

Abstract
Carbonic anhydrases (CA) catalyze the reversible conversion of CO2 to HCO3−. Some bicarbonate transporters bind CA, forming a complex called a transport metabolon, to maximize the coupled catalytic/transport flux. SLC26A6, a plasma membrane Cl−/HCO3− exchanger with a suggested role in pancreatic HCO3− secretion, was found to bind the cytoplasmic enzyme CAII. Mutation of the identified CAII binding (CAB) site greatly reduced SLC26A6 activity, demonstrating the importance of the interaction. Regulation of SLC26A6 bicarbonate transport by protein kinase C (PKC) was investigated. Angiotensin II (AngII), which activates PKC, decreased Cl−/HCO3− exchange in cells coexpressing SLC26A6 and AT1a-AngII receptor. Activation of PKC reduced SLC26A6/CAII association in immunoprecipitates. Similarly, PKC activation displaced CAII from the plasma membrane, as monitored by immunofluorescence. Finally, mutation of a PKC site adjacent to the SLC26A6 CAB site rendered the transporter unresponsive to PKC. PKC therefore reduces CAII/SLC26A6 interaction, reducing bicarbonate transport rate. Taken together, our data support a mechanism for acute regulation of membrane transport: metabolon disruption.
Keywords: bicarbonate transport, carbonic anhydrase, cystic fibrosis, protein kinase C, SLC26A6
Introduction
Bicarbonate is a membrane-impermeable molecule central to cellular, blood and whole-body pH homeostasis. Transmembrane movement of bicarbonate is facilitated by integral membrane transport proteins (Sterling and Casey, 2002). Many factors influence the rate of HCO3− transport. However, interactions between bicarbonate transporters (BTs) and carbonic anhydrase (CA) enzymes have emerged as critical to bicarbonate flux (Vince and Reithmeier, 1998; Sterling et al, 2001, 2002a; Gross et al, 2002; Loiselle et al, 2004). CAs catalyze the reversible conversion between CO2 and HCO3−, and thus both produce and consume the transport substrate of BT. Localization of CA to the surface of a BT maximizes the rate of bicarbonate transport by maximizing the transmembrane bicarbonate gradient local to the BT. The complex of a BT with a CA has been called a bicarbonate transport metabolon and is central to the physiology of these transporters. Other transport metabolons, consisting of a transporter and the enzyme that metabolizes the transport substrate, have yet to be identified, but there are many examples where the cellular physiology suggests they occur.
Three separate groups of proteins facilitate bicarbonate transport: SLC4a Cl−/HCO3− exchangers, SLC4a sodium-coupled transporters and certain members of the SLC26a family (McMurtrie et al, 2004). Within each of these groups there are now examples of CA/BT interactions, representing bicarbonate transport metabolons (Vince and Reithmeier, 1998; Sterling et al, 2001, 2002b; Gross et al, 2002; McMurtrie et al, 2004). SLC4a Cl−/HCO3− exchangers bind both cytosolic CAII and extracellular CAIV to maximize bicarbonate transport activity (Vince and Reithmeier, 1998; Sterling et al, 2001, 2002a). Functional and physical interactions between the human sodium-coupled NBC1 (SLC4a4) Na+/HCO3− cotransporter and CAII have also been found (Gross et al, 2002). A member of the SLC26A family, DRA (SLC26A3), also requires cytosolic CAII to manifest full HCO3− transport, but no evidence was found for CAII binding by DRA (Sterling et al, 2002b). The present report is the first identification of an SLC26a BT that interacts physically with CA.
In all, 14 mammalian CA isoforms, CAI-XIV (Schwartz, 2002), catalyze the reversible hydration/dehydration of CO2 and HCO3−, respectively. Ubiquitous cytoplasmic CAII is a 29 kDa polypeptide characterized by high enzymatic activity and great susceptibility to inhibition by sulfonamides such as acetazolamide (ACTZ) (Spicer et al, 1990). CAII is strongly associated with the inner leaflet of the apical plasma membrane in human pancreatic duct cells, where it may couple to some HCO3− transport proteins (Mahieu et al, 1994; Alvarez et al, 2001b).
Members of the SLC4A anion exchanger (AE) family (Sterling and Casey, 2002) and the SLC26A anion transport gene family (Everett and Green, 1999), mediate anion exchange at the plasma membrane of mammalian cells. The SLC26A family (SLC26A1–A11) transports a range of anionic substrates (Mount and Romero, 2004). SLC26A6 has been cloned from both humans (SLC26A6) (Lohi et al, 2000; Waldegger et al, 2001) and mice (Slc26a6, also known as CFEX) (Knauf et al, 2001).
Substrate anion specificity and transport stoichiometry of anion exchange by SLC26A6 have been studied. Using an anion exchange mechanism, SLC26A6 transports a variety of anions, including sulfate, formate, oxalate, nitrate, and iodide (Mount and Romero, 2004). While the most physiologically relevant transport is Cl−/HCO3− and Cl−/OH− exchange (Ko et al, 2002; Alvarez et al, 2004; Chernova et al, 2005), the anion transport stoichiometry of SLC26A6 is controversial. Mouse Slc26a6 has been reported to facilitate Cl−/HCO3− exchange by an electrogenic mechanism (Ko et al, 2002; Xie et al, 2002). In contrast, a recent report found that Cl−/HCO3− exchange by both mouse Slc26a6 and the human ortholog, SLC26A6, was electroneutral (Chernova et al, 2005).
