

Abstract
Soluble guanylyl cyclase (sGC) is the major cellular receptor for the intercellular messenger nitric oxide (NO) and mediates a wide range of physiological effects through elevation of intracellular cGMP levels. Critical to our understanding of how NO signals are decoded by receptive cells and translated into a useful physiological response is an appreciation of the molecular and kinetic details of the mechanism by which NO activates sGC. It is known that NO binds to a haem prosthetic group on the receptor and triggers a conformational change that increases the catalysis of cGMP synthesis by several hundred-fold. The haem is covalently attached to sGC at His-105 of the β1 subunit, and it was thought previously that activation of sGC by NO occurs in two steps: binding of NO to the haem to form a biliganded state and then rupture of the bond to His-105 triggering an increase in catalytic activity. A recent investigation of the kinetics of sGC activation [Zhao, Y., Brandish, P. E., Ballou, D. P. & Marletta, M. A. (1999) Proc. Natl. Acad. Sci. USA, 96, 14753–14758], however, proposed an additional mechanism by which NO regulates sGC activity, namely, by influencing the rate of cleavage of the His-105 bond. The existence of a second (unidentified) NO-binding site on the enzyme was hypothesized and suggested to be fundamental to cellular NO-signal transduction. Here, we show that it is unnecessary to postulate any such additional mechanism because the results obtained are predicted by the simpler model of sGC activation with a single NO-binding event.
Nitric oxide (NO) is a freely diffusible intercellular signaling molecule that mediates a wide variety of physiological effects in the vasculature, central and peripheral nervous systems, and elsewhere (1–4). At high levels, NO is also a cytotoxic agent, and is implicated in several clinical conditions, ranging from acute disorders such as septic shock and stroke (5, 6) to long-term degenerative diseases such as multiple sclerosis and cancer (7, 8). Because of its importance in health and disease, the regulation of NO synthesis has been studied extensively. Much less is understood about how NO signals are decoded and translated into downstream physiological effects. The best known NO receptor is the enzyme soluble guanylyl cyclase (sGC), the activity of which results in cGMP accumulation in target cells, but many of the basic properties of sGC activation in cells remain unclear.
In its molecular makeup, sGC exists as an αβ-heterodimer, but only two isoforms (α1β1, α2β1) so far have been shown to exist at the protein level (9, 10). Most enzymological studies to date have been carried out on the widely expressed α1β1 isoform, and it has become clear that sensitivity of the enzyme to NO is conferred by a single haem moiety that is associated with histidine residue 105 (His-105) of the β1 subunit. Because haem-free sGC could be activated by protoporphyrin IX, which resembles five-coordinate nitrosyl haem structurally, it was hypothesized that active sGC required a five-coordinate nitrosyl haem complex (11). These and other observations led to the formulation of a two-step model in which NO binds to the sGC-haem, forming the six-coordinate complex, and then the bond joining the haem to His-105 breaks, resulting in the five-coordinate species (12). This second step triggers a conformational change that propagates to the active site, enhancing catalytic efficiency.
Recently, a study by Zhao and et al. (13) investigated the subsecond kinetics of sGC activation by NO by using stopped-flow spectroscopy to follow changes in the absorption peak (Soret band) of the haem moiety. As well as providing a clear kinetic description of NO binding, this study made the assertion that the rate of transition from the six-coordinate to the five-coordinate sGC also depended on NO concentration. This assertion was taken to indicate a previously uncharacterized mechanism for regulation of sGC activity by NO whereby the ligand not only determined the amount of occupied enzyme but also determined how quickly the enzyme is activated. Various additional reactions of NO with sGC were considered to account for this finding, including the presence of a second (non-haem) NO-binding site. If correct, this interpretation represents a significant departure from the classical view of sGC activation, with important implications for NO-signal transduction in cells. Reflecting its potential significance, subsequent publications have proposed mechanisms that could account for a second NO-binding step (14, 15), and the model of Zhao et al. has been adopted in a theoretical treatment of NO and sGC kinetics in vivo (16).
