

Abstract
Aminoacyl-tRNA synthetases catalyze the first step of protein synthesis. It was shown recently that human tyrosyl-tRNA synthetase (TyrRS) can be split into two fragments having distinct cytokine activities, thereby linking protein synthesis to cytokine signaling pathways. Tryptophanyl-tRNA synthetase (TrpRS) is a close homologue of TyrRS. A natural fragment, herein designated as mini TrpRS, was shown by others to be produced by alternative splicing. Production of this fragment is reported to be stimulated by IFN-γ, a cytokine that also stimulates production of angiostatic factors. Mini TrpRS is shown here to be angiostatic in a mammalian cell culture system, the chicken embryo, and two independent angiogenesis assays in the mouse. The full-length enzyme is inactive in the same assays. Thus, protein synthesis may be linked to the regulation of angiogenesis by a natural fragment of TrpRS.
Aminoacyl-tRNA synthetases catalyze the first step of protein synthesis that consists of the aminoacylation of tRNAs. Recently, we demonstrated that one of the tRNA synthetases—human tyrosyl-tRNA synthetase (TyrRS)—has novel cytokine functions in addition to its role in protein synthesis (1). This demonstration established a link between protein synthesis and signal transduction. Under apoptotic conditions in culture, full-length TyrRS is secreted, and two distinct cytokines can then be generated by an extracellular protease such as leukocyte elastase (1). Whereas the full-length enzyme is inactive in assays for a variety of cytokine activities, the sequestered cytokines are released by splitting the native enzyme into the NH2-terminal catalytic fragment (known as mini TyrRS) and an extra COOH domain that is appended to the catalytic core of the enzyme (Fig. 1). The extra COOH domain of human TyrRS has cytokine activities like those of mature human endothelial monocyte-activating polypeptide II. On the other hand, human mini TyrRS binds strongly to the CXC-chemokine receptor CXCR1 and, like IL-8, functions as a chemoattractant for polymorphonuclear leukocytes (PMNs) (1).
Figure 1.

Schematic representation of human TyrRS and TrpRS constructs used in this study. Shaded regions of full-length TyrRS and TrpRS represent COOH- and NH2-terminal appended domains, respectively. Numbers on the left and right correspond to the NH2- and COOH-terminal residues relative to the human full-length enzymes, respectively. The COOH domain of TyrRS has sequence similarity to mature endothelial monocyte-activating polypeptide II (45). The NH2 domain of TrpRS has sequence similarity to the extra domains of human GluProRS (a fusion of glutamyl- and prolyl-tRNA synthetases), MetRS, GlyRS, and HisRS (8, 10–15).
The catalytic core domain of tryptophanyl-tRNA synthetase (TrpRS) is a close homologue of the catalytic domain of TyrRS (2–4). As shown in Fig. 1, mammalian TrpRS have an NH2-terminal extension that is absent from lower eukaryotic or prokaryotic TrpRS (5–7). The NH2-terminal extension contains an additional 58–78 amino acids compared with yeast (Saccharomyces cerevisiae) or eubacterial TrpRS (Pyrococcus abyssi), respectively. Residues 12–65 of the NH2-terminal extension of human TrpRS are homologous to the WHEP-TRP conserved domain protein family (pfam00458) (8). This domain has a helix-turn-helix architecture (9) and also is present in the human tRNA synthetases for glycine, histidine, and methionine (10–13). Three repeats of this domain are inserted into the linker region of the fused glutamyl-prolyl-tRNA synthetase (9, 14, 15). Interestingly, in histidyl-tRNA synthetase, this domain is a major antigenic epitope for autoantibodies found in myositis patients (13).
In normal cells, human TrpRS exists as two forms; the major form is the full-length protein, and the other is a truncated TrpRS (mini TrpRS) in which most of the extra NH2-terminal domain is deleted because of alternative splicing of the pre-mRNA (16, 17), with Met-48 being deduced as the NH2-terminal residue of mini TrpRS (16). The expression of human mini TrpRS is highly stimulated in human cells by the addition of IFN-γ (18). Stimulation by IFN-γ also induces production of the angiostatic chemokines IP-10 and MIG (19, 20) and attenuates expression of the angiogenic chemokine IL-8 (21). Bovine full-length and truncated TrpRS, in which the extra NH2-terminal domain is deleted by proteolysis (5), are highly expressed in the pancreas and secreted into the pancreatic juice (22, 23). The existence of various truncated forms of TrpRS as well as the close similarity between mammalian TyrRS and mammalian TrpRS raised the possibility that truncated TrpRSs have a function in addition to aminoacylation (5, 24). We hypothesized that mammalian TrpRS may have signal transduction activities specific to angiogenesis.
