

Abstract
Transforming growth factor-β (TGF-β) inhibits osteoblast differentiation through inhibition of the function of Runx2 (Cbfa1) by Smad3. The mechanism through which TGF-β/Smad3 inhibits Runx2 function has not been characterized. We show that TGF-β induces histone deacetylation, primarily of histone H4, at the osteocalcin promoter, which is repressed by TGF-β, and that histone deacetylation is required for repression of Runx2 by TGF-β. This repression occurs through the action of the class IIa histone deacetylases (HDAC)4 and 5, which are recruited through interaction with Smad3 to the Smad3/Runx2 complex at the Runx2-binding DNA sequence. Accordingly, HDAC4 or 5 is required for efficient TGF-β-mediated inhibition of Runx2 function and is involved in osteoblast differentiation. Our results indicate that class IIa HDACs act as corepressors for TGF-β/Smad3-mediated transcriptional repression of Runx2 function in differentiating osteoblasts and are cell-intrinsic regulators of osteoblast differentiation.
Keywords: CBFA1, chromatin remodeling, mesenchymal differentiation, osteoblast, transcription
Introduction
Mesenchymal differentiation along the osteoblast, myocyte or adipocyte lineages presents an accessible, physiological system to study how transforming growth factor-β (TGF-β) represses cell differentiation and regulates transcription. As these programs progress, cells sequentially express genes that characterize the differentiated osteoblast, myocyte or adipocyte. TGF-β inhibits the progression of differentiation through functional repression of key transcription factors (Alliston et al, 2001; Liu et al, 2001, 2004; Choy and Derynck, 2003).
During osteoblast differentiation, cells activate the Runx2 (Cbfa1) transcription factor and subsequently express osteocalcin and Osterix. Induction of Runx2 expression is critical in osteoblast differentiation, as it is required for the expression of osteocalcin and other proteins that allow osteoblasts to mineralize collagen I-rich matrix to form bone. Without Runx2, osteoblasts do not differentiate and cannot form bone (Harada and Rodan, 2003).
Osteoblasts secrete TGF-β into the bone matrix and respond to it, thereby enabling autocrine regulation. TGF-β stimulates preosteoblast proliferation, while inhibiting full differentiation. Accordingly, TGF-β inhibits expression of osteocalcin and other markers of osteoblast function (Alliston and Derynck, 2000). A key event in this inhibition is the repression of Runx2 function by TGF-β, thereby downregulating the expression of Runx2 target genes, such as osteocalcin and Runx2 (Alliston et al, 2001).
The Smads act as effectors of the TGF-β-induced changes in gene expression. Binding of TGF-β to its TβRI/TβRII receptor complex at the cell surface activates Smad2 and 3 through phosphorylation, which then form complexes with Smad4 and translocate into the nucleus. These complexes associate with transcription factors and bind DNA to regulate gene expression. This versatility in transcription factor interactions, together with the ability of activated Smads to recruit the coactivators CBP or p300, is the basis for the context-dependent activation of gene expression in response to TGF-β (Feng and Derynck, 2005).
Much less is known about repression of transcription by TGF-β and it is not known what defines whether a target gene is to be activated or repressed by TGF-β. Corepressors such as TGIF, Evi-1, c-Ski, and SnoN can interact with Smads and recruit histone deacetylases (HDAC) into the transcription machinery; however, these inhibit Smad-activated transcription and do not confer repression by TGF-β (Feng and Derynck, 2005). Only few examples of TGF-β-mediated repression have been studied. TGF-β represses myogenesis through association of Smad3 with bHLH transcription factors, such as MyoD, thus interfering with MyoD/E protein dimerization and interaction of MyoD with E-box sequences (Liu et al, 2001). Smad3 also associates with mouse embryo fibroblast (MEF)2, thereby blocking the interaction of MEF2 with the coactivator GRIP1, which is required for MEF2's functions as transcription coactivator for myogenic bHLH proteins (Liu et al, 2004). HDACs are not involved in either mechanism. In the repression of c-myc by TGF-β, a complex of Smad3 with p107-E2F4/5-DP1 associates with Smad4 and translocates into the nucleus, where it binds c-myc regulatory sequences (Chen et al, 2002). While p107 and E2F4 can form a complex with HDAC1 independently of TGF-β (Ferreira et al, 1998), no evidence for HDAC-dependent repression by TGF-β was reported. Finally, BMP4 can repress the activity of Nkx3.2. Nkx3.2 binds HDAC1 and binding of BMP-activated Smad1 to another Nkx3.2 segment stabilizes the Nkx3.2/HDAC1 interaction (Kim and Lassar, 2003). In neither case were Smads shown to recruit HDACs.
The association of Smads with coactivators, such as CBP/p300, Mediator components or SMIF, may induce chromatin remodeling, as histone acetylation has been correlated with transcriptional activation (Berger, 2002), and CBP and p300 are acetyltransferases (Kouzarides, 2000). Conversely, recruitment of HDACs at sites of Smad-mediated transcriptional repression may lead to deacetylation of histones. No evidence for Smad-mediated changes in histone acetylation or chromatin structure has been reported.
We reported that TGF-β inhibits Runx2 function through direct interaction of Smad3 with Runx2 at Runx2-binding DNA sequences of osteoblast differentiation genes, for example, the osteocalcin and runx2 genes, without decreasing DNA binding of Runx2. This inhibition results in transcription repression at the osteocalcin promoter and inhibition of osteoblast differentiation (Alliston et al, 2001). We now address the mechanism through which TGF-β/Smad3 represses Runx2 function and osteocalcin expression. The class IIa HDACs HDAC4 and/or 5 were directly recruited by Smad3 to Runx2, thereby forming a stable complex of Smad3, Runx2, and HDAC(s) at the Runx2-binding DNA sequence. This mechanism is in contrast to the repression of myogenic transcription by TGF-β/Smad3, which does not involve HDAC recruitment. Decreasing the functional levels of these HDACs enhanced osteoblast differentiation, suggesting an intrinsic role of class IIa HDACs, in cooperation with Smads, in osteoblast differentiation.
Results
Repression of Runx2 by TGF-β requires HDAC activity
To examine whether repression of Runx2 by TGF-β at the Runx2-binding site OSE2 (Alliston et al, 2001) involves HDAC activity, we examined the effect of trichostatin A (TSA), an inhibitor of class I and class II HDACs (Yoshida et al, 1990). As reported (Alliston et al, 2001), Runx2 activated transcription from the 6OSE2 promoter and this activation was repressed by TGF-β (Figure 1A). Increasing TSA levels overcame this repression and reversed it to activation in response to TGF-β. TSA also increased the reporter activity without added TGF-β (Figure 1A), likely a reflection of a balance between acetylation and deacetylation and/or autocrine TGF-β signaling. TSA did not significantly affect the activation of transcription from the 3TP or Smad7 promoters (Figure 1B and C). TSA also inhibited the repression of endogenous osteocalcin mRNA levels by TGF-β in ROS17/2.8 osteosarcoma cells (Figure 1D). These results indicate that class I or II HDAC activity is required for repression of Runx2 function by TGF-β, and suggest a correlation of transcriptional repression with histone deacetylation.
Figure 1.
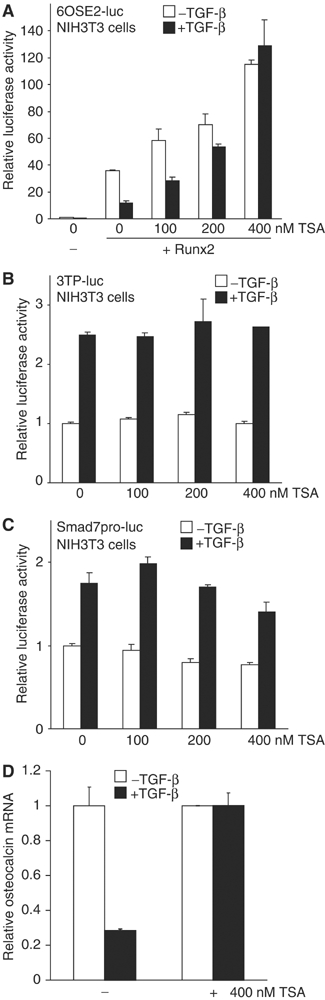
Effect of TSA on Runx2-mediated transcription from OSE2 sequences. (A) TSA reverses TGF-β-induced repression of Runx2 activity at 6OSE2-luc in NIH3T3 cells. Cells were transfected with a Runx2 plasmid and the 6OSE2-luc reporter. After 24 h, cells were treated with TSA for 16 h in the presence or absence of TGF-β, and the luciferase activity was measured. (B, C) TSA does not affect activation of transcription by TGF-β at the 3TP or Smad7 promoters. (D) TSA inhibits TGF-β-induced repression of endogenous osteocalcin mRNA expression in the ROS17/2.8 cells, as assessed by real-time PCR.