In this article, we show, by using classical receptor theory, that the kinetics reported by Zhao et al. are predictable from the simple two-step model, rendering unnecessary the postulate that there exists an additional regulatory site for NO on sGC.
Methods
Model for sGC Activation Based on Classical Receptor Theory.
There is a direct analogy between the traditional two-step model for sGC activation by NO and the del Castillo-Katz model for receptor activation (17). Although originally formulated to describe activation of ligand-gated ion channels, this model applies to any scenario where the binding of an agonist to its receptor causes a conformational change in the protein. It provides an explicit description of the complex relationship between the microscopic rate constants for receptor activation and the macroscopic (experimentally measured) kinetics of a population of receptors (18).
In the model, a receptor exists in three states: agonist free (R), agonist bound (AR), and active (AR*). These states correspond to the unbound, six-coordinate, and five-coordinate NO-sGC species, respectively (Fig. 1). When agonist is added to free receptor, the rate of change of the concentration of receptor in each state over time will be described by two differential equations:
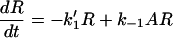 |
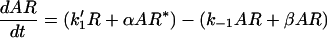 |
and AR* = R0 − R − AR
Figure 1.

The analogy between a classical receptor model (del Castillo-Katz) (a) and the mechanism of sGC activation (b). k1, k−1, α, and β are the microscopic constants for the forward and backward transitions. sGC, unbound sGC; NO-sGC6, six-coordinate bound sGC; NO-sGC5*, five-coordinate active sGC; H, His-105.
where k1′ = k1A
and A = concentration of agonist.
At t = 0, R = R0 and AR = AR* = 0
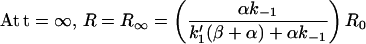 |
The solution to the system is the sum of two exponential terms:
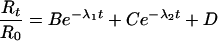 |
1 |
Thus, on addition of agonist to the receptor, the two exponential terms will become smaller with time at a rate determined by the values of λ1 and λ2 until, at equilibrium, the fraction of free receptor equals D.
The rate constants λ1 and λ2 are solutions of the quadratic.
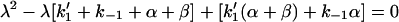 |
which are:
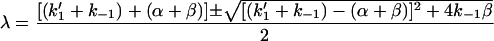 |
where the alternatives + gives rise to λ1 and − gives rise to λ2.
The limits of λ1 and λ2 are (k1′ + k−1) and (α + β), respectively.
From initial and boundary conditions, the values of B, C, and D are found:
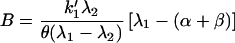 |
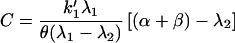 |
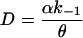 |
where θ = k1′(β + α) + αk−1
B + C + D = 1, such that before addition of agonist, all receptors are unbound.
The kinetics for activation of sGC predicted by this model were simulated and compared with existing experimental data. Additional details describe the fraction of total sGC in the bound and active states with time, the deactivation kinetics, and equilibrium constants, and are published as supporting information on the PNAS web site, www.pnas.org.
Simulations were carried out with a simple custom-written program using Microsoft visual basic 6.0. A copy is available from T.C.B. on request.
Results
Zhao et al. (13) examined the kinetics of sGC activation by NO by measuring the change in sGC-haem absorbance at 431 nm after rapid mixing of purified sGC and NO over a range of concentrations, including substoichiometric ones. The absorbance at 431 nm is proportional to the concentration of unbound sGC, and therefore, a decrease in absorption with time corresponds to the progressive disappearance of free sGC as NO binds and triggers the conformational change that leads to activation. The study was, in part, an extension and reinterpretation of an earlier publication by the group (19).
Zhao et al. (13) reported that disappearance of free sGC occurred in three exponential phases, but concluded that the intermediate phase was artifactual, being attributable to low levels of denatured protein. Thus, functional sGC progressed to equilibrium in two exponential phases, the first being complete within ≈0.01 s and the second taking ≈10 s, when the NO concentration used was in the low micromolar range (13, 19). Kinetic data closely resembling the published results are illustrated in Fig. 2a. Zhao et al. went on to measure the rate constants for these two phases and demonstrated that both of the observed rate constants increased with increasing NO concentration (13, 19), in a manner similar to that shown in Fig. 2b.