Slc26a6, identified in the brush border membrane of renal proximal tubule cells, has been reported as the candidate protein mediating Cl−/formate exchange, contributing to renal NaCl reabsorption (Knauf et al, 2001). SLC26A6 is also abundant in small intestine, where it performs apical Cl−/HCO3− exchange (Wang et al, 2002). The phenotype of an Slc26a6 knockout mouse supports a role of the transporter in renal formate and oxalate transport and significant contribution to duodenal bicarbonate secretion (Wang et al, 2005). SLC26A6 is also present in the heart and stomach (Petrovic et al, 2002; Alvarez et al, 2004).
In pancreatic duct, which secretes a fluid containing up to 140 mM HCO3−, SLC26A6 has a major role in apical HCO3− secretion, stimulated by the cystic fibrosis gene product, CFTR (Ko et al, 2002; Simpson et al, 2005). Failure of bicarbonate secretion leads to the pancreatic insufficiency prevalent in cystic fibrosis (CF) (Steward et al, 2005).
Protein kinase C (PKC) inhibits pancreatic bicarbonate secretion. The pancreas has a local renin–angiotensin system and, in exocrine pancreas, angiotensin II (AngII) receptor subtypes AT1 and AT2 are found in pancreatic ducts (Tsang et al, 2004). AngII inhibits anion secretion in pancreatic cells, which is blocked upon PKC inhibition, suggesting that AngII acts through PKC (Cheng et al, 1999). In addition, substance P inhibits an AE on the luminal membrane of pancreatic ducts, in a PKC-dependent manner. This PKC-inhibited AE is blocked by stilbene disulfonates (Hegyi et al, 2005), in a mode consistent with SLC26A6. The effects of PKC on SLC26A6 have not, however, been studied previously.
Little is known about mechanisms that regulate SLC26A6 transport activity. A recent report showed reciprocal regulatory interaction between CFTR and the SLC26A transporters SLC26A3 and SLC26A6 when expressed in HEK293 cells (Ko et al, 2004). SLC26A3 markedly activated CFTR, facilitated by PDZ ligands and binding of the SLC26A STAS domain to the CFTR R domain (Ko et al, 2004). SLC26A family members share a conserved C-terminal cytosolic domain, which, interestingly, is also found in bacterial anti-sigma factor antagonists (Aravind and Koonin, 2000). The STAS domain is named for sulfate transporters and bacterial anti-sigma factor antagonists. Expression of recombinant STAS domain activates CFTR (Ko et al, 2004). In addition, CFTR and SLC26A transporters colocalize to the luminal membrane of native pancreatic duct cells.
This manuscript provides a possible molecular mechanism for the observation that PKC inhibits pancreatic bicarbonate secretion. We examined the role of the bicarbonate transport metabolon in the regulation of SLC26A6 Cl−/HCO3−exchange activity. We found that SLC26A6 is the first member of the SLC26A family confirmed to bind CAII, and is thus the first shown to form a transport metabolon. The binding site for CAII on SLC26A6 is adjacent to the PKC phosphorylation site responsible for inhibition of SLC26A6 transport. PKC disruption of CAII binding is coincident with a decrease in transport rate of SLC26A6. Taken together, our data support a new model for regulation of membrane transport: metabolon disruption.
Results
Physical and functional interactions between CAII and the human SLC26A6 Cl−/HCO3− exchanger
C-terminal cytoplasmic domains of some BTs bind CAII to activate bicarbonate transport. However, interactions between CAII and any SLC26A family member have not been reported. Comparison of the amino-acid sequences of cytoplasmic C-terminal regions of SLC26A6 and SLC26A3 revealed that SLC26A6 has consensus CAII binding (CAB) sites in its C-terminal tail but that SLC26A3, which does not bind CAII (Sterling et al, 2002b), does not (Figure 1A). Seven potential CAB sites, consisting of a hydrophobic residue followed by four amino acids, with at least two acidic residues, were identified at the extreme C-terminus of the SLC26A6 protein (Vince and Reithmeier, 1998) (Figure 1A). To examine whether SLC26A6 cytoplasmic domain could bind CAII, a glutathione S-transferase (GST) fusion protein corresponding to SLC26A6 amino acids Q497–D633 (GSTA6–Q497–D633) was constructed. GST alone and GSTA6–Q497–D633 were resolved by SDS–PAGE and blotted onto a membrane. The blot was overlaid with CAII-containing lysates and the presence of CAII adherent to the blot assessed by immunoblotting. The blot showed specific binding of CAII to GSTA6–Q497–D633 (Figure 1B). Quantification of GST present in the GST and GSTA6–Q497–D633 bands (Figure 1C) and the amount of CAII bound to these bands revealed that the CAII/GST and CAII/GSTA6–Q497–D633 ratios were 0.05±0.01 units and 1.41±0.60 units, respectively (n=3, P<0.05). GSTA6–Q497–D633 thus binds 28 times more CAII than does GST alone.
Figure 1.
