

Abstract
Bcl-2 family proteins regulate cell death through the mitochondrial apoptotic pathway. Here, we show that the Drosophila Bax-like Bcl-2 family protein Drob-1 maintains mitochondrial function to protect cells from neurodegeneration. A pan-neuronal knockdown of Drob-1 results in lower locomotor activity and a shorter lifespan in adult flies. Either the RNAi-mediated downregulation of Drob-1 or overexpression of Drob-1 antagonist Buffy strongly enhances the polyglutamine-induced accumulation of ubiquitinated proteins and subsequent neurodegeneration. Furthermore, ectopic expression of Drob-1 suppresses the neurodegeneration and premature death of flies caused by expanded polyglutamine. Drob-1 knockdown decreases cellular ATP levels, and enhances respiratory inhibitor-induced mitochondrial defects such as loss of membrane potential (Δψm), morphological abnormalities, and reductions in activities of complex I+III and complex II+III, as well as cell death. Taken together, these results suggest that Drob-1 is essential for neuronal cell function, and that Drob-1 protects neurons from expanded polyglutamine-mediated neurodegeneration through the regulation of mitochondrial homeostasis.
Keywords: Bcl-2, cell death, mitochondrial function, neurodegeneration, polyglutamine disease
Introduction
A balance between cell proliferation and apoptosis is important for the normal development of multicellular organisms. Superfluous or damaged cells must be removed by apoptosis, while cells required for the subsequent stages of development must be protected by cell survival factors (reviewed in Baehrecke, 2002). The Bcl-2 family of proteins, which includes both anti- and proapoptotic members, plays key regulatory roles in apoptosis. Accumulating evidence in mammalian systems suggests that antiapoptotic Bcl-2 family proteins (e.g., Bcl-2 and Bcl-xL) prevent the mitochondrial release of cytochrome c, which is required for the formation of the Apaf-1 apoptosome and therefore caspase activation (Zou et al, 1997). Conversely, in response to cell-death stimuli, proapoptotic Bcl-2 proteins (e.g., Bax and Bak) facilitate the release of cytochrome c and other death-promoting factors from the mitochondria by forming pores or channels, or by altering the mitochondrial membrane permeability and the structural architecture of the mitochondria (Martinou and Green, 2001; Scorrano et al, 2002). Thus, the antideath Bcl-2 family members counteract the function of the prodeath family members.
Bcl-2 family proteins are conserved throughout evolution (Igaki and Miura, 2004). CED-9, a Bcl-2 family protein in the nematode Caenorhabditis elegans, plays an essential role in preventing programmed cell death; however, the mechanisms by which Bcl-2 family proteins prevent cell death may not be conserved between C. elegans and mammals. A major role of CED-9 is to sequester the caspase-activating protein CED-4 to the mitochondria and to inhibit CED-4 function; this is different from the major role of mammalian Bcl-2 family proteins that control mitochondrial cytochrome c release. The Drosophila genome encodes Apaf-1 and Bcl-2 family proteins that are structurally and functionally related to their mammalian orthologs (Igaki and Miura, 2004). However, cytochrome c may not be required to promote cell death in Drosophila S2 cells (Dorstyn et al, 2002, 2004; Zimmermann et al, 2002), suggesting that Drosophila Bcl-2 family proteins play another role that may not be involved in cytochrome c release. Drosophila has two Bcl-2 family proteins, Drob-1/Debcl/dBorg-1/dBok-1 (Brachmann et al, 2000; Colussi et al, 2000; Igaki et al, 2000; Zhang et al, 2000) and Buffy/dBorg-2 (Brachmann et al, 2000; Quinn et al, 2003). Drob-1 and Buffy share BH1, BH2, BH3, and weak BH4 homology regions and the C-terminal transmembrane region, and structurally belong to the Bax subfamily. Drob-1 has been shown to be a proapoptotic protein based on the observations that (i) ectopic expression of Drob-1 in fly eyes or Drosophila S2 cells results in cell death, and that (ii) the functional knockdown of Drob-1 by RNAi leads to the inhibition of cell death in embryos (Brachmann et al, 2000; Colussi et al, 2000; Igaki et al, 2000). In contrast to the proapoptotic function of Drob-1, Buffy can function as an antiapoptotic factor by inhibiting Drob-1 function (Quinn et al, 2003).
The aim of the present study was to elucidate the in vivo role of Drob-1 in Drosophila. The RNAi-mediated knockdown of Drob-1 in embryos prevented most cell deaths, supporting its proapoptotic role. Unexpectedly, a pan-neuronal knockdown of Drob-1 caused lower locomotor behavior activity and a shorter lifespan, and it also enhanced polyglutamine-mediated neurodegeneration. The inhibition of Drob-1 function by RNAi or by overexpressing Buffy caused a reduction in cellular ATP levels and an increase in the accumulation of ubiquitinated proteins. In the presence of respiratory inhibitors, Drob-1 knockdown enhanced abnormalities in mitochondrial morphology, loss of Δψm, and cell death, and decreased complex I+III and complex II+III activities. Furthermore, the overexpression of Drob-1 protected cells from polyglutamine-mediated neurodegeneration. These findings indicate that Drob-1 functions as an antiapoptotic protein in neuronal cells by regulating mitochondrial homeostasis.
Results
Drob-1 positively regulates programmed cell death during embryogenesis
To elucidate the physiological role of Drob-1 in Drosophila, we established Drob-1 ‘knockdown' transgenic lines bearing an inverted-repeat (IR) construct of drob-1 (UAS-drob-1-IR) (Supplementary Figure 1A). The functionality of the UAS-drob-1-IR transgene was confirmed by crossing UAS-drob-1-IR flies to UAS-drob-1debcl (Colussi et al, 2000) flies using an eye-specific GAL4 driver GMR-GAL4. The expression of the drob-1-IR transgene completely blocked the small-eye phenotype caused by Drob-1 overexpression (Supplementary Figure 1B–E). On the other hand, knockdown of Drob-1 did not affect the ablated eye phenotype induced by overexpression of other proapoptotic proteins such as Rpr, Hid, Grim, and Dmp53 (data not shown).
To examine the phenotypes caused by the knockdown of Drob-1, the UAS-drob-1-IR transgene was expressed in selected tissues or at selected stages using different GAL4 drivers. As a control, we used a transgenic fly bearing an IR construct of lac-Z (UAS-lacZ-IR), which showed no detectable phenotypes with GAL4 drivers used. Ubiquitous expression of the drob-1-IR transgene at the entire developmental stages using da-GAL4 driver resulted in a larval lethality (Supplementary Table 1). TUNEL analysis of stage 12–13 da>drob-1-IR embryos showed a remarkable reduction in the number of dying cells (Supplementary Figure 1I–K) compared with control da>lacZ-IR embryos (Supplementary Figure 1F–H), consistent with previous studies using drob-1 dsRNA (Brachmann et al, 2000; Colussi et al, 2000). Knockdown of Drob-1 in the nervous system using elav-GAL4 caused a semilethal phenotype at the larval–pupal polyphasic stages (Supplementary Table 1). On the other hand, drob-1-IR expression targeted to the developing retina or photoreceptor neurons using GMR-GAL4 or sev-GAL4 had no effect on eye morphology (data not shown). These observations indicate that the developmental programmed cell death is positively regulated by Drob-1, and that Drob-1 may play different roles at different developmental stages or in different cell types such as neurons.
Pan-neuronal Drob-1 knockdown results in lower locomotor activity and a shorter lifespan
Bcl-2 family proteins play an important role in the nervous system (reviewed in Lossi and Merighi, 2003; Becker and Bonni, 2004). To elucidate the physiological role of Drob-1 in neurons, we examined the effect of a pan-neuronal knockdown of Drob-1 on locomotor behavior and lifespan. The lifespans of two independent pan-neuronal Drob-1 knockdown fly lines (elav>drob-1-IR#3 and elav>drob-1-IR#10) were markedly shorter than those of control elav>lacZ-IR flies (Figure 1A) (mean lifespans: elav>lacZ-IR, 71.1±3.9 days; elav>drob-1-IR#3, 46.2±2.1 days; elav>drob-1-IR#10, 44.6±1.8 days; Supplementary Table 2). The frequency of locomotion in elav>drob-1-IR flies (at days 13–16) was significantly reduced compared with that in control flies (Figure 1B). These data reveal that Drob-1 is essential for neuronal cells to maintain flies' normal locomotion and lifespan.
Figure 1.