TGF-β induces histone deacetylation at the endogenous osteocalcin promoter
We used chromatin immunoprecipitation (ChIP) assays to assess whether TGF-β induces histone deacetylation at the OSE2 sequence of the osteocalcin promoter. Histones H3 and H4 are acetylated at the osteocalcin promoter in osteoblasts and ROS17/2.8 cells, in which osteocalcin is expressed (Shen et al, 2003). In ROS17/2.8 cells, histone H3 and H4 acetylation was detected at the osteocalcin promoter segment that spans the OSE2 sequence, consistent with the transcription from this promoter (Figure 2A). TGF-β reduced histone H4 acetylation, with little effect on histone H3 acetylation, thus correlating histone deacetylation with repression of transcription from the OSE2-containing promoter by TGF-β. Histone H4 deacetylation was apparent at 30 min after adding TGF-β and persisted for the next 30 min (Figure 2B and D) up to 4 h (Figure 2A). The TGF-β-induced histone deacetylation suggests rapid recruitment of HDAC activity to the OSE2 sequence in response to TGF-β.
Figure 2.
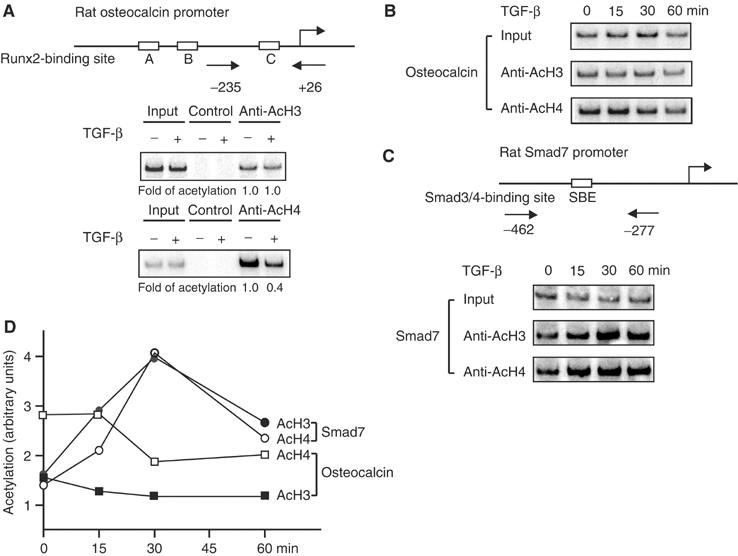
TGF-β-induced chromatin changes at the osteocalcin and Smad7 promoters. (A) TGF-β induces deacetylation of histone H4 at the osteocalcin promoter in ROS17/2.8 cells. Chromatin from cells treated with or without TGF-β for 4 h was immunoprecipitated with anti-acetylated histone H3 (anti-AcH3) or H4 (anti-AcH4), or control beads, and analyzed with PCR using primers spanning the −235 to +26 sequence in the osteocalcin promoter, including the OSE2 sequence. (B) Rapid deacetylation of histone H4 upon TGF-β treatment. ROS17/2.8 cells were treated with TGF-β, and ChIP assays were performed as in panel A. (C) TGF-β stimulates histone H3 and H4 acetylation at the Smad7 promoter, as assessed by ChIP assays using primers that span the −462 to −277 sequence that includes the SBE sequence. (D) The levels of histone H3 and H4 acetylation from panels B and C were quantified.
We also addressed the effect of TGF-β at the Smad7 promoter, which is activated by TGF-β (Hua et al, 2000), in the same ROS17/2.8 cells. Using primers that span the Smad-binding sequence, we showed increased histone acetylation, starting at 15 min after TGF-β addition (Figure 2C and D), correlating histone acetylation with Smad7 gene activation by TGF-β. The kinetics of TGF-β-induced H3 and H4 acetylation at the Smad7 promoter, and the time course of histone deacetylation at the osteocalcin promoter are consistent with the rapid translocation of the Smad complex into the nucleus, and assembly into transcription complexes.
Class IIa HDAC4 and 5 are corepressors in the repression of the osteocalcin promoter by TGF-β
The TGF-β-induced histone deacetylation at the OSE2 element of the osteocalcin promoter raises the question as to which HDAC mediates the repression. We tested class I and class II HDACs for their effects on Runx2-mediated transcription from the OSE2-containing osteocalcin promoter starting at −147. Class I HDAC1, 2, and 3, and class IIb HDAC6 did not repress transcription from the −147OC-luc reporter. In contrast, class IIa HDAC4, 5, and 7 repressed Runx2-mediated transcription, and this repression was enhanced by adding TGF-β (Figure 3A). Full repression by HDAC4 or 5 required their catalytic activity (data not shown). Similar results were obtained using the 6OSE2 reporter (data not shown). These results implicate class IIa HDACs in the repression of Runx2-mediated transcription by TGF-β. As HDAC7 is primarily expressed in T lymphocytes (Dequiedt et al, 2003), we focused on HDAC4 and 5, which are more widely expressed, including, in mesenchymal cells (Verdin et al, 2003). A role of other class IIa HDACs that are expressed in osteoblasts can, however, not be excluded.
Figure 3.
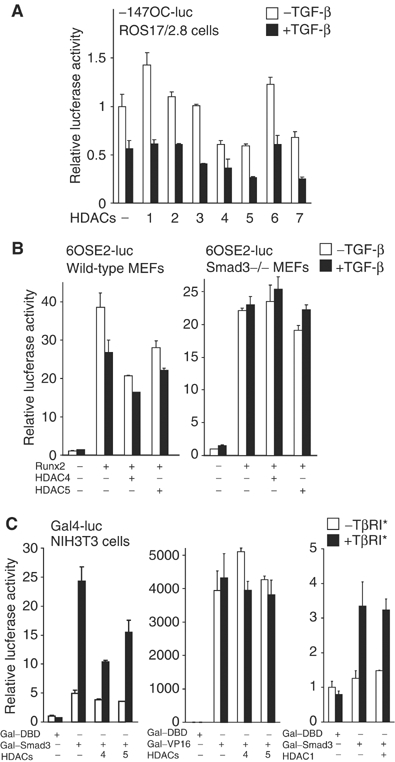
Class IIa HDACs are corepressors in TGF-β-mediated repression of Runx2-dependent transcription. (A) Effects of individual HDACs on Runx2-mediated transcription in the absence or presence of TGF-β. ROS17/2.8 cells were transfected with −147OC-luc reporter and individual HDAC plasmids, with or without TGF-β. (B) Wild-type or Smad3−/− MEFs were transfected with 6OSE2-luc and the indicated plasmids, with or without TGF-β. (C) HDAC4 and 5 repress Gal–Smad3-mediated transcription. NIH3T3 cells were transfected with a plasmid encoding Gal4–DNA-binding domain (Gal–DBD), Gal–Smad3 or Gal–VP16, without or with HDAC4, 5 or 1 plasmid, as well as the Gal4-luc reporter with five tandem Gal4-binding sites. An activated TβRI receptor was coexpressed to activate TGF-β signaling.
Since the repression of Runx2 at the osteocalcin promoter by TGF-β is mediated by Smad3 (Alliston et al, 2001), we tested the role of Smad3 in the repression of Runx2-mediated transcription by HDAC4 or 5. TGF-β, and HDAC4 and HDAC5, repressed Runx2 activity in wild-type MEFs, but not in Smad3-negative MEFs (Figure 3B). Thus, repression of Runx2 at the OSE2 sequence by class IIa HDACs is Smad3-dependent.
Since the repression by HDAC4 or 5 depends on Smad3, we assessed whether they target its transactivation function. We used a Gal–Smad3 fusion, which binds to Gal4-binding sites, and activates transcription in response to TGF-β. HDAC4 and 5 repressed the transactivation function of Gal–Smad3, while HDAC1 did not (Figure 3C). HDAC4 and 5, however, did not affect Gal–VP16-driven transcription. In addition, HDAC4 and 5 repressed the transcription from tandem Smad-binding elements in the 4SBE-luc reporter, which is mediated by Smad3/4 (data not shown). These results indicate that HDAC4 and 5 repress the transactivation function of Smad3.
Expression of HDAC4 and 5 in mesenchymal cells and osteoblasts
For HDAC4 and 5 to physiologically regulate Runx2 activity, they need to be expressed in differentiating osteoblasts. HDAC4 and 5 mRNAs are expressed in muscle and brain (Grozinger et al, 1999), but their expression in developing bone was not examined. We thus isolated mouse cDNAs for HDAC4 and 5 and evaluated their expression in bone sections of E17.5 day embryos using in situ hybridization.
Runx2 was expressed in the bones of the hand, humerus, and head (Figure 4A–C). In the hand, HDAC4 localized in the bone, muscle, and connective tissue. Differentiating osteoblasts, identified by their Runx2 expression, expressed HDAC4 mRNA (Figure 4A). HDAC5 was also expressed in the bones and surrounding muscle and connective tissue. Runx2 and HDAC5 were coexpressed in osteoblasts of the humerus and frontonasal bone (Figure 4B and C). HDAC5 was expressed as high in osteoblasts as in the brain and muscle, reported to express high HDAC5 levels. Osteoblasts, in which HDAC5 and Runx2 expression colocalized, also expressed osteocalcin. The patterns of HDAC5 and Runx2 expression were not identical, as Runx2 was also expressed in less mature osteoblasts. The colocalization of HDAC4/5 and Runx2 in the most mature cells, in which TGF-β can repress Runx2 function, supports their role as mediators of TGF-β repression.