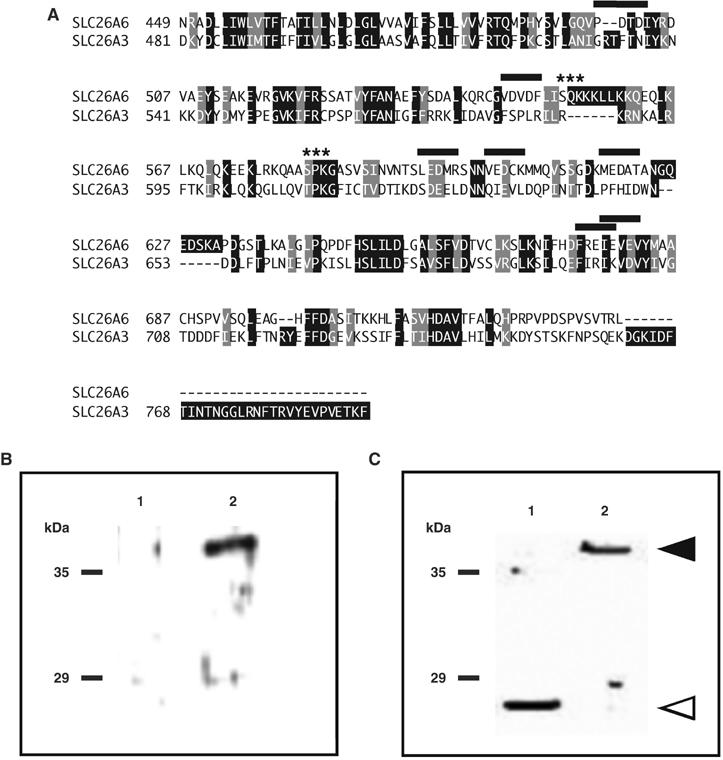
SLC26A6 C-terminal region interacts with CAII. (A) Amino-acid sequence alignment of human bicarbonate transport proteins, SLC26A6 (GenBank AF279265), and SLC26A3 (GenBank L02785). Shading indicates sequence identity (black) and sequence similarity (gray). Potential CAII-binding sites, consisting of a hydrophobic residue followed by four residues, two of which are acidic, are indicated (black overline). Asterisks mark consensus PKC phosphorylation sites (Expert Protein Analysis System, http://ca.expasy.org/). (B–C) In all, 10 μg of either GST alone (1) or GSTA6-Q497D633 (2) were resolved by SDS–PAGE on 10% acrylamide gels and transferred to PVDF membranes. Blots were incubated with a lysate of HEK293 cells, which endogenously express CAII. Blots were then probed with anti-CAII antibody (B) or anti-GST antibody (C). Arrowheads indicate the migration positions of GST alone (open) and GSTA6-Q496-D633 (filled).
Binding of CAII to the Q497–D633 amino-acid region of the SLC26A6 C-terminus was further assessed by a solid-phase binding assay. CAII was immobilized on microtiter plates and incubated with different concentrations of GST and GSTA6–Q497–D633. GST associated with the plate was detected with an anti-GST antibody. GSTA6–Q497–D633 exhibited saturable binding to immobilized CAII, with a half-maximal binding occurring at 141±7 nM (calculated from data corrected for GST binding alone, not shown), whereas GST alone bound nonspecifically, as indicated by the linearity of binding and lack of saturation (Figure 2A). We conclude that the Q497–D633 region may mediate the interaction of SLC26A6 with CAII.
Figure 2.
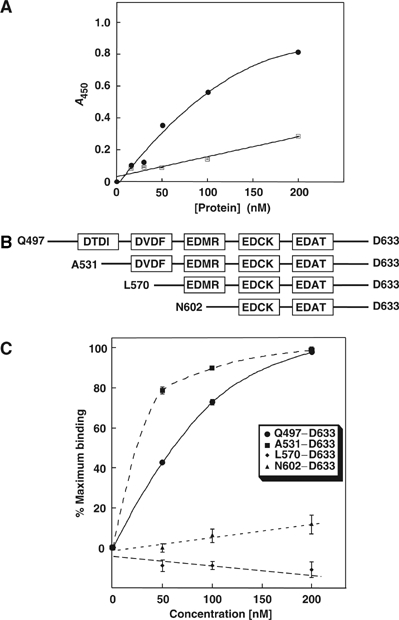
Identification of the CAII-binding site in the C-terminal region of SLC26A6. (A) CAII, immobilized on a microtiter dish, was incubated with various concentrations of GST (squares) or GSTA6–Q496–D633 (circles). Bound GST, or GST-fusion protein was detected by an enzyme-linked immunosorbant assay. (B) GST fusion proteins correspond to the entire SLC26A6 C-terminus (Q497-D633) and regions progressively truncated from the N-terminus, as indicated in the diagram. Truncation mutants were designed to include different consensus CAII-binding motifs (boxes). (C) Plate-immobilized CAII was incubated with various concentrations of SLC26A6 C-terminal GST-fusion proteins Q497–D633 (•), A531–D633 (▪), L570–D633 (♦) and N602–D633 (▴), and binding was monitored. GST-alone binding has been subtracted.
To identify the CAB site in the C-terminal tail, we focused on five potential CAB sites in the Q497–D633 region. A series of GST fusion proteins were constructed, spanning the Q497–D633 region, but sequentially deleting the consensus CAB sites (Figure 2B). Microtiter dish-binding assays revealed that GSTA6–Q497–D633 and GSTA6–A531–D633 bound CAII, but no binding was detected for GSTA6–L570–D633 or GSTA6–N602–D633 (Figure 2C). We conclude that the SLC26A6 D546–F549 region binds CAII.
