

Abstract
Rat cortical astrocytes in pure culture are functionally coupled to neighboring cells via connexin (Cx) 43 gap junctions under ordinary conditions. Small fluorescent molecules such as Lucifer yellow (LY) pass between cell interiors via gap junctions, but do not enter the cells when externally applied. Subjecting rat and mouse cortical astrocytes to “chemical ischemia” by inhibition of glycolytic and oxidative metabolism induced permeabilization of cells to Lucifer yellow and ethidium bromide before loss of membrane integrity determined by dextran uptake and lactate dehydrogenase release. The gap junction blockers octanol and 18α-glycyrrhetinic acid markedly reduced dye uptake, suggesting that uptake was mediated by opening of unapposed hemichannels. Extracellular La3+ also reduced dye uptake and delayed cell death. The purinergic blocker, oxidized ATP, was ineffective. Astrocytes isolated from mice with targeted deletion of the Cx43 coding DNA exhibited greatly reduced dye coupling and ischemia-induced dye uptake, evidence that dye uptake is mediated by Cx43 hemichannels. Dye coupling was reduced but not blocked by metabolic inhibition. Blockade of lipoxygenases or treatment with free radical scavengers reduced dye uptake by rat astrocytes, suggesting a role for arachidonic acid byproducts in hemichannel opening. Furthermore, permeabilization was accompanied by reduction in ATP levels and dephosphorylation of Cx43. Although hemichannel opening would tend to collapse electrochemical and metabolic gradients across the plasma membrane of dying cells, healthy cells might rescue dying cells by transfer of ions and essential metabolites via Cx43 gap junctions. Alternatively, dying astrocytes might compromise the health of neighboring cells via Cx43 gap junctions, thereby promoting the propagation of cell death.
Keywords: astroglia‖dye uptake‖dye coupling‖Cx43‖phosphorylation
Gap junctions connect the cytoplasmic domains of contacting cells, allowing ionic and metabolic exchange between them and mediating metabolic cooperation thought to optimize the functioning of many tissues (1, 2). Gap junctions are formed of connexins, a family of homologous protein subunits (3–5), and their channels are connexin dodecamers formed of hexameric hemichannels, one from each of the coupled cells (6). Hemichannels are formed in the endoplasmic reticulum and inserted into the cell membrane and subsequently dock with a hemichannel in the adjacent cell (7). The channel diameter and selectivity, while varying among junctions formed of different connexins, is ≈1.2 nm, admitting molecules of nominally 1 kDa. Because open hemichannels in nonjunctional membrane were thought likely to have permeability properties similar to those of the intercellular channels, it was generally assumed that, under physiological conditions, “unapposed” hemichannels were closed to prevent metabolic stress and death caused by the collapse of ionic gradients, loss of small metabolites, and influx of Ca2+ (see, for example, ref. 1). However, putative opening of hemichannels has now been observed in numerous preparations, as indicated by (i) permeabilization of the cells to molecules to which gap junctions are permeable without permeabilization to larger molecules, indicative of loss of membrane integrity, (ii) block of this permeability by blockers of gap junctions, (iii) absence of the permeabilization in cells not expressing connexins, and (iv) patch clamp recording of single channel activity with appropriate properties.
Opening of endogenous hemichannels appears to occur in astrocytes in primary culture (8) under nearly physiological conditions. (Findings with respect to other cell types are discussed elsewhere, in text that is published as supporting information on the PNAS web site, www.pnas.org) Opening of hemichannels by strong depolarization and/or bathing in low Ca2+ solutions was previously reported in astrocytes (9). Connexin (Cx) 43 in the brain is primarily found in astrocytes (10). Although cortical astrocytes may also express Cx26, Cx30, Cx40, Cx43, and Cx45 in vivo or in vitro (11–13), gap junctions between cultured cortical astrocytes have single channel conductance and other biophysical properties (14, 15) like those observed in Cx43 transfectants (16–18).
In the central nervous system, astrocytes play an important role in maintaining the composition of the extracellular milieu, which may be facilitated by gap junctions between them (19, 20). Under pathological conditions, such as hypoxia/reoxygenation, arachidonic acid byproducts cause reduction in gap junction-mediated coupling of astrocytes (21). In addition, metabolic inhibition of astrocytes leads to dephosphorylation of their Cx43 (21), an effect that is mediated in part by calcineurin, a protein phosphatase, and that may contribute to the reduction in gap junctional communication observed under those conditions (22). Consistent with a general homeostatic role, blocking astrocyte gap junctions increases neuronal vulnerability to an oxidative stress (23). Conversely, open gap junctions could mediate propagation and increase in spatial extent of cell injury, as has been observed in an in vitro model using a glioma cell line (24). Cx43 hemichannels in astrocytes may open under essentially normal conditions and release ATP and NAD+, which may serve a paracrine signaling function and participate in propagation of Ca2+ waves (8, 25).
