreases vasculature in placentas
Compared to DMSO, ITE did not alter the relative thicknesses of the labyrinth or basal zones (Fig. 2A‒C ). However, ITE decreased the relative area of the labyrinth zone but increased (P < 0.05) the relative area of the basal zone (Fig. 2D ). ITE caused placental lesions with a decrease in the number of blood vessels and enlarged maternal sinusoids (Fig. 2E and F ). ITE also induced the formation of oedematous stroma and patches of densely packed death cells and immune cell infiltration in the labyrinth zone (Fig. 2E and F ). No significant morphological changes were detected in the basal zone. Semiquantitative analysis of CD31 immunostaining revealed that ITE reduced the vascular area density by 18% and the CD31 staining intensity by 26% in the labyrinth zone (Fig. 2G ). The TUNEL assay showed that abundant positive staining was present in rat placentas treated with DNase I (a positive control, Fig. 2H ) but not in those treated without DNase I (Fig. 2I ). A sporadic distribution of apoptotic cells was observed in a few placental tissue sections from the ITE group, primarily in the basal zone (Fig. 2J ). These data suggest that ITE mainly affects cells in the labyrinth zone, primarily endothelial cells and the vasculature, leading to decreased cellular growth in the labyrinth zone.
Figure 2. ITE decreases vascular density and induces apoptosis in rat placentas.
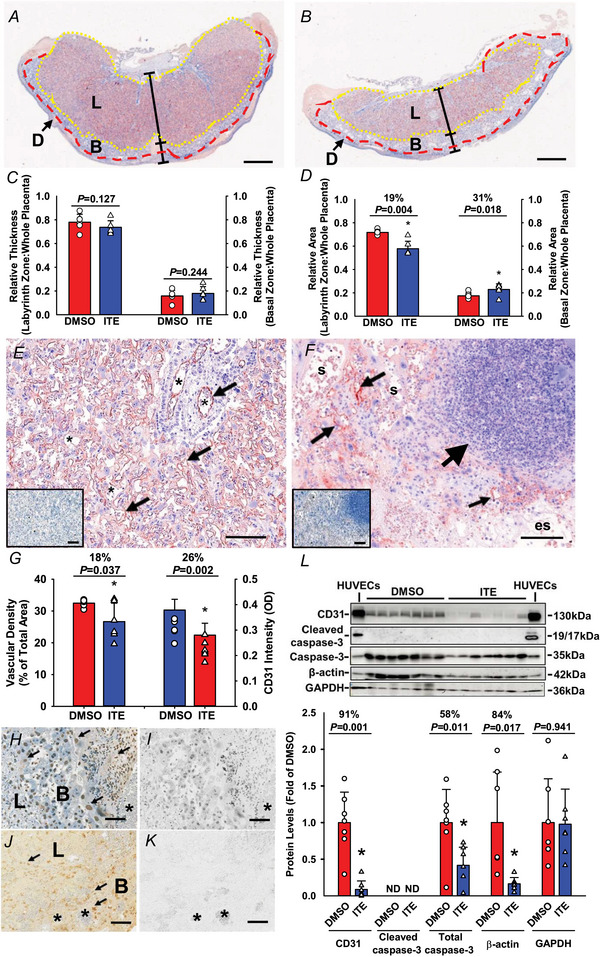
Pregnant rats were treated with ITE or DMSO from GD10 to GD19. Placentas were collected on GD20. The placental tissue sections were subjected to CD31 immunostaining followed by haematoxylin counterstaining. A and B, representative images of rat placentas from the ITE and DMSO groups, respectively. The reddish colour indicates CD31‐positive staining. Blue: haematoxylin counterstaining; D: decidua basalis. L: labyrinth zone. B: basal zone. Bar = 1 mm. C and D, the relative thicknesses and areas of the L and B zones were quantified. Ee‒Gg, vascular density and CD31 staining intensity in the L zone. E and F, representative images of CD31 immunostaining in the L zone from the ITE (E) and DMSO (F) groups. The insets in E and F are adjacent tissue sections treated with normal goat IgG as a control. Small arrows: vascular endothelial cells. Large arrow: an area with a density of dead cells and immune cell infiltration. s: maternal sinusoids; es: oedematous stoma. G, vascular area density and CD31 staining intensity were quantified. H‒K, cell apoptosis. The tissue sections were subjected to a TUNEL assay. H and Ii, representative images of two adjacent placental sections from the DMSO group. The sections were pretreated with DNase I (a positive control) (H) or without DNase I (a negative control) (I) followed by slight counterstaining with haematoxylin. J and K, representative images of two adjacent placental sections from the ITE with (J) or without (K; a negative control) recombinant terminal deoxynucleotidyl transferase. Arrows: apoptotic cells. *: blood vessels. Brownish nuclear 3,3′‐diaminobenzidine staining was used to stain apoptotic cells; blue: haematoxylin counter‐staining. *P < 0.05 versus DMSO. Bar = 100 μm. n = 6 placentas from different dams/group. L, Western blotting analysis for CD31 and cleaved caspase‐3. Data normalized to GAPDH are expressed as the means ± SD fold of the control. HUVECs at the caspase‐3 band were treated with staurosporine (200 nM; a positive control for cleaved caspase‐3). The Mann‒Whitney rank‐sum test or Student's t test was performed to compare differences between DMSO and ITE. *P < 0.05 versus DMSO. n = 7 placentas from different dams/group. [Colour figure can be viewed at wileyonlinelibrary.com]
Western blotting data are shown in Fig. 2L . Compared to DMSO, ITE reduced CD31 protein levels by 91% in the placenta, consistent with decreases in vascular density and CD31 immunostaining intensity (Fig. 2G ). ITE did not induce the formation of cleaved caspase‐3 but decreased total caspase‐3. ITE reduced β‐actin but not GAPDH levels.