Since CAII binds the C-terminus of SLC26A6 with high affinity, we examined the influence of CAII on SLC26A6 HCO3− transport activity. Rates of pHi alkalinization (Cl− efflux phase of assays) were assessed for SLC26A6-transfected cells before and 10 min after treatment with the membrane-permeant CA inhibitor, ACTZ, 150 μM (Figure 3A). ACTZ, which has no direct effects on the BTs so far examined (Cousin and Motais, 1976), reduced the rate of pHi change associated with HCO3− movement by 55±3% (n=3, P<0.05) (Figure 3B). As a control, two consecutive Cl−/HCO3− exchange assays were performed in the absence of ACTZ. In this case, the rates of pHi change in the consecutive assays were indistinguishable (not shown). The anion exchange inhibitor, 4,4′-diisothiocyanatostilbene-2,2′-disulfonic acid, disodium salt (DIDS) (1 mM), reduced the rate of transport by 92±1% (n=3, P<0.05), as reported previously (Xie et al, 2002; Lohi et al, 2003) (Figure 3B). It is important to note that HEK293 cells endogenously express CAII (Sterling et al, 2001). Results measured during HCO3− efflux (Cl− influx phase of assays) were essentially the same as those found during cellular alkalinization (Figure 3B, black versus white bars). These data suggest that functional CAII is required for full SLC26A6 transport activity. All transport rates have been corrected for background activity of HEK293 cells transfected with empty vector.
Figure 3.
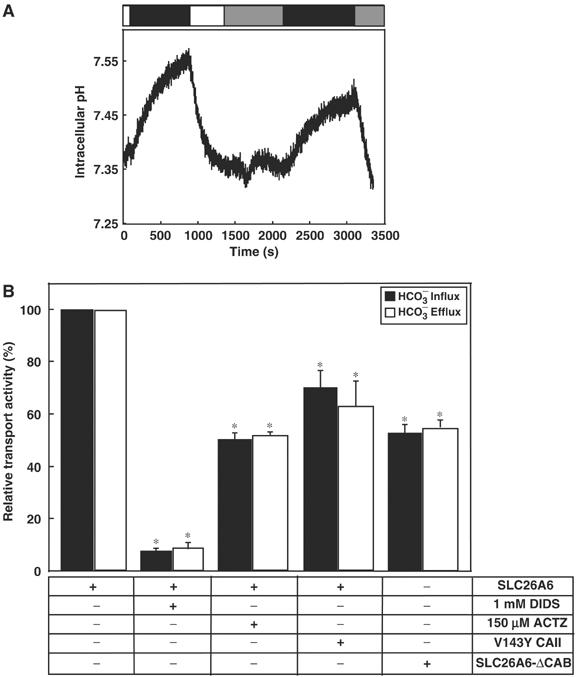
Inhibition of SLC26A6 Cl−/HCO3− exchange activity by manipulation of CA. (A) HEK293 cells, individually transfected with SLC26A6, SLC26A6-ΔCAB, or cotransfected with SLC26A6 and V143Y CAII cDNAs, were loaded with BCECF-AM. Cells were perfused alternately with Cl−-containing (open bar) and Cl−-free (black bar) Ringer's. In some experiments, cells were incubated with 1 mM DIDS between the first and second cycles of buffer switching. SLC26A6-transfected HEK293 cells were switched from Cl−-containing to Cl−-free Ringer's buffer and the process was repeated, but in the presence of the membrane-permeant CA inhibitor 150 μM ACTZ (gray bar). (B) Cl−/HCO3− exchange activity, relative to WT SLC26A6, for SLC26A6 or SLC26A6-ΔCAB expressed alone or coexpressed with functionally inactive V143Y CAII (n=4–6). *P<0.05. Rates were measured during HCO3− influx (black bars) and HCO3− efflux (white bars).
HEK293 cells express sufficient endogenous CAII to maximize HCO3− transport of overexpressed transporters (Sterling et al, 2001). We thus used a dominant-negative approach to study the effect of CAII on Cl−/HCO3− exchange activity by SLC26A6. The functionally inactive V143Y CAII mutant was transiently overexpressed in HEK293 cells. V143Y CAII is expressed approximately 20-fold over the level of endogenous CAII and V143Y CAII retains its ability to bind CAB sites (Sterling et al, 2001). Overexpression of V143Y CAII, in the presence of endogenous wild-type (WT) CAII, displaces functional WT CAII from cytoplasmic binding sites (Sterling et al, 2001). Figure 3B shows that overexpression of V143Y CAII decreased the rate of SLC26A6 bicarbonate transport rate by 30±6% (n=5, P<0.05). These data suggest that the presence of cytosolic CAII is not sufficient to activate SLC26A6 transport; CAII needs to localize to the SLC26A6 CAB site.
The CAB site, identified as 546DVDF549 (Figure 2C), was mutated to 546NVNF549 in full-length SLC26A6 to generate SLC26A6-ΔCAB. The transport activity of the mutant was dramatically reduced in comparison to WT SLC26A6, by 50±5% (n=3, P<0.05) (Figure 3B). Mutations sometimes reduce the efficiency of cell-surface processing of proteins. However, the reduced transport rate could not be explained by a reduction in cell surface expression since SLC26A6-ΔCAB expression at the cell surface was indistinguishable from WT SLC26A6 (Supplementary Figure 1; 27±5 and 28±5% of protein at the surface for WT and SLC26A6-ΔCAB, respectively). Taken together, our data demonstrate that CAII binds human SLC26A6 at 546DVDF549, which activates bicarbonate transport. This is the first demonstration that an SLC26A family member forms a bicarbonate transport metabolon.