Materials and Methods
Protein Production and Biochemical Analysis.
Human full-length and truncated TrpRS genes including a COOH-terminal six-histidine tag were cloned into plasmid pET20b and overexpressed in Escherichia coli strain BL 21 (DE3) (Novagen) by induction with 1 mM isopropyl β-D-thiogalactopyranoside for 4 h. Full-length human TrpRS encodes residues 1–471, mini TrpRS residues 48–471, T1-TrpRS residues 74–471, and T2-TrpRS residues 94–471. Schematic diagrams of the proteins used in this study are shown in Fig. 1. Using the procedures described by the manufacturers, the proteins were purified on a nickel affinity column [His⋅Bind resin (Novagen) or Ni-NTA agarose (Qiagen, Chatsworth, CA)] from the supernatant of lysed cells. Endotoxin was removed from the protein solutions by phase separation using Triton X-114 (25) and was determined to be <0.01 endotoxin units/ml by a Limulus amebocyte lysate gel-clot assay (E-Toxate, Sigma). Protein concentration was determined by the Bradford assay with BSA (Sigma) as a standard (Bio-Rad). All truncated TrpRS variants, except T2-TrpRS, were functional in tryptophan-dependent pyrophosphate-ATP exchange (26, 27).
PMN elastase cleavage of human full-length TrpRS was performed at a protease:protein ratio of 1:3,000 in PBS (pH 7.4) at 37°C. Cleavage products were evaluated by SDS/12.5% PAGE and Western blot analysis with anti-His6-tag antibodies (Invitrogen). Edman degradation was performed on the PMN elastase cleavage products to determine their NH2-terminal sequences by using an ABI model 494 sequencer at the Protein and Nucleic Acid Core Facility of The Scripps Research Institute.
In Vitro Human Umbilical Vein Endothelial Cell (HUVEC) Assays.
HUVECs were obtained from Clonetics (San Diego) and maintained in EGM-2 BulletKit medium (Clonetics) in an atmosphere of 5% CO2 in air at 37°C according to the instructions of the supplier. Human vascular endothelial growth factor-165 (VEGF165) (Biosource International, Camarillo, CA) and human IP-10 (R&D Systems) were also used in several experiments.
HUVEC migration assays were performed by using a modified Boyden chamber (48-well chamber) (NeuroProbe, Cabin John, MD) with polycarbonate membranes (8.0 μm pore size) (Costar) as follows (28, 29). The wells were coated with 25 μg/ml human fibronectin (Biosource International) in PBS overnight and allowed to air-dry. HUVECs were suspended in DMEM (GIBCO/BRL) containing 0.1% BSA (Sigma) and added to the upper chamber at 2 × 105 cells per well. A chemotactic stimulus, VEGF165 (0.5 nM), was placed in the lower chamber, and cells were allowed to migrate for 6 h at 37°C in a 5% CO2 incubator. For inhibition assays, full-length TrpRS (500 nM) or mini TrpRS (500 nM) was added to the upper and lower chambers and also to the HUVECs 30 min before placement in the chamber. After incubation, nonmigrant cells were removed from the upper face of the membrane with a cotton swab. Migrant cells (those attached to the lower face) were fixed in methanol and visualized with the Hemacolor stain set (EM Diagnostic Systems, Gibbstown, NJ). Migrating cells were counted in high-power fields.
HUVEC proliferation assays were performed with a CellTiter 96 Aqueous One Solution cell proliferation assay kit (Promega). Tissue culture-treated plates (96-well) (Corning) were coated with 0.1% gelatin (Sigma) overnight. HUVECs were seeded at a density of 5 × 103 cells per well in DMEM (GIBCO/BRL) containing heat-inactivated FBS (10%, Sigma), penicillin (100 units/ml, Sigma), and streptomycin (100 μg/ml, Sigma) in the gelatinized plates on day 0. The following day, the cells were treated with the proliferation stimulus (VEGF165, 2 nM). After 72 h of incubation, the extent of HUVEC proliferation was measured according to the methods supplied with the kit. Inhibition of proliferation was performed by adding inhibitors (full-length TrpRS or mini TrpRS, 2 μM) to the cells 30 min before the addition of the proliferation stimulus. Inhibition assays were presented as percentages of net proliferation of VEGF (2 nM).