The present work describes changes in permeability of the plasma membrane and of gap junction channels in cultured astrocytes subjected to metabolic inhibition or “chemical ischemia” induced by block of oxidative and glycolytic metabolism. We found that metabolic inhibition permeabilized astrocytes to gap junction permeant molecules and that uptake of these molecules was blocked by gap junction blockers and La3+. This permeabilization preceded loss of membrane integrity measured by uptake of dextran and loss of lactate dehydrogenase (LDH). Block of hemichannels by extracellular application of La3+ delayed the loss of membrane integrity induced by chemical inhibition. Metabolic inhibition also permeabilized wild-type mouse astrocytes, but astrocytes of Cx43 deficient mice were resistant. Dye coupling was reduced but not blocked by metabolic inhibition. Pretreatment of astrocytes with nordihydroguaiaretic acid, a lipoxygenase blocker, or free radical scavengers substantially reduced dye uptake induced by metabolic inhibition, suggesting that hemichannel opening is mediated by arachidonic acid byproducts and arguing for specificity of the response (see supporting text). Furthermore, ATP levels and phosphorylation of Cx43 were decreased by metabolic inhibition before permeabilization, but the data do not demonstrate a causal relationship. Although prolonged opening of Cx43 hemichannels would dissipate electrochemical and metabolic gradients across the plasma membrane, open gap junctions between cells may allow nonpermeabilized cells to protect or “rescue” permeabilized cells; conversely, open gap junctions may allow for propagation of cell death from cells with open hemichannels to unaffected neighboring cells.
Materials and Methods
Reagents and drug treatments are listed in supporting text.
Cell Cultures.
Astrocytes from neocortex of rat embryos (embryonic day 19) were prepared and maintained in culture as previously described (26). Astrocytes were plated on 60-mm Primaria plastic culture dishes (Nunc Clone) with or without glass coverslips (4 × 106 cells per plate 24 h before the experiment), or on 96-multiwell plates (50 × 103 cells in 0.1 ml of culture medium per well 24 h before the experiment) and kept at 37°C in a 5% CO2/95% air atmosphere at nearly 100% relative humidity. All figures except Fig. 4 used rat astrocytes that were from the first or second passage of confluent primary cultures.
Figure 4.

Cx43 deficient astrocytes are not permeabilized by metabolic inhibition. Confluent cultures of mouse cortical astrocytes were subjected to metabolic inhibition. (A) Astrocytes of wild-type mice (circles) showed progressive uptake of LY (measured as the percentage of fields with 10 or more labeled cells of ≈30 cells) during metabolic inhibition reaching 100% after ≈140 min. Cx43del/del astrocytes showed no LY uptake during 360 min metabolic inhibition (triangles). (B) Cx43fl/fl astrocytes (circles) showed progressive LY uptake similar to that of wild type astrocytes. Cx43fl/fl,hGFAP-cre astrocytes (triangles) showed reduced and delayed dye uptake during metabolic inhibition, reaching a plateau of ≈30% after ≈120 min. Each point represents the mean value obtained from 10–20 fields in each of three experiments ±SD.
Generation of Astrocytes from Cx43del/del and Cx43fl/fl, Human Glial Fibrillary Acidic Protein (hGFAP)-cre Mutant Mice (Fig. 4 Only).
Cx43del/del mice were obtained by interbreeding heterozygous animals carrying a Cx43del allele with a lacZ gene in place of the Cx43 coding region (27). At birth, astrocytes cultures were prepared from each brain as described for rat astrocytes, and the donor animal was genotyped by PCR analysis of tail-tip DNA as described (27).
Mice with astrocyte-specific inactivation of the Cx43 gene were generated by crossing mice of the Cx43fl strain, in which the Cx43 coding region is flanked by loxP sites (27), and mice from the TgN25Mes strain, carrying a cre recombinase transgene under the human glial fibrillary acidic protein promoter (hGFAP-cre; ref. 28). F2 litters were analyzed for absence of the wild-type Cx43 allele and presence of the Cx43fl allele and hGFAP-cre transgene as described (27, 28). In Cx43fl/fl, hGFAP-cre astrocyte cultures (purity: 90–95%), at least 90% of the cells lacked Cx43 immunoreactivity after 4 weeks in culture (D.S. and M.T., unpublished results). Primary cultures astrocytes from newborn wild-type BALB/c or Cx43 deficient mice of either gender were prepared and maintained as described above for rat astrocytes.
Dye Uptake.
Stock solutions of fluorescent dyes were prepared in 150 mM LiCl [110
μM Lucifer yellow (LY)/2.5 mM
4′,6-diamidino-2-phenylindole (DAPI)/25 mM dextran-LY, 10 kDa]
or in PBS (5 mM ethidium bromide (EtdBr)/2.5 mM propidium
iodide). For visualization of dye uptake in Figs.