Figure 4.
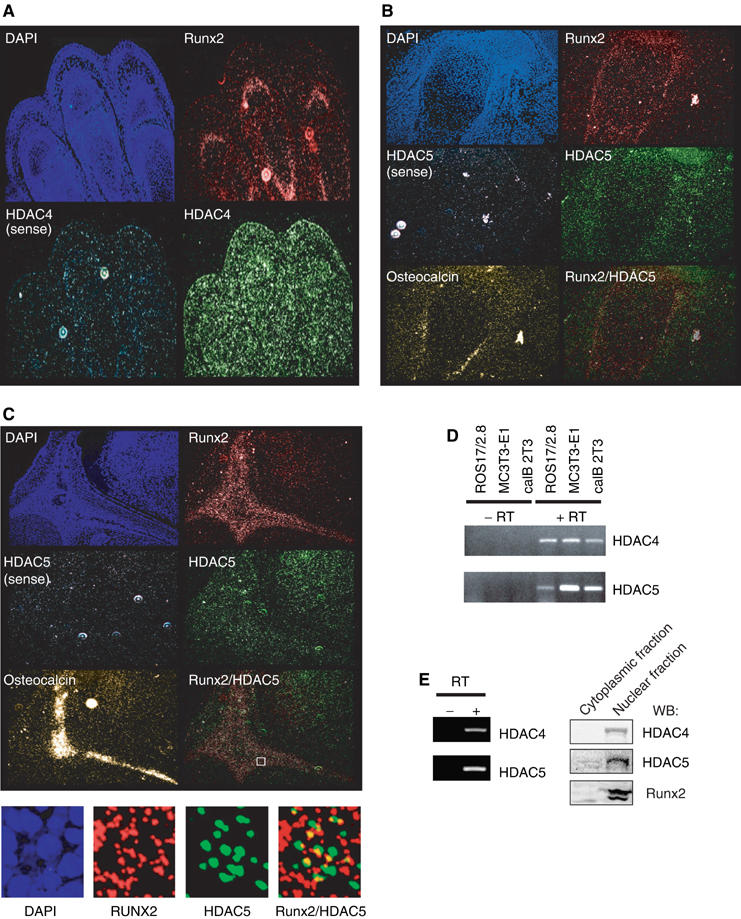
Localization of HDAC4 and 5 mRNA by in situ hybridization. Hybridization of 35S-labeled riboprobes in E17.5 mouse bone sections was visualized using dark-field microscopy to localize mRNAs of HDAC4, 5, Runx2, or osteocalcin. Counterstaining with DAPI visualized the nuclei. HDAC4 mRNA was localized in the digits of the front paw (A), and HDAC5 mRNA was localized in the humerus (B), and frontonasal bone of the head (C). Sense control probes show the specificity for HDAC4 or 5. Colocalization of Runx2 and HDAC5 mRNA is shown by superimposing the staining, taking into account the scattering of the activated silver grains (B, C). A higher magnification of a framed field is shown in the lower panel. (D) Detection by RT–PCR of HDAC4 and 5 mRNA in ROS17/2.8, MC3T3-E1, and caIB 2T3 cells. (E) Detection of mRNA or protein of HDAC4 and 5 by RT–PCR or Western blotting, respectively, in primary calvarial osteoblasts.
ROS17/2.8, MC3T3-E1 and caIB 2T3 cells, which are used to study osteoblast differentiation and express Runx2 (Alliston et al, 2001), also expressed HDAC4 and 5 mRNA (Figure 4D). In addition, primary osteoblasts derived from calvariae that develop through intramembranous ossification (not involving chondrocytes) expressed HDAC4 and 5 mRNA and protein (Figure 4E).
Smad3 interacts with HDAC4 and 5
The requirement for Smad3 in the repression of Runx2 by HDAC4 or 5 (Figure 3B) and the repression of Smad3's transactivation function by HDAC4 and 5 (Figure 3C) suggested an interaction of HDAC4 and 5 with Smad3. Accordingly, HDAC4 and 5 associated with Smad3 in transfected cells (Figure 5A and B) and at endogenous levels in ROS17/2.8 cells, in the absence or presence of added TGF-β (Figure 5C). The interaction of HDAC4 or 5 with Smad3 in the absence of added TGF-β is likely to result from: (a) the distribution of HDAC4 and 5 in both the cytoplasm and nucleus, with HDAC4 being more cytoplasmic and HDAC5 predominantly nuclear (Grozinger and Schreiber, 2000), (b) the well-documented autocrine TGF-β signaling in cultured cells, and (c) the presence of Smad3 in both compartments even in the absence of added TGF-β.
Figure 5.
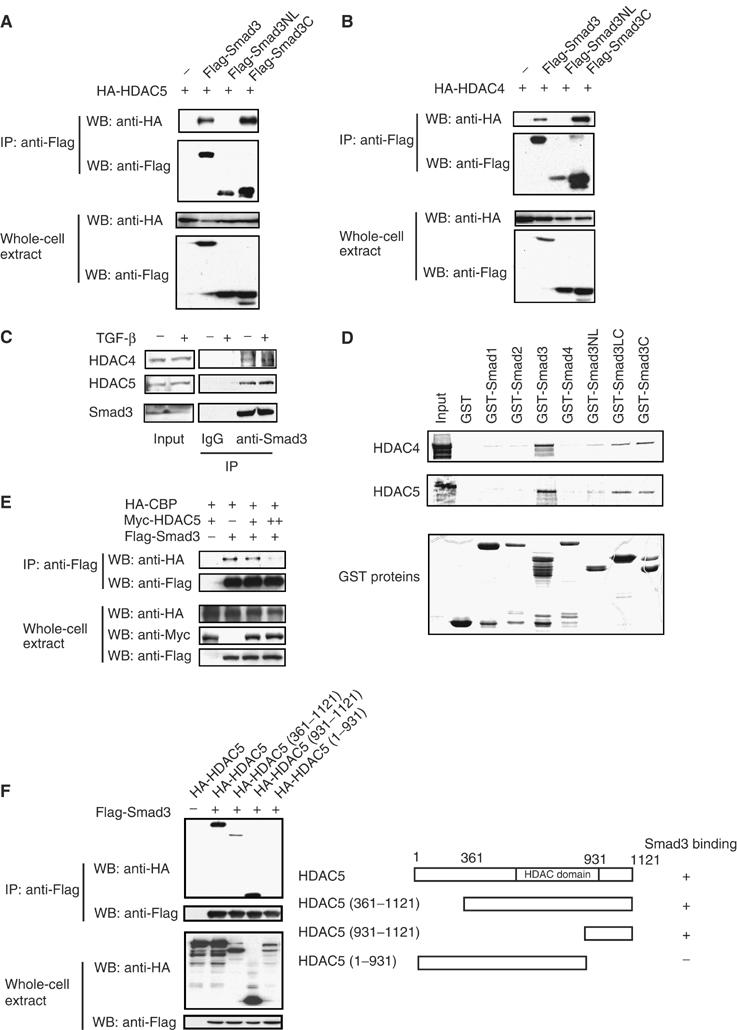
Interaction of Smad3 with HDAC4 and 5. (A, B) COS cells were transfected with the indicated plasmids and processed for immunoprecipitation with anti-Flag antibodies. Smad3NL contains the MH1 domain and linker segment, while Smad3C contains the MH2 domain. (C) Interaction of endogenous Smad3 and HDAC4 or 5. ROS17/2.8 cells were treated with or without TGF-β for 3 h before lysis and immunoprecipitation with anti-Smad3 or IgG, and immunoblotting for HDAC4, 5, or Smad3. (D) Interaction of Smad3 with HDAC4 or 5 in vitro. 35S-labeled, in vitro translated HDAC4 or 5 was adsorbed to GST or GST-Smads (or derivatives) bound to glutathione-Sepharose, and analyzed by SDS–PAGE and autoradiography. The Coomassie Blue-stained gel shows the purified GST proteins. (E) HDAC5 decreases the binding of Smad3 to CBP. 293T cells expressing the indicated proteins were subject to immunoprecipitation with anti-Flag antibodies. (F) Mapping of the HDAC5 segment that interacts with Smad3. COS cells expressing Flag-tagged Smad3 and HA-tagged HDAC5 or mutants were processed for immunoprecipitation assays.