In Vivo Angiogenesis Assays.
Three different assays for angiogenesis were used to examine TrpRS for activities in vivo (30–34). The chicken chorioallantoic membrane (CAM) assay was performed as described (35, 36) with 10-day chick embryos obtained from McIntyre Poultry (Lakeside, CA). Cortisone acetate-treated 5-mm filter disks soaked with 20-μl samples of VEGF165 (1 pmol) in PBS were placed onto the CAMs. Inhibitors and control samples [PBS alone, IP-10 (120 pmol), mini TrpRS (60 pmol), or full-length TrpRS (60 pmol)] were added topically to the filter disks for 3 consecutive days. After 72 h, the CAM tissue associated with the filter disk was harvested and photographed at ×10 magnification on an Olympus model SZH10 stereomicroscope. Angiogenesis was quantified by analyzing the number of branching blood vessels within the area of each disk.
Mouse matrigel angiogenesis assays were performed as described with the following modifications (34). Athymic wehi mice were s.c. implanted with 400 μl of growth factor-depleted matrigel (Becton Dickinson) containing 8 pmol of VEGF. Intraperitoneal injections of mini TrpRS (1 nmol) or full-length TrpRS (1 nmol) were administered on days 2, 3, and 4. On day 5, the mice were intravenously injected with the fluorescein-labeled endothelial binding lectin Griffonia (Bandeiraea) Simplicifolia I, isolectin B4 (Vector Laboratories), and the matrigel plugs were resected. Fluorescent images of sectioned plugs were taken with a fluorescent Olympus model SZH10 confocal stereomicroscope. The fluorescein content of each plug was quantified by spectrophotometric analysis after grinding the plug in RIPA buffer (10 mM sodium phosphate, pH 7.4/150 mM sodium chloride/1% Nonidet P-40/0.5% sodium deoxycholate/0.1% SDS).
Mouse retinal angiogenesis assays were performed by the following method. Immediately after birth (P0), retinal vasculature is virtually absent in the mouse. By 2 weeks postnatal (P14), the retina has attained an adult pattern of retinal vessels coincident with the onset of vision. Physiological vascularization of the retina occurs during this period via a stereotypical, biphasic developmental pattern of angiogenesis. Initially, spoke-like peripapillary vessels grow radially from the central retinal artery and vein, becoming progressively interconnected by a capillary plexus that forms between them. This superficial retinal vascular layer (primary layer) grows in area, volume, and complexity, centrifugally, as a monolayer within the nerve fiber layer during the first 9 days following birth. The second phase of retinal vessel formation begins between postnatal days 8 (P8) and 10 (P10), when collateral branches sprout from capillaries of the superficial plexus and penetrate into the retina where their tips branch and anastamose laterally to form a planar, deep vascular layer (secondary layer) that is in place by P12–P14. Qualitative evaluation of the effects of compounds on angiogenesis can be done by simply photographing the primary and secondary layers and determining the percentage of eyes in which formation of the deep layer is completely or partially inhibited (see accompanying article for a complete description of this model).
Mini TrpRS (5 pmol in 0.5 μl) or full-length TrpRS (5 pmol in 0.5 μl) were injected intravitreally into neonatal mice on postnatal day 7 or 8, and the retinas were harvested on P12 or P13. The blood vessels were visualized by staining the retina with a rabbit anti-mouse collagen IV antibody (Chemicon, 1:200 dilution in blocking buffer: 10% goat serum/3% BSA in PBS) for 18 h at 4°C. An Alexa Fluor 594-conjugated goat anti-rabbit IgG antibody (Molecular Probes, 1:200 dilution in blocking buffer) was incubated with the retina for 2 h at 4°C. Angiostatic activity was evaluated based on the effect of injected proteins on formation of the deep (secondary) retinal vascular layer and compared with intravitreal injection of 0.5 μl of PBS. None of the proteins used in this study produced an adverse effect on the primary layer.
Results
Identification of TrpRS Fragments with Signal Transduction Activity.