1, 2F, 5B,
and 6, 0.5 μl of stock solutions were applied at room temperature
through a Gilson pipetter with 10-μl pipette tip over the area of the
culture plate to be evaluated, and, 2 min later, the dye was washed
away and replaced with recording medium [HCO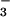 -free
F-12 medium buffered with 20 mM Hepes (pH 7.2)]. This timing
produced little background labeling. In some experiments, a gap
junction channel blocker, 18α-glycyrrhetinic acid (AGA) or octanol,
was applied for 5 min just before dye application. In permeabilized
cells, EtdBr labeling was seen as fluorescence of the nuclei; LY and
Texas red also preferentially labeled nuclei, but in addition labeled
cytoplasmic components. Retained dye was visualized on an inverted
microscope (Nikon Diaphot) equipped with xenon arc lamp illumination,
UV excitation filter, and a Nikon BV-1A filter (for green emission) or
an Omega XF34 filter (for red emission). For Fig.
2 A–F, astrocytes
plated on 96-multiwell plates were used to quantify uptake of EtdBr
(100 μM in PBS), by using a Perkin–Elmer LS 50B luminescence
spectrometer collecting light from nearly an entire well (518 nm
excitation and 605 nm emission). EtdBr fluoresces strongly only when
bound to DNA or RNA and did not need to be washed off for the
measurements.
-free
F-12 medium buffered with 20 mM Hepes (pH 7.2)]. This timing
produced little background labeling. In some experiments, a gap
junction channel blocker, 18α-glycyrrhetinic acid (AGA) or octanol,
was applied for 5 min just before dye application. In permeabilized
cells, EtdBr labeling was seen as fluorescence of the nuclei; LY and
Texas red also preferentially labeled nuclei, but in addition labeled
cytoplasmic components. Retained dye was visualized on an inverted
microscope (Nikon Diaphot) equipped with xenon arc lamp illumination,
UV excitation filter, and a Nikon BV-1A filter (for green emission) or
an Omega XF34 filter (for red emission). For Fig.
2 A–F, astrocytes
plated on 96-multiwell plates were used to quantify uptake of EtdBr
(100 μM in PBS), by using a Perkin–Elmer LS 50B luminescence
spectrometer collecting light from nearly an entire well (518 nm
excitation and 605 nm emission). EtdBr fluoresces strongly only when
bound to DNA or RNA and did not need to be washed off for the
measurements.
Figure 1.

Metabolic inhibition permeabilizes astrocytes to small positively and negatively charged fluorescent dyes; gap junction blockers prevent dye uptake. (Top) Phase contrast images. (Middle) EtdBr fluorescence. (Bottom) LY fluorescence. Before metabolic inhibition, confluent astrocyte cultures bathed with EtdBr (B) and LY for 2 min did not retain either dye (B and C, respectively). Astrocytes treated with 5 ng/ml antimycin A plus 0.3 mM iodoacetic acid for 75 min became permeable to both EtdBr and LY, as indicated by uptake of the dyes applied in the bath for 2 min (E, F). Sister cultures treated for the last 5 min of 75 min metabolic inhibition with either 1 mM octanol (G–I) or 35 μM AGA (J–L) did not take up either EtdBr (H and K) or LY (I and L).
Figure 2.

Progressive EtdBr uptake is induced by metabolic inhibition, but not by membrane depolarization or low extracellular calcium. Uptake is sensitive to a gap junction blocker but not to a blocker of P2X ATP receptors. Astrocyte monolayers in 96-well plates were treated with metabolic inhibitors for different periods of time with EtdBr applied for the last 15 min before fluorescence was measured. (A–D) A progressive increase in fluorescence intensity and number of labeled cells was observed from time 0 to 15, 60 and 90 min of metabolic inhibition. (E) Time course of EtdBr uptake, measured as fluorescence, by astrocytes during metabolic inhibition. Each point represents the average value for five wells in each of three independent experiments corrected for initial control values and normalized to the maximal value measured at 120 min and plotted ±SD. (F) Uptake of EtdBr measured after 75 min metabolic inhibition was markedly reduced by 5 min prior treatment with 35 μM 18α-glycyrrhetinic acid (AGA), but not with 300 μM oxidized ATP (o-ATP). Uptake of EtdBr in astrocytes bathed for 2 h with a solution containing 55 mM K+, zero extracellular Ca2+ plus 5 mM EGTA (0 mM Ca2+), or both high K+ and zero Ca2+ was similar to that observed in astrocytes maintained under control conditions. Each point represents the normalized value obtained in three experiments ±SD.
Figure 5.