Smad3 is composed of an MH1 domain that binds DNA and an MH2 domain that contains the transactivation function, connected by a linker (Feng and Derynck, 2005). HDAC4 and 5 interacted strongly with the MH2 domain of Smad3, but not with the MH1 domain and linker segment (Figure 5A and B). The interactions with the MH2 domain may explain the repression of the transactivation of Gal–Smad3 by HDAC4 or 5 (Figure 3C). HDAC4 or 5 also interacted with GST-fused Smad3 or the MH2 domain of Smad3, but not with GST itself. HDAC4 and 5 did not detectably interact with GST-fused Smad1, 2, or 4 (Figure 5D). These results suggest that HDAC4 and 5 interact directly and specifically with the transactivation domain of Smad3. Since Smad3 interacts through this domain with CBP/p300 (Feng et al, 1998), we examined whether HDAC5 affected the interaction of Smad3 with CBP. Increasing HDAC5 levels decreased this interaction (Figure 5E), which may explain in part its ability to repress the transactivation function of Smad3 (Figure 3C).
To map the region in HDAC5 that associates with Smad3, we generated truncated forms of HDAC5. Full-length HDAC5 and the C-terminal HDAC5 segment from amino acid 931 to 1121 interacted with Smad3, whereas the segment from amino acid 1 to 931 did not (Figure 5F). Similar results were obtained for HDAC4 (data not shown).
TGF-β/Smad3 stabilizes HDAC5 complex formation at the osteocalcin promoter
Since HDAC4 and 5 are structurally similar and both interacted with Smad3, we focused on HDAC5, rather than duplicating all experiments with HDAC4 and 5, in further characterization of the interactions. HDAC5 interacted more strongly with Smad3 than HDAC4, likely a consequence of the predominance of HDAC5 and 4 in the nucleus and cytoplasm, respectively, but HDAC4 and 5 otherwise behaved similarly (data not shown).
Endogenous or tagged HDAC5 co-precipitated with endogenous or tagged Runx2, respectively, and this interaction was stronger in response to TGF-β (Figure 6A and B). These data suggest that activated Smad3 recruits HDAC5 to Runx2 and stabilizes the HDAC5/Runx2 complex. Accordingly, Smad3/4, but not Smad3 alone, enhanced Runx2 association with HDAC5, and TGF-β signaling further enhanced complex formation (Figure 6C). Conversely, Runx2 enhanced the interaction between Smad3 and HDAC5 (Figure 6D). In addition, sequential immunoprecipitations showed that Runx2, Smad3, and HDAC5 form a ternary complex in vivo (Figure 6E). These data suggest that the three proteins stabilize each other's interactions, resulting in a stable complex of Smad3, HDAC5, and Runx2.
Figure 6.
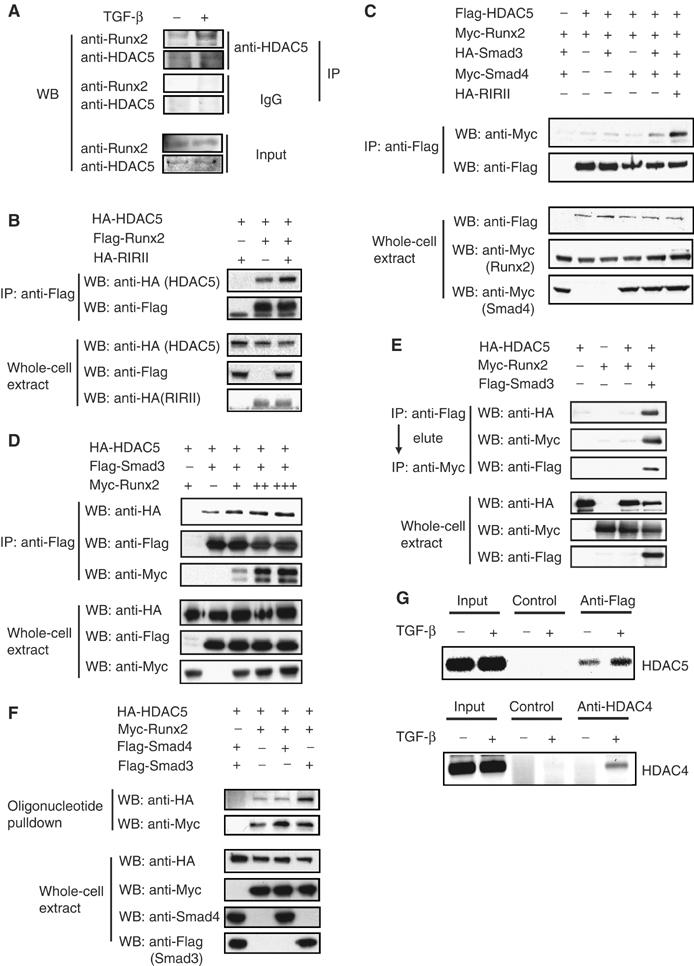
Effects of TGF-β/Smad3 on repressor complex formation. (A) TGF-β induces interaction of endogenous Runx2 and HDAC5. ROS 17/2.8 cells were incubated for 4 h with or without added TGF-β. Immunoprecipitation assays were performed with anti-HDAC5 antibodies. (B, C) TGF-β and Smad3/4 enhance the interaction of Runx2 with HDAC5. COS cells expressing the indicated proteins with or without an active TGF-β receptor chimera were processed for immunoprecipitations and Western blotting with indicated antibodies. (D) Runx2 enhances the interaction of Smad3 with HDAC5. Increasing levels of Runx2 were coexpressed with Smad3 and HDAC5, and lysates were subject to immunoprecipitation and Western blotting. (E) Runx2, Smad3, and HDAC5 form a ternary complex. 293T cells expressing the indicated plasmids were subject to immunoprecipitation with anti-Flag. Protein complexes were eluted from the antibody and subjected to immunoprecipitation with anti-Myc and Western blotting. (F) Effects of Smad3 on recruitment of HDAC5 to the OSE2 sequence. COS cells expressing the indicated proteins were subjected to DNA precipitation assay using biotinylated 2xOSE2 oligonucleotide. Protein–DNA complexes were analyzed by Western blotting. (G) Recruitment of HDAC4 or 5 to the endogenous osteocalcin promoter in response to TGF-β. ROS17/2.8 cells or ROS17/2.8 cells expressing Flag-tagged HDAC5 were treated with or without TGF-β for 4 h and ChIP assays were performed using anti-HDAC4, anti-Flag for HDAC5 or control beads, and analyzed with PCR using primers spanning the −235 to +26 sequence in the osteocalcin promoter, including the OSE2 sequence (Figure 2A).
To examine protein interactions at the OSE2 sequence, we conducted DNA affinity precipitations using biotinylated oligonucleotides. Runx2 bound the OSE2 sequence, and binding of HDAC5 was not detected in the absence of Runx2 (Figure 6F; Alliston et al, 2001). HDAC5 bound to the OSE2 sequence in the presence of Runx2, and this binding was enhanced when Smad3, but not Smad4, was coexpressed, indicating that Smad3 stabilizes the HDAC5 interaction with Runx2 at the promoter and assists in the recruitment of HDAC5. TGF-β-induced recruitment of endogenous HDAC5 through Smad3 to the osteocalcin promoter could not be unambiguously shown using ChIP assays, consistent with the general lack of success in detecting HDAC5 interactions by ChIP. We therefore expressed tagged HDAC5 in ROS17/2.8 cells and did ChIP assays using epitope antibody. HDAC5 was recruited to the endogenous osteocalcin promoter and this recruitment was enhanced upon addition of TGF-β (Figure 6G). Endogenous HDAC4 was recruited to osteocalcin promoter in response to TGF-β (Figure 6G).
HDAC4 and 5 mediate TGF-β-induced repression of osteocalcin expression
To evaluate the role of class IIa HDACs in TGF-β/Smad3-mediated gene repression, we tested whether the segment of HDAC5 (amino acids 931–1121) that interacts with Smad3 (Figure 5F) interferes with the interaction of Smad3 with HDAC5. Increasing levels of HDAC5(931–1121) decreased the association of Smad3 with HDAC5 (Figure 7A) or HDAC4 (data not shown). Thus, HDAC5(931–1121) may be used to interfere with the recruitment of class IIa HDACs to Smad3 in a dominant-negative manner. This interference with HDAC binding to Smad3 may primarily occur in the cytoplasm, since HDAC5(931–1121) contains the nuclear export signal and was mostly localized in the cytoplasm (data not shown).
Figure 7.