Mini TrpRS is a truncated version of TrpRS, missing 47 NH2-terminal residues, that is produced by alternative splicing (IFN-γ induction) (18). Alone, neither TrpRS nor mini TrpRS induced migration of HUVECs. However, when the cells were induced with VEGF165 and treated with mini TrpRS, migration was inhibited (Fig. 2A). In contrast, full-length TrpRS had no effect on VEGF165-stimulated HUVEC chemotaxis. HUVEC proliferation induced by VEGF165 was also inhibited by mini TrpRS, whereas full-length TrpRS had no effect (Fig. 2B).
Figure 2.

Activities of human TrpRS constructs on HUVECs. (A) Inhibition of HUVEC migration by human TrpRS constructs. Average number of cells per high-power field migrating in response to stimulation by cell medium, VEGF (0.5 nM), VEGF (0.5 nM) + full-length TrpRS (500 nM), VEGF (0.5 nM) + mini TrpRS (500 nM). (B) Inhibition of endothelial cell proliferation by human TrpRS constructs. Human TrpRS (2 μM) were assayed on HUVEC cells in the presence of VEGF165 (2 nM) in a 72-h proliferation experiment. Cell proliferation is presented as the percentage of net VEGF165-induced (2 nM) proliferation. Values represent the mean ± standard deviation from five experiments.
In bovine pancreas, fragments of TrpRS are present in pancreatic secretions (22, 23), raising the possibility that proteolytic forms of TrpRS, in addition to the alternative spliced variant, might have biological activity. Because PMN elastase can release the active cytokines mini TyrRS and the endothelial monocyte-activating polypeptide II-like COOH domain from full-length TyrRS (1), we examined PMN elastase cleavage of full-length TrpRS. PMN elastase digestion of recombinant full-length TrpRS (54 kDa) produced two fragments: 47- and 44-kDa fragments designated T1-TrpRS and T2-TrpRS, respectively. These fragments were similar in size to mini TrpRS (49 kDa) (Fig. 3). Sequencing showed that the NH2 termini were SNHGP and SAKGI, demonstrating that the fragments began at Ser-71 and Ser-94, respectively. Western blot analysis with antibodies directed against the COOH-terminal His6-tag of the recombinant protein indicated that both fragments had the His6-tag at their COOH termini. Therefore, only the NH2 terminus of TrpRS was removed. To check the activity of the elastase cleavage product, T1-TrpRS that is closest in size to mini TrpRS, we prepared the recombinant protein and tested it in HUVEC migration and cell proliferation assays. T1-TrpRS blocked VEGF165-induced HUVEC chemotaxis and inhibited VEGF165-induced HUVEC proliferation (data not shown).
Figure 3.
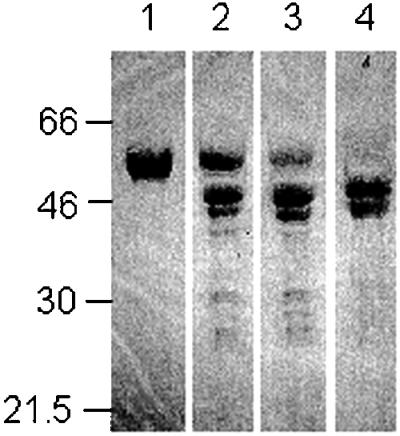
Proteolytic cleavage of human TrpRS. A protease:protein ratio of 1:3,000 was used. The cleavage was done at 37°C in PBS (pH 7.4) for 0 min (lane 1), 15 min (lane 2), 30 min (lane 3), or 60 min (lane 4). Samples were analyzed on SDS/12.5% polyacrylamide gels. Molecular size markers are given to the left (in kilodaltons).
Angiostatic Activity of TrpRS Fragments in CAM Assay.
Chick CAM assays were performed to test for angiostatic activity of TrpRS. VEGF165-induced angiogenesis in CAMs was clearly inhibited by mini TrpRS or T1-TrpRS (Fig. 4), whereas human full-length TrpRS had no observable angiostatic activity (data not shown). As a control, the angiostatic CXC-chemokine IP-10 was used to inhibit angiogenesis stimulated by VEGF165. It caused a similar inhibition of angiogenesis, as did mini TrpRS and T1-TrpRS.
Figure 4.

Activity of human TrpRS in the chicken chorioallantoic membrane assay. The data are presented as the mean number of blood vessel branches ± standard deviations from 5–8 embryos in each sample. (A) Angiostatic activity of human TrpRS fragments. PBS, VEGF165 (1 pmol), VEGF165 (1 pmol) + IP10 (120 pmol), VEGF165 (1 pmol) + full-length TrpRS (60 pmol), VEGF165 (1 pmol) + mini TrpRS (60 pmol), or VEGF165 (1 pmol) + T1-TrpRS (60 pmol). VEGF165 was added on day 0; IP10 and TrpRS were added for three consecutive days starting at day 1. (B) Digital images of representative filter discs showing angiogenesis (CAM assays) stimulated by VEGF165 and inhibited by mini TrpRS.