Regulation of human SLC26A6 Cl−/HCO3− exchange activity by AngII
PKC-coupled signaling cascades inhibit luminal bicarbonate secretion in pancreatic duct, a process likely mediated by SLC26A6 (Hegyi et al, 2005). AT1a AngII receptors activate PKC (Thomas et al, 1996). The effect of AngII-dependent signaling on SLC26A6 Cl−/HCO3− exchange was investigated in HEK293 cells transiently cotransfected with SLC26A6 and AT1a AngII receptor cDNAs (Figure 4A). SLC26A6 caused changes of intracellular pH, associated with the coupled exchange of Cl− for HCO3− across the plasma membrane (Figure 4A). Average Cl−/HCO3− exchange rate measured during HCO3− influx decreased by 38±8% upon AngII treatment, compared with control (1.29±0.18 versus 0.81±0.15 mM/min, n=5, P<0.05) (Figure 4A and B). Two consecutive Cl− removal/re-addition pulses under control conditions produced similar results, indicating that exposure of cells to a second Cl− removal/re-addition pulse did not affect transport activity (not shown). Since AngII stimulation induces PKC activation, we examined the effect of the broad-spectrum PKC inhibitor chelerythrine (CHE, 10 μM) on Cl−/HCO3− exchange by SLC26A6, in cells also expressing AT1a receptor (Figure 4B). CHE blocked the AngII-induced decrease of Cl−/HCO3− exchange activity by SLC26A6 (2.10±0.14 versus 2.31±0.16 mM/min, n=3) (Figure 4B), indicating that AngII effects were mediated by PKC.
Figure 4.
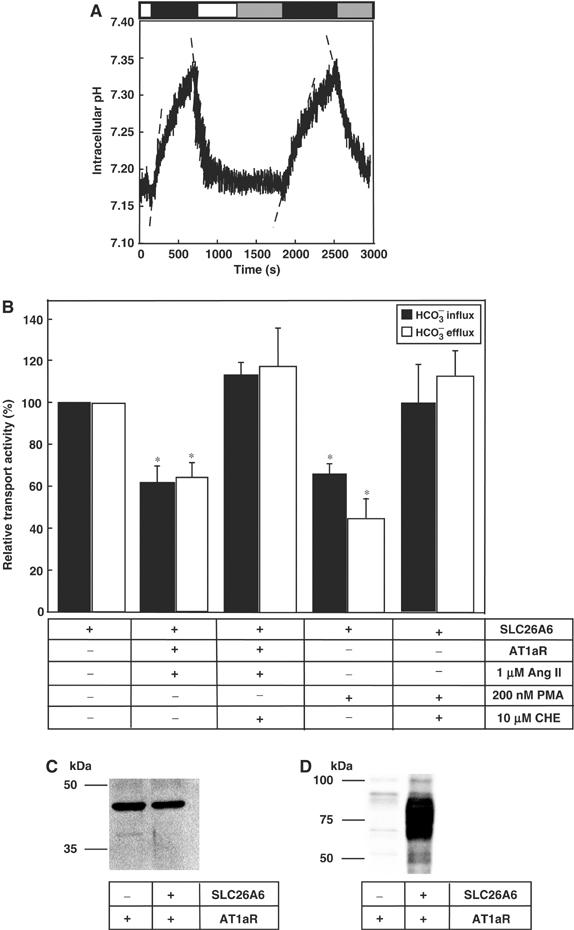
PKC activation inhibits SLC26A6 Cl−/HCO3− exchange activity. (A) HEK293 cells were cotransfected with human SLC26A6 and AT1a-AngII receptor, cDNAs. At 48 h after transfection, Cl−/HCO3− anion exchange assays were performed before, and 10 min after exposure to AngII (1 μM, gray bar). HEK293 cells were exposed to medium containing Cl− (open bar) or Cl−-free medium (black bar), to drive the exchange of Cl− for HCO3−. Initial rates of change of pHi during the first 100 s were estimated by linear regression (dashed line). (B) Mean values of anion transport activity relative to cells expressing SLC26A6 alone. Cells expressing AT1a receptors were exposed to AngII (1 μM) either in the absence or presence of the PKC inhibitor, CHE (10 μM). SLC26A6-expressing cells were also exposed to 10 min treatment with PMA, either in the presence or the absence of the PKC inhibitor, CHE (10 μM). Rates were measured during HCO3− influx (black bars) and HCO3− efflux (white bars). *P<0.05, n=4–5. (C) HEK293 cells were either transfected with AT1aR cDNA, or cotransfected with SLC26A6 and AT1aR cDNAs. Samples were analyzed by SDS–PAGE, transferred to PVDF membranes, and probed with either anti-AT1aR antibody (C) or anti-SLC26A6 antibody (D).
Phorbol 12-myristate 13-acetate ester (PMA), a nonhydrolysable diacyl glycerol mimetic, also activates PKC. The effect of 200 nM PMA was tested in HEK293 cells transiently transfected with SLC26A6. PMA treatment induced a 35±5% decrease in the Cl−/HCO3− exchange activity of SLC26A6 (n=4, P<0.05). The PKC inhibitor, CHE (10 μM), prevented the PMA-induced decrease of Cl−/HCO3− exchange activity (102±14%, n=4, P<0.05) (Figure 4B). These results suggest that the decrease in SLC26A6 Cl−/HCO3− exchange activity upon treatment with AngII is mediated by PKC.
Figure 4C shows similar expression levels for AT1a receptors in HEK293 cells transfected with AT1a alone or cotransfected with both AT1a and SLC26A6 cDNAs. Similarly, strong immunoreactivity was present in lysates of HEK293 cells transfected with SLC26A6 cDNA, and probed with anti-SLC26A6 C-terminus antibody (Figure 4D). SLC26A6 migration as a broad band was previously found for SLC26A6 protein expressed in MDCK cells, suggesting that the protein might be heterogeneously glycosylated (Waldegger et al, 2001).
Does the phosphorylation of PKC affect the binding of CAII to the SLC26A6 C-terminus?