Gap junctions between metabolically inhibited astrocytes are dye permeable at times when Cx43 is largely dephosphorylated. (A, D, and G) Phase views. (B, E, and H) EtdBr fluorescence. (C, F, I, and J–L) LY fluorescence. After 75 min metabolic inhibition, astrocytes showed EtdBr and LY uptake (B and C) that was prevented by extracellular application of 0.1 mM La3+ (E and F). In the same culture plate treated with La3+, EtdBr and LY coinjected into one cell diffused to several neighboring cells (H and I). Dye coupling in control medium (J) was not significantly different from dye coupling after 20 min exposure to 0.1 mM La3+ (K). Dye coupling of cells subjected to metabolic inhibition for 75 min with La3+ for the last 20 min was reduced (L). Western blot analysis of phosphorylation of Cx43 (M) showed that, under control conditions (lane control) and after treatment with La3+ for 20 min (lane La3+), the two phosphorylated forms (P3 and P2) were most abundant (compare with Fig. 6). Metabolic inhibition for 75 min (lane met. inh.) caused marked dephosphorylation that was not affected by addition of La3+ added for the last 20 min (lane met. inh./La3+).
For time-lapse recording of dye uptake, a microincubator adapted to fit on the stage of an Olympus IX-70 inverted microscope was used with a fluorescence imaging system and ×60 objective. Temperature was maintained at 37°C, and inflow of humidified air into the chamber was maintained. Cells were bathed in 1 μM EtdBr, and images were acquired with a OlymPix 2000 cooled digital camera and UltraView software (Perkin–Elmer). Fluorescence exposure times were ≈10 ms, with 30 s between images. Typically, a phase contrast image was acquired for each fluorescence image.
In experiments with mouse astrocytes, dye uptake was considered positive for a field that included ≈30 cells, if visual inspection through a fluorescence microscope revealed 10 or more labeled cells.
Results
Metabolic Inhibition Induces Uptake of Positively and Negatively Charged Fluorescent Molecules Through a Pathway Sensitive to Gap Junction Blockers.
Cx43 gap junctions are known to be permeable to 4′,6-diamidino-2-phenylindole (DAPI; charge +2), EtdBr (charge +1), and LY (charge −2; ref. 29). In confluent astrocyte cultures in control medium, application of these molecules for 2 min did not label cells (Fig. 1B, EtdBr; Fig. 1C, LY). In cultures treated with both antimycin A (5 ng/ml) and iodoacetic acid (0.3 mM) for 75 min, most cells became labeled when EtdBr (Fig. 1 E) and LY (Fig. 1F) were coapplied for 2 min. Addition of gap junction blockers, 1 mM octanol (Fig. 1 H and I) or 35 μM AGA (Fig. 1 K and L), 5 min before dye application completely inhibited the uptake of EtdBr (Fig. 1 H and K) and LY (Fig. 1 I and L).
The time course of EtdBr uptake during metabolic inhibition is shown in Fig. 2. A gradual increase in staining intensity and number of stained cells was observed (Fig. 2 A–D). After 75 min metabolic inhibition, all nuclei were stained. Full field measurements of fluorescence showed a gradual increase to reach a maximal level by ≈75 min of metabolic inhibition (Fig. 2E). Although EtdBr uptake after 75 min metabolic inhibition was greatly reduced by 35 μM AGA, applied 5 min before the dye (Fig. 2F), the uptake was not affected by 300 μM oxidized ATP (Fig. 2F). The ineffectiveness of oxidized ATP indicates that EtdBr did not enter through P2X receptors (30).
In spectrophotometric measurements of fluorescence of individual cells during metabolic inhibition (Fig. 3A), each cell initially showed a low basal rate of dye uptake. After about 40 min, dye uptake markedly increased until it reached a plateau at about 90 min (Fig. 3A, small filled circles). Although the points are averages of 10 cells, each cell's uptake had a similar time course, but a slightly different latency of the increase in rate of uptake.
Figure 3.

EtdBr uptake induced by metabolic inhibition precedes membrane breakdown. (A) Time course of EtdBr uptake by confluent astrocytes during metabolic inhibition averaged for 10 cells ± SD (fluorescence intensity, left ordinate) and LDH release to the extracellular medium (right ordinate). LDH release from cultures was measured in 96-well plates by using a kit from Promega and expressed as the percentage of total LDH in sister cultures. EtdBr uptake accelerated after about 45 min metabolic inhibition and reached a plateau at about 90 min (small filled circles, frequent measurements, mean ± SD for 10 cells, representative of three experiments). In astrocytes with the same treatment but in the presence of La3+ (small open circles), EtdBr uptake increased slowly until about 140 min and then accelerated. LDH release above baseline (see Materials and Methods) was detected after 120 min metabolic inhibition and was near maximal after 150 min (large filled circles). In the presence of La3+, LDH release was delayed by about 30 min (large open circles). Each data point represents the average ±SD of six wells in each of three experiments. (B) Astrocytes subjected to 75 min metabolic inhibition were impermeable to dextran-LY (10 kDa), although they were permeable to EtdBr (not shown). (C) After 120 min metabolic inhibition, astrocytes were permeable to dextran-LY.