Mouse Matrigel Plug Assay.
To extend these studies into a mammalian system, the activity of TrpRS was examined in a mouse matrigel plug assay (34). On i.p. injection, matrigel containing an angiogenic stimulus forms a plug into which blood vessels can migrate. Introduction of VEGF165 into the matrigel plug induced angiogenesis (Fig. 5). VEGF165-induced angiogenesis was blocked by i.p. injections of mini TrpRS (Fig. 5). A similar inhibition of angiogenesis was seen when mini TrpRS was included directly into the matrigel plug as when it was delivered by i.p. injection. No effect was seen on injection of full-length TrpRS (data not shown).
Figure 5.

Activity of human TrpRS in a murine matrigel model of angiogenesis. Athymic wehi mice were s.c. implanted with 400 μl of growth factor-depleted matrigel containing an angiogenic stimulus. Intraperitoneal injections (i.p.) of mini TrpRS were given on days 2, 3, and 4. On day 5, the mice were intravenously injected with the fluorescein-labeled endothelial binding lectin Griffonia (Bandeiraea) Simplicifolia I, isolectin B4. The plugs were resected and solubilized, and the fluorescein content was quantitated by spectrometry. (A) Relative fluorescein content of resected matrigel plugs treated with medium, VEGF165 (8 pmol), VEGF (8 pmol) + i.p. injected mini TrpRS (1 nmol), VEGF165 (8 pmol) + mini TrpRS (1 nmol, included in matrigel). (B) Fluorescent images of resected matrigel plugs by using confocal microscopy. Stimulation of angiogenesis by VEGF165 and inhibition of VEGF165-stimulated angiogenesis by mini TrpRS. In the absence of VEGF, the images are similar to those shown on the right.
Inhibition of Vascularization of Mouse Retina.
Because TrpRS potently inhibited VEGF165-induced angiogenesis in the CAMs and mouse matrigel assay, it was of interest to examine the angiostatic activity of TrpRS on endogenous vascularization and angiogenesis in the mouse retina (37). Intravitreous application of mini TrpRS during neovascularization and angiogenesis inhibited the formation of new blood vessels in the deep (secondary) vascular layer without any adverse effect on previously formed vessels in the superficial (primary) vascular layer (Fig. 6). Complete inhibition of the secondary layer was observed in 8.2% (n = 73) of PBS-treated eyes (data not shown), 11.0% of full-length TrpRS-treated eyes (n = 18), and 28% of mini TrpRS-treated eyes (n = 75). The effect of full-length TrpRS was similar to that of the control injections of PBS, where no significant inhibition occurred.
Figure 6.

Fragments of TrpRS inhibit angiogenesis in a postnatal mouse retinal model. Representative images of superficial (primary layer) and deep (secondary layer) retinal vasculature observed after intravitreal injection of mini TrpRS (Upper) or full-length TrpRS (Lower). On postnatal day 8, eyes were treated by intravitreous injection of PBS, human full-length TrpRS (5 pmol), or mini TrpRS (5 pmol), and retinas were harvested on postnatal day 12 or 13. Blood vessels were visualized after staining with anti-collagen IV antibodies. Qualitative image analysis of retinal vessel formation between P8 and P12 showed complete inhibition of the secondary layer in 28% of eyes treated with mini TrpRS. Full-length TrpRS had no effect on secondary layer formation. Note that the primary layer was unaffected by either mini or full-length TrpRS.
Discussion
The natural mini TrpRS fragment of TrpRS is active as an inhibitor in several distinct and unrelated assays, including HUVEC migration and proliferation, chick chorioallantoic membrane, mouse matrigel, and mouse retinal angiogenesis assays. The full-length protein is inactive, thus suggesting that alternative splicing or cleavage is essential for activation of angiostatic activity. That the full-length protein is inactive is consistent with the angiostatic activity of a TrpRS fragment being part of a specific biological mechanism and not an epiphenomenon associated with TrpRS per se.