Figure 2.

The kinetics for disappearance of unbound sGC after addition of agonist, as predicted by the two-step model. (a) The fraction of total receptor in the unbound (agonist-free) state (R) plotted against time, with 1, 3, and 10 μM NO added at time 0. (b) The variation of the observed rate constants λ1 (solid line) and λ2 (dashed line) for the two exponential phases of the decline in fraction of total sGC in the unbound state, with increasing NO concentration. Values of microscopic constants used: k1 = 8 × 107 M−1⋅s−1; k−1 = 200 s−1; α = 0.066 s−1; β = 1.3 s−1. See refs. 13 and 19 for comparison with experimental data.
These results have here been reexamined by testing the experimental data against an algebraic solution to a model that describes the activation of sGC by NO. The model is based on that of del Castillo-Katz for the two-step activation of classical neurotransmitter receptors (Fig. 1 and Methods), and has been used to simulate the kinetics for progression of agonist and receptor to equilibrium (i.e., from t = 0 to t = ∞). The data shown in Fig. 2 a and b are derived from the model. The “microscopic” rate constants for the individual transitions were chosen by selecting for values giving kinetics similar to the published experimental data outlined above (13, 19), demonstrating that it is possible to mimic these data accurately by using the model. For a direct assessment of the accuracy of the simulations against experimental data, Fig. 2a should be compared with figure 1 in ref. 19 and figure 2c in ref. 13, and Fig. 2b should be compared with figure 2 a and b in ref. 19 and figure 2d in ref. 13.
The finding that the rate constant for the second kinetic phase depends on NO concentration is a direct prediction of the model. The rate constants for both phases are the solutions of a quadratic equation (see Methods) and are related to the NO concentration (unless agonist binding is irreversible). The precise relationship is shown in Fig. 2b. It should be noted that the predicted relationship between the rate constant for the first phase and NO concentration is effectively linear (cf. data points in figure 2a in ref. 19), whereas the relationship for the second phase is clearly nonlinear. At higher concentrations of NO, the rate constant for the second phase will tend to a maximal value (= α + β; see Methods), where an increase in NO will have no further effect.
The model also can be used to predict the fraction of receptor in the other states, as well as other aspects of sGC function, in particular, the deactivation kinetics and equilibrium constants (Fig. 3). The rate of deactivation can be found by simulating an abrupt removal of free NO after an equilibrium position has been reached (see supporting information). Using the values of the microscopic constants selected by fitting to activation data at 10°C (19), the resulting rate of deactivation has been derived (Fig. 3). The predicted half-time for deactivation is 10.5 s, a value similar to estimates of around 18 s obtained at 20°C (20, 21), but nearly 20-fold faster than another estimate (22). The predicted EC50 value for NO activation of sGC is 120 nM, which is in good agreement with published estimates in the 80–250 nM range (10, 19).
Figure 3.

Simulation of activation and deactivation kinetics for sGC. The fraction of total sGC in each state (NO-free, R, large-dash line; NO-bound, AR, small-dash line; active, AR*, solid line) over time is shown after the addition of 1 μM NO at t = 0. The rate of deactivation is shown as the decline in active sGC (AR*) after an instantaneous removal of free NO (arrow). The value of microscopic constants has been selected to replicate data obtained at 10°C (19) for activation. Deactivation and activation proceed in two exponential phases. The first phase of deactivation (see Inset) is apparent as a lag of ≈0.001 s with a rate constant λ3 = 201 s−1 (essentially equal to the loss of bound 3GC with rate constant k−1) followed by an exponential decline with a rate constant λ4 = 0.0656 s−1 (essentially equal to the rate-limiting step, the backwards transition with constant α). For details, see supporting information on the PNAS web site. The rate of deactivation closely resembles that of ref. 20, but note that, in that study, the estimate for kobs (≡ λ4) of 0.04 s−1 was obtained at 20°C. The disparity with ref. 22 (k = 3.6 × 10−3 s−1) cannot readily be accounted for, except by methodological differences. Equilibrium constants are as follows: KA = 2.5 × 10−6 M, E = 19.6, and EC50 = 120 nM, where KA = dissociation constant, E = efficacy constant, and EC50 = KA/(1 + E); see ref. 18.