Because, in other cell systems, strong depolarization or exposure to low extracellular calcium concentrations induces opening of hemichannels (see supporting text), we studied whether these conditions result in permeabilization of astrocytes. Extracellular application of 60 mM K+ depolarizes cultured astrocytes from about −75 mV to about −10 mV (31); we depolarized astrocyte cultures by replacing 55 mM NaCl in the bathing solution with 55 mM KCl. Astrocytes incubated in 55 mM K+,Ca2+-free PBS (pH 7.3) containing 5 mM EGTA, or Ca2+-free plus 55 mM K+, for up to 2 h did not become permeable to EtdBr (Fig. 2J) or LY (not shown).
Coapplication of EtdBr and dextran-LY (10 kDa) after 75 min metabolic inhibition demonstrated that the cells were not permeable to dextran-LY (Fig. 3B), although they had become permeable to EtdBr (not shown, see Fig. 2D). After 90 min of metabolic inhibition, astrocytes were clearly permeable to dextran-LY (Fig. 3C), indicating membrane breakdown. Consistent with this interpretation, onset of LDH release from the cells started after about 90 min of metabolic inhibition and increased progressively to reach a plateau at about 120 min (Fig. 3A, large filled circles). It has been reported that hemichannel currents can be blocked by extracellular application of La3+ (32). Thus, we determined the effect of La3+ (0.05 mM) on the time course of dye uptake and LDH release during metabolic inhibition. La3+ applied concurrently with metabolic inhibitors maintained EtdBr uptake at near basal levels until about the time that LDH was being released (Fig. 3A, small open circles). Furthermore, La3+ delayed LDH released by about 30 min (Fig. 3A, large open circles). The delay of LDH release by La3+ suggests that open hemichannels provided an additional stress on the cells, so that they lost membrane integrity sooner than if the hemichannels had been blocked by La3+.
Astrocytes Deficient in Cx43 Are Resistant to Permeabilization by Metabolic Inhibition.
If astrocytes deficient in Cx43 were resistant to permeabilization by metabolic inhibition, it would be strong evidence that Cx43 hemichannels mediate the dye uptake. To evaluate this possibility, we used astrocytes of normal mice and two types of Cx43-deficient mice, conventional “knockout” Cx43del/del mice (in which the Cx43 coding sequence was replaced by lacZ) and tissue-specific “knockout” Cx43fl/fl, hGFAP-cre mice (in which Cx43 expression was absent in at least 90% of the astrocytes). Virtually 100% of astrocytes from wild-type and Cx43fl/fl mice were dye coupled for LY. Consistent with previous reports that astrocyte gap junctions are mainly formed by Cx43 (14, 15), cells from Cx43del/del mice were not dye coupled, and cells from Cx43fl/fl, hGFAP-Cre mice were coupled at a low incidence (about 10%, not illustrated). Astrocytes from wild-type mice showed uptake of extracellularly applied LY after 20 min metabolic inhibition, indicating membrane permeabilization, but cells from Cx43del/del mice remained impermeable to LY for 360 min metabolic inhibition (Fig. 4A). (Rat astrocytes exhibited membrane breakdown after ≈120 min metabolic inhibition, Fig. 3; mouse astrocytes appear to be more resistant.) Dye uptake during metabolic inhibition of cells from Cx43fl/fl mice (Fig. 4B) was comparable to that of wild-type astrocytes, but cells of Cx43fl/fl, hGFAP-cre mice showed reduced and delayed dye uptake (Fig. 4B). The residual permeabilization measured in the cultures of Cx43fl/fl, hGAFP-cre astrocytes might have been due to the fact that a fraction of these cells still express some Cx43, because they failed to express GFAP (and may have been nonastrocytic contaminating cells) or because of incomplete action of the cre recombinase. The incidence of coupling of wild-type astrocytes subjected to metabolic inhibition remained as high as in control cells for at least 360 min. However, after a period of metabolic inhibition sufficient to induce membrane permeabilization, injected dye was rapidly lost from the cells. In Cx43-deficient cells, fluorescence of microinjected dye did not decline during metabolic inhibition, consistent with absence of permeabilization (not illustrated). LY uptake observed in Cx43fl/fl, hGFAP-cre astrocytes treated with metabolic inhibitors for 240 min was prevented by 35 μM AGA applied 5 min before LY application (not illustrated).
Dye Coupling Between Astrocytes Is Reduced but Not Blocked by Metabolic Inhibition.