Although dose–response studies were not carried out with mini TrpRS, the accompanying article describes such investigations with the smaller T2-TrpRS (vide supra). For example, T2-TrpRS was shown active as an angiogenesis inhibitor in a dose–response titration in the range of 0.1 to 10 nM (using T2-TrpRS directly in the mouse matrigel plug) (37). Interestingly, even when administered by i.p. injection, mini TrpRS can arrest VEGF165-stimulated blood vessel development in the mouse (Fig. 5). This observation shows that free mini TrpRS is sufficiently stable in vivo to act as an angiostatic factor at sites distal to that where it is produced. Although little is known about the secretion of TrpRS, previous results demonstrate secretion of TyrRS from cultured cells during apoptosis (1), and preliminary studies showed secretion of TrpRS under similar conditions (K.W., unpublished observations). Collectively, these results suggest a role for TrpRS in angiogenic pathways of natural systems. Further validation of that role requires identification of the relevant endothelial cell receptor.
In some cell lines, the angiostatic mini TrpRS is highly expressed in the presence of the anti-tumorigenic IFN-γ (18, 38–41), where mini TrpRS can be produced by alternative splicing of the pre-mRNA (16, 17). As stated above, stimulation by IFN-γ also induces production of angiostatic chemokines. Removal of the NH2-terminal domain from human TrpRS by pre-mRNA alternative splicing or by PMN elastase cleavage generates angiostatic versions of TrpRS. Recently, PMN elastase was shown to be present in human colorectal carcinoma with particular enrichment at the tumor–host interface (42). Also, breast and non-small cell lung cancer cells are known to secrete PMN elastase in vitro (43, 44). Possibly, human TrpRS secreted from apoptotic cells may be cleaved by PMN elastase at the tumor–host interface. The cleaved enzyme could then act as an angiostatic factor to attenuate tumor invasion.
Acknowledgments
We thank Dr. Lois Smith for providing helpful comments on the manuscript. We thank Dr. Jie Leng and Mauricio Rosenfeld for assistance with the chick CAM assays. We also thank Drs. Allan C. Shaw and Just Justesen for rabbit polyclonal antibodies against human TrpRS. This work was supported by National Institutes of Health Grants GM23562 (to P.S.) and EY12599 (to M.F.), National Cancer Institute Grant CA92577 (to P.S.), a fellowship from the National Foundation for Cancer Research (to P.S.), and the Robert Mealey Program for the Study of Macular Degenerations (M.F.). K.W. was supported by a Japan Society for the Promotion of Science Postdoctoral Fellowship for Research Abroad (1997–1999).
Abbreviations
- TyrRS
tyrosyl-tRNA synthetase
- TrpRS
tryptophanyl-tRNA synthetase
- PMN
polymorphonuclear leukocytes
- HUVEC
human umbilical vein endothelial cell
- VEGF165
human vascular endothelial growth factor-165
- CAM
chorioallantoic membrane
References
- 1.Wakasugi K, Schimmel P. Science. 1999;284:147–151. doi: 10.1126/science.284.5411.147. [DOI] [PubMed] [Google Scholar]
- 2.Doublie S, Bricogne G, Gilmore C, Carter C W., Jr Structure. 1995;3:17–31. doi: 10.1016/s0969-2126(01)00132-0. [DOI] [PubMed] [Google Scholar]
- 3.Ribas de Pouplana L, Frugier M, Quinn C L, Schimmel P. Proc Natl Acad Sci USA. 1996;93:166–170. doi: 10.1073/pnas.93.1.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brown J R, Robb F T, Weiss R, Doolittle W F. J Mol Evol. 1997;45:9–16. doi: 10.1007/pl00006206. [DOI] [PubMed] [Google Scholar]