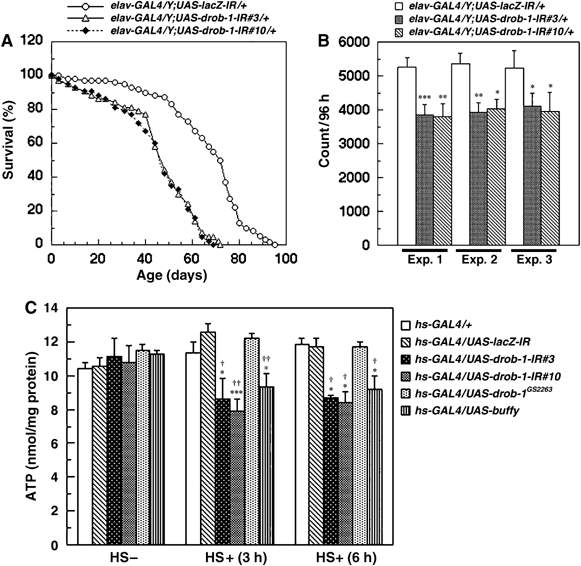
RNAi-mediated knockdown of Drob-1 results in a shorter lifespan, lower locomotor activity, and ATP depletion. (A) Expression of the UAS-drob-1-IR or UAS-lacZ-IR transgene was targeted to cells in the peripheral and central nervous systems using an elav-GAL4 driver. Two independent elav>drob-1-IR fly lines (#3 and #10) show significantly shorter lifespans than do control flies (elav>lacZ-IR). (B) Locomotor activity was analyzed using the DAM system as described in Materials and methods. Two independent elav>drob-1-IR fly lines (#3 and #10) show lower locomotion activity for 96 h (13–16 days after eclosion) than do control flies (elav>lacZ-IR). Each bar in the graph shows the mean±s.e.m. of 32 flies (n=32) of each genotype. Three independent experiments (Exp. 1–3) were performed. *P<0.05, **P<0.01, and ***P<0.005 relative to control by Student's t-test. (C) ATP levels in hs-GAL4/+, hs-GAL4/UAS-lacZ-IR, hs-GAL4/UAS-drob-1-IR#3, hs-GAL4/UAS-drob-1-IR#10, hs-GAL4/UAS-drob-1GS2263, and hs-GAL4/UAS-buffy adult flies (4 days after eclosion) 3 or 6 h after treatment with or without heat shock (twice at 37°C for 30 min with a 30 min interval) were measured as described in Materials and methods. Each value shows the mean±s.e.m. of three independent experiments. *P<0.05, **P<0.01, and ***P<0.005 for each value as compared with hs-GAL4/+, †P<0.05, ††P<0.01, and †††P<0.005 for each value as compared with hs-GAL4/UAS-lacZ-IR by Student's t-test. GS2263 is a fly line that can overexpress untagged Drob-1 in a GAL4-dependent manner.
Downregulation of Drob-1 leads to cellular ATP depletion
Progressive mitochondrial dysfunction is thought to be an important pathogenic mechanism that leads to irreversible damage in neuronal cells (reviewed in Orth and Schapira, 2001). Mitochondrial dysfunction is also involved in the aging process in both invertebrates and vertebrates, including humans (reviewed in Lenaz et al, 2000, 2002; Golden et al, 2002; Pollack et al, 2002; Tsang and Lemire, 2003). We therefore analyzed the ATP levels in Drob-1 knockdown flies as a marker of mitochondrial metabolism. The ATP levels in Drob-1 knockdown flies were significantly less than those in control fly lines (Figure 1C). On the other hand, overexpression of Drob-1 did not affect the ATP levels (Figure 1C). Another Drosophila Bcl-2 family protein, Buffy, can bind to and inactivate Drob-1 (Quinn et al, 2003). We found that overexpression of Buffy reduced the ATP levels in adult flies (Figure 1C). We confirmed that the levels of drob-1 mRNA were markedly reduced, but the levels of buffy were unaffected in flies expressing drob-1-IR (data not shown). The buffy mRNA levels were also unaffected in flies overexpressing Drob-1 (data not shown). These results suggest that the inactivation of Drob-1 induces changes in mitochondrial energy metabolism and leads to ATP depletion.
Downregulation of Drob-1 enhances polyglutamine-induced toxicity
Since we found that Drob-1 plays a crucial role in neurons, we next asked whether the reduction of Drob-1 would affect neurodegeneration. We investigated the role of Drob-1 in the pathogenesis of a fly model of polyglutamine disease, since the expression of expanded polyglutamine decreases the cellular concentrations of ATP (Sanchez et al, 2003). A fly model of Machado–Joseph disease (MJD), generated by overexpression of a truncated form of the human MJD protein with an expanded polyglutamine stretch (MJDtr-Q78), shows progressive neural degeneration (Warrick et al, 1998). Consistent with previous findings (Warrick et al, 1998, 1999), we found that the targeted expression of expanded polyglutamine in neurons using the elav-GAL4 driver resulted in early adult death (Figure 2A and B). Flies expressing both drob-1-IR and MJDtr-Q78 in their neurons showed a significantly shorter lifespan than did the flies expressing MJDtr-Q78 alone, suggesting that the downregulation of Drob-1 increases the neural toxicity caused by expanded polyglutamine (Figure 2A and B).
Figure 2.
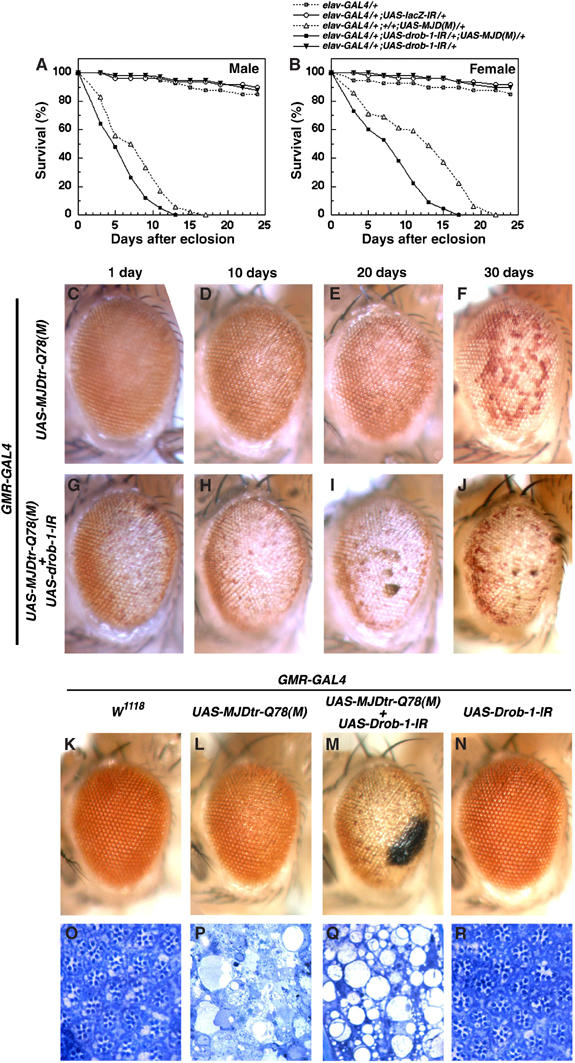
RNAi-mediated knockdown of Drob-1 enhances polyglutamine-induced neuronal toxicity. (A, B) Expression of the UAS-drob-1-IR or UAS-MJDtr-Q78 transgene was targeted to the nervous system using the elav-GAL4 driver. We used two control fly lines: one bears only the elav-GAL4 transgene and the other bears both the UAS-lacZ-IR and elav-GAL4 transgenes. The elav>drob-IR, MJDtr-Q78(M) flies died earlier than the elav>MJDtr-Q78(M) flies. (C–J) Light micrographs of the eyes of flies 1 day (C, G), 10 days (D, H), 20 days (E, I), and 30 days (F, J) after eclosion are shown. (K–R) Eyes (K–N) and tangential sections of the eyes (O–R) of flies 1 day after eclosion are shown. (K, O) Control flies bearing only the driver GMR-GAL4 show normal eyes. Genotypes are as follows: GMR-GAL4/CySM1; UAS-MJDtr-Q78(M)/+ (C–F), GMR-GAL4/UAS-drob-1-IR; UAS-MJDtr-Q78(M)/+ (G–J), GMR-GAL4/CySM1 (K, O), GMR-GAL4/CySM1; UAS-MJDtr-Q78(M)/+ (L, P), GMR-GAL4/UAS-drob-1-IR; UAS-MJDtr-Q78(M)/+ (M, Q), and GMR-GAL4/UAS-drob-1-IR; TM3, Sb/+ (N, R).
The expression of pathogenic human expanded polyglutamine proteins (e.g., MJD protein and huntingtin) in Drosophila compound eyes elicits late-onset degeneration and the loss of photoreceptor neurons. The ectopic expression of MJDtr-Q78 led to late-onset degeneration in adult eyes (Figure 2C–F) (Jackson et al, 1998; Warrick et al, 1998). We found that the coexpression of drob-1-IR with MJDtr-Q78 remarkably accelerated the onset of the neurodegeneration (Figure 2G–J and Supplementary Figure 2). As shown in Figure 2K–N (external eyes) and Figure 2O–R (tangential sections of the eyes), the knockdown of Drob-1 strongly enhanced the polyglutamine-induced lack of pigment and severe loss of retinal structure in day 1 flies (Figure 2L, M, P, and Q). The GMR>drob-1-IR eye was normal as compared with the control eye bearing only the promoter transgene, GMR-GAL4 (Figure 2K, N, O, and R). The overexpression of Buffy also enhanced the polyglutamine-induced neurodegeneration (Figure 3D and E) and lethality (Figure 3A and B and Supplementary Table 2), suggesting that Buffy acts as a proneurodegenerative factor by inactivating Drob-1. Indeed, the eyes coexpressing Drob-1, Buffy, and MJDtr-Q78 showed significantly weaker degenerative eye phenotype than the eyes expressing Buffy and MJDtr-Q78 (Supplementary Figure 3), supporting the notion that Buffy enhances neurodegeneration by suppressing Drob-1.
Figure 3.