ITE dysregulates transcriptome in placentas
Compared to DMSO, ITE up‐ and downregulated 409 and 907 genes in female (F) placentas, respectively, among which 69 were located on the X chromosome (Fig. 3A and B ; Supporting information, Table S3). ITE up‐ and downregulated 571 and 1449 genes in male (M) placentas, respectively, among which 114 were located on the X chromosome, and none were on the Y chromosome. ITE commonly induced 926 differentially expressed genes (DEGs) in F and M placentas (Supporting information, Table S3). Among these DEGs, 188 and 731 were up‐ and downregulated, respectively, in both ITE‐F and ITE‐M; six (LOC690483, Ugt2b15, LOC100910565, AABR07057442.1, U1 and Rfxapl1) were downregulated in M but upregulated in F; one (ZFP280B) was downregulated in F but upregulated in M; and 136 were located on the X chromosome (Supporting information, Table S3).
Figure 3. ITE dysregulates the transcriptome in rat placentas.
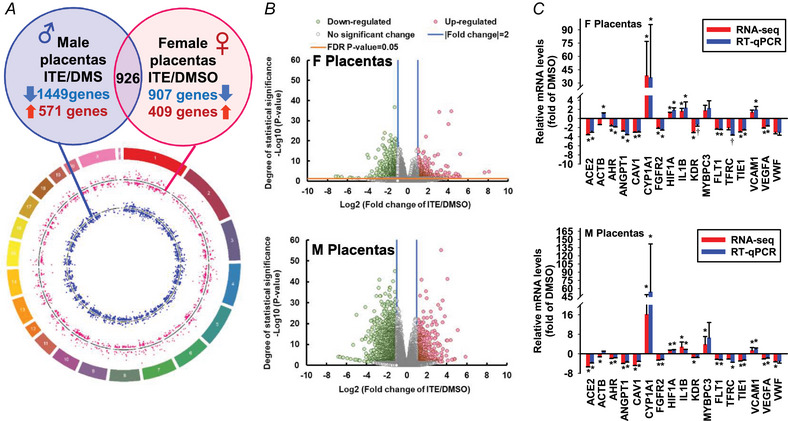
A, circos plot illustrating the chromosomal position of DEGs in F (red dots) and M (blue dots) placentas. Each dot represents one gene. The numbers and letters in the outer ring indicate the chromosomal location. For each scatter plot track, dots outside and inside the centreline are up‐ and downregulated genes, respectively. B, volcano plots showing DEGs in F and M placentas. Grey dots: no significant difference; red and green dots: >2‐fold up‐ and downregulation, respectively (FDR‐adjusted P < 0.05) in the ITE group vs. the DMSO group. n = 13, 16, 8, and 13 for the DMSO‐F, DMSO‐M, ITE‐F, and ITE‐M groups, respectively. C, RT‐qPCR validation of ITE‐dysregulated genes in F and M placentas. *Means differ (FDR‐adjusted P < 0.05) from DMSO; †means differ (0.1 > FDR‐adjusted P > 0.05) from DMSO. n = 4/fetal sex/group. F, female; M, male. [Colour figure can be viewed at wileyonlinelibrary.com]
The RT‐qPCR and RNA‐seq data were strongly correlated (r = 0.999 and 0.982 for F and M placentas, respectively, P < 0.001) (Fig. 3C ). In F placentas, ITE upregulated CYP1A1, HIF1A and IL1B but downregulated ACE2, AHR, ANGPT1, CAV1, FGFR2, SFLT1, TIE1 and VEGFA (Fig. 3C ). ITE did not alter the expression of MYBPC3, TFRC or VWF. RNA‐seq, but not RT‐qPCR, revealed that ITE downregulated ACTB and KDR. RT‐qPCR, but not RNA‐seq analysis, showed that ITE upregulated VCAM1. In M placentas (Fig. 3C ), ITE upregulated CYP1A1, HIF1A, IL1B, MYBPC3 and VCAM1 but downregulated ACE2, AHR, ANGPT1, CAV1, FGFR2, SFLT1, TFRC, TIE1, VEGFA and VWF. RNA‐seq, but not RT‐qPCR, revealed that ITE attenuated the expression of ACTB and KDR in M placentas. Both RNA‐seq and RT‐qPCR analyses revealed that ITE robustly increased the levels of CYP1A1 and decreased the levels of AhR in F and M placentas, indicating the activation of the AhR pathway.
Bioinformatics analysis of the ITE‐induced placental DEGs revealed that 637 biological functions were enriched (Fig. 4A ; Supporting information, Table S4). Of these 637 functions, 363 (e.g. cancer, hypertension, angiogenesis, diabetes mellitus, vasculogenesis, leukocyte migration, inflammatory response, and vaso‐occlusion) were commonly enriched in F and M placentas; 137 each were uniquely enriched in F (e.g. hypertensive heart disease, abnormal morphology of vasculature, perinatal death, and proliferation of endothelial cells) and M (e.g. cellular infiltration by blood cells, quantity of cytokine, necrosis, and apoptosis) placentas.
Figure 4. ITE dysregulates pathways in rat placentas.
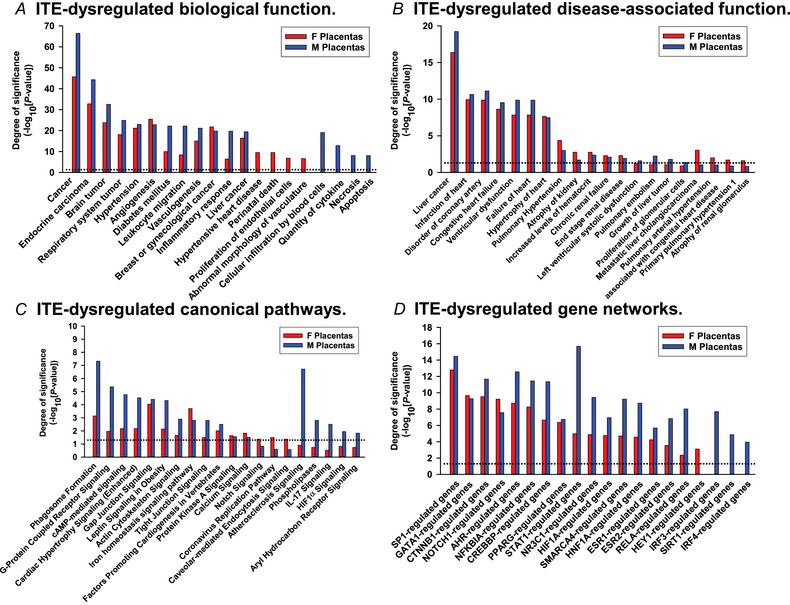
A, biological functions. B, disease‐associated biological functions. C, canonical pathway‐associated genes. D, gene networks. Significant enrichment was determined using IPA software (P < 0.05, Fisher's exact test). Dotted line: P = 0.05. F, female; M, male. [Colour figure can be viewed at wileyonlinelibrary.com]
The ITE‐induced placental DEGs were enriched in 249 disease‐associated functions (Fig. 4B ; Supporting information, Table S5). Of these 249 functions, 132 were commonly enriched in the F and M placentas, while 53 and 62 were enriched only in the F and M placentas, respectively. For example, disease‐associated functions, including liver cancer, coronary artery disorders, congenital heart failure, failure of heart, pulmonary hypertension, and atrophy of kidney were commonly enriched in F and M placentas (Fig. 4B ). However, pulmonary embolism and growth of liver tumour were only enriched in F placentas, and pulmonary artery hypertension and atrophy of renal glomerulus were only enriched in M placentas (Fig. 4B ).