- 5.Lemaire G, Gros C, Epely S, Kaminski M, Labouesse B. Eur J Biochem. 1975;51:237–252. doi: 10.1111/j.1432-1033.1975.tb03924.x. [DOI] [PubMed] [Google Scholar]
- 6.Epely S, Gros C, Labouesse J, Lemaire G. Eur J Biochem. 1976;61:139–146. doi: 10.1111/j.1432-1033.1976.tb10004.x. [DOI] [PubMed] [Google Scholar]
- 7.Scheinker V S, Beresten S F, Mazo A M, Ambartsumyan N S, Rokhlin O V, Favorova O O, Kisselev L L. Eur J Biochem. 1979;97:529–540. doi: 10.1111/j.1432-1033.1979.tb13141.x. [DOI] [PubMed] [Google Scholar]
- 8.Bateman A, Birney E, Durbin R, Eddy S R, Howe K L, Sonnhammer E L. Nucleic Acids Res. 2000;28:263–266. doi: 10.1093/nar/28.1.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jeong E J, Hwang G S, Kim K H, Kim M J, Kim S, Kim K S. Biochemistry. 2000;39:15775–15782. doi: 10.1021/bi001393h. [DOI] [PubMed] [Google Scholar]
- 10.Tsui F W, Siminovitch L. Nucleic Acids Res. 1987;15:3349–3367. doi: 10.1093/nar/15.8.3349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frolova L Y, Sudomoina M A, Grigorieva A Y, Zinovieva O L, Kisselev L. Gene. 1991;109:291–296. doi: 10.1016/0378-1119(91)90624-k. [DOI] [PubMed] [Google Scholar]
- 12.Ge Q, Trieu E P, Targoff I N. J Biol Chem. 1994;269:28790–28797. [PubMed] [Google Scholar]
- 13.Hirakata M, Suwa A, Takeda Y, Matsuoka Y, Irimajiri S, Targoff I N, Hardin J A, Craft J. Arthritis Rheum. 1996;39:146–151. doi: 10.1002/art.1780390119. [DOI] [PubMed] [Google Scholar]
- 14.Fett R, Knippers R. J Biol Chem. 1991;266:1448–1455. [PubMed] [Google Scholar]
- 15.Rho S B, Lee K H, Kim J W, Shiba K, Jo Y J, Kim S. Proc Natl Acad Sci USA. 1996;93:10128–10133. doi: 10.1073/pnas.93.19.10128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tolstrup A B, Bejder A, Fleckner J, Justesen J. J Biol Chem. 1995;270:397–403. doi: 10.1074/jbc.270.1.397. [DOI] [PubMed] [Google Scholar]
- 17.Turpaev K T, Zakhariev V M, Sokolova I V, Narovlyansky A N, Amchenkova A M, Justesen J, Frolova L Y. Eur J Biochem. 1996;240:732–737. doi: 10.1111/j.1432-1033.1996.0732h.x. [DOI] [PubMed] [Google Scholar]
- 18.Shaw A C, Rossel Larsen M, Roepstorff P, Justesen J, Christiansen G, Birkelund S. Electrophoresis. 1999;20:984–993. doi: 10.1002/(SICI)1522-2683(19990101)20:4/5<984::AID-ELPS984>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 19.Kaplan G, Luster A D, Hancock G, Cohn Z A. J Exp Med. 1987;166:1098–1108. doi: 10.1084/jem.166.4.1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Farber J M. Biochem Biophys Res Commun. 1993;192:223–230. doi: 10.1006/bbrc.1993.1403. [DOI] [PubMed] [Google Scholar]
- 21.Gusella G L, Musso T, Bosco M C, Espinoza-Delgado I, Matsushima K, Varesio L. J Immunol. 1993;151:2725–2732. [PubMed] [Google Scholar]
- 22.Sallafranque M L, Garret M, Benedetto J P, Fournier M, Labouesse B, Bonnet J. Biochim Biophys Acta. 1986;882:192–199. doi: 10.1016/0304-4165(86)90155-8. [DOI] [PubMed] [Google Scholar]
- 23.Favorova O O, Zargarova T A, Rukosuyev V S, Beresten S F, Kisselev L L. Eur J Biochem. 1989;184:583–588. doi: 10.1111/j.1432-1033.1989.tb15053.x. [DOI] [PubMed] [Google Scholar]
- 24.Kisselev L, Frolova L, Haenni A L. Trends Biochem Sci. 1993;18:263–267. doi: 10.1016/0968-0004(93)90178-p. [DOI] [PubMed] [Google Scholar]
- 25.Aida Y, Pabst M J. J Immunol Methods. 1990;132:191–195. doi: 10.1016/0022-1759(90)90029-u. [DOI] [PubMed] [Google Scholar]