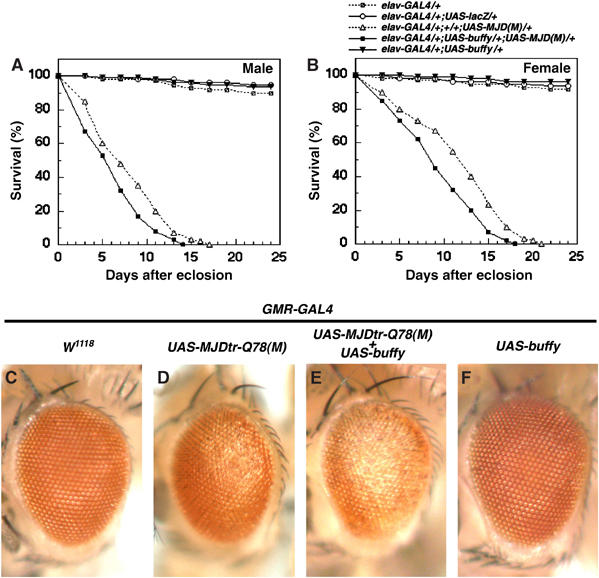
Buffy, a Drob-1 antagonist, enhances polyglutamine-induced neurodegeneration. (A, B) Longevity curve of each genotype indicated is shown. (C–F) Adult eyes of 1-day-old flies with the following genotypes are shown: (C) GMR-GAL4/CySM1, (D) GMR-GAL4/CySM1; UAS-MJDtr-Q78/+, (E) GMR-GAL4/UAS-buffy; UAS-MJDtr-Q78/+, and (F) GMR-GAL4/UAS-buffy.
Overexpression of Drob-1 suppresses expanded polyglutamine-induced neurodegeneration
Overexpression of HA-tagged Drob-1 in the Drosophila compound eye causes a small-eye phenotype (Colussi et al, 2000; Igaki et al, 2000); however, nontagged Drob-1 does not (Brachmann et al, 2000). We used nontagged Drob-1 (UAS-drob-1GS2263) to examine whether Drob-1 could function as a protective factor against neurodegeneration induced by expanded polyglutamine. The pan-neuronal moderate expression of Drob-1 slightly expanded the lifespan (mean lifespans: elav>lacZ, 72.3±1.3; elav>drob-1GS2263, 78.3±2.3; P<0.05) (Figure 4A and Supplementary Table 2). On the other hand, the pan-neuronal overexpression of Buffy caused a shorter lifespan (mean lifespan: elav>buffy, 63.6±4.7; P<0.05) (Figure 4A and Supplementary Table 2). The ectopic expression of Drob-1 at moderate levels, which showed no detectable phenotype in the eye on its own (Figure 4F and I), suppressed the MJDtr-Q78-induced neurodegeneration (Figure 4D, E, G, and H). Similar results were also obtained using flies bearing the GMR-drob-1 transgene, which did not cause the rough-eye phenotype by itself (Supplementary Figure 4). In addition, Drob-1 markedly rescued the early adult death of flies expressing MJDtr-Q78 in their neurons (Figure 4B and C and Supplementary Table 2). Together, these results suggest that Drob-1 can protect neurons from polyglutamine-mediated neurodegeneration.
Figure 4.
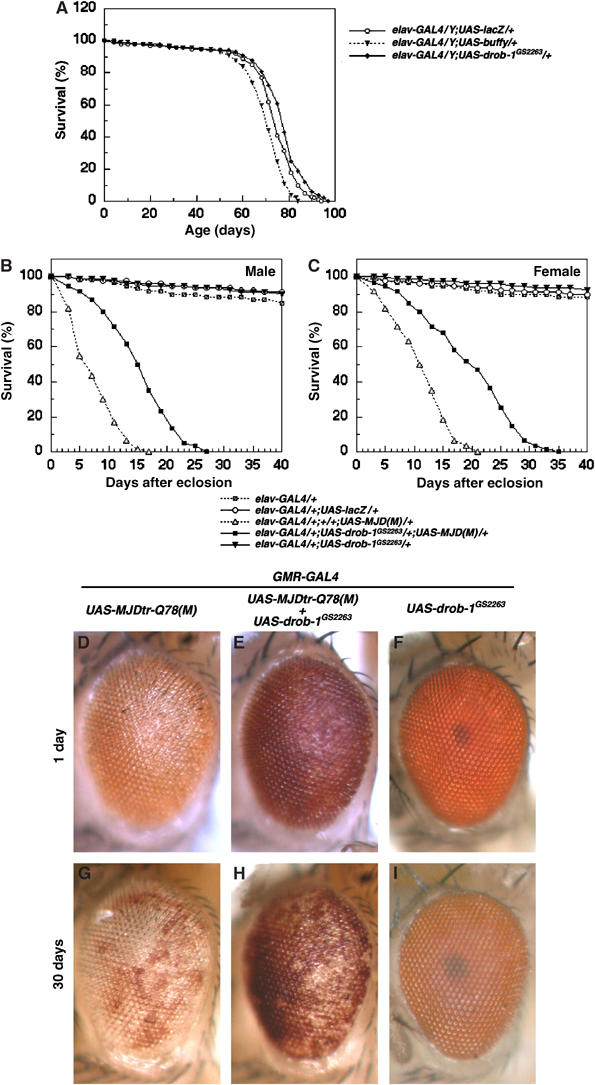
Drob-1 protects neurons from polyglutamine-induced toxicity. (A–C) Longevity curve of each genotype indicated is shown. (D–I) External eye phenotypes of (D, G) GMR-GAL4/+; UAS-MJDtr-Q78/+, (E, H) GMR-GAL4/UAS-drob-1GS2263; UAS-MJDtr-Q78/+, and (F, I) GMR-GAL4/UAS-drob-1GS2263, 1 day (D–F) or 30 days (G–I) after eclosion are shown. GS2263 is a fly line that can overexpress untagged Drob-1 in a GAL4-dependent manner.
Drob-1 is required to reduce the accumulation of undegraded proteins caused by expanded polyglutamine
The overexpression of expanded polyglutamine reduces proteasome activity and increases the accumulation of undegraded proteins in S2 cells and flies (Kanuka et al, 2003). We found that the downregulation of Drob-1 or overexpression of Buffy resulted in an increase of ubiquitinated proteins in Drosophila heads (Figure 5A). In addition, coexpression of MJDtr-Q78 with drob-1-IR or buffy enhanced the accumulation of ubiquitinated proteins (Figure 5A). We therefore pursued the role of Drob-1 in the accumulation of ubiquitinated proteins and subsequent cell death in Drosophila S2 cells. The accumulation of ubiquitinated proteins caused by proteasome inhibitors such as lactacystin or MG-132 was accelerated by knocking down of Drob-1 (Figure 5B and Supplementary Figure 5A). The proteasome inhibitors caused a severe reduction in cell viability that was greatly enhanced by Drob-1 knockdown (Figure 5C and Supplementary Figure 4B). On the other hand, the knockdown of Buffy, which may activate Drob-1 function, significantly suppressed the proteasome inhibition-induced cell death (Figure 5C and Supplementary Figure 5B). In contrast, the knockdown of either Drob-1 or Buffy did not affect the cell death induced by tunicamycin (Figure 5D). These observations suggest that Drob-1 protects cells from cytotoxicity induced by the disruption of proteasome function. Supporting this idea, the downregulation of Drob-1, but not of Buffy, induced cellular ATP depletion (Figure 5E) that may cause a suppression of proteasome function (Beal et al, 1993; Sanchez et al, 2003).
Figure 5.
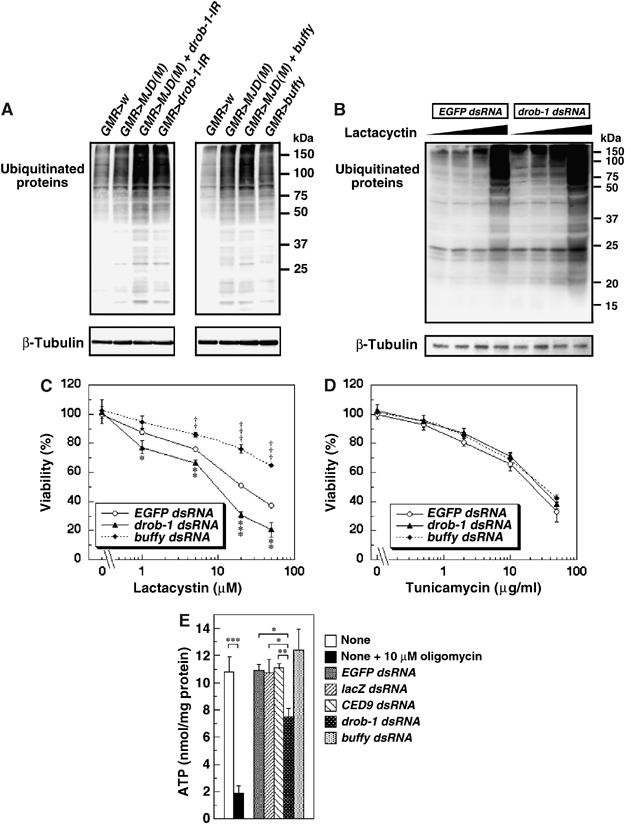
Knockdown of Drob-1 enhances the accumulation of ubiquitinated proteins. (A) Fly heads from each line of the indicated genotype (1 day after eclosion) were subjected to immunoblotting with an anti-ubiquitin antibody and anti-β-tubulin antibody. (B) S2 cells were cultured with either EGFP dsRNA or drob-1 dsRNA for 48 h, and incubated with or without lactacystin (1, 5, and 20 μM) for an additional 24 h as described in Materials and methods. The cell lysate was subjected to Western analysis using the anti-ubiquitin and anti-β-tubulin antibodies. (C, D) S2 cells were transfected with EGFP, drob-1, or buffy dsRNA for 48 h, and left untreated or treated with lactacystin, a proteasome inhibitor (1, 5, 20, and 50 μM) (B), or tunicamycin, an inhibitor of N-glycosylation that induces the rapid unfolded protein response (UPR) (0.5, 2, 10, and 50 μg/ml) (C), for 24 h. Cell viability was determined by cell death assay as described in Materials and methods. Mean±s.e.m., n=3, *P<0.05, **P<0.005, and ***P<0.0005 for cells with drob-1 dsRNA, †P<0.05, ††P<0.005, and †††P<0.0005 for cells with buffy dsRNA as compared with control cells (with EGFP dsRNA) by paired Student's t-test. (E) Cells were cultured with or without 10 μM oligomycin, an inhibitor of mitochondrial ATP synthase, for 1 h, or with EGFP dsRNA, lacZ dsRNA, ced-9 dsRNA, drob-1 dsRNA, or buffy dsRNA for 72 h. Cellular ATP content was measured as described in Materials and methods. Each value shows the mean±s.e.m. of four (n=4) independent experiments. *P<0.05, **P<0.01, and ***P<0.005 by paired Student's t-test.