The 95 canonical pathways enriched in the ITE‐induced placental DEGs included phagosome formation, G‐protein coupled receptor signalling, cAMP‐mediated signalling, and gap junction signalling (Fig. 4C , Supporting information, Table S6). Of these 95 pathways, 35 were commonly enriched in both F and M placentas. These pathways included phagosome formation, G‐protein coupled receptor signalling, cardiac hypertrophy signalling (enhanced), lipopolysaccharide/interleukin (IL)‐1‐mediated inhibition of RXR functions, gap junction signalling, tight junction signalling, protein kinase A signalling, and calcium signalling pathways. Moreover, three pathways (notch signalling, coronavirus replication pathway, and caveolar‐mediated endocytosis signalling) were enriched only in F placentas, while 55 pathways, including atherosclerosis signalling, phospholipases, IL‐17 signalling, HIFα signalling, and aryl hydrocarbon receptor signalling, were enriched only in M placentas.
Upstream regulator analysis identified 96 enriched gene networks, of which 81 were commonly enriched in F and M placentas and 1 and 14 were uniquely enriched in F and M placentas, respectively (Fig. 4D ; Supporting information, Table S7). These common gene networks included NOTCH1‐, AHR‐, HIF1A‐, ESR1‐, ESR2‐ and CREBBP‐regulated genes, in which ESR1‐ (90 vs. 116 in F and M placentas) and CREBBP‐ (86 vs. 108) regulated genes contained the most abundant numbers of target genes (Supporting information, Table S7). Additionally, HEY1‐regulated genes were uniquely enriched in F placentas; IRF3‐, SIRT1‐ and IRF4‐regulated genes were only enriched in M placentas (Fig. 4D ).
ITE dysregulates immune cell infiltration in placentas
CYBERSORTx analysis predicted that the top eight immune cells with a fraction ≥2.8% in DMSO‐treated placentas were dendritic cells resting, NK cells resting, macrophages M0, B cells naïve, T cells follicular helper, macrophages M2, T cells CD4 memory resting, and eosinophils (Supporting information, Tables S8 and S9). The percentages of six subtypes of cells (monocytes, plasma cells, T cells CD4 naïve, T cells regulatory, T cells CD4 memory activated, and mast cells resting) ranged from 1.3% to 2.1%. Eight subtypes of cells (macrophages M1, mast cells activated, neutrophils, NK cells activated, dendritic cells activated, T cells gamma delta, T cells CD8, and B cells memory) accounted for less than 1% or were absent. Additionally, the fractions of macrophages M2 in NT‐M and ITE‐M were 44% and 38% greater than those in the NT‐F and ITE‐F, respectively (Supporting information, Table S9).
ITE‐dysregulated immune cell infiltration in placentas is shown in Supporting information Fig. S1 and Table S9. Compared with DMSO, ITE increased the fractions of plasma cells (2.1‐fold that of DMSO), monocytes (2.7‐fold), macrophages M0 (1.2‐fold), macrophages M2 (2.3‐fold) and eosinophils (1.7‐fold), whereas it decreased the fractions of T cells follicular helper (0.6‐fold), NK cells resting (0.8‐fold) and dendritic cells resting (0.6‐fold) in rat placentas (Supporting information, Table S9). Within each placental sex, ITE‐induced changes in immune cell fractions were similar to those found in combined F and M placentas (Supporting information, Fig. S1), although changes in three cell subtypes (plasma cells and eosinophils in F or M cells and macrophages M2 in F cells) did not reach significance. This loss of significance is due to the reduced sample size for each placenta sex.
Discussion
Our study provides novel evidence that ITE induces PE‐like phenotypes in rats. Additionally, ITE disrupts the transcriptome and immune cell infiltration in rat placentas in a fetal sex‐specific manner. Collectively, our findings suggest that the over‐regulation of endogenous AhR ligands may contribute to the pathophysiology of PE.
In contrast to previous reports showing that a single dose of ITE had no adverse effect on fetuses or placentas in rats (Wu et al., 2014) or mice (Henry et al., 2006), our study reveals that multiple doses of ITE reduce fetal and placental weights, along with decreased placental vascular density and blood flow in rats, mirroring the effects of TCDD in animals (Hernández‐Ochoa et al., 2009; Ishimura et al., 2009; Ivnitski‐Steele & Walker, 2005; Peterson et al., 1993). Thus, like TCDD, repeated exposure to endogenous AhR ligands can impair vascular growth in rat placentas.
Our finding that ITE induces PE‐like phenotypes in rats implies that over‐regulation of endogenous AhR ligands may be a common contributor to the development of hypertension in general, considering that AhR ligands also induce hypertension in male mice (Chang et al., 2017) and rats (Masaki et al., 2021) and are involved in pulmonary arterial hypertension in humans (Masaki et al., 2021). Although it was previously believed that PE does not affect vascular growth in human placentas (Burton et al., 1996; Li, Zhao et al., 2015), more recent reports using the more comprehensive 3D imaging analysis demonstrate that PE reduces placental vascular growth (Hermans et al., 2023; Shen et al., 2022). Thus, the ITE‐reduced placental vascular growth likely contributes to the elevated blood pressure and decreased uteroplacental blood flow induced by ITE in rats. In addition, the ITE‐decreased placental growth is associated with reduced total caspase‐3 but without the formation of cleaved caspase‐3. These data suggest that total, but not cleaved, caspase‐3 may mediate the ITE‐decreased placental growth since total caspase‐3 has been implicated in participating in cell function beyond apoptosis (e.g. muscle and renal cell growth) (Eskandari & Eaves, 2022).