Discussion
In measuring the rate constants of the two phases of decline in free sGC concentration on exposure to NO, Zhao et al. implied that these values correspond to the microscopic rate constants for NO binding and subsequent scission of the His-105 bond (k1 and β in Fig. 1). On this basis, it was reasoned that if the rate constant for bond cleavage (β) increased with increasing NO concentration, the ligand must in some way drive the conversion. In fact, the authors measured the rate at which a population of sGC comes to equilibrium between three states and, in this article, we show that the two kinetic phases observed experimentally do not equate to the two steps undertaken by individual receptors during activation.
There is a complex relationship between the rate constants describing the individual mechanistic steps in the activation of a receptor by a ligand (“microscopic” constants) and the observed kinetic phases on mixing ligand with a population of receptors (18). This problem has been extensively investigated for neurotransmitter receptors (23), and we have used classical receptor theory (specifically, the del Castillo-Katz model) as the basis for a quantitative description of sGC-activation kinetics. This model has been solved previously with matrix notation (see ref. 24 and references therein), a method that can be readily extended to more complex models, but a simple algebraic solution does not seem to have been published before. By using the model, we have reexamined the findings of Zhao et al. (13) and have demonstrated that the basis for the authors' hypothesis that NO binds twice to sGC during activation is not justified, as the finding that both of the observed kinetic phases show NO concentration-dependency is a direct prediction of the simpler model with a single binding event (Fig. 2b). From the model, it is apparent that the measured rate constants for the observed kinetic phases correspond to λ1 and λ2 in Eq. 1 (see Methods) and do not equate to any meaningful physical property of the receptor.
The model also allows the prediction of other properties of the receptor, such as the rate of deactivation and EC50 for NO activation. By using values for the microscopic rate constants selected by trial and error to result in activation kinetics closely resembling published experimental data (19), it was found that the corresponding predictions for deactivation rate and EC50 were consistent also with some published data (10, 19–21) but not others (22). A note of caution is that these values may be just one combination of many that can adequately simulate experimental data. Our aim has not been to determine reliable estimates for the true rate constants, but merely to demonstrate that the traditional model for NO activation is not incompatible with present experimental findings. It is desirable to carefully design experiments to address specific predictions of this model and to test it explicitly.
Experimental Limitations.
To test the model experimentally in a reliable way, conditions must exist where the free concentration of agonist is maintained at a constant level throughout the course of the experiment. This approach is technically difficult for sGC, as micromolar concentrations of receptor are required to obtain a satisfactory absorbance signal, but the affinity of sGC for NO lies in the submicromolar range. Mixing of substoichiometric concentrations of NO with sGC will lead to a rapid drop in the free NO concentration as receptors (reversibly) bind and sequester agonist. This fact can account for the discrepancy in the data of Marletta and coworkers (13, 19) and another study by Makino et al. (25), who also examined sGC activation kinetics. In contrast to Zhao et al., it was observed that the second kinetic phase of free sGC decline did not vary when NO concentration was decreased. At the NO concentrations used (10 μM and 60 μM), it was likely that Makino et al. were measuring only supramaximal responses, where an increase in NO concentration would have little effect on the value of λ2. In contrast, Zhao et al. used NO concentrations that ranged from substoichiometric to large excess, and so the full range of λ2 values would have been revealed. The value of constants obtained when using stoichiometric (or lower) levels of NO must be interpreted with caution.
The Value of Applying the Model.