Downregulation of Drob-1 enhances mitochondrial dysfunction
Expanded polyglutamine protein causes ATP loss, a mitochondrial dysfunction (Sanchez et al, 2003). The mitochondrial complex II enzyme activity is selectively decreased in the striatum in Huntington's disease (HD) patients (reviewed in Browne and Beal, 2004). In addition, mitochondrial membrane depolarization may be involved in neuronal cell death in HD (Panov et al, 2002; Ruan et al, 2004). These studies suggest that an impairment of mitochondrial respiratory function may play a role in the pathogenesis of polyglutamine diseases. We therefore examined in S2 cells whether the knockdown of Drob-1 affects mitochondrial membrane depolarization and cell death induced by mitochondrial respiratory chain inhibitors such as rotenone, a complex I inhibitor, or 3-nitropropionic acid (3-NP), a mitochondrial complex II inhibitor that is used for generating animal models of HD (Beal et al, 1993; Browne and Beal, 2004). JC-1 is widely used to measure the mitochondrial depolarization in live cell. JC-1 monomer exhibits green fluorescence and JC-1 aggregate (J-aggregate) at high concentration exhibits red fluorescence (Smiley et al, 1991). It allows us to label the mitochondria as well as to observe membrane potential (Δψm). Although TMRM or TMRE would be more appropriate for quantitative assay of membrane potential, J-aggregate formation increases linearly with applied membrane potential in a limited range and was therefore used for a qualitative analysis of Δψm. Treatment with either rotenone or 3-NP reduced the presence of red J-aggregates, indicating a relative decrease of Δψm, in a dose-dependent manner (Figure 6). The knockdown of Drob-1 strongly enhanced the rotenone- or 3-NP-induced reduction of Δψm (Figure 6). Moreover, the downregulation of Drob-1 enhanced cell death induced by either rotenone or 3-NP (Figure 7A and B). The knockdown of Drob-1, without complex I or complex II inhibition, did not affect Δψm and cell viability (Figures 6 and 7). On the other hand, the knockdown of Buffy significantly suppressed rotenone- or 3-NP-induced cell death (Figure 7A and B). In contrast, no difference was seen in the cell death induced by other mitochondrial inhibitors such as antimycin A (complex III), KCN (complex IV), or oligomycin (complex V) in S2 cells treated with control, drob-1, or buffy dsRNA (Figure 7C–E). We further analyzed morphological and biochemical alterations in mitochondria in Drob-1 knockdown S2 cells following the treatment with rotenone or 3-NP. In analyzing with transmission electron microscopy, we observed three types of previously described mitochondrial morphologies: normal mitochondria, swollen and higher electron-dense mitochondria, and swollen and lower electron-dense mitochondria with disrupted outer membrane (Ghadially, 1982; Angermuller et al, 1998; Sesso et al, 2004). It has been reported that the morphology of mitochondria with ruptured membrane is a sign of the very early stage of apoptosis, and it might be involved in mitochondrial permeability transition and loss of Δψm (Angermuller et al, 1998; Sesso et al, 2004). We found that Drob-1 knockdown significantly increased the percentage of mitochondria showing abnormal morphologies in S2 cells with intact nuclei (Figure 8A–G). In addition, the downregulation of Drob-1 significantly enhanced the reduction of activities of mitochondrial respiratory chain complex I+III (NADH-cytochrome c oxidoreductase) and complex II+III (succinate-cytochrome c oxidoreductase) in S2 cells treated with rotenone or 3-NP (Figure 8H and I). Together, these data suggest that Drob-1 plays an important role in the maintenance of mitochondrial homeostasis, and may protect cells from neurodegeneration caused by mitochondrial dysfunction through a defect in complex I or complex II.
Figure 6.
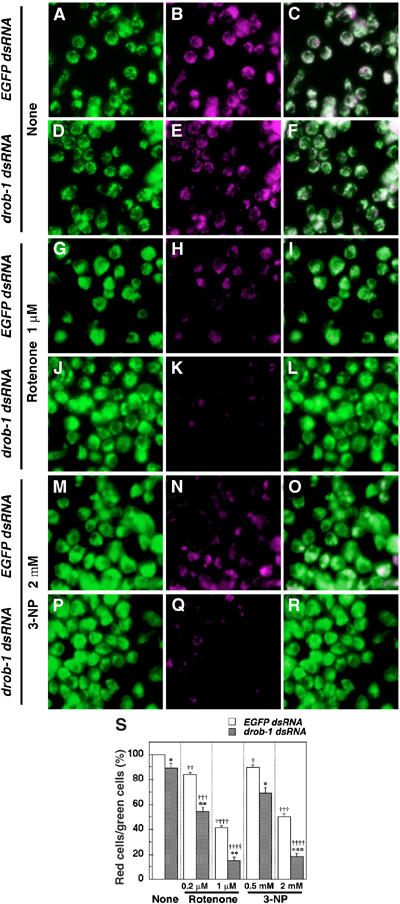
Knockdown of Drob-1 enhances rotenone- or 3-NP-induced loss of mitochondrial membrane potential (Δψm) in S2 cells. (A–R) Cells were cultured with either EGFP dsRNA or drob-1 dsRNA for 48 h, and then incubated with or without rotenone (0.2 and 1 μM) or 3-NP (0.5 and 2 mM) for an additional 24 h. After the treatment, cells were labeled with Δψm-sensitive dye JC-1 and imaged with confocal microscope as described in Materials and methods. Total (inactive+active) and active mitochondria are labeled green JC-1 monomers (A, D, G, J, M, and P) and red J-aggregates (B, E, H, K, N, and Q) fluorescence, respectively (see Materials and methods). Overlay image of green and red JC-1 fluorescence is shown in panels C, F, I, L, O, and R. (S) Quantitative analysis of Δψm was performed by calculating the percentage of the number of red J-aggregates fluorescence-labeled cells out of that of total cells (green JC-1 monomers fluorescence-labeled cells) as described in Materials and methods. Each value shows the mean±s.e.m. of three independent experiments. Approximately 300 cells were analyzed per each condition in each experiment. *P<0.05, **P<0.01, and ***P<0.005 for each value as compared with EGFP dsRNA-treated cells under each experimental condition, †P<0.05, ††P<0.01, †††P<0.005, and ††††P<0.001 for each value as compared with control (cells treated with EGFP dsRNA only) by Student's t-test.
Figure 7.
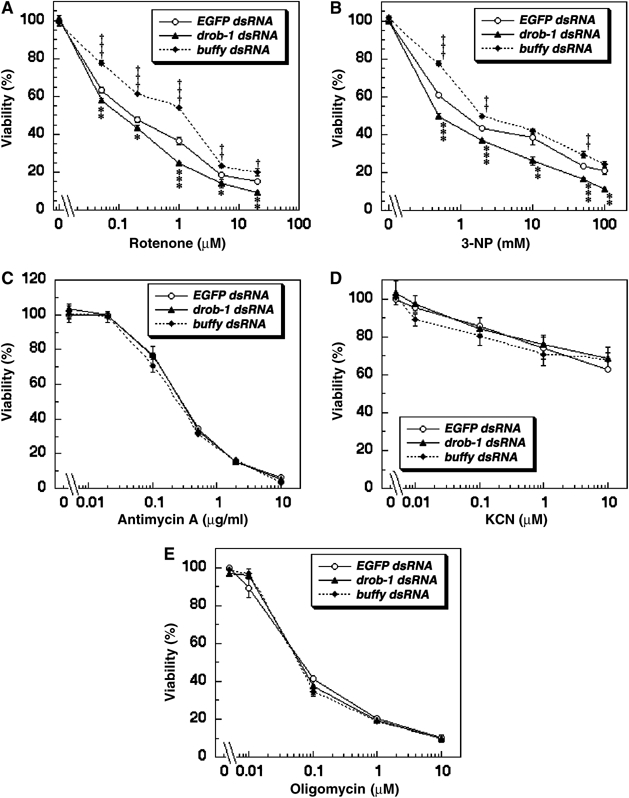
Knockdown of Drob-1 specifically enhances rotenone- or 3-NP-induced cell death in S2 cells. (A–E) Cells were transfected with EGFP, drob-1, or buffy dsRNA for 48 h, and left untreated or treated with rotenone (0.05, 0.2, 1, 5, and 20 μM) (A), 3-NP (0.5, 2, 10, 50, and 100 mM) (B), antimycin A (0.02, 0.1, 0.5, 2, and 10 μg/ml) (C), KCN (0.01, 0.1, 1, and 10 μM) (D), or oligomycin (0.01, 0.1, 1, and 10 μM) (E). Mean±s.e.m., n=3, *P<0.05, **P<0.005, and ***P<0.0005 for cells with drob-1 dsRNA, †P<0.05, ††P<0.005, and †††P<0.0005 for cells with buffy dsRNA as compared with control cells (with EGFP dsRNA) by paired Student's t-test.