The ITE‐induced transient increase in blood pressure observed in pregnant rats contrasts with previous reports that repeated treatment with 3MC and FICZ elevated blood pressure for up to 4–8 weeks in male mice (Chang et al., 2017) and rats (Masaki et al., 2021). The distinct potential of different AhR ligands for AhR activation may partially explain these discrepancies (Denison et al., 2011). However, sex‐ and pregnancy state‐specific regulation of blood pressure by AhR ligands is more likely to contribute to these discrepancies since sex steroid hormones (e.g. oestrogen and androgen) can interact with the AhR pathway (Denison et al., 2011; Pocar et al., 2005). For example, increased oestrogen during late pregnancy could antagonize AhR ligand‐elevated blood pressure by interfering with AhR transcriptional activity (Denison et al., 2011; Pocar et al., 2005) and enhancing vasodilatation (Li et al., 2022). In addition, downregulation of AhR in both female and male placentas induced by ITE (Fig. 3C ) may imply a similar decrease in AhR expression in vascular smooth muscle, which may contribute to the desensitization of ITE's action on blood pressure after GD17. Moreover, although it failed to elevate blood pressure in near‐term rats, it is noteworthy that ITE decreases uteroplacental blood flow, possibly due to the reduced placental vasculature.
Our study uncovered sexual dimorphisms in ITE‐dysregulated transcriptome in placentas. Intriguingly, only 5% and 0% of the DEGs are located on the X chromosome in female and male rat placentas, respectively (Fig. 3 and Supporting information, Table S3), and none of the DEGs are located on the Y chromosome. Thus, genes on sex chromosomes may not significantly contribute to the ITE‐induced transcriptomic differences between female and male placentas, as observed with PE‐induced transcriptomic differences (Zhou et al., 2019).
In rat placentas, a single dose of TCDD has been shown to induce 182 DEGs using representational difference analysis and microarray technology (Mizutani et al., 2004). Our study identifies a new set of ITE‐induced fetal sex‐specific DEGs, thereby expanding the list of AhR ligand‐induced DEGs in placentas. Furthermore, of 182 TCDD‐induced DEGs (Mizutani et al., 2004), 29 overlap those identified in ITE‐treated rat placentas, even though only eight exhibit the same regulatory trends (upregulated: Cxcl10, Cxcl2, Cyp1a1, Cyp1b1, Mx1, Mx2 and Tgfbi; downregulated: Gata1), suggesting differential regulation between ITE and TCDD in the placental transcriptome.
Notably, male rat placentas had 54% more ITE‐induced DEGs than their female counterparts, implying a greater susceptibility or adaptability of male placentas to endogenous AhR ligand stimuli. Additionally, the majority of these DEGs are downregulated in male (72%) and female (69%) placentas and are critical to vascular function. For instance, decreases in angiotensin II (ANG II, a potent vasoconstrictor) receptors (Agtr1a and Agtr2) and Ace2 (Fig. 3 and Supporting information, Table S3) may reduce ANG II activity, decreasing ITE‐elevated blood pressure (Fig. 1C ). Moreover, decreased expression of key endothelial regulators (e.g. Angpt1, Cav1, Fgfr2, Kdr, Tie1 and Vegfa; Fig. 3 and Supporting information, Table S3) may contribute to the reduced placental vascular density.
Our bioinformatics analysis reveals that ITE differentially dysregulates many biological processes, disease‐associated functions, pathway‐associated genes, and gene networks between female and male placentas. Top biological processes included those relevant to cancer, tumour, hypertension, vascular formation and growth angiogenesis, and inflammatory response. ITE also dysregulates common genes between female and male placentas, primarily enriched in liver cancer, cardiovascular diseases and kidney disease and associated with many common signalling pathways (e.g. cardiac hypertrophy signalling (enhanced) and gap junction signalling). Notably, ESR1 and CREBBP emerge as the two most prominent hub genes in the ITE‐dysregulated gene network (Supporting information, Table S7). Conversely, several ITE‐dysregulated processes and pathways are enriched uniquely in female and male placentas. For example, hypertensive heart disease is only enriched in female placentas, while cellular infiltration by blood cells is predicted solely in male placentas. Moreover, notch signalling and caveolar‐mediated endocytosis signalling are enriched in female placentas, while atherosclerosis signalling, IL‐17 signalling and HIF1α signalling are enriched in male placentas.
The immune cell distribution in human placentas has been predicted using single‐cell RNA‐seq (Pique‐Regi et al., 2019), indicating the presence of T‐cells resting and activated, NK cells, monocytes, and macrophages 1/2 with the most abundant amount of T‐cells‐activated (∼15–30% of total immune cells). Our current CIBERSORTx analysis similarly predicts relatively high proportions of NK cells resting (15.5%), monocytes (2.1%), and macrophages M2 (5.6%) in vehicle control‐treated rat placentas (Supporting information, Table S8). However, our study shows that dendritic cells resting account for 29.8% of the total immune cells, with macrophages M0 and NK cells resting for another 31% of the total immune cells. Thus, together with the observation that mast cells activated, neutrophils, NK cells activated, and dendritic cells activated were absent in rat placentas (Supporting information, Fig. S1, Tables S8 and S9), these data imply that distinct distributions of immune cells exist between human and rat placentas and that near‐term placenta in rats may represent a state of strong tissue repair.