Because wild-type rat or mouse astrocytes treated with metabolic inhibitors did not retain microinjected LY but did show (transient) dye coupling, we were concerned that the intercellular movement of dye might be due to leakage from the injected cell and uptake by neighboring cells rather than passage through gap junctions. To prevent leakage of the microinjected dyes, we blocked hemichannels with extracellular La3+ (0.1 mM), as previously described (32). We found that EtdBr and LY uptake by metabolically inhibited rat astrocytes (Fig. 5 B and C) was blocked by incubation in PBS containing 0.1 mM La3+ (Fig. 5 E and F). In the presence of La3+, microinjected EtdBr (Fig. 5H) and LY (Fig. 5I) were retained by the metabolically inhibited cells and diffused to several neighboring cells (21). La3+ application had no effect on LY or EtdBr coupling of control astrocytes (Fig. 5 J and K; mean number of LY coupled cells 18.0 ± 6.7 for control, 15.4 ± 7.3 for La3+, n = 20, P > 0.2). The number of cells to which dye spread was reduced by metabolic inhibition (Fig. 5L; mean number of coupled cells 4.2 ± 3.5 for metabolic inhibition in the presence of La3+, n = 16, P < 0.001, for comparison with control or lanthanum-treated alone). Reduced coupling during metabolic inhibition has been reported by others (33).
We used Western blotting (21) to examine the effect of La3+ treatment on metabolic inhibition and dephosphorylation of Cx43. Astrocytes incubated for 20 min in control medium with 0.1 mM La3+ showed Cx43 phosphorylation that was very similar to that of cells incubated under control conditions (Fig. 5M, lanes control and La3+). Astrocytes subjected to 75 min of metabolic inhibition showed marked dephosphorylation of Cx43 (lane met. inh.), and treatment with 0.1 mM La3+ starting 20 min before completing 75 min metabolic inhibition had no effect on the dephosphorylation (lane met. inh./La3+). We conclude that La3+ treatment has little if any effect on phosphorylation state and that metabolic inhibition that greatly reduces Cx43 phosphorylation does not block gap junctional communication.
Discussion
We demonstrate here that cortical astrocytes subjected to metabolic inhibition become permeable to gap junction-permeant molecules, both positively and negatively charged. We ascribe this increase in permeability to opening of nonjunctional Cx43 hemichannels, because it is blocked by gap junction blockers, because it occurs before loss of membrane integrity, and because it is not observed in astrocytes lacking Cx43. Furthermore, because it is not blocked by oxidized ATP, it is not due to entry through P2X receptors, which are permeable to cations and small molecules, including LY (30). Membrane depolarization and low extracellular calcium are reported to induce hemichannel opening in astrocytes (9), as well as a number of other cell types (see supporting text), but we did not find these treatments to be effective in our cultures. We did find that permeabilization was reduced or prevented by blockers of lipoxygenases and by free radical scavengers, implicating products of arachidonic acid production in the permeabilization process and suggesting a specific gating response (Fig. 6, which is published as supporting information on the PNAS web site). We also found that metabolic inhibition reduces but does not block dye coupling of astrocytes, although there is a marked reduction in ATP levels and in phosphorylation of Cx43 (Fig. 7, which is published as supporting information on the PNAS web site).
In a reconstituted system, Cx43 hemichannels are opened by dephosphorylation and closed by mitogen-activated protein (MAP) kinase-mediated phosphorylation (34). Moreover, inhibition of protein dephosphorylation prevents opening of hemichannels in Novikoff cells induced by low extracellular Ca2+ (22, 35). During metabolic inhibition, we found that there was a marked increase in dephosphorylated Cx43 by the time of onset of EtdBr uptake (Fig. 7). Thus, it is possible that dephosphorylation of Cx43 leads to opening of astrocyte hemichannels. However, dephosphorylation was not complete, leaving the possibility that open hemichannels and/or cell–cell channels were not dephosphorylated. An issue still to be examined is whether block of lipoxygenases and antioxidants, which reduces hemichannel opening (Fig. 6), affects Cx43 phosphorylation. Single channel recording from inside-out patches during application of kinases and phosphatases should clarify the role of phosphorylation in hemichannel gating.
In brain slices subjected to ischemia, Cx43 is dephosphorylated about 1 h after ATP levels reach a minimum (33), but, in cultured astrocytes, we found that the reduction in ATP levels induced by metabolic inhibition preceded or occurred simultaneously with dephosphorylation of Cx43 (Fig. 7). Thus, Cx43 appears to be dephosphorylated by different mechanisms in these two systems. In astrocyte cultures, Cx43 dephosphorylation induced by hypoxia is mediated by calcineurin (22). In brain slices, inhibition of ATP synthesis and dephosphorylation of Cx43 do not block intercellular diffusion of LY, although coupling is reduced (33). Similarly, we observed that astrocyte permeabilization induced by metabolic inhibition did not block intercellular diffusion of LY or EtdBr. These findings are in agreement with the recent demonstration that gap junctions allow propagation and spatial amplification of cell death induced by metabolic inhibition in an astrocytoma cell line (24). Conversely and particularly in focal ischemia, open gap junctions might allow cells that had not been permeabilized to rescue their compromised neighbors. Opening of hemichannels, which are permeable to the anion LY and cation EtdBr, could contribute to tissue damage by permitting collapse of Na+/K+ gradients, influx of toxic amounts of Ca2+, and loss of essential metabolites or transmitters, such as amino acids, ATP, and reduced glutathione. Although hemichannel opening during metabolic inhibition would appear to be deleterious, it may represent an exaggeration of a normal response, such as the release of ATP or NAD+ for generation of cyclic-ADP ribose and Ca2+ waves (8, 25). The inhibitory effect of lipoxygenase block and free radical scavengers on hemichannel opening is consistent with a regulated mechanism of paracrine signaling.