PKC activation decreased SLC26A6 Cl−/HCO3− exchange activity (Figure 4). To determine the phosphorylation site responsible for this regulation, we analyzed the C-terminal cytoplasmic domain of SLC26A6 and found five consensus PKC phosphorylation sites, SQK (553–555), SPK (582–584), TLK (636–638), SLK (667–669), and TKK (706–708) (ExPASy Molecular Biology Server, http://www.expasy.org/tools/scanprosite). Interestingly, S553 and S582 are close to the SLC26A6 CAB site (546DVDF549), which led us to consider that phosphorylation by PKC might decrease CAII/SLC26A6 interaction. To investigate this possibility, HEK293 cells were transfected with WT SLC26A6, or PKC consensus site mutants SLC26A6-S553A and SLC26A6-S582A. Transport activity of SLC26A6-S553A and SLC26A6-S582A did not differ significantly from WT SLC26A6, indicating that PKC consensus site mutants were fully functional (Figure 5A). Upon PKC activation with PMA, WT and S582A SLC26A6 were inhibited to a similar extent (Figure 5A). However, SLC26A6 ΔCAB and S553A had no significant response to PMA. These data suggest that both S553 and the CAB site have indispensable roles in regulation of SLC26A6 by PKC.
Figure 5.
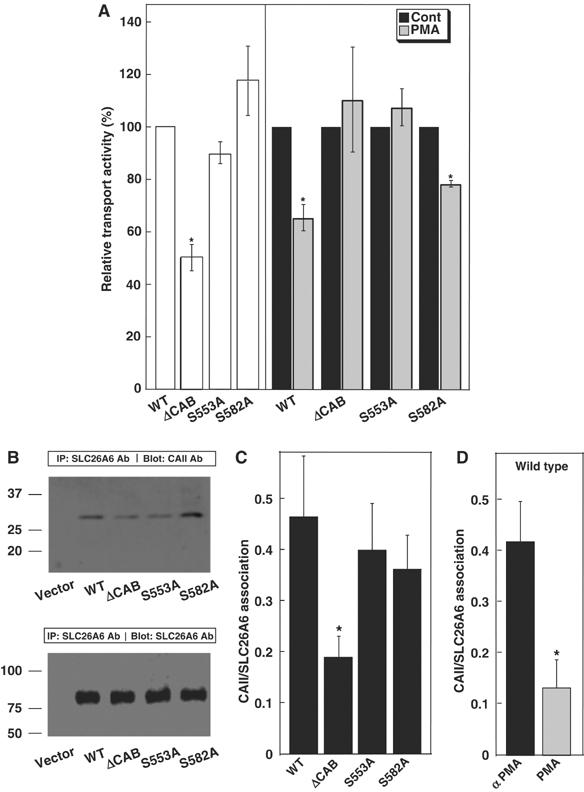
Identification of the PKC-responsive site in the SLC26A6 C-terminal region. (A) HEK293 cells transfected with WT SLC26A6 cDNA or the indicated mutants were switched from Cl−-containing to Cl−-free Ringer's buffer and the process was repeated after 10 min incubation with 200 nM PMA. Open bars indicate Cl−/HCO3− exchange activity, relative to WT SLC26A6 for samples not treated with PMA. Transport activity before (black bars) and after (grey bars) PMA treatment was normalized to the activity of associated with each cell type, under control conditions. *P<0.05, n=4. (B) HEK293 cells were transfected with vector alone, WT, ΔCAB, S553A or S582A SLC26A6, as indicated. Cell lysates wre immunoprecipitated with anti-SLC26A6 antibody and immunoprecipitates were probed for associated CAII on immunoblots probed with anti-CAII antibody (upper panel). The amount of SLC26A6 present in each sample was assessed on parallel blots probed with anti-SLC26A6 antibody (lower panel). (C) Immunoprecipitation of CAII with SLC26A6 variants was calculated as (amount of CAII/amount of SLC26A6). (D) The effect of PMA on CAII/SLC26A6 association was measured as in panels B and C, except that SLC26A6-expressing cells were incubated with either 200 nM αPMA (black bar) or PMA (grey bar) for 1 h prior to cell lysis. *P<0.05.
The association of endogenously expressed CAII with SLC26A6 was further assessed in immunoprecipitates (Figure 5B and C). The amount of CAII bound to SLC26A6 was quantified on immunoblots and normalized to the amount of SLC26A6 expressed in each case. WT SLC26A6 and the PKC site mutants S553A and S582A bound CAII to the same extent, indicating that S553 and S582 are not required for CAII binding. However, SLC26A6-ΔCAB bound only about one-third as much CAII, confirming that the 546DVDF549 sequence mutated in SLC26A6-ΔCAB forms the major or only CAB site in SLC26A6. Using the same methodology, the influence of PKC on SLC26A6/CAII interaction was measured (Figure 5D). Treatment of WT SLC26A6-expressing cells with PMA resulted in a decrease of CAII binding to the same low level as SLC26A6-ΔCAB. This suggests that PKC phosphorylation disrupts the binding of CAII to SLC26A6. Consistent with this, treatment of SLC26A6-expressing cells with PMA, but not the inactive isomer α-PMA, led to phosphorylation of SLC26A6 (Supplementary Figure 2).
Differences in transport activity of the SLC26A6 mutants could be explained by alterations of expression level or efficiency of protein processing to the cell surface. However, immunoblots showed similar expression levels for WT SLC26A6, SLC26A6-ΔCAB, SLC26A6-S553A, and SLC26A6-S582A mutants, when normalized by endogenous β-actin expression (Supplementary Figure 3). Differences in the degree of cell surface processing of SLC26A6 variants do not explain these findings, since there were no significant differences (Supplementary Figure 1A and B; 27±5, 28±5, 34±3, and 28±5% of protein at surface for WT, SLC26A6-ΔCAB, SLC26A6-S553A, and SLC26A6-S582A, respectively). Similarly, PMA did not affect the level of SLC26A6 expression at the plasma membrane (Supplementary Figure 1C).