Endogenous AhR ligands, including ITE, regulate the proliferation and differentiation of immune cells (e.g. Th1, Th17, Treg and dendritic cells) in mice (Rothhammer & Quintana, 2019; Stockinger et al., 2014). Similarly, our data have shown that ITE alters the infiltration of innate (e.g. monocytes, macrophages 0/2, NK cells resting, dendritic cells resting, and eosinophils) and adaptive (e.g. plasma cells and T cells follicular helper) immune cells in rat placentas. Our observation that ITE increases monocytes and plasma cells is in agreement with previous findings in the PE‐maternal circulation (Faas et al., 2014; Miller et al., 2022). Notably, ITE decreases Tregs by ∼3‐fold (Supporting information, Table S9), although this decrease does not reach significance, which is consistent with the reduction in Tregs in circulation and tissues in PE (Faas et al., 2014; Miller et al., 2022). The novel observation that ITE increases the number of macrophages 0/2 is important, as macrophages 2 may participate in placental tissue repair after ITE‐induced tissue damage. Moreover, in contrast to decreased eosinophils in PE maternal circulation (Mtali et al., 2019), ITE increases eosinophils in rat placentas. Thus, ITE likely partially mimics PE‐induced alterations in immune cell distribution. Additionally, ITE appears to maintain immune responses by increasing monocytes, plasma cells and eosinophils, while decreasing Treg levels in the placenta. However, caution should be taken when interpreting these data since the signature matrix LM22 is based on a human dataset, and humans and rats share only 25% of their genomes, although they share almost all disease‐associated genes (Gibbs et al., 2004). Additional limitations include the inability to distinguish fetal and maternal origins of immune cells and the inability to classify these immune cells into subsets within each subtype. We also did not verify the distributions of these immune cells in the placenta using other approaches.
The mechanisms governing the sexual dimorphisms of ITE‐dysregulated placental responses remain unclear. Sex steroid hormones may partly control these dimorphisms because of the differential expression of aromatase between female and male placentas (Sathishkumar et al., 2012) and the differential distributions of circulating levels of gonadal hormones between female and male fetuses (Furuhashi et al., 1982).
In conclusion, our study demonstrates that the over‐regulation of endogenous AhR ligands induces PE‐like phenotypes in rats along with defects in placental vascular growth. These adverse effects are associated with sexually dimorphic alternations of the transcriptome and immune cell infiltration in the placenta. These findings suggest that the AhR ligand pathway could represent a promising therapeutic and sex‐specific target for PE‐associated vascular dysfunctions. Finally, the limitations of our current study should be recognized. First, this study is limited by the relatively small sample number, especially in measuring the ITE‐altered maternal cardiovascular function. Second, the lack of fetal sex typing restricts the analysis of the sexual dimorphism in ITE‐induced morphological and biochemical changes in fetal placentas. Third, the ITE‐disrupted immune cell filtration in placentas is not confirmed using other approaches (e.g. immunohistochemistry or flow cytometry). Fourth, this is mainly a descriptive study without further exploring the pathophysiological mechanisms underlying ITE‐associated maternal and fetal dysfunction. Fifth, all the conclusions of the current study are drawn exclusively from a rat model. Further studies are needed to verify these changes induced by ITE using other animal (e.g. non‐human primates) or human tissue and cells (e.g. human placentas and HUVECs) models.
Additional information
Competing interests
The authors have no conflict of interest.
Author contributions
W.L., K.W., S.K. and J.Z. conceived this study. Y.J.Z., C.Z., W.L., K.W., S.K. and J.Z. designed this study. Y.J.Z., C.Z., S.Y.Z., J.S.M. and H.H.L. acquired and analysed the data. Y.J.Z. and C.Z. drafted the manuscript. W.L., K.W., S.K. and J.Z. critically revised the manuscript. All authors have read and approved the final version of this manuscript and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This study was supported by the American Heart Association award 19CDA34660348 (C.Z.). The project was also supported by the Translational Basic and Clinical Pilot Award (C.Z., S.K. and J.Z.) from the UW Institute for Clinical and Translational Research and the Clinical and Translational Science Award program through the NIH National Centre for Advancing Translational Sciences, grant UL1TR002373.
Translational perspective.
To date, the aetiology of preeclampsia (PE) is unknown, and there are limited therapeutic tools for PE‐associated endothelial dysfunction because the mechanisms underlying PE are poorly understood. Herein, we have reported that an endogenous aryl hydrocarbon receptor (AhR) ligand induces PE‐like phenotypes in pregnant rats. These phenotype changes are associated with fetal sex‐specific dysregulation of the transcriptome in placentas. These data suggest a novel mechanism underlying PE. The AhR ligand‐dysregulated genes and pathways might represent promising therapeutic and fetal sex‐specific targets for preeclampsia‐induced impairment of vascular functions in placentas.
Supporting information
Peer Review History
Acknowledgements
We thank Ms Lori Uttech‐Hanson from the Office of Grant Writing and Collaborative Project Development, University of Wisconsin–Madison, for English editing. We are grateful to Qi‐zi He, M.D., Ph.D., a pathologist at the Department of Pathology, Shanghai First Maternity and Infant Hospital, Tongji University School of Medicine, Shanghai, China, for evaluating the pathology of rat placental sections.
Biographies
Yingjie Zhao is an Associate Professor at the Department of Rheumatology, Qilu Hospital of Shandong University. He obtained his B.S. in Medicine and M.S. in Rheumatology and Clinical Immunology from Qingdao University, and his Ph.D. from Shandong University, China. He completed his postdoctoral training at the Department of Obstetrics and Gynecology, University of Wisconsin‐Madison. His research focuses on the immune mechanism of pregnancies in lupus and other autoimmune diseases.

Chi Zhou is an Assistant Professor at the School of Animal and Comparative Biomedical Sciences at the University of Arizona. After obtaining her Ph.D. in Reproductive Physiology from the University of Alberta, Canada, she completed her postdoctoral training at the Department of Obstetrics and Gynecology, University of Wisconsin‐Madison. Her current research interests are to elucidate the cellular and molecular mechanisms underlying endothelial dysfunction in complicated pregnancies.

Handling Editors: Laura Bennet & Rebecca Simmons
The peer review history is available in the Supporting Information section of this article (https://doi.org/10.1113/JP287503#support‐information‐section).
Y.‐J. Zhao and C. Zhou contributed equally to this work.