When examining sGC deactivation rates and equilibrium constants, different groups have reported divergent data. Estimates of the half-time for the disappearance of active sGC after the removal of free NO range from 5 s (20) to 3 min (22). Similarly, estimates of the potency (EC50) of NO vary from ≤250 nM (19) to ≈80 nM (10, 26). Therefore, it seems that discrepancies may exist in the behavior of sGC as purified by different groups. These discrepancies may be attributable to methodological differences in purification protocols and/or strategies for measuring the different constants.
As the del Castillo-Katz model can be explicitly solved (see Methods and supporting information), it can be applied easily to purified sGC. Testing the predictions of this clearly delineated model on all features of sGC function will allow estimates for the values of microscopic constants to be constrained by demanding a common fit to measured rates of activation and deactivation and the potency of NO at steady-state. Hopefully, this strategy will resolve some of the discrepancies in the behavior of different sGC preparations.
Limitations of the Model.
The principal concern with the model is the applicability of information gained from studies on purified sGC to the receptor in vivo. Recent evidence has revealed that the NO receptor behaves substantially differently within intact cells from its behavior in the test tube. In particular, the rate of deactivation is ≈25-fold faster than even the highest estimate for purified sGC (27), and the receptor exhibits a desensitizing profile of catalytic activity (28). The mechanistic details of this discrepancy in behavior are unknown at present. Nevertheless, rigorous testing of the model using purified sGC will provide a firm basis for the later expansion of the model to incorporate the features of sGC function in a physiological context.
A second issue is how reasonable estimates for the microscopic rate constants for each transition are to be obtained. To a certain extent, iteration to fit experimental data are satisfactory, but unlike classical receptors where single channel recording has revolutionized modeling (29), changes in only a population of sGC can be measured at present. New technology would be required to probe the activity of the NO receptor in greater detail.
As a final note, it should perhaps be clearly stated that the model of Zhao et al. with a second NO-binding event is not disproved by the simpler model. Indeed, NO dependency for the second step would, of course, be predicted if NO did drive the six- to five-coordinate transition. The issue, however, is that the data available at present do not demand any extra complexity for interpretation than the simple single-binding-event model. Thus, to propose the existence of additional NO-binding sites at this stage is unwarranted.
Supplementary Material
Acknowledgments
We thank Prof. D. Colquhoun for helpful discussions. Work in this laboratory is funded by The Wellcome Trust.
Abbreviation
- sGC
soluble guanylyl cyclase
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Moncada S, Palmer R M, Higgs E A. Pharmacol Rev. 1991;43:109–142. [PubMed] [Google Scholar]
- 2.Garthwaite J, Boulton C L. Annu Rev Physiol. 1995;57:683–706. doi: 10.1146/annurev.ph.57.030195.003343. [DOI] [PubMed] [Google Scholar]
- 3.Sanders K M, Ward S M. Am J Physiol. 1992;262:G379–G392. doi: 10.1152/ajpgi.1992.262.3.G379. [DOI] [PubMed] [Google Scholar]
- 4.Patel S, Robb-Gaspers L D, Stellato K A, Shon M, Thomas A P. Nat Cell Biol. 1999;1:467–471. doi: 10.1038/70249. [DOI] [PubMed] [Google Scholar]