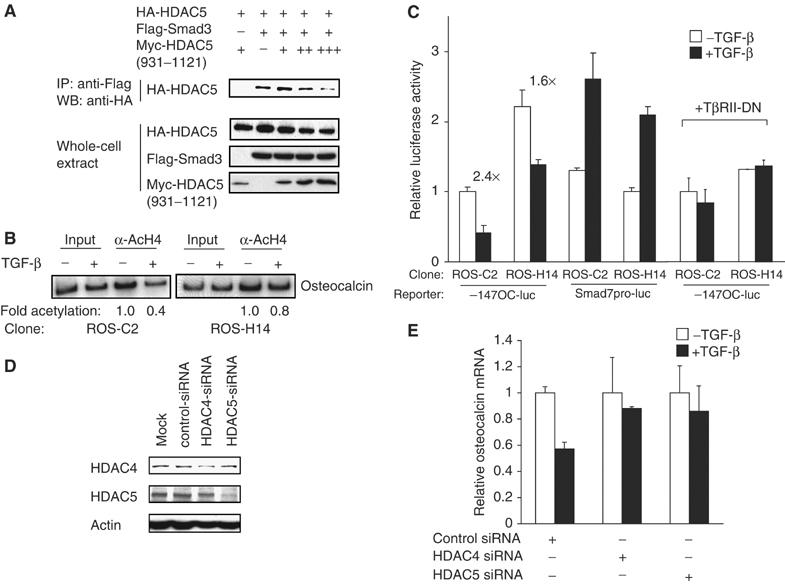
HDAC4 and 5 are required for repression of Runx2 function by TGF-β. (A) HDAC5(931–1121) interferes with the interaction of Smad3 with HDAC5. COS cells expressing Smad3, HDAC5, and increasing amounts of HDAC5(931–1121) were processed for immunoprecipitations and Western blotting. (B) HDAC5(931–1121) inhibits TGF-β-induced deacetylation of histone H4 at the osteocalcin promoter. ROS17/2.8 cells expressing HDAC5(931–1121) (ROS-H14 cells) or harboring empty vector (ROS-C2 cells) were treated with or without TGF-β, and subjected to ChIP assays with anti-acetylated histone H4 antibodies. The PCR primers spanned the −235 to +26 segment of the osteocalcin promoter (Figure 2A). (C) HDAC5(931–1121) decreases TGF-β repression of the osteocalcin promoter, but does not significantly affect transcription from the Smad7 promoter. ROS17/2.8 cells expressing HDAC5(931–1121) (ROS-H14 cells) or control ROS-C2 cells were transfected with −147OC-luc or Smad7pro-luc, with or without expression of a dominant-negative TGF-β receptor II (TβRII-DN), with or without added TGF-β, and luciferase activities were measured. The numbers above the bar graphs show the fold repression. (D) Effectiveness of siRNA-mediated depletion of HDAC4 and 5. Endogenous HDAC4 or 5 in the presence or absence of siRNAs was visualized by Western blotting. (E) Effects of HDAC4 or 5 siRNA on repression of osteocalcin gene expression by TGF-β. ROS17/2.8 cells were transfected with HDAC4 or 5 siRNA, and osteocalcin mRNA expression was measured by real-time PCR.
We generated ROS17/2.8 cells that express HDAC5(931–1121) and named them ROS-H14. We examined the effect of HDAC5(931–1121) on deacetylation at the osteocalcin promoter, since TGF-β induces histone H4 deacetylation (Figure 2A). TGF-β-induced histone H4 deacetylation was attenuated in ROS-H14 cells, when compared with control cells (Figure 7B), strongly suggesting a critical role of class IIa HDACs in TGF-β-induced histone deacetylation of the osteocalcin promoter.
Transcription from the OSE2-containing osteocalcin promoter was tested in ROS-H14 cells with or without adding TGF-β. ROS-H14 cells had higher transcription than control ROS17/2.8 cells (Figure 7C), consistent with autocrine TGF-β stimulation and derepression of this promoter by the Smad3-interacting HDAC5(931–1121). ROS-H14 cells showed an attenuated repression in response to TGF-β compared with control cells, reminiscent of the effect of TSA on repression (Figure 1A). The derepression of the osteocalcin promoter seen in ROS-H14 cells was inhibited by a dominant-negative type II TGF-β receptor that blocks autocrine TGF-β signaling. Under TβRII-DN expression, which blocks Smad3 recruitment to Runx2 at the promoter (Alliston et al, 2001), control and ROS-H14 cells showed a similar transcription level that was not affected by adding TGF-β (Figure 7C). The effect of HDAC5(931–1121) on repressible transcription from the osteocalcin promoter was selective, since ROS-H14 cells differed only minimally from control cells in transcription activation of the Smad7 promoter by TGF-β. The slightly decreased transcription from the Smad7 promoter in ROS-H14 cells may result from interference of HDAC5(931–1121) with the interaction of Smad3 with Smad4 or p300/CBP. These results suggest that interaction of Smad3 and HDAC4/5 mediates the repression of the osteocalcin gene by TGF-β.
We also conducted siRNA experiments in ROS17/2.8 cells to assess the requirement of HDAC4 and 5 for repression of osteocalcin mRNA expression by TGF-β (Figure 7D and E). The repression by TGF-β was not changed by treatment with control siRNA (Figure 7E). However, both HDAC4- and 5-specific siRNAs significantly decreased the repression of osteocalcin mRNA levels by TGF-β (Figure 7E), consistent with the decreased HDAC4 or 5 expression (Figure 7D). These results strongly suggest that HDAC4 and 5 mediate the repression of Runx2-mediated transcription of the osteocalcin gene.
Class IIa HDACs regulate osteoblast differentiation and its inhibition by TGF-β
Finally, we evaluated the roles of HDAC4/5 in osteoblast differentiation in cell culture. We have shown that TGF-β represses osteoblast differentiation through Smad3. Thus, Smad3 inhibited osteoblast differentiation and cooperated with TGF-β in repressing differentiation, while interference with Smad3 signaling enhanced differentiation and repressed the inhibition by TGF-β (Alliston et al, 2001). Since Smad3 repressed Runx2 function by recruiting class IIa HDAC4 and 5, we evaluated their role in osteoblast differentiation. caIB 2T3 cells were used since, unlike ROS17/2.8 cells, they undergo progressive osteoblast differentiation in culture and their differentiation is regulated by endogenous TGF-β/Smad3 signaling (Alliston et al, 2001).
Retrovirally infected caIB 2T3 cells, selected for HDAC5(931–1121) expression (Figure 8A), were evaluated for osteoblast differentiation, with or without added TGF-β. Interference with HDAC4/5 recruitment decreased the downregulation of osteocalcin mRNA by TGF-β (Figure 8B), consistent with decreased histone H4 deacetylation at the osteocalcin promoter in the presence of HDAC5(931–1121) (Figure 7B). siRNA-mediated downregulation of HDAC4 or 5 levels (Figure 8A) also decreased the downregulation of osteocalcin mRNA expression by TGF-β (Figure 8C), similarly to what we observed in ROS17/2.8 cells (Figure 7E). Cells expressing HDAC5(931–1121) showed enhanced differentiation in the absence of exogenous TGF-β, as assessed by calcified matrix deposition, and increased resistance to the inhibition by TGF-β (Figure 8D). These results resemble the enhanced osteoblast differentiation and inhibition of the repression of differentiation by TGF-β in the presence of a dominant-negative Smad3 mutant (Alliston et al, 2001). These observations are consistent with a role of class IIa HDACs as Smad3-associated corepressors and the role of Smad3 as intrinsic TGF-β-dependent inhibitor of Runx2 function and osteoblast differentiation.
Figure 8.
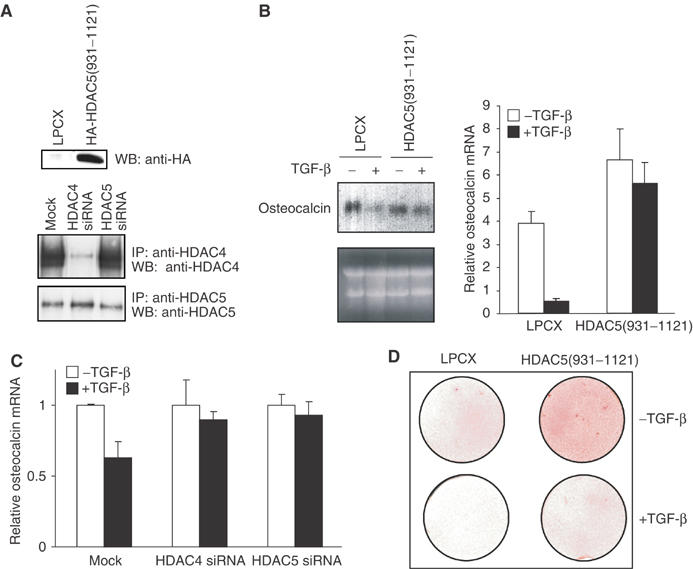
Class IIa HDACs contribute to the inhibition of osteoblast differentiation by TGF-β. (A) caIB 2T3 cells were infected with LPCX or LPCX-HA-HDAC5(931–1121) (upper panel) or transfected with HDAC4 or HDAC5 siRNAs (lower panel). Western analysis validated expression of HA-HDAC5(931–1121) (upper panel) or reduction of HDAC4 or 5 levels (lower panel). (B) HDAC5(931–1121) expression inhibits the reduction of osteocalcin mRNA expression by TGF-β in caIB 2T3 cells at day 6 in differentiation medium. Ethidium bromide staining shows similar RNA loading for Northern hybridization. Osteocalcin mRNA was quantified using real-time PCR and normalized to RPL19 mRNA expression, which is not affected by TGF-β. (C) HDAC4- or 5-specific siRNAs inhibit the repression of osteocalcin mRNA expression by TGF-β in caIB 2T3 cells at day 6 in differentiation medium. Real-time PCR values were normalized against RPL19 mRNA. (D) HDAC5 (931–1121) reduces the inhibition of matrix mineralization by TGF-β, as assessed by Alizarin Red staining on day 11 of differentiation.