This article was first published as a preprint. Zhao Y‐j, Zhou C, Wei Y‐y, Zhang S‐y, Mishra JS, Li H‐h, Lei W, Wang K, Kumar S, Zheng J. 2023. An Endogenous Aryl Hydrocarbon Receptor Ligand Induces Preeclampsia‐like Phenotypes: Transcriptome, Phosphoproteome, and Cell Functions. bioRxiv. https://doi.org/10.1101/2023.12.20.572271
This is an Editor's Choice article from the 15 January 2025 issue.
Data availability statement
Supporting data are available within the article and its online supplementary files (Fig. S1 and Tables S1–S9) at Figshare: https://doi.org/10.6084/m9.figshare.26750551. RNA‐seq data have been deposited in the NCBI Gene Expression Omnibus (GEO) database (GEO accession number GSE250296).
References
- Agbor, L. N. , Elased, K. M. , & Walker, M. K. (2011). Endothelial cell‐specific aryl hydrocarbon receptor knockout mice exhibit hypotension mediated, in part, by an attenuated angiotensin II responsiveness. Biochemical Pharmacology, 82(5), 514–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blesson, C. S. , Chinnathambi, V. , Hankins, G. D. , Yallampalli, C. , & Sathishkumar, K. (2015). Prenatal testosterone exposure induces hypertension in adult females via androgen receptor‐dependent protein kinase Cδ‐mediated mechanism. Hypertension, 65(3), 683–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton, G. J. , Reshetnikova, O. S. , Milovanov, A. P. , & Teleshova, O. V. (1996). Stereological evaluation of vascular adaptations in human placental villi to differing forms of hypoxic stress. Placenta, 17(1), 49–55. [DOI] [PubMed] [Google Scholar]
- Chang, C. C. , Hsu, Y. H. , Chou, H. C. , Lee, Y. G. , & Juan, S. H. (2017). 3‐methylcholanthrene/Aryl‐hydrocarbon receptor‐mediated hypertension through eNOS inactivation. Journal of Cellular Physiology, 232(5), 1020–1029. [DOI] [PubMed] [Google Scholar]
- Denison, M. S. , Soshilov, A. A. , He, G. , DeGroot, D. E. , & Zhao, B. (2011). Exactly the same but different: Promiscuity and diversity in the molecular mechanisms of action of the aryl hydrocarbon (dioxin) receptor. Toxicological Sciences, 124(1), 1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eskandari, E. , & Eaves, C. J. (2022). Paradoxical roles of caspase‐3 in regulating cell survival, proliferation, and tumorigenesis. Journal of Cell Biology, 221(6), e202201159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faas, M. M. , Spaans, F. , & De Vos, P. (2014). Monocytes and macrophages in pregnancy and pre‐eclampsia. Frontiers in Immunology, 5, 298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuhashi, N. , Suzuki, M. , Fukaya, T. , Kono, H. , Shinkawa, O. , Tachibana, Y. , & Takahashi, T. (1982). Concentrations of luteinizing hormone–human chorionic gonadotropin, beta subunit of human chorionic gonadotropin, follicle‐stimulating hormone, estradiol, cortisol, and testosterone in cord sera and their correlations. American Journal of Obstetrics and Gynecology, 143(8), 918–921. [DOI] [PubMed] [Google Scholar]
- Gibbs, R. A. , Weinstock, G. M. , Metzker, M. L. , Muzny, D. M. , Sodergren, E. J. , Scherer, S. , Scott, G. , Steffen, D. , Worley, K. C. , Burch, P. E. , Okwuonu, G. , Hines, S. , Lewis, L. , DeRamo, C. , Delgado, O. , Dugan‐Rocha, S. , Miner, G. , Morgan, M. , Hawes, A. , … Collins, F. (2004). Genome sequence of the Brown Norway rat yields insights into mammalian evolution. Nature, 428, 493–521. [DOI] [PubMed] [Google Scholar]
- Gopalakrishnan, K. , Mishra, J. S. , Chinnathambi, V. , Vincent, K. L. , Patrikeev, I. , Motamedi, M. , Saade, G. R. , Hankins, G. D. , & Sathishkumar, K. (2016). Elevated testosterone reduces uterine blood flow, spiral artery elongation, and placental oxygenation in pregnant rats. Hypertension, 67(3), 630–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutiérrez‐Vázquez, C. , & Quintana, F. J. (2018). Regulation of the immune response by the aryl hydrocarbon receptor. Immunity, 48(1), 19–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry, E. C. , Bemis, J. C. , Henry, O. , Kende, A. S. , & Gasiewicz, T. A. (2006). A potential endogenous ligand for the aryl hydrocarbon receptor has potent agonist activity in vitro and in vivo. Archives of Biochemistry and Biophysics, 450(1), 67. [DOI] [PubMed] [Google Scholar]
- Hermans, S. , Pilon, J. , Eschweiler, D. , Stegmaier, J. , Severens‐Rijvers, C. A. H. , Al‐Nasiry, S. , van Zandvoort, M. , & Kapsokalyvas, D. (2023). Definition and quantification of three‐dimensional imaging targets to phenotype pre‐eclampsia subtypes: An exploratory study. International Journal of Molecular Sciences, 24(4), 3240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernández‐Ochoa, I. , Karman, B. N. , & Flaws, J. A. (2009). The role of the aryl hydrocarbon receptor in the female reproductive system. Biochemical Pharmacology, 77(4), 547–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishimura, R. , Kawakami, T. , Ohsako, S. , & Tohyama, C. (2009). Dioxin‐induced toxicity on vascular remodeling of the placenta. Biochemical Pharmacology, 77(4), 660. [DOI] [PubMed] [Google Scholar]
- Ivnitski‐Steele, I. , & Walker, M. K. (2005). Inhibition of neovascularization by environmental agents. Cardiovascular Toxicology, 5(2), 215–226. [DOI] [PubMed] [Google Scholar]
- Jiang, Y. Z. , Wang, K. , Fang, R. , & Zheng, J. (2010). Expression of aryl hydrocarbon receptor in human placentas and fetal tissues. Journal of Histochemistry and Cytochemistry, 58(8), 679–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khankin, E. V. , Royle, C. , & Karumanchi, S. A. (2010). Placental vasculature in health and disease. Seminars in Thrombosis and Hemostasis, 36(03), 309–320. [DOI] [PubMed] [Google Scholar]