Supplementary Material
Acknowledgments
We gratefully acknowledge the technical assistance of Ms. Teresa Vergaras. We thank Dr. Albee Messing (Madison, WI) for generously providing mice of the TgN25Mes strain. This work was supported by a Fondo Nacional de Ciencia y Tecnología grant (8990008 to J.C.S.), National Institutes of Health grants (NS 07512 to M.V.L.B.; NS 36706 to F.F.B.), a German Research Association grant through SFB 284 and SFB 400 (to K.W.), and a travel fellowship from Consejo Nacional de Investigaciones Científicas y Técnicas (CONICYT)/Deutscher Akademischer Austauschdienst (DAAD) (to E.A.E.).
Abbreviations
- Cx
connexin
- LDH
lactate dehydrogenase
- hGFAP
human glial fibrillary acidic protein
- LY
Lucifer yellow
- AGA
18α-glycyrrhetinic acid
References
- 1.Bennett M V L, Barrio L C, Bargiello T A, Spray D C, Hertzberg E, Sáez J C. Neuron. 1991;6:305–320. doi: 10.1016/0896-6273(91)90241-q. [DOI] [PubMed] [Google Scholar]
- 2.Simon A M, Goodenough D A. Trends Cell Biol. 1998;8:477–483. doi: 10.1016/s0962-8924(98)01372-5. [DOI] [PubMed] [Google Scholar]
- 3.Bennett M V L, Zheng X, Sogin M L. Soc Gen Physiol Ser. 1994;49:223–233. [PubMed] [Google Scholar]
- 4.O'Brien J, Bruzzone R, White T W, al-Ubaidi M R, Ripps H. J Neurosci. 1998;18:7625–7637. doi: 10.1523/JNEUROSCI.18-19-07625.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Willecke, K., Eiberger, J., Degen, J., Eckardt, D., Romualdi, A., Gueldenagel, M., Deutsch, U. & Soehl, G. (2002) Biol. Chem., in press. [DOI] [PubMed]
- 6.Yeager M, Nicholson B J. Curr Opin Struct Biol. 1996;6:183–192. doi: 10.1016/s0959-440x(96)80073-x. [DOI] [PubMed] [Google Scholar]
- 7.Musil L S, Goodenough D A. J Cell Biol. 1991;115:1357–1374. doi: 10.1083/jcb.115.5.1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Verderio C, Bruzzone S, Zocchi E, Fedele E, Schenk U, De Flora A, Matteoli M. J Neurochem. 2001;78:646–657. doi: 10.1046/j.1471-4159.2001.00455.x. [DOI] [PubMed] [Google Scholar]
- 9.Hofer A, Dermietzel R. Glia. 1998;24:141–154. doi: 10.1002/(sici)1098-1136(199809)24:1<141::aid-glia13>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 10.Dermietzel R, Traub O, Hwang T K, Beyer E, Bennett M V L, Spray D C, Willecke K. Proc Natl Acad Sci USA. 1989;86:10148–10152. doi: 10.1073/pnas.86.24.10148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dermietzel R, Gao Y, Scemes E, Vieira D, Urban M, Kremer M, Bennett M V L, Spray D C. Brain Res Brain Res Rev. 2000;32:45–56. doi: 10.1016/s0165-0173(99)00067-3. [DOI] [PubMed] [Google Scholar]
- 12.Nagy J I, Rash J E. Brain Res Brain Res Rev. 2000;32:29–44. doi: 10.1016/s0165-0173(99)00066-1. [DOI] [PubMed] [Google Scholar]
- 13.Rash J E, Yasumura T, Dudek F E, Nagy J I. J Neurosci. 2001;21:1983–2000. doi: 10.1523/JNEUROSCI.21-06-01983.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dermietzel R, Hertberg E L, Kessler J A, Spray D C. J Neurosci. 1991;11:1421–1432. doi: 10.1523/JNEUROSCI.11-05-01421.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Giaume C, Fromaget C, el Aoumari A, Cordier J, Glowinski J, Gros D. Neuron. 1991;6:133–143. doi: 10.1016/0896-6273(91)90128-m. [DOI] [PubMed] [Google Scholar]
- 16.Kwak B R, Sáez J C, Wilders R, Chanson M, Fishman G I, Hertzberg E L, Spray D C, Jongsma H J. Pflügers Arch. 1995;430:770–778. doi: 10.1007/BF00386175. [DOI] [PubMed] [Google Scholar]