Figure 8.
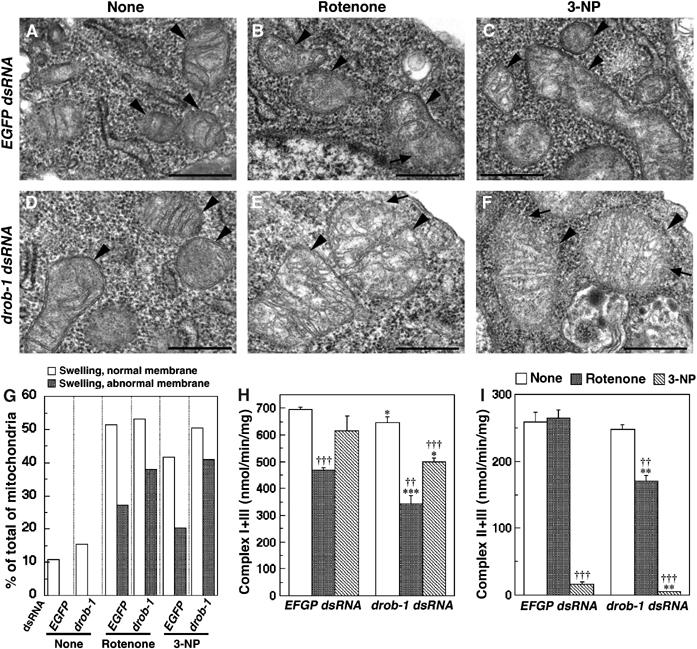
Knockdown of Drob-1 enhances rotenone- or 3-NP-induced mitochondrial morphological abnormalities and decreases activities of respiratory chain complexes in S2 cells. (A–F) Electron microscopy images of mitochondria in EGFP dsRNA- or drob-1 dsRNA-treated S2 cells with or without the treatment with rotenone or 3-NP (arrowheads: mitochondria). Rotenone or 3-NP induced swollen and lower electron-dense mitochondria with disrupted outer membranes, signs of degeneration and derangement of the membranes (arrows). Scale bars correspond to 500 nm. (G) Quantification of mitochondrial morphological abnormalities. Mitochondria were classified into three types based on their morphologies: normal mitochondria, swollen and higher electron-dense mitochondria, and swollen and lower electron-dense mitochondria with abnormal membrane structures. Approximately 150 mitochondria in at least 15 cells were analyzed in an experimental group. (H, I) Mitochondrial complex I+III and complex II+III activities. Mean±s.e.m., n=3, *P<0.05, **P<0.005, and ***P<0.0005 for cells with drob-1 dsRNA compared with cells with EGFP dsRNA, †P<0.05, ††P<0.005, and †††P<0.0005 for cells with rotenone or 3-NP as compared with cells without drugs (none) by paired Student's t-test.
Discussion
In this report, we have shown that Drob-1 can either promote (cell death during embryogenesis) or inhibit (polyglutamine-induced neurodegeneration) cell death, depending on a variety of conditions. In addition, we have shown that Buffy also has dual functions, namely a survival function (for cell death during embryogenesis) (Quinn et al, 2003) and a proapoptotic function (for polyglutamine-induced neurodegeneration).
Certain members of the Bcl-2 family proteins can function as both anti- and prodeath factors. Antiapoptotic Bcl-2 and Bcl-xL can be converted into proapoptotic proteins when they are cleaved by caspases or by other proteases (Cheng et al, 1997; Clem et al, 1998). The resulting C-terminal fragments have a ‘Bax-like' prodeath activity that induces cytochrome c release from mitochondria and forms pores in synthetic membranes (Kirsch et al, 1999; Basanez et al, 2001). C. elegans CED-9 also exhibits prodeath as well as antideath activity (Hengartner and Horvitz, 1994). Proapoptotic Bax and Bak may promote or inhibit neuronal death depending on the specific death stimulus, neuron subtype, and stage of postnatal development (Lewis et al, 1999; Fannjiang et al, 2003). Bax promotes the survival of trigeminal ganglia neurons during development in mice that are deficient in NGF or TrkA, while it promotes the death of superior cervical ganglia neurons in the same models (Middleton and Davies, 2001). Bax potently protects mice and cultured hippocampal neurons from Sindbis virus-induced apoptosis, whereas it promotes the death of Sindbis virus-infected dorsal root ganglia neurons (Lewis et al, 1999). Bak protects hippocampal neurons from the cell death caused by excitotoxicity or viral infection; however, as mice mature, Bak function is converted from anti- to prodeath in virus-infected spinal cord neurons (Fannjiang et al, 2003). Bak also protects mice from kainate-induced seizures, suggesting a possible role in regulating synaptic activity (Fannjiang et al, 2003). Drob-1 has also been shown to have a protective activity against serum-deprivation- or CED-3-induced S2 cell death (Brachmann et al, 2000). Thus, individual Bcl-2 family proteins can have a pro- or antiapoptotic function, depending on the cellular context or specific stimulus. These findings, combined with our observations in this study, suggest that the dual-function nature of Bcl-2 family proteins may be evolutionarily conserved from nematodes to mammals.
Mitochondrial function (i.e., the production of ATP, regulation of apoptosis, and production of reactive oxygen species (ROS)) is crucial for the maintenance of postmitotic tissues (e.g., muscles and brain) in normal aging, and plays a role in degenerative diseases in humans and in animal models (reviewed in Lenaz et al, 2000, 2002; Orth and Schapira, 2001; Golden et al, 2002; Pollack et al, 2002; Tsang and Lemire, 2003). During normal aging and the progression of human degenerative diseases, a decrease in the total number of cells in some postmitotic tissues (e.g., heart, skeletal muscle, and brain) is associated with a reduction in mitochondrial metabolic activity (reviewed in Lenaz et al, 2000, 2002; Orth and Schapira, 2001; Pollack et al, 2002). We have shown that Drob-1 plays an important role in the survival of postmitotic neurons under both physiological and pathological conditions. Importantly, our finding that downregulation of Drob-1 results in a decrease in cellular ATP levels suggests that Drob-1 may be involved in the maintenance of mitochondrial metabolism (Figures 1 and 5). In addition, Drob-1 protects cells from stresses that cause mitochondrial dysfunction (Figures 6 and 7). Thus, our results suggest that Drob-1 may regulate the homeostasis of neurons and the aging process by maintaining mitochondrial metabolism. The ubiquitin–proteasome system plays a crucial role in preventing the polyglutamine-induced accumulation of unfolded proteins (reviewed in Ciechanover and Brundin, 2003). This system acts in an ATP-dependent manner. Inhibition of the mitochondrial respiratory chain by the complex I inhibitor rotenone reduces the ubiquitin-proteasomal activity in both rat primary dopaminergic neurons and human SH-SY5Y neuroblastoma cells (Hoglinger et al, 2003; Shamoto-Nagai et al, 2003). Expanded polyglutamine protein has been reported to cause mitochondrial dysfunction, ATP loss, a defect in complex II enzyme activity, and subsequent inhibition of the ATP-dependent ubiquitin–proteasome system (Beal et al, 1993; Kanuka et al, 2003; Sanchez et al, 2003). Our results, combined with these previous findings, led us to propose a model in which Drob-1 suppresses polyglutamine-induced ATP depletion, thereby facilitating the subsequent activation of the ubiquitin–proteasome system, which protects neurons from cell death and degeneration. Buffy can antagonize this survival function of Drob-1 in neurons (Figure 9).
Figure 9.