- Kurien, B. T. , Everds, N. E. , & Scofield, R. H. (2004). Experimental animal urine collection: a review. Laboratory Animals, 38(4), 333–361. [DOI] [PubMed] [Google Scholar]
- Li, Y. , Han, B. , Salmeron, A. G. , Bai, J. , & Chen, D. B. (2022). Estrogen‐induced uterine vasodilation in pregnancy and preeclampsia. Journal of Maternal Fetal and Neonatal Medicine, 4(1), 52–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Y. , Wang, K. , Zou, Q. Y. , Jiang, Y. Z. , Zhou, C. , & Zheng, J. (2017). ITE suppresses angiogenic responses in human artery and vein endothelial cells: Differential roles of AhR. Reproductive Toxicology, 74, 181–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Y. , Wang, K. , Zou, Q. Y. , Magness, R. R. , & Zheng, J. (2015). 2,3,7,8‐Tetrachlorodibenzo‐p‐dioxin differentially suppresses angiogenic responses in human placental vein and artery endothelial cells. Toxicology, 336, 70–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Y. , Zhao, Y. J. , Zou, Q. Y. , Zhang, K. , Wu, Y. M. , Zhou, C. , Wang, K. , & Zheng, J. (2015). Preeclampsia does not alter vascular growth and expression of CD31 and vascular endothelial cadherin in human placentas. Journal of Histochemistry and Cytochemistry, 63(1), 22–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Y. , Zhou, C. , Lei, W. , Wang, K. , & Zheng, J. (2020). Roles of aryl hydrocarbon receptor in endothelial angiogenic responses†. Biology of Reproduction, 103(5), 927–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund, A. K. , Agbor, L. N. , Zhang, N. , Baker, A. , Zhao, H. , Fink, G. D. , Kanagy, N. L. , & Walker, M. K. (2008). Loss of the aryl hydrocarbon receptor induces hypoxemia, endothelin‐1, and systemic hypertension at modest altitude. Hypertension, 51(3), 803–809. [DOI] [PubMed] [Google Scholar]
- Lund, A. K. , Goens, M. B. , Nuñez, B. A. , & Walker, M. K. (2006). Characterizing the role of endothelin‐1 in the progression of cardiac hypertrophy in aryl hydrocarbon receptor (AhR) null mice. Toxicology and Applied Pharmacology, 212(2), 127–135. [DOI] [PubMed] [Google Scholar]
- Masaki, T. , Okazawa, M. , Asano, R. , Inagaki, T. , Ishibashi, T. , Yamagishi, A. , Umeki‐Mizushima, S. , Nishimura, M. , Manabe, Y. , Ishibashi‐Ueda, H. , Shirai, M. , Tsuchimochi, H. , Pearson, J. T. , Kumanogoh, A. , Sakata, Y. , Ogo, T. , Kishimoto, T. , & Nakaoka, Y. (2021). Aryl hydrocarbon receptor is essential for the pathogenesis of pulmonary arterial hypertension. Proceedings of the National Academy of Sciences, USA, 118(11), e2023899118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller, D. , Motomura, K. , Galaz, J. , Gershater, M. , Lee, E. D. , Romero, R. , & Gomez‐Lopez, N. (2022). Cellular immune responses in the pathophysiology of preeclampsia. Journal of Leukocyte Biology, 111(1), 237–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra, J. S. , Te Riele, G. M. , Qi, Q. R. , Lechuga, T. J. , Gopalakrishnan, K. , Chen, D. B. , & Kumar, S. (2019). Estrogen receptor‐β mediates estradiol‐induced pregnancy‐specific uterine artery endothelial cell angiotensin type‐2 receptor expression. Hypertension, 74(4), 967–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizutani, T. , Yoshino, M. , Satake, T. , Nakagawa, M. , Ishimura, R. , Tohyama, C. , Kokame, K. , Kangawa, K. , & Miyamoto, K. (2004). Identification of 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin (TCDD)‐inducible and ‐suppressive genes in the rat placenta: induction of interferon‐regulated genes with possible inhibitory roles for angiogenesis in the placenta. Endocrine Journal, 51(6), 569–577. [DOI] [PubMed] [Google Scholar]
- Moll, W. (2003). Structure adaptation and blood flow control in the uterine arterial system after hemochorial placentation. European Journal of Obstetrics, Gynecology, and Reproductive Biology, 110(Suppl 1), S19–S27. [DOI] [PubMed] [Google Scholar]
- Mtali, Y. S. , Lyimo, M. A. , Luzzatto, L. , & Massawe, S. N. (2019). Hypertensive disorders of pregnancy are associated with an inflammatory state: Evidence from hematological findings and cytokine levels. BioMed Central Pregnancy Childbirth, 19(1), 237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen, L. P. , & Bradfield, C. A. (2008). The search for endogenous activators of the aryl hydrocarbon receptor. Chemical Research in Toxicology, 21(1), 102–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osikoya, O. , Ahmed, H. , Panahi, S. , Bourque, S. L. , & Goulopoulou, S. (2019). Uterine perivascular adipose tissue is a novel mediator of uterine artery blood flow and reactivity in rat pregnancy. The Journal of Physiology, 597(15), 3833–3852. [DOI] [PubMed] [Google Scholar]
- Pang, L. P. , Li, Y. , Zou, Q. Y. , Zhou, C. , Lei, W. , Zheng, J. , & Huang, S. A. (2017). ITE inhibits growth of human pulmonary artery endothelial cells. Experimental Lung Research, 43(8), 283–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson, R. E. , Theobald, H. M. , & Kimmel, G. L. (1993). Developmental and reproductive toxicity of dioxins and related compounds: Cross‐species comparisons. Critical Reviews in Toxicology, 23(3), 283–335. [DOI] [PubMed] [Google Scholar]
- Pique‐Regi, R. , Romero, R. , Tarca, A. L. , Sendler, E. D. , Xu, Y. , Garcia‐Flores, V. , Leng, Y. , Luca, F. , Hassan, S. S. , & Gomez‐Lopez, N. (2019). Single cell transcriptional signatures of the human placenta in term and preterm parturition. eLife, 8, e52004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platten, M. , Nollen, E. A. A. , Röhrig, U. F. , Fallarino, F. , & Opitz, C. A. (2019). Tryptophan metabolism as a common therapeutic target in cancer, neurodegeneration and beyond. Nature Reviews Drug Discovery, 18(5), 379–401. [DOI] [PubMed] [Google Scholar]