Taken together, we propose that phosphorylation of S553 in the SLC26A6 C-terminal tail displaces CAII from its binding site in the cytoplasmic domain of SLC26A6.
Colocalization of SLC26A6 and CAII in cells
Localization of SLC26A6 protein and endogenous CAII was assessed in cells treated with PMA or the biologically inactive isomer, 4-α-phorbol 12-myristate 13-acetate esters (α-PMA). As expected, SLC26A6 had a pericellular (plasma membrane) localization after PMA and α-PMA treatments (Figure 6A). In α-PMA-treated cells, CAII localized mainly to the plasma membrane (Figure 6A, upper middle panel). However, treatment with PMA displaced CAII from the plasma membrane (Figure 6B and C, lower middle panel). Specificity of the CAII and SLC26A6 signals was shown by the absence of signal in samples treated with secondary antibody and no primary antibody (not shown). As a control, colocalization of CAII with the α1a adrenergic receptor (α1aR), expressed in transfected HEK293 cells, was also assessed by immunocytochemistry (not shown).
Figure 6.
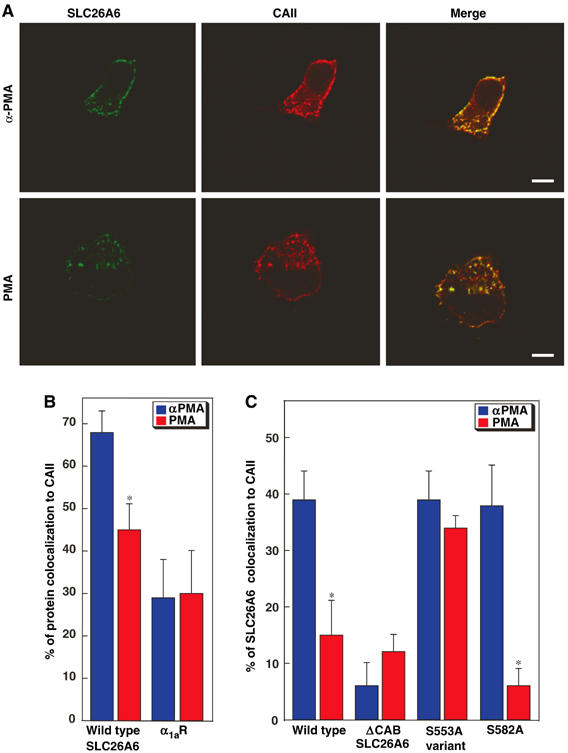
Effect of PKC activation on CAII cellular localization. HEK293 cells, transfected with an SLC26A6 variant or with the α1a adrenergic receptor (α1aR), were plated on glass slides. Cells were incubated for 1 h with either PMA, or the biologically inactive α-PMA isomer. (A) In WT-SLC26A6, transfected cells were stained with rabbit anti-SLC26A6 antibody, followed by Alexa Fluor 488-conjugated chicken anti-rabbit IgG secondary antibody (SLC26A6, green) or with goat anti-CAII antibody, followed by Alexa Fluor 594-conjugated chicken anti-goat IgG (CAII, red). Colocalization of CAII and SLC26A6 is yellow (merge). Images were collected with a Zeiss LSM 510 laser-scanning confocal microscope. Scale bar=10 μm. (B) Images were analyzed with MetaMorph® Software to quantify the degree of CAII colocalization with either SLC26A6 or α1aR, in cells treated with PMA (red bars) or αPMA (blue bars). *P<0.05 (n=7–20 cells). (C) Colocalization of SLC26A6 variants with endogenous CAII. Values in this panel were corrected for background colocalization represented by the value of α1aR. *P<0.05.
Quantitative analysis revealed that SLC26A6/CAII colocalization was significantly less in PMA-treated cells than in αPMA-treated cells, 52±8% and 70±5% overlap, respectively (Figure 6B, P<0.05), indicating that PMA treatment reduced SLC26A6/CAII colocalization. α1aR localizes to the plasma membrane, but colocalization with CAII was approximately one-third (29±6%, Figure 6B) that for SLC26A6, and treatment with PMA did not affect colocalization of α1aR and CAII (30±7%, Figure 6B).
We further examined the CAII/SLC26A6, by confocal microscopy of the SLC26A6 mutants ΔCAB, S553A, and S582A (Supplementary Figure 4). Quantitative analysis revealed that ΔCAB-SLC26A6 colocalizes with CAII to a very low extent, consistent with a failure to bind CAII (Figure 6C). S582A-SLC26A6 colocalized with CAII to a similar extent as WT-SLC26A6 and responded to PMA similarly to WT-SLC26A6 (Supplementary Figures 4B and 6C). In contrast, S553A-SLC26A6 bound CAII like WT, but failed to respond to PKC activation (Figure 6C). Taken together, activation of PKC using PMA caused a displacement of CAII from SLC26A6 at the plasma membrane. Phosphorylation of S553 of SLC26A6 by PKC displaces CAII from the surface of SLC26A6.