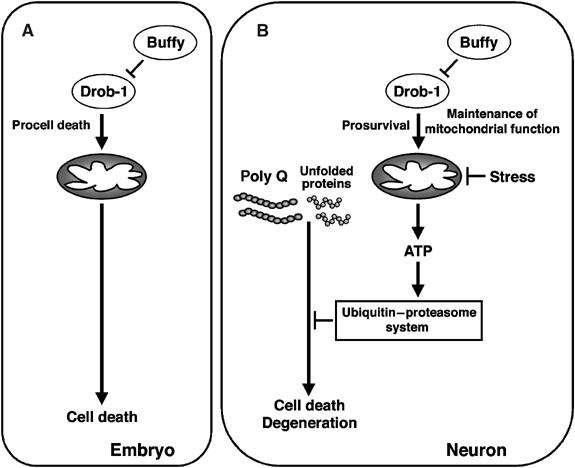
A model for the context-dependent dual function of Drob-1. Drob-1 plays an important role in facilitating programmed cell death during embryogenesis (A). Drob-1 can also protect neurons from polyglutamine-, unfolded protein-, or mitochondrial inhibition-induced pathological cell death and degeneration (B). Drob-1 may regulate mitochondrial ATP homeostasis thereby attenuating the toxicity caused by disruption of the ubiquitin–proteasome system. Buffy can antagonize both the pro- and anti-cell-death function of Drob-1.
We have shown that downregulation of Drob-1 induces ATP depletion and a shorter lifespan in flies. In C. elegans, mitochondrial complex II deficiency causes a shorter lifespan, hypersensitivity to oxidative stress, energy depletion, ROS overproduction, and CED-3- and CED-4-dependent supernumerary cell death (Ishii et al, 1998; Senoo-Matsuda et al, 2001, 2003). In the complex II-deficient C. elegans mutant mev-1, the shorter lifespan is partially rescued by a loss-of-function mutation of CED-3, suggesting that the supernumerary apoptosis may contribute to shortening the lifespan in C. elegans (Senoo-Matsuda et al, 2003). Interestingly, the shorter lifespan in the mev-1 mutant may be associated with a decrease in the mitochondrial localization of CED-9 and its downregulation (Senoo-Matsuda et al, 2003). In mammals, Bcl-xL can prevent the perturbation of mitochondrial ATP/ADP exchange caused by growth factor deprivation, and can maintain oxidative phosphorylation under the growth-factor-withdrawal condition (Vander Heiden et al, 1999). The proapoptotic Bcl-2 family protein Bad is required to assemble the mitochondria-based glucokinase complex, which regulates glycolysis (Danial et al, 2003). Thus, the regulation of mitochondrial homeostasis may be an evolutionarily conserved role of Bcl-2 family proteins.
Our findings also suggest that Bcl-2 family proteins may play a crucial role in the pathogenesis of polyglutamine diseases. It would be greatly informative to determine whether Bcl-2 family proteins also play a crucial role in mammalian systems that can be a therapeutic target for neurodegenerative disorders. Further study of Drob-1 should increase our understanding of the universal roles of Bcl-2 family proteins and may contribute to the development of new therapeutic applications, not only for polyglutamine diseases, but also for other abnormal-protein-accumulating neurodegenerative diseases, such as Alzheimer's disease, Parkinson's disease, and amyotrophic lateral sclerosis.
Materials and methods
Fly stocks
Fly culture and crosses were carried out at 25°C. Canton-S or white1118 was used as a wild-type strain. da-GAL4, GMR-GAL4, and elav-GAL4 fly lines were used as driver strains. All general fly stocks and GAL4 lines including UAS-reaper, GMR-reaper, and GMR-hid were obtained from Drosophila stock centers. We also used UAS-MJDtr-Q78 (Warrick et al, 1998), UAS-lacZ-IR (Kennerdell and Carthew, 2000), UAS-buffy (Quinn et al, 2003), UAS-drob-1debcl (Colussi et al, 2000), GMR-Dmp53 (Ollmann et al, 2000), and GMR-drob-1 (Igaki et al, 2000) flies. UAS-drob-1GS2263 was a kind gift from Toshiro Aigaki. The UAS-lacZ fly line was a gift from Yasushi Hiromi.
Generation of RNAi transgenic fly lines
IR transgenic fly lines of drob-1 were generated using a modified transformation vector, pUAST-D13 (a kind gift from Ryu Ueda). A 500-bp-long cDNA fragment was amplified by PCR and inserted as an IR into pUAST-D13. In all cases, IRs were constructed in a head-to-head orientation. Transgenic flies were generated by general P-element-mediated transformation.
Plasmids and dsRNAs
pUAST-HA-drob-1 and pBSSK-HA-drob-1 were described previously (Igaki et al, 2000). A driver plasmid that expresses GAL4 under control of the actin5C promoter (pWAGAL4) was a kind gift from Yasushi Hiromi. pCaspeR-hs-lacZ was described previously (Hisahara et al, 1998). dsRNAs for drob-1, buffy, or EGFP were synthesized and the cells were treated with dsRNA essentially as described previously (Igaki et al, 2002).
ATP assay, immunoblotting, and imaging of mitochondrial membrane potential (Δψm) using RNAi in Drosophila S2 cells
S2 cells were cultured in six-well plates (5 × 105 cells/well). The cells were washed with serum-free medium and treated with 20 μg/ml dsRNA in the serum-free medium for 30 min. Two volumes of Schneider's medium containing 10% FCS were then added to the medium, and the cells were cultured for 48 or 72 h. For immunoblotting or imaging of Δψm, cells were treated with various amounts of the indicated inhibitors for an additional 24 h. And then, cells were subjected to the assays.
Cell-death assay using RNAi in Drosophila S2 cells
For the cell death assay, S2 cells were cultured in 24-well plates (1 × 105 cells/well) and were cotransfected using CellFectin (Invitrogen) with a driver plasmid pWAGAL4 and pCaspeR-hs-lacZ, a reporter plasmid that encodes β-galactosidase under control of the hsp70 promoter, together with 25 ng of EGFP or drob-1 dsRNA. At 48 h after the transfection, the cells were left untreated or treated with various amounts of the indicated inhibitors at 26°C for 24 h. Cells were then heat-shocked at 37°C for 2 h as described (Hisahara et al, 1998), and cultured at 26°C for another 24 h. The cells were lysed in 300 μl of 1 × Reporter lysis buffer (Promega) and each lysate was assayed for β-galactosidase activity in a reaction mixture containing 1 mg/ml o-nitrophenyl-β-D-galactopyranoside, as described (Igaki et al, 2000).
Longevity assay
For the longevity assay, more than 100 flies of each sex were collected for each genotype within 24 h after eclosion and maintained at 25°C. Flies were transferred to fresh food every 2 or 3 days and the numbers of dead flies were counted.
Locomotor activity assay
Male flies of each genotype were collected within 24 h after eclosion and entrained to a 12 h light:12 h dark cycle (LD12:12) at 25°C for at least 3 days. Flies were transferred to glass tubes for Drosophila activity monitoring (DAM) system (Trikinetics, Waltham, MA) interfaced with an Apple computer and locomortor activity was recorded under LD12:12 at 25°C for 14 days.
Assay for ATP levels
Heat-shocked hs-GAL4/UAS-lacZ-IR, hs-GAL4/UAS-drob-1-IR, hs-GAL4/UAS-drob-1GS2263, or hs-GAL4/UAS-buffy adult flies were homogenized in 1 × Reporter lysis buffer (Promega), and were quickly frozen. The frozen fly samples were boiled for 15 min to destroy ATPase activity, then spun at 17 800 g for 5 min and the supernatant was diluted 100-fold with the same buffer. S2 cells were lysed in 0.5 ml of 1 × Reporter lysis buffer (Promega), and the lysate was quickly frozen in a dry ice/methanol bath. After thawing on ice, the cells were diluted 100-fold with the same buffer. The cellular ATP content in fly tissues or S2 cells was quantified by a luciferin- and luciferase-based assay using an ATP Determination Kit (Molecular Probes). Luminofluorescence was measured using the Wallac ARVO SX 1420 Multilabel Counter (Perkin Elmer Life Sciences), and the data were normalized to the protein content.
Histology
Flies were prepared for semithin sections and the sections were subjected to toluidine blue staining as described (Kanuka et al, 1999). For the light microscopic images of adult eyes, flies were anesthetized and examined with a Nikon SMZ1000 microscope (Nikon) equipped with an AxioCam digital camera (Carl Zeiss).
Immunoblotting
For adult heads, 12 fly heads were carefully dissected from anesthetized flies (1 day after eclosion) and lysed in 48 μl of SDS sample buffer. The S2 cells or adult head lysates were then separated by 10% SDS–PAGE and subjected to immunoblotting using an anti-ubiquitin mouse monoclonal antibody (1:250; Stressgen), an anti-β-tubulin mouse monoclonal antibody (1:500; CHEMICON), and an anti-mouse IgG-HRP antibody (1:1000; Promega). Signals were visualized using ECL plus (Amersham).
Imaging of Δψm
Δψm was analyzed using 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolyl-carbocyanine iodide (JC-1; Molecular Probes), a lipophilic cationic fluorescence dye. JC-1 is driven into mitochondria in a membrane potential-dependent manner. At high mitochondrial membrane potentials, JC-1 accumulates sufficiently in the mitochondria to form aggregates that fluoresce red. At lower mitochondrial potentials, less dye enters mitochondria resulting in monomers that fluoresce green (Smiley et al, 1991). This assay allowed one to quantify the percentage of highly energized mitochondria (with both red and green fluorescence) and depolarized mitochondria (with green fluorescence only). S2 cells were incubated with 5 μg/ml JC-1 (made up as a 1 mg/ml stock in dimethyl sulfoxide) for 10 min at room temperature in the dark. Then, cells were washed three times with PBS and live cell imaging was performed with a Zeizz LSM 510 META laser scanning confocal microscopy system. The ratio of number of cells with highly energized red mitochondria to total number of cells with green mitochondria was calculated. Approximately 500 cells were analyzed per experimental condition.