- 26.Kisselev L L, Favorova O O, Nurbekov M K, Dmitriyenko S G, Engelhardt W A. Eur J Biochem. 1981;120:511–517. doi: 10.1111/j.1432-1033.1981.tb05729.x. [DOI] [PubMed] [Google Scholar]
- 27.Jorgensen R, Sogaard T M, Rossing A B, Martensen P M, Justesen J. J Biol Chem. 2000;275:16820–16826. doi: 10.1074/jbc.275.22.16820. [DOI] [PubMed] [Google Scholar]
- 28.Yoshida A, Anand-Apte B, Zetter B R. Growth Factors. 1996;13:57–64. doi: 10.3109/08977199609034566. [DOI] [PubMed] [Google Scholar]
- 29.Masood R, McGarvey M E, Zheng T, Cai J, Arora N, Smith D L, Sloane N, Gill P S. Blood. 1999;93:1038–1044. [PubMed] [Google Scholar]
- 30.Friedlander M, Brooks P C, Shaffer R W, Kincaid C M, Varner J A, Cheresh D A. Science. 1995;270:1500–1502. doi: 10.1126/science.270.5241.1500. [DOI] [PubMed] [Google Scholar]
- 31.Friedlander M, Theesfeld C L, Sugita M, Fruttiger M, Thomas M A, Chang S, Cheresh D A. Proc Natl Acad Sci USA. 1996;93:9764–9769. doi: 10.1073/pnas.93.18.9764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brooks P C, Silletti S, von Schalscha T L, Friedlander M, Cheresh D A. Cell. 1998;92:391–400. doi: 10.1016/s0092-8674(00)80931-9. [DOI] [PubMed] [Google Scholar]
- 33.Nicolaou K C, Trujillo J I, Jandeleit B, Chibale K, Rosenfeld M, Diefenbach B, Cheresh D A, Goodman S L. Bioorg Med Chem. 1998;6:1185–1208. doi: 10.1016/s0968-0896(98)00090-x. [DOI] [PubMed] [Google Scholar]
- 34.Eliceiri B P, Paul R, Schwartzberg P L, Hood J D, Leng J, Cheresh D A. Mol Cell. 1999;4:915–924. doi: 10.1016/s1097-2765(00)80221-x. [DOI] [PubMed] [Google Scholar]
- 35.Brooks P C, Montgomery A M, Rosenfeld M, Reisfeld R A, Hu T, Klier G, Cheresh D A. Cell. 1994;79:1157–1164. doi: 10.1016/0092-8674(94)90007-8. [DOI] [PubMed] [Google Scholar]
- 36.Brooks P C, Montgomery A M, Cheresh D A. Methods Mol Biol. 1999;129:257–269. doi: 10.1385/1-59259-249-X:257. [DOI] [PubMed] [Google Scholar]
- 37.Otani A, Slike B M, Dorrell M, Hood J, Kinder K, Ewalt K L, Cheresh D, Schimmel P, Friedlander M. Proc Natl Acad Sci USA. 2002;99:178–183. doi: 10.1073/pnas.012601899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rubin B Y, Anderson S L, Xing L, Powell R J, Tate W P. J Biol Chem. 1991;266:24245–24248. [PubMed] [Google Scholar]
- 39.Fleckner J, Rasmussen H H, Justesen J. Proc Natl Acad Sci USA. 1991;88:11520–11524. doi: 10.1073/pnas.88.24.11520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fleckner J, Martensen P M, Tolstrup A B, Kjeldgaard N O, Justesen J. Cytokine. 1995;7:70–77. doi: 10.1006/cyto.1995.1009. [DOI] [PubMed] [Google Scholar]
- 41.Reano A, Richard M H, Denoroy L, Viac J, Benedetto J P, Schmitt D. J Invest Dermatol. 1993;100:775–779. doi: 10.1111/1523-1747.ep12476463. [DOI] [PubMed] [Google Scholar]
- 42.Bjornland K, Buo L, Scott H, Konttinen Y, Johansen H T, Aasen A O. Int J Oncol. 1998;12:535–540. doi: 10.3892/ijo.12.3.535. [DOI] [PubMed] [Google Scholar]
- 43.Yamashita J I, Ogawa M, Ikei S, Omachi H, Yamashita S I, Saishoji T, Nomura K, Sato H. Br J Cancer. 1994;69:72–76. doi: 10.1038/bjc.1994.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yamashita J, Tashiro K, Yoneda S, Kawahara K, Shirakusa T. Chest. 1996;109:1328–1334. doi: 10.1378/chest.109.5.1328. [DOI] [PubMed] [Google Scholar]
- 45.Kleeman T A, Wei D, Simpson K L, First E A. J Biol Chem. 1997;272:14420–14425. doi: 10.1074/jbc.272.22.14420. [DOI] [PubMed] [Google Scholar]