Discussion
We have shown that TGF-β inhibits osteoblast differentiation through Smad3-mediated repression of Runx2 function (Alliston et al, 2001). We now address the mechanism of Smad3-mediated repression of Runx2 at the osteocalcin promoter. Our results lead to several conclusions. (1) TGF-β-activated Smad3 regulates chromatin remodeling, correlating repression and activation by Smad3 with histone deacetylation and acetylation, respectively. (2) At the osteocalcin promoter, TGF-β-activated Smad3 represses Runx2 through direct recruitment of class IIa HDACs, such as HDAC4 and 5. (3) We provide evidence that the control of chromatin remodeling through acetylation and deacetylation regulates the progression of osteoblast differentiation. (4) Class IIa HDACs act as cell-intrinsic regulators of osteoblast differentiation.
Linking histone acetylation and deacetylation to TGF-β-induced transcription regulation
Transcription levels depend on the dynamic state of histone acetylation. Histone acetylation correlates generally with transcriptionally active genes, while repression correlates with histone deacetylation by corepressors (Berger, 2002). This notion has been applied to the regulation of gene expression by extracellular signals. Thus, activation of nuclear receptors induces histone acetylation to activate genes through recruitment of p300 or CBP with their intrinsic acetylase activities, yet can also result in class I HDAC recruitment, with consequent histone deacetylation and repression (Glass and Rosenfeld, 2000). Thus, nuclear receptors can act as platforms for acetylases and deacetylases, from which chromatin remodeling is initiated. Class II HDACs have not been shown to initiate chromatin remodeling.
Smads have not been shown to be linked to chromatin remodeling. Such role is, however, suggested by their ability to recruit the CBP or p300 acetyltransferases, or corepressors that associate with class I HDACs (e.g., Evi-1, c-Ski, and SnoN) and inhibit transcription activation (Feng and Derynck, 2005). We now show that TGF-β induces, through Smad3 interaction with regulatory DNA sequences, changes in histone acetylation of target genes. In the same cells, TGF-β promotes histone acetylation to activate Smad7 expression, and deacetylation to repress osteocalcin transcription. The acetylation of the Smad7 promoter and equally rapid deacetylation of the osteocalcin promoter occur with a similar timing as TGF-β-induced DNA binding of Smads (Inman et al, 2002). Thus, similarly to nuclear receptors, Smads function as ligand-activated platforms that mediate histone acetylation and deacetylation and initiate chromatin remodeling. Unlike nuclear receptors, however, Smad3 recruits class IIa HDACs, thereby bypassing a requirement for additional corepressors. Whether Smads mediate histone acetylation or deacetylation, and activate or repress transcription, depends on the regulatory sequences and interactions with other transcription factors. Thus, in contrast to the OSE2 sequence of the osteocalcin promoter, Smad3 can cooperate with Runx1 at the regulatory sequences of the germline Igα gene promoter to enhance histone H4 acetylation (data not shown) and activate transcription (Zhang and Derynck, 2000; Alliston et al, 2001).
Smad3-mediated recruitment of class IIa HDACs effects repression
Little is known about the mechanisms of Smad-mediated repression, and Smads have not been shown to recruit HDACs into transcription complexes. Thus, TGF-β/Smad3 blocks MyoD and MEF2 functions through interference with protein interactions (Liu et al, 2001, 2004), resulting in HDAC-independent repression. In the repression of c-myc transcription by TGF-β, Smad3 interacts with E2F4/5-p107 at c-myc regulatory sequences, but HDACs were not shown to be involved (Chen et al, 2002). In BMP-mediated repression of Nkx3.2 function, Smad1 stabilizes the interaction of HDAC1 with Nkx3.2, but was not reported to interact with HDAC1 (Kim and Lassar, 2003).
We now show that class IIa HDACs are recruited by Smad3 into a complex at a Runx2-binding site in the osteocalcin promoter, where Runx2 stabilizes the Smad3/HDAC interaction and Smad3 stabilizes the Runx2/HDAC association. Thus, the interaction of Smad3 and Runx2 at the promoter is a prerequisite for efficient class IIa HDAC recruitment and TGF-β-dependent repression. As class IIa HDACs and Smads both shuttle between nucleus and cytoplasm, HDAC/Smad complexes may form prior to nuclear translocation and binding to Runx2 at promoter sequences. The low-level interaction of HDAC4 with Runx2 in the absence of added TGF-β, yet in the presence of autocrine TGF-β/Smad3 signaling (intrinsic in most cells), was recently reported by Vega et al (2004).
We found that HDAC4 and 5, which are both expressed in mesenchymal cells and osteoblasts, mediate, possibly in combination with other class IIa HDACs, the repression of Runx2 by TGF-β. Since both HDACs interacted similarly with Smad3, we assume that Smad3 can interact with all class IIa HDACs to mediate TGF-β-induced transcription repression in different cell types. Thus, HDAC7 could mediate TGF-β-induced repression in T lymphocytes through cooperation of Smad3 with Runx transcription factors (Pardali et al, 2000; Zhang and Derynck, 2000). In contrast to class IIa HDACs, class I HDACs and the class IIb HDAC6 did not cooperate with TGF-β in the repression of Runx2.
While we provide the first evidence for HDAC recruitment by Smads, an uncharacterized deacetylase activity was shown to interact with the MH1 domain of Smad3 (Liberati et al, 2001). The interactions of HDAC4 and 5 with the MH2 domain of Smad3 suggest that class IIa HDAC recruitment does not account for this activity.
A controlled balance between histone acetylation and deacetylation in osteoblast differentiation
The role of histone modifications in regulating transcription suggests that a signal-mediated balance between histone acetylation and deacetylation on specific genes controls differentiation. In osteoblast differentiation, histone H3 and H4 acetylation has been correlated with osteocalcin gene transcription. Thus, vitamin D3 enhances osteocalcin gene expression through increased histone H3/H4 acetylation at its promoter (Shen et al, 2003). We now show that TGF-β represses osteocalcin expression by promoting histone deacetylation through recruitment of class IIa HDACs by Smad3, and that this event contributes to the inhibition of osteoblast maturation. These observations support the notion that a controlled balance of histone acetylation and deacetylation regulates osteoblast differentiation.
HDAC4 and 5 have been shown to regulate myogenic differentiation through their association with MEF2 (Lu et al, 2000; McKinsey et al, 2000), but no effects of HDAC4/5 binding to MEF2 on the histone acetylation status of myogenic regulatory sequences were shown. During the preparation of this manuscript, HDAC4 was shown to be made in prehypertrophic chondrocytes and regulate chondrocyte hypertrophy by inhibiting the activity of Runx2, thus affecting endochondral bone formation (Vega et al, 2004). We now show that HDAC4 and/or 5 regulate Runx2 activity, osteocalcin promoter acetylation, and osteoblast differentiation, depending on TGF-β/Smad3 signaling. Consistent with these results, HDAC4 and 5 are expressed in developing bone, primary osteoblasts, and preosteoblast-like cell lines. In (pre)osteoblasts, HDAC4 and 5 are coexpressed with Runx2 (Figure 4), while Smad3 and TGF-β receptors are also expressed in preosteoblasts (Sakou et al, 1999). Thus, the coexpression of these effectors in preosteoblasts supports an autocrine regulation of Runx2 function and chromatin remodeling in osteogenic differentiation.
Interference with the HDAC4/5 interaction with Smad3 blocked the repression of osteocalcin expression by TGF-β and enhanced osteoblast differentiation. Since HDAC5(931–1121) interfered with the association of HDAC4 and 5 with Smad3, it likely also disrupts possible interactions of other class IIa HDACs, such as HDAC9 (Zhou et al, 2001), whose expression in osteoblasts was not evaluated. The striking effect of the dominant-negative, as well as siRNA-mediated, interference on differentiation thus identifies class IIa HDACs as cell-intrinsic regulators of osteoblast differentiation. These effects were strikingly similar to the effects of interference with Smad3 function, which enhances osteoblast differentiation and inhibits the repression of osteocalcin expression and osteoblast differentiation by TGF-β (Alliston et al, 2001). It is therefore likely that the cell-intrinsic regulation by class IIa HDACs is linked to their role in autocrine TGF-β/Smad3 repression of Runx2 function. Further studies will clarify in detail the roles of class IIa HDACs in osteoblast differentiation.
Materials and methods
Plasmids
The reporter plasmids 6OSE2-luc and −147OC-luc (Alliston et al, 2001), 3TP-Luc (Carcamo et al, 1995) and Smad7Pro-Luc (Nagarajan et al, 1999) were described. The Gal4-Luc (FR-Luc) reporter was from Stratagene.