Mitochondrial isolation
S2 cells were cultured with 25 μg/ml EGFP dsRNA or drob-1 dsRNA for 72 h, and incubated with or without 0.05 μM rotenone or 0.5 mM 3-NP for an additional 16 h. Cells were then harvested and homogenized in isolation buffer (210 mM mannitol, 70 mM sucrose, 0.1 mM EDTA, and 5 mM Tris–HCl, pH 7.4). Mitochondria were isolated by differential centrifugation and suspended in Tris–EDTA buffer (0.1 mM EDTA and 50 mM Tris–HCl, pH 7.4) as described (Senoo-Matsuda et al, 2001).
Mitochondrial complex I and complex II assay
The activities of complex I+III and complex II+III in mitochondria isolated from S2 cells were measured as described (Senoo-Matsuda et al, 2001).
Transmission electron microscopy
S2 cells were cultured with 25 μg/ml EGFP dsRNA or drob-1 dsRNA for 72 h, and incubated with or without 0.05 μM rotenone or 0.5 mM 3-NP for an additional 16 h. Cells were fixed with 2.5% glutaraldehyde in 0.1 M Sorenson's buffer (pH 7.2) for 12 h. The samples were then postfixed with 1% OsO4 in 0.1 M Sorenson's buffer (pH 7.2) for 1 h. Enblock staining was performed using 1% tannic acid. After dehydration, samples were embedded in Lx-112 (Ladd Research Industries Inc.). Semithin 1 μm and thin 60 nm sections were cut on the MT-7000 urtramicrotome. Thin sections were stained with uranyl acetate and lead citrate and examined under a JEOL JEM-1200 EXII transmission electron microscope operating at 80 kV.
Statistical analysis
Data are given as means±s.e.m. Student's t-tests were performed on all quantitative analyses.
Supplementary Material
Supplementary Table 1
Supplementary Figure 1
Supplementary Table 2
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure 5
Acknowledgments
We are grateful to T Aigaki, N Bonini, R Carthew, Y Hiromi, C Kopczynski, H Richardson, and R Ueda for materials and flies, the Berkeley Drosophila Genome Project for providing information, and the Bloomington Stock Center for fly stocks. We also thank K Mikoshiba for kind support. We thank N Ishii for invaluable advice, K Yasuda, E Schon, S Krisuna, and A Naini for technical support and sharing of their laboratory for mitochondrial respiratory activity studies, and K Brown for technical support for electron microscopy studies. We thank J Takahashi and S Osawa for technical support. Finally, we thank L Johnston for kind support. This work was supported in part by grants from the Japanese Ministry of Education, Science, Sports, Culture, and Technology (to MM), the RIKEN Bioarchitect Research Grant (to MM), and the RIKEN Special Postdoctoral Researchers Program Grant (to NS-M). This study was also supported in part by the Naito Foundation and Takeda Science Foundation. TI was supported in part by a fellowship of Yamanouchi Foundation for Research on Metabolic Disorders, and is a research fellow of the Human Frontier Science Program. NS-M was a research fellow of the Special Postdoctoral Researchers Program, RIKEN, and is a postdoctoral fellow of the Japan Society for the Promotion of Science.
References
- Angermuller S, Kunstle G, Tiegs G (1998) Pre-apoptotic alterations in hepatocytes of TNFalpha-treated galactosamine-sensitized mice. J Histochem Cytochem 46: 1175–1183 [DOI] [PubMed] [Google Scholar]
- Baehrecke EH (2002) How death shapes life during development. Nat Rev Mol Cell Biol 3: 779–787 [DOI] [PubMed] [Google Scholar]
- Basanez G, Zhang J, Chau BN, Maksaev GI, Frolov VA, Brandt TA, Burch J, Hardwick JM, Zimmerberg J (2001) Pro-apoptotic cleavage products of Bcl-xL form cytochrome c-conducting pores in pure lipid membranes. J Biol Chem 276: 31083–31091 [DOI] [PubMed] [Google Scholar]
- Beal MF, Hyman BT, Koroshetz W (1993) Do defects in mitochondrial energy metabolism underlie the pathology of neurodegenerative diseases? Trends Neurosci 16: 125–131 [DOI] [PubMed] [Google Scholar]
- Becker EB, Bonni A (2004) Cell cycle regulation of neuronal apoptosis in development and disease. Prog Neurobiol 72: 1–25 [DOI] [PubMed] [Google Scholar]
- Brachmann CB, Jassim OW, Wachsmuth BD, Cagan RL (2000) The Drosophila bcl-2 family member dBorg-1 functions in the apoptotic response to UV-irradiation. Curr Biol 10: 547–550 [DOI] [PubMed] [Google Scholar]
- Browne SE, Beal MF (2004) The energetics of Huntington's disease. Neurochem Res 29: 531–546 [DOI] [PubMed] [Google Scholar]
- Cheng EHY, Kirsch DG, Clem RJ, Ravi R, Kastan MB, Bedi A, Ueno K, Hardwick JM (1997) Conversion of Bcl-2 to a Bax-like death effector by caspases. Science 278: 1966–1968 [DOI] [PubMed] [Google Scholar]
- Ciechanover A, Brundin P (2003) The ubiquitin proteasome system in neurodegenerative diseases: sometimes the chicken, sometimes the egg. Neuron 40: 427–446 [DOI] [PubMed] [Google Scholar]
- Clem RJ, Cheng EHY, Karp CL, Kirsch DG, Ueno K, Takahashi A, Kastan MB, Griffin DE, Earnshaw WC, Veliuona MA, Hardwick JM (1998) Modulation of cell death by Bcl-xL through caspase interaction. Proc Natl Acad Sci USA 95: 554–559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colussi PA, Quinn LM, Huang DCS, Coombe M, Read SH, Richardson H, Kumar S (2000) Debcl a proapoptotic Bcl-2 homologue, is a component of the Drosophila melanogaster cell death machinery. J Cell Biol 148: 703–710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danial NN, Gramm CF, Scorrano L, Zhang CY, Krauss S, Ranger AM, Datta SR, Greenberg ME, Licklider LJ, Lowell BB, Gygi SP, Korsmeyer SJ (2003) BAD and glucokinase reside in a mitochondrial complex that integrates glycolysis and apoptosis. Nature 424: 952–956 [DOI] [PubMed] [Google Scholar]
- Dorstyn L, Mills K, Lazebnik Y, Kumar S (2004) The two cytochrome c species, DC3 and DC4, are not required for caspase activation and apoptosis in Drosophila cells. J Cell Biol 167: 405–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorstyn L, Read S, Cakouros D, Huh JR, Hay BA, Kumar S (2002) The role of cytochrome c in caspase activation in Drosophila melanogaster cells. J Cell Biol 156: 1089–1098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fannjiang Y, Chong-HyKim C, Huganir RL, Zou S, Lindsten T, Thompson CB, Mito T, Traystman RJ, Larsen T, Griffin DE, Mandir AS, Dawson TM, Dike S, Sappington AL, Kerr DA, Jonas EA, Kaczmarek LK, Hardwick JM (2003) BAK alters neuronal excitability and can switch from anti- to pro-death function during postnatal development. Dev Cell 4: 575–585 [DOI] [PubMed] [Google Scholar]
- Ghadially FN (1982) Ultrastructurel Pathology of the Cell and Matrix, 2nd edn. London, UK: Butterworths [Google Scholar]
- Golden TR, Hinerfeld DA, Melov S (2002) Oxidative stress and aging: beyond correlation. Aging Cell 1: 117–123 [DOI] [PubMed] [Google Scholar]
- Hengartner MO, Horvitz HR (1994) Activation of C. elegans cell death protein CED-9 by an amino-acid substitution in a domain conserved in Bcl-2. Nature 369: 318–320 [DOI] [PubMed] [Google Scholar]
- Hisahara S, Kanuka H, Shoji SI, Yoshikawa S, Okano H, Miura M (1998) Caenorhabditis elegans anti-apoptotic gene ced-9 prevents ced-3-induced cell death in Drosophila cells. J Cell Sci 111: 667–673 [DOI] [PubMed] [Google Scholar]
- Hoglinger GU, Carrard G, Michel PP, Medja F, Lombes A, Ruberg M, Friguet B, Hirsch EC (2003) Dysfunction of mitochondrial complex I and the proteasome: interactions between two biochemical deficits in a cellular model of Parkinson's disease. J Neurochem 86: 1297–1307 [DOI] [PubMed] [Google Scholar]
- Igaki T, Kanuka H, Inohara N, Sawamoto K, Nunez G, Okano H, Miura M (2000) Drob-1, a Drosophila member of the Bcl-2/CED-9 family that promotes cell death. Proc Natl Acad Sci USA 97: 662–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igaki T, Miura M (2004) Role of Bcl-2 family members in invertebrates. Biochim Biophys Acta 1644: 73–81 [DOI] [PubMed] [Google Scholar]