- Pocar, P. , Fischer, B. , Klonisch, T. , & Hombach‐Klonisch, S. (2005). Molecular interactions of the aryl hydrocarbon receptor and its biological and toxicological relevance for reproduction. Reproduction, 129(4), 379–389. [DOI] [PubMed] [Google Scholar]
- Rana, S. , Lemoine, E. , Granger, J. P. , & Karumanchi, S. A. (2019). Preeclampsia: Pathophysiology, Challenges, and Perspectives. Circulation Research, 124(7), 1094–1112. [DOI] [PubMed] [Google Scholar]
- Roberts, J. M. , Rich‐Edwards, J. W. , McElrath, T. F. , Garmire, L. , & Myatt, L. (2021). Subtypes of preeclampsia: Recognition and determining clinical usefulness. Hypertension, 77(5), 1430–1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothhammer, V. , & Quintana, F. J. (2019). The aryl hydrocarbon receptor: An environmental sensor integrating immune responses in health and disease. Nature Reviews Immunology, 19(3), 184–197. [DOI] [PubMed] [Google Scholar]
- Sathishkumar, K. , Balakrishnan, M. , Chinnathambi, V. , Chauhan, M. , Hankins, G. D. , & Yallampalli, C. (2012). Fetal sex‐related dysregulation in testosterone production and their receptor expression in the human placenta with preeclampsia. Journal of Perinatology, 32(5), 328–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen, X. , Wang, C. , Yue, X. , Wang, Q. , Xie, L. , Huang, Z. , Huang, X. , Li, J. , Xu, Y. , Chen, L. , Lye, S. , Wei, Y. , & Wang, Z. (2022). Preeclampsia associated changes in volume density of fetoplacental vessels in Chinese women and mouse model of preeclampsia. Placenta, 121, 116–125. [DOI] [PubMed] [Google Scholar]
- Song, J. , Clagett‐Dame, M. , Peterson, R. E. , Hahn, M. E. , Westler, W. M. , Sicinski, R. R. , & DeLuca, H. F. (2002). A ligand for the aryl hydrocarbon receptor isolated from lung. Proceedings of the National Academy of Sciences, USA, 99(23), 14694–14699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stejskalova, L. , Vecerova, L. , Peréz, L. M. , Vrzal, R. , Dvorak, Z. , Nachtigal, P. , & Pavek, P. (2011). Aryl hydrocarbon receptor and aryl hydrocarbon nuclear translocator expression in human and rat placentas and transcription activity in human trophoblast cultures. Toxicological Sciences, 123(1), 26–36. [DOI] [PubMed] [Google Scholar]
- Stockinger, B. , Meglio, P. D. , Gialitakis, M. , & Duarte, J. H. (2014). The aryl hydrocarbon receptor: Multitasking in the immune system. Annual Review of Immunology, 32(1), 403–432. [DOI] [PubMed] [Google Scholar]
- Walisser, J. A. , Bunger, M. K. , Glover, E. , & Bradfield, C. A. (2004). Gestational exposure of Ahr and Arnt hypomorphs to dioxin rescues vascular development. Proceedings of the National Academy of Sciences, USA, 101(47), 16677–16682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, K. , Li, Y. , Jiang, Y. Z. , Dai, C. F. , Patankar, M. S. , Song, J. S. , & Zheng, J. (2013). An endogenous aryl hydrocarbon receptor ligand inhibits proliferation and migration of human ovarian cancer cells. Cancer Letters, 340(1), 63–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, Y. , Chen, X. , Zhou, Q. , He, Q. , Kang, J. , Zheng, J. , Wang, K. , & Duan, T. (2014). ITE and TCDD differentially regulate the vascular remodeling of rat placenta via the activation of AhR. PLoS ONE, 9(1), e86549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, J. , Merialdi, M. , Platt, L. D. , & Kramer, M. S. (2010). Defining normal and abnormal fetal growth: promises and challenges. American Journal of Obstetrics and Gynecology, 202(6), 522–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, N. (2011). The role of endogenous aryl hydrocarbon receptor signaling in cardiovascular physiology. Journal of Cardiovascular Disease Research, 2(2), 91–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, Y. J. , Zhou, C. , Wei, Y. Y. , Li, H. H. , Lei, W. , Boeldt, D. S. , Wang, K. , & Zheng, J. (2022). Differential distribution of tryptophan‐metabolites in fetal and maternal circulations during normotensive and preeclamptic pregnancies. Reproductive Sciences, 29(4), 1278–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, C. , Freel, C. , Mills, O. , Yang, X. R. , Yan, Q. , & Zheng, J. (2023). MicroRNA‐29 differentially mediates preeclampsia‐dysregulated cellular responses to cytokines in female and male fetal endothelial cells. The Journal of Physiology, 601(16), 3631–3645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, C. , Yan, Q. , Zou, Q. Y. , Zhong, X. Q. , Tyler, C. T. , Magness, R. R. , Bird, I. M. , & Zheng, J. (2019). Sexual dimorphisms of preeclampsia‐dysregulated transcriptomic profiles and cell function in fetal endothelial cells. Hypertension, 74(1), 154–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, C. , Zou, Q. Y. , Li, H. , Wang, R. F. , Liu, A. X. , Magness, R. R. , & Zheng, J. (2017). Preeclampsia downregulates microRNAs in fetal endothelial cells: Roles of miR‐29a/c‐3p in endothelial function. Journal of Clinical Endocrinology and Metabolism, 102(9), 3470–3479. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Peer Review History
Data Availability Statement
Supporting data are available within the article and its online supplementary files (Fig. S1 and Tables S1–S9) at Figshare: https://doi.org/10.6084/m9.figshare.26750551. RNA‐seq data have been deposited in the NCBI Gene Expression Omnibus (GEO) database (GEO accession number GSE250296).