- 17.Moreno A P, Sáez J C, Fishman G I, Spray D C. Circ Res. 1994;74:1050–1057. doi: 10.1161/01.res.74.6.1050. [DOI] [PubMed] [Google Scholar]
- 18.Bukauskas F F, Bukauskiene A, Bennett M V L, Verselis V K. Biophys J. 2001;81:137–152. doi: 10.1016/S0006-3495(01)75687-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen K C, Nicholson C. Biophys J. 2000;78:2776–2797. doi: 10.1016/S0006-3495(00)76822-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hansson E, Muyderman H, Leonova J, Allansson L, Sinclair J, Blomstrand F, Thorlin T, Nilsson M, Ronnback L. Neurochem Int. 2000;37:317–329. doi: 10.1016/s0197-0186(00)00033-4. [DOI] [PubMed] [Google Scholar]
- 21.Martínez A D, Sáez J C. Brain Res Brain Res Rev. 2000;32:250–258. doi: 10.1016/s0165-0173(99)00086-7. [DOI] [PubMed] [Google Scholar]
- 22.Li W E, Nagy J I. Eur J Neurosci. 2000;12:2644–2650. doi: 10.1046/j.1460-9568.2000.00162.x. [DOI] [PubMed] [Google Scholar]
- 23.Blanc E M, Bruce-Keller A J, Mattson M P. J Neurochem. 1998;70:958–970. doi: 10.1046/j.1471-4159.1998.70030958.x. [DOI] [PubMed] [Google Scholar]
- 24.Lin J H, Weigel H, Cotrina M L, Liu S, Bueno E, Hansen A J, Hansen T W, Goldman S, Nedergaard M. Nat Neurosci. 1998;1:494–500. doi: 10.1038/2210. [DOI] [PubMed] [Google Scholar]
- 25.Cotrina M L, Lin J H, Alves-Rodrigues A, Liu S, Li J, Azmi-Ghadimi H, Kang J, Naus C C, Nedergaard M. Proc Natl Acad Sci USA. 1998;95:15735–15740. doi: 10.1073/pnas.95.26.15735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Martínez A D, Sáez J C. Brain Res. 1999;816:411–423. doi: 10.1016/s0006-8993(98)01016-6. [DOI] [PubMed] [Google Scholar]
- 27.Theis M, de Wit C, Schlaeger T M, Eckardt D, Krüger O, Döring B, Risau W, Deutsch U, Pohl U, Willecke K. Genesis. 2001;29:1–13. doi: 10.1002/1526-968x(200101)29:1<1::aid-gene1000>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 28.Zhuo L, Theis M, Alvarez-Maya I, Brenner M, Willecke K, Messing A. Genesis. 2001;31:85–94. doi: 10.1002/gene.10008. [DOI] [PubMed] [Google Scholar]
- 29.Elfgang C, Eckert R, Lichtenberg-Frate H, Butterweck A, Traub O, Klein R A, Hulser D F, Willecke K. J Cell Biol. 1995;129:805–817. doi: 10.1083/jcb.129.3.805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ballerini P, Rathbone M P, Di Iorio P, Renzetti A, Giuliani P, D'Alimonte I, Trubiani O, Caciagli F, Ciccarelli R. NeuroReport. 1996;7:2533–2537. doi: 10.1097/00001756-199611040-00026. [DOI] [PubMed] [Google Scholar]
- 31.Harold D E, Walz W. Neuroscience. 1992;47:203–211. doi: 10.1016/0306-4522(92)90133-m. [DOI] [PubMed] [Google Scholar]
- 32.John S A, Kondo R, Wang S Y, Goldhaber J I, Weiss J N. J Biol Chem. 1999;274:236–240. doi: 10.1074/jbc.274.1.236. [DOI] [PubMed] [Google Scholar]
- 33.Cotrina M L, Kang J, Lin J H, Bueno E, Hansen T W, He L, Liu Y, Nedergaard M. J Neurosci. 1998;18:2520–2537. doi: 10.1523/JNEUROSCI.18-07-02520.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim D Y, Kam Y, Koo S K, Joe C O. J Biol Chem. 1999;274:5581–5587. doi: 10.1074/jbc.274.9.5581. [DOI] [PubMed] [Google Scholar]
- 35.Liu T F, Li H Y, Atkinson M M, Johnson R G. Methods Find Exp Clin Pharmacol. 1995;17:23–28. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}