- Igaki T, Yamamoto-Goto Y, Tokushige N, Kanda H, Miura M (2002) Down-regulation of DIAP1 triggers a nobel Drosophila cell death pathway mediated by Dark and DRONC. J Biol Chem 277: 23103–23106 [DOI] [PubMed] [Google Scholar]
- Ishii N, Fujii M, Hartman PS, Tsuda M, Yasuda K, Senoo-Matsuda N, Yanase S, Ayusawa D, Suzuki K (1998) A mutation in succinate dehydrogenase cytochrome b causes oxidative stress and ageing in nematodes. Nature 394: 694–697 [DOI] [PubMed] [Google Scholar]
- Jackson GR, Salecker I, Dong X, Yao X, Arnheim N, Faber PW, MacDonald ME, Zipursky SL (1998) Polyglutamine-expanded human huntingtin transgenes induce degeneration of Drosophila photoreceptor neurons. Neuron 21: 633–642 [DOI] [PubMed] [Google Scholar]
- Kanuka H, Kuranaga E, Hiratou T, Igaki T, Nelson B, Okano H, Miura M (2003) Cytosol–endoplasmic reticulum interplay by Sec61alpha translocon in polyglutamine-mediated neurotoxicity in Drosophila. Proc Natl Acad Sci USA 100: 11723–11728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanuka H, Sawamoto K, Inohara N, Matsuno K, Okano H, Miura M (1999) Control of the cell death pathway by Dapaf-1, a Drosophila Apaf-1/CED-4-related caspase activator. Mol Cell 4: 757–769 [DOI] [PubMed] [Google Scholar]
- Kennerdell JR, Carthew RW (2000) Heritable gene silencing in Drosophila using double-stranded RNA. Nat Biotechnol 18: 896–898 [DOI] [PubMed] [Google Scholar]
- Kirsch DG, Doseff A, Chau BN, Lim DS, de Souza-Pinto NC, Hansford R, Kastan MB, Lazebnik YA, Hardwick JM (1999) Caspase-3-dependent cleavage of Bcl-2 promotes release of cytochrome c. J Biol Chem 274: 21155–21161 [DOI] [PubMed] [Google Scholar]
- Lenaz G, Bovina C, D'Aurelio M, Fato R, Formiggini G, Genova ML, Giuliano G, Merlo Pich M, Paolucci U, Parenti Castelli G, Ventura B (2002) Role of mitochondria in oxidative stress and aging. Ann NY Acad Sci 959: 199–213 [DOI] [PubMed] [Google Scholar]
- Lenaz G, D'Aurelio M, Merlo Pich M, Genova ML, Ventura B, Bovina C, Formiggini G, Parenti Castelli G (2000) Mitochondrial bioenergetics in aging. Biochim Biophys Acta 1459: 397–404 [DOI] [PubMed] [Google Scholar]
- Lewis J, Oyler GA, Ueno K, Fannjiang Y, Chau BN, Vornov J, Korsmeyer SJ, Zou S, Hardwick JM (1999) Inhibition of virus-induced neuronal apoptosis by Bax. Nat Med 5: 832–835 [DOI] [PubMed] [Google Scholar]
- Lossi L, Merighi A (2003) In vivo cellular and molecular mechanisms of neuronal apoptosis in the mammalian CNS. Prog Neurobiol 69: 287–312 [DOI] [PubMed] [Google Scholar]
- Martinou JC, Green DR (2001) Breaking the mitochondrial barrier. Nat Rev Mol Cell Biol 2: 63–67 [DOI] [PubMed] [Google Scholar]
- Middleton G, Davies AM (2001) Populations of NGF-dependent neurones differ in their requirement for BAX to undergo apoptosis in the absence of NGF/TrkA signalling in vivo. Development 128: 4715–4728 [DOI] [PubMed] [Google Scholar]
- Ollmann M, Young LM, Di Como CJ, Karim F, Belvin M, Robertson S, Whittaker K, Demsky M, Fisher WW, Buchman A, Duyk G, Friedman L, Prives C, Kopczynski C (2000) Drosophila p53 is a structural and functional homolog of the tumor suppressor p53. Cell 101: 91–101 [DOI] [PubMed] [Google Scholar]
- Orth M, Schapira AH (2001) Mitochondria and degenerative disorders. Am J Med Genet 106: 27–36 [DOI] [PubMed] [Google Scholar]
- Panov AV, Gutekunst CA, Leavitt BR, Hayden MR, Burke JR, Strittmatter WJ, Greenamyre JT (2002) Early mitochondrial calcium defects in Huntington's disease are a direct effect of polyglutamines. Nat Neurosci 5: 731–736 [DOI] [PubMed] [Google Scholar]
- Pollack M, Phaneuf S, Dirks A, Leeuwenburgh C (2002) The role of apoptosis in the normal aging brain, skeletal muscle, and heart. Ann NY Acad Sci 959: 93–107 [DOI] [PubMed] [Google Scholar]
- Quinn L, Coombe M, Mills K, Daish T, Colussi P, Kumar S, Richardson H (2003) Buffy, a Drosophila Bcl-2 protein, has anti-apoptotic and cell cycle inhibitory functions. EMBO J 22: 3568–3579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruan Q, Lesort M, MacDonald ME, Johnson GV (2004) Striatal cells from mutant huntingtin knock-in mice are selectively vulnerable to mitochondrial complex II inhibitor-induced cell death through a non-apoptotic pathway. Hum Mol Genet 13: 669–681 [DOI] [PubMed] [Google Scholar]
- Sanchez I, Mahlke C, Yuan J (2003) Pivotal role of oligomerization in expanded polyglutamine neurodegenerative disorders. Nature 421: 373–379 [DOI] [PubMed] [Google Scholar]
- Scorrano L, Ashiya M, Buttle K, Weiler S, Oakes SA, Mannella CA, Korsmeyer SJ (2002) A distinct pathway remodels mitochondrial cristae and mobilizes cytochrome c during apoptosis. Dev Cell 2: 55–67 [DOI] [PubMed] [Google Scholar]
- Senoo-Matsuda N, Hartman PS, Akatsuka A, Yoshimura S, Ishii N (2003) A complex II defect affects mitochondrial structure, leading to ced-3- and ced-4-dependent apoptosis and aging. J Biol Chem 278: 22031–22036 [DOI] [PubMed] [Google Scholar]
- Senoo-Matsuda N, Yasuda K, Tsuda M, Ohkubo T, Yoshimura S, Nakazawa H, Hartman PS, Ishii N (2001) A defect in the cytochrome b large subunit in complex II causes both superoxide anion overproduction and abnormal energy metabolism in Caenorhabditis elegans. J Biol Chem 276: 41553–41558 [DOI] [PubMed] [Google Scholar]
- Sesso A, Marques MM, Monteiro MM, Schumacher RI, Colquhoun A, Belizario J, Konno SN, Felix TB, Botelho LA, Santos VZ, Da Silva GR, Higuchi Mde L, Kawakami JT (2004) Morphology of mitochondrial permeability transition: morphometric volumetry in apoptotic cells. Anat Rec A Discov Mol Cell Evol Biol 281: 1337–1351 [DOI] [PubMed] [Google Scholar]
- Shamoto-Nagai M, Maruyama W, Kato Y, Isobe K, Tanaka M, Naoi M, Osawa T (2003) An inhibitor of mitochondrial complex I, rotenone, inactivates proteasome by oxidative modification and induces aggregation of oxidized proteins in SH-SY5Y cells. J Neurosci Res 74: 589–597 [DOI] [PubMed] [Google Scholar]
- Smiley ST, Reers M, Mottola-Hartshorn C, Lin M, Chen A, Smith TW, Steele GD Jr, Chen LB (1991) Intracellular heterogeneity in mitochondrial membrane potentials revealed by a J-aggregate-forming lipophilic cation JC-1. Proc Natl Acad Sci USA 88: 3671–3675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsang WY, Lemire BD (2003) The role of mitochondria in the life of the nematode, Caenorhabditis elegans. Biochim Biophys Acta 1638: 91–105 [DOI] [PubMed] [Google Scholar]
- Vander Heiden MG, Chandel NS, Schumacker PT, Thompson CB (1999) Bcl-xL prevents cell death following growth factor withdrawal by facilitating mitochondrial ATP/ADP exchange. Mol Cell 3: 159–167 [DOI] [PubMed] [Google Scholar]
- Warrick JM, Chan HY, Gray-Board GL, Chai Y, Paulson HL, Bonini NM (1999) Suppression of polyglutamine-mediated neurodegeneration in Drosophila by the molecular chaperone HSP70. Nat Genet 23: 425–428 [DOI] [PubMed] [Google Scholar]
- Warrick JM, Paulson HL, Gray-Board GL, Bui QT, Fischbeck KH, Pittman RN, Bonini NM (1998) Expanded polyglutamine protein forms nuclear inclusions and causes neural degeneration in Drosophila. Cell 93: 939–949 [DOI] [PubMed] [Google Scholar]
- Zhang H, Holzgreve W, De Geyter C (2000) Evolutionarily conserved Bok proteins in the Bcl-2 family. FEBS Lett 480: 311–313 [DOI] [PubMed] [Google Scholar]
- Zimmermann KC, Ricci JE, Droin NM, Green DR (2002) The role of ARK in stress-induced apoptosis in Drosophila cells. J Cell Biol 156: 1077–1087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou H, Henzel WJ, Liu X, Lutschg A, Wang X (1997) Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c-dependent activation of caspase-3. Cell 90: 405–413 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1
Supplementary Figure 1
Supplementary Table 2
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure 5