Expression vectors for HDAC1–7 (Grozinger et al, 1999; Miska et al, 1999; Fischle et al, 2001) were provided by S Schreiber, T Kouzarides, and E Verdin. To generate tagged HDAC4 or 5, or deletion mutants of HDAC5, PCR-generated coding regions were inserted into the EcoRI/XbaI sites of pXF1F or pXF1H (Feng et al, 1998). To generate a retroviral vector expressing HA-tagged HDAC5(931–1131), the PCR-generated coding region was inserted into the HpaI site of LPCX (Choy et al, 2000). N-terminally Flag-tagged Smad3 and derivatives were expressed from pRK5 (Zhang et al, 1998). Expression plasmids for Flag-tagged Runx2 (Alliston et al, 2001), GST-fused Smads or Smad fragments (Zhang et al, 1998), HA-RIRII, that is, a chimera of the TβRII and TβRI cytoplasmic domains (Feng and Derynck, 1996), and Gal–Smad3 (Feng et al, 1998) were described. Details of plasmid constructions will be provided upon request.
Cell culture and transfections
Primary osteoblasts were isolated from the calvariae of 2-day-old mice as described (Alliston et al, 2001). NIH3T3 and ROS17/2.8 cells were grown in DMEM with 10% FBS. Smad3−/− and wt MEFs were cultured in DMEM with 20% FBS. Cells were plated at 2 × 105 cells/well in six-well plates and transfected using Effectene reagents (Qiagen). At 1 day after transfection, cells were transferred to medium with 0.2% FBS with or without 1–5 ng/ml TGF-β for 16 h. Luciferase activities were assayed for and normalized to β-galactosidase from a cotransfected β-galactosidase plasmid.
Chromatin immunoprecipitations
To examine the changes in acetylation on the indicated genes, ROS17/2.8 cells were treated with or without 5 ng/ml TGF-β, and ChIP assays were carried out using anti-acetylated H3 or H4 antibodies (Upstate Biotechnologies) as described (Liu et al, 2004). To detect HDAC4 at the osteocalcin promoter, ROS17/2.8 cells were treated with 5 ng/ml TGF-β for 4 h, followed by ChIP assay using anti-HDAC4 antibody (Abcam). For HDAC5, ROS17/2.8 cells transfected to express Flag-tagged HDAC5 were subjected to ChIP assay using anti-Flag antibody (Sigma). The PCR primers were: rat osteocalcin promoter, 5′-GCAGCTCCGGGAAGAGGTCTG-3′ (forward) and 5′-GCTAGGTCTGCACCGAGTTGC-3′ (reverse); rat Smad7 promoter, 5′-CTCTGTAGACCTGGGAGAGGGTGG-3′ (forward) and 5′-CCCTCCGCTCGGCTGGTTCCACT-3′ (reverse).
Immunoprecipitations and immunoblotting
Transfected COS cells were lysed by sonication in 25 mM Tris–HCl, pH 7.5, 150 mM NaCl, 1% Triton X-100, 10% glycerol, and protease inhibitor cocktail. Lysates were subjected to immunoprecipitation with anti-Flag M2 agarose (Sigma), followed by Western blotting using anti-Flag (Sigma), anti-HA (Covance), or anti-Myc (Roche Molecular Biochemicals) antibodies. To show a Runx2/Smad3/HDAC5 complex, lysates were subjected to immunoprecipitation with anti-Flag M2 agarose (Sigma) and immune complexes were eluted with Flag peptide (Sigma). Eluates were immunoprecipitated with anti-Myc (Covance) followed by Western blotting. To detect endogenous protein interactions, ROS17/2.8 cells were cultured for 3 h in DMEM with 0.2% FBS with or without 10 ng/ml TGF-β. Cells were harvested and lysed by sonication in HKMG buffer (Chen et al, 2001). The lysates were precleared with IgG and immunoprecipitated with rabbit anti-Smad3 (Zymed), anti-HDAC5 (Abcam), or IgG (Zymed), followed by Western blotting using mouse anti-Smad2/3 (BD Transduction Laboratories), rabbit anti-HDAC4 (Cell Signaling Technologies), rabbit anti-HDAC5 (Cell Signaling Technologies), or rabbit anti-Runx2 (Santa Cruz Biotech).
GST adsorption and DNA precipitation assays
Binding of 35S-labeled HDAC4 and 5 to GST fusion proteins of Smads was determined as described (Liu et al, 2001). For DNA precipitation assays, transfected COS cells expressing Runx2, HDAC5, Smad3, and/or Smad4 were lysed by sonication in HKMG buffer (Chen et al, 2001) and lysates were processed as described (Liu et al, 2004) using biotinylated double-strand 2xOSE2 oligonucleotide (Alliston et al, 2001).
In situ hybridization
Mouse HDAC4 and 5 cDNAs were generated by PCR amplication of mouse brain cDNA using primers for human HDAC4 (bp 19–627) or 5 (bp 1210–1532) (Grozinger et al, 1999). The cDNA segments to generate osteocalcin and Runx2 cRNAs have been described (Ferguson et al, 1999). In situ hybridization was performed using 35S-labeled cRNAs as described (Ferguson et al, 1999). Sections were counterstained with DAPI to visualize the nuclei. Dark-field illumination was used to reveal in situ labeling.
RNA isolation and RT–PCR
RNA was isolated from ROS17/2.8, MC3T3-E1, or caIB 2T3 cells (Alliston et al, 2001), and used as a template for reverse trancriptase and random hexamer primers. For PCR amplification of HDAC4 and 5 sequences, the primer sequences were: HDAC4, 5′-CAGATGGACTTTCTGGCCG-3′ (forward) and 5′-GAGCTGCTGCAGCTTCTG-3′ (reverse); HDAC5, 5′-TCGCACCTCACCGCCTCCCCGAAGCTG-3′ (forward) and 5′-AGAGGTCGGTGCCTCGGGAGCTTACCCACCGT -3′ (reverse).
RNA interference
dsRNAi to target rat or mouse HDAC4 or 5 expression was synthesized by Dharmacon or Qiagen. The coding sequences were: HDAC4, 5′-GAACAUAUCAAGCAGCAGCdTdT-3′; rat HDAC5, 5′-GGAUGGCACUGUUAUUAGUdTdT-3′; mouse HDAC5, 5′-CGGCCUCGGAACCCAACUU. dsRNAi to GFP or Renilla luciferase (Dharmacon) was used as control. ROS17/2.8 cells were transfected with dsRNAi using Oligofectamine (Invitrogen) three times with 48-h intervals, transferred to DMEM with 0.2% FBS with or without 2 ng/ml TGF-β for 24 h, and RNA was prepared using RNeasy (Qiagen). mRNAs were quantified by real-time PCR using TaqMan probes and the ABI Prism 7000 system (Applied Biosystems), and normalized against GAPDH or RPL19 mRNA. Primer and probe sequences for osteocalcin, GAPDH, and RPL19 mRNAs are available upon request. For Western analysis, ROS17/2.8 cells, transfected with dsRNAi twice with 48-h intervals, were analysed using rabbit anti-HDAC4 or -HDAC5 antibodies (Cell Signaling Technologies).
Generation of stable cell lines
ROS17/2.8 cells were transfected with pRK5-HA-HDAC5(931–1121) or empty pRK5, and a puromycin resistance gene using Effectene reagents (Qiagen), and selected in 2 μg/ml puromycin for 1–2 weeks. caIB 2T3 cell populations expressing HA-tagged HDAC5 (931–1121) were infected using a LPCX-based retroviral vector (Choy et al, 2000). Expression levels of HDAC5(931–1121) were verified using Western blotting.
Osteoblast differentiation
Infected caIB 2T3 cells were grown in α-MEM, 10% FBS, 2 μg/ml puromycin, and transfected with dsRNAi twice at 48-h intervals using Oligofectamine (Invitrogen). Lysates were immunoprecipitated using rabbit anti-HDAC4 and -HDAC5 (Cell Signaling Technologies), followed by Western analysis. For differentiation, cells were transferred upon confluence into α-MEM, 2% FBS, 100 μg/ml ascorbic acid, 5 mM β-glycerophosphate with or without 1 ng/ml TGF-β. mRNA was analyzed after 6 days (Alliston et al, 2001) and mineralization was assayed at day 11 using Alizarin Red-S (Beck et al, 1998).
Acknowledgments
We thank T Kouzarides, S Schreiber and E Verdin for plasmids, and D Hu for help with in situ hybridization. This research was supported by grants RO1-CA63101 and P60 DE13058 to RD and a Hulda Irene Duggan Arthritis Investigator award to TA.