- 5.Titheradge M A. Biochim Biophys Acta. 1999;1411:437–455. doi: 10.1016/s0005-2728(99)00031-6. [DOI] [PubMed] [Google Scholar]
- 6.Iadecola C. Trends Neurosci. 1997;20:132–139. doi: 10.1016/s0166-2236(96)10074-6. [DOI] [PubMed] [Google Scholar]
- 7.Kroncke K D, Fehsel K, Kolb-Bachofen V. Clin Exp Immunol. 1998;113:147–156. doi: 10.1046/j.1365-2249.1998.00648.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tamir S, Tannenbaum S R. Biochim Biophys Acta. 1996;1288:F31–F36. doi: 10.1016/0304-419x(96)00021-2. [DOI] [PubMed] [Google Scholar]
- 9.Hobbs A J. Trends Pharmacol Sci. 1997;18:484–491. doi: 10.1016/s0165-6147(97)01137-1. [DOI] [PubMed] [Google Scholar]
- 10.Russwurm M, Behrends S, Harteneck C, Koesling D. Biochem J. 1998;335:125–130. doi: 10.1042/bj3350125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ignarro L J, Wood K S, Wolin M S. Proc Natl Acad Sci USA. 1982;79:2870–2873. doi: 10.1073/pnas.79.9.2870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sharma V S, Magde D. Methods. 1999;19:494–505. doi: 10.1006/meth.1999.0892. [DOI] [PubMed] [Google Scholar]
- 13.Zhao Y, Brandish P E, Ballou D P, Marletta M A. Proc Natl Acad Sci USA. 1999;96:14753–14758. doi: 10.1073/pnas.96.26.14753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lawson D M, Stevenson C E, Andrew C R, Eady R R. EMBO J. 2000;19:5661–5671. doi: 10.1093/emboj/19.21.5661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sampath V, Zhao X J, Caughey W S. J Biol Chem. 2001;276:13635–13643. doi: 10.1074/jbc.M006588200. [DOI] [PubMed] [Google Scholar]
- 16.Condorelli P, George S C. Biophys J. 2001;80:2110–2119. doi: 10.1016/S0006-3495(01)76184-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.del-Castillo J, Katz B. Proc R Soc London B. 1957;146:369–381. doi: 10.1098/rspb.1957.0018. [DOI] [PubMed] [Google Scholar]
- 18.Colquhoun D. Br J Pharmacol. 1998;125:924–947. doi: 10.1038/sj.bjp.0702164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stone J R, Marletta M A. Biochemistry. 1996;35:1093–1099. doi: 10.1021/bi9519718. [DOI] [PubMed] [Google Scholar]
- 20.Kharitonov V G, Russwurm M, Magde D, Sharma V S, Koesling D. Biochem Biophys Res Commun. 1997;239:284–286. doi: 10.1006/bbrc.1997.7470. [DOI] [PubMed] [Google Scholar]
- 21.Margulis A, Sitaramayya A. Biochemistry. 2000;39:1034–1039. doi: 10.1021/bi992040p. [DOI] [PubMed] [Google Scholar]
- 22.Brandish P E, Buechler W, Marletta M A. Biochemistry. 1998;37:16898–16907. doi: 10.1021/bi9814989. [DOI] [PubMed] [Google Scholar]
- 23.Colquhoun D, Sakmann B. Neuron. 1998;20:381–387. doi: 10.1016/s0896-6273(00)80982-4. [DOI] [PubMed] [Google Scholar]
- 24.Colquhoun D, Hawkes A G. In: Single-Channel Recording. Sakmann B, Neher E, editors. New York: Plenum; 1995. pp. 589–633. [Google Scholar]
- 25.Makino R, Matsuda H, Obayashi E, Shiro Y, Iizuka T, Hori H. J Biol Chem. 1999;274:7714–7723. doi: 10.1074/jbc.274.12.7714. [DOI] [PubMed] [Google Scholar]
- 26.Schmidt K, Desch W, Klatt P, Kukovetz W R, Mayer B. Naunyn-Schmiedeberg's Arch Pharmacol. 1997;355:457–462. doi: 10.1007/pl00004969. [DOI] [PubMed] [Google Scholar]
- 27.Bellamy T C, Garthwaite J. J Biol Chem. 2001;276:4287–4292. doi: 10.1074/jbc.M006677200. [DOI] [PubMed] [Google Scholar]
- 28.Bellamy T C, Wood J, Goodwin D A, Garthwaite J. Proc Natl Acad Sci USA. 2000;97:2928–2933. doi: 10.1073/pnas.97.6.2928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Colquhoun D, Hawkes A G. In: Single-Channel Recording. Sakmann B, Neher E, editors. New York: Plenum; 1995. pp. 397–482. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.