References
- Alliston T, Choy L, Ducy P, Karsenty G, Derynck R (2001) TGF-β-induced repression of CBFA1 by Smad3 decreases cbfa1 and osteocalcin expression and inhibits osteoblast differentiation. EMBO J 20: 2254–2272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alliston TN, Derynck R (2000) Transforming growth factor-β in skeletal development and maintenance. In Skeletal Growth Factors, Canalis E (ed), pp 233–249. Philadelphia: Lippincott Williams & Wilkins Press [Google Scholar]
- Beck GR Jr, Sullivan EC, Moran E, Zerler B (1998) Relationship mineralization in differentiating MC3T3-E1 osteoblasts. J Cell Biochem 68: 269–280 [DOI] [PubMed] [Google Scholar]
- Berger SL (2002) Histone modifications in transcriptional regulation. Curr Opin Genet Dev 12: 142–148 [DOI] [PubMed] [Google Scholar]
- Carcamo J, Zentella A, Massagué J (1995) Disruption of transforming growth factor β signaling by a mutation that prevents transphosphorylation within the receptor complex. Mol Cell Biol 15: 1573–1581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CR, Kang Y, Massagué J (2001) Defective repression of c-myc in breast cancer cells: a loss at the core of the transforming growth factor β growth arrest program. Proc Natl Acad Sci USA 98: 992–999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CR, Kang Y, Siegel PM, Massagué J (2002) E2F4/5 and p107 as Smad cofactors linking the TGF β receptor to c-myc repression. Cell 110: 19–32 [DOI] [PubMed] [Google Scholar]
- Choy L, Derynck R (2003) Transforming growth factor-β inhibits adipocyte differentiation by Smad3 interacting with CCAAT/enhancer-binding protein (C/EBP) and repressing C/EBP transactivation function. J Biol Chem 278: 9609–9619 [DOI] [PubMed] [Google Scholar]
- Choy L, Skillington J, Derynck R (2000) Roles of autocrine TGF-β receptor and Smad signaling in adipocyte differentiation. J Cell Biol 149: 667–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dequiedt F, Kasler H, Fischle W, Kiermer V, Weinstein M, Herndier BG, Verdin E (2003) HDAC7, a thymus-specific class II histone deacetylase, regulates Nur77 transcription and TCR-mediated apoptosis. Immunity 18: 687–698 [DOI] [PubMed] [Google Scholar]
- Feng XH, Derynck R (1996) Ligand-independent activation of transforming growth factor (TGF) β signaling pathways by heteromeric cytoplasmic domains of TGF-β receptors. J Biol Chem 271: 13123–13129 [DOI] [PubMed] [Google Scholar]
- Feng XH, Derynck R (2005) Specificity and versatility in TGF-β signaling through Smads. Ann Rev Cell Dev Biol, in press [DOI] [PubMed] [Google Scholar]
- Feng XH, Zhang Y, Wu RY, Derynck R (1998) The tumor suppressor Smad4/DPC4 and transcriptional adaptor CBP/p300 are coactivators for Smad3 in TGF-β-induced transcriptional activation. Genes Dev 12: 2153–2163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson C, Alpern E, Miclau T, Helms JA (1999) Does adult fracture repair recapitulate embryonic skeletal formation? Mech Dev 87: 57–66 [DOI] [PubMed] [Google Scholar]
- Ferreira R, Magnaghi-Jaulin L, Robin P, Harel-Bellan A, Trouche D (1998) The three members of the pocket proteins family share the ability to repress E2F activity through recruitment of a histone deacetylase. Proc Natl Acad Sci USA 95: 10493–10498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischle W, Dequiedt F, Fillion M, Hendzel MJ, Voelter W, Verdin E (2001) Human HDAC7 histone deacetylase activity is associated with HDAC3 in vivo. J Biol Chem 276: 35826–35835 [DOI] [PubMed] [Google Scholar]
- Glass CK, Rosenfeld MG (2000) The coregulator exchange in transcriptional functions of nuclear receptors. Genes Dev 14: 121–141 [PubMed] [Google Scholar]
- Grozinger CM, Hassig CA, Schreiber SL (1999) Three proteins define a class of human histone deacetylases related to yeast Hda1p. Proc Natl Acad Sci USA 96: 4868–4873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grozinger CM, Schreiber SL (2000) Regulation of histone deacetylase 4 and 5 and transcriptional activity by 14-3-3-dependent cellular localization. Proc Natl Acad Sci USA 97: 7835–7840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada S, Rodan GA (2003) Control of osteoblast function and regulation of bone mass. Nature 423: 349–355 [DOI] [PubMed] [Google Scholar]
- Hua X, Miller ZA, Benchabane H, Wrana JL, Lodish HF (2000) Synergism between transcription factors TFE3 and Smad3 in transforming growth factor-β-induced transcription of the Smad7 gene. J Biol Chem 275: 33205–33208 [DOI] [PubMed] [Google Scholar]
- Inman GJ, Nicolas FJ, Hill CS (2002) Nucleocytoplasmic shuttling of Smads 2, 3, and 4 permits sensing of TGF-β receptor activity. Mol Cell 10: 283–294 [DOI] [PubMed] [Google Scholar]
- Kim DW, Lassar AB (2003) Smad-dependent recruitment of a histone deacetylase/Sin3A complex modulates the bone morphogenetic protein-dependent transcriptional repressor activity of Nkx3.2. Mol Cell Biol 23: 8704–8717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouzarides T (2000) Acetylation: a regulatory modification to rival phosphorylation? EMBO J 19: 1176–1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberati NT, Moniwa M, Borton AJ, Davie JR, Wang XF (2001) An essential role for Mad homology domain 1 in the association of Smad3 with histone deacetylase activity. J Biol Chem 276: 22595–22603 [DOI] [PubMed] [Google Scholar]
- Liu D, Black BL, Derynck R (2001) TGF-β inhibits muscle differentiation through functional repression of myogenic transcription factors by Smad3. Genes Dev 15: 2950–2966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D, Kang JS, Derynck R (2004) TGF-β-activated Smad3 represses MEF2-dependent transcription in myogenic differentiation. EMBO J 23: 1557–1566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, McKinsey TA, Zhang CL, Olson EN (2000) Regulation of skeletal myogenesis by association of the MEF2 transcription factor with class II histone deacetylases. Mol Cell 6: 233–244 [DOI] [PubMed] [Google Scholar]
- McKinsey TA, Zhang CL, Lu J, Olson EN (2000) Signal-dependent nuclear export of a histone deacetylase regulates muscle differentiation. Nature 408: 106–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miska EA, Karlsson C, Langley E, Nielsen SJ, Pines J, Kouzarides T (1999) HDAC4 deacetylase associates with and represses the MEF2 transcription factor. EMBO J 18: 5099–5107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagarajan RP, Zhang J, Li W, Chen Y (1999) Regulation of Smad7 promoter by direct association with Smad3 and Smad4. J Biol Chem 274: 33412–33418 [DOI] [PubMed] [Google Scholar]
- Pardali E, Xie XQ, Tsapogas P, Itoh S, Arvanitidis K, Heldin CH, ten Dijke P, Grundstrom T, Sideras P (2000) Smad and AML proteins synergistically confer transforming growth factor β1 responsiveness to human germ-line IgA genes. J Biol Chem 275: 3552–3560 [DOI] [PubMed] [Google Scholar]
- Sakou T, Onishi T, Yamamoto T, Nagamine T, Sampath T, ten Dijke P (1999) Localization of Smads, the TGF-β family intracellular signaling components during endochondral ossification. J Bone Miner Res 14: 1145–1152 [DOI] [PubMed] [Google Scholar]
- Shen J, Hovhannisyan H, Lian JB, Montecino MA, Stein GS, Stein JL, Van Wijnen AJ (2003) Transcriptional induction of the osteocalcin gene during osteoblast differentiation involves acetylation of histones H3 and H4. Mol Endocrinol 17: 743–756 [DOI] [PubMed] [Google Scholar]
- Vega RB, Matsuda K, Oh J, Barbosa AC, Yang X, Meadows E, McAnally J, Pomajzl C, Shelton JM, Richardson J, Karsenty G, Olson E (2004) Histone deacetylase 4 controls chondrocyte hypertrophy during skeletogenesis. Cell 119: 555–566 [DOI] [PubMed] [Google Scholar]
- Verdin E, Dequiedt F, Kasler HG (2003) Class II histone deacetylases: versatile regulators. Trends Genet 19: 286–293 [DOI] [PubMed] [Google Scholar]
- Yoshida M, Kijima M, Akita M, Beppu T (1990) Potent and specific inhibition of mammalian histone deacetylase both in vivo and in vitro by trichostatin A. J Biol Chem 265: 17174–17179 [PubMed] [Google Scholar]
- Zhang Y, Derynck R (2000) Transcriptional regulation of the transforming growth factor-β-inducible mouse germ line Ig α constant region gene by functional cooperation of Smad, CREB, and AML family members. J Biol Chem 275: 16979–16985 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Feng XH, Derynck R (1998) Smad3 and Smad4 cooperate with c-Jun/c-Fos to mediate TGF-β-induced transcription. Nature 394: 909–913 [DOI] [PubMed] [Google Scholar]
- Zhou X, Marks PA, Rifkind RA, Richon VM (2001) Cloning and characterization of a histone deacetylase, HDAC9. Proc Natl Acad Sci USA 98: 10572–10577 [DOI] [PMC free article] [PubMed] [Google Scholar]