Discussion
The data presented here support a new mechanism for regulation of membrane transport. BTs bind the cytosolic enzyme, CAII, at their cytosolic surface (Sterling et al, 2001; Dahl et al, 2003; Loiselle et al, 2004; Pushkin et al, 2004). Localization of CAII, which produces the transport substrate HCO3−, activates the bicarbonate transport rate by maximizing the local concentration of HCO3− at the transport site. The complex of a BT with CA has been termed a bicarbonate transport metabolon (Sterling et al, 2001). Here we have shown for the first time that residues 546–549 of SLC26A6 form an essential part of a cytoplasmic CAII-binding site. The observation that mutation of the CAII-binding site significantly reduced SLC26A6 transport activity is consistent with a bicarbonate transport metabolon, where the CAII/transporter association maximizes the HCO3− transport flux. In SLC26A6-expressing cells, PKC activation resulted in (1) phosphorylation of SLC26A6, (2) reduction of SLC26A6 transport activity and (3) displacement of CAII from the cytosolic surface of the plasma membrane. Finally, the SLC26A6/CAII interaction was disrupted by PKC-mediated phosphorylation of the SLC26A6 C-terminal region. From these observations, we propose that PKC inhibits SLC26A6 HCO3− transport by disruption of the bicarbonate transport metabolon (displacement of CAII from the surface of SLC26A6) (Figure 7). This model was confirmed by the insensitivity of the CAB site mutant and the S553A mutant to PKC activation. We have thus determined the mechanism by which a phosphorylation event induces regulation of transport. Transport metabolon disruption, displacement of an enzyme that catalyzes production of a transport substrate from the binding site on the surface of the transporter, represents a new mechanism for regulation of membrane transporter function. While we present data only for the bicarbonate transport metabolon, metabolon disruption may represent a mechanism for acute regulation of transport activity by other transporters of cellular metabolites.
Figure 7.
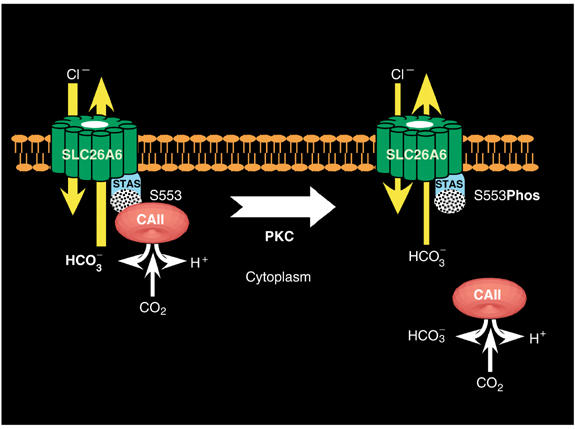
Regulation of SLC26A6 bicarbonate transport by metabolon disruption. CAII binds the CAB site (stippled) within the STAS domain of SLC26A6. Localization of CAII to the CAB site maximizes the local HCO3− concentration at the SLC26A6 transport site, thereby maximizing transport rate. PKC phosphorylates SLC26A6 at S553, which displaces CAII from the CAB site. Isolation of CAII from the surface of SLC26A6 reduces the local concentration of HCO3−, reducing the transport rate. Arrows on the SLC26A6 image represent the movement of Cl− and HCO3−, where the arrow width indicates the relative rate in each case.
The C-terminal cytoplasmic domains of SLC4A Cl−/HCO3− exchangers and Na+/HCO3− cotransporters bind CAII, which activates their bicarbonate transport rate by maximizing the local concentration of HCO3− at the transport site (Alvarez et al, 2003; Loiselle et al, 2004; Pushkin et al, 2004). This is the first demonstration that an SLC26A family transporter binds CAII to activate HCO3− transport, which demonstrates that CAII binding is found among all three groups of BTs that are known. Binding affinity of SLC26A6 for CAII (141 nM) was comparable to that seen previously for AE1 (20 nM) and NBC3 (101 nM) (Vince and Reithmeier, 1998; Loiselle et al, 2004). In contrast, the C-terminal region of SLC26A3 does not contain a consensus CAB motif. Interestingly, binding of CAII by the kidney variant of the NBC1 Na+/HCO3− cotransporter is enhanced following protein kinase A treatment of NBC1. However, a concomitant increase in transport flux was not observed (Gross et al, 2002). Thus, there is precedent for modulation of CAII/bicarbonate interaction by phosphorylation, but this is the first example where modulation of the metabolon serves to regulate transport. Additional support for the idea of regulation of membrane transport by metabolon modulation comes from studies of the interactions between the NHE1 Na+/H+ exchanger and CAII (Li et al, 2002). NHE1 bound CAII at an unidentified site in the NHE1 cytoplasmic domain to activate Na+/H+ exchange. Phosphorylation of NHE1 cytoplasmic domain by a heart cell lysate induced phosphorylation at multiple unidentified sites and a concomitant increase in NHE1 CAII binding and transport activity, which is suggestive of metabolon modulation.
CAII/BT interaction is established to occur electrostatically between the positive N-terminal region of CAII and an acidic motif on the BT. It is thus surprising that PKC-mediated addition of a phosphate group adjacent to the SLC26A6 CAB site results in displacement of CAII/SLC26A6 interaction. In the absence of a high-resolution structure for the complex of CAII with a BT, it is not possible to provide a firm explanation for how this occurs. However, we can speculate that interactions between CAII and a BT are more extensive than the two minimal motifs (basic CAII tail and acidic BT tail). In fact, these two regions are likely too small to represent the complete interacting surface. The identified PKC site (S553) is close to, but not co-incident with, the CAB site. The addition of phosphate by PKC may result in an electrostatic repulsion at a site adjacent to the CAB site.
Our data may have significance for the regulation of pancreatic bicarbonate secretion, particularly in CF, where bicarbonate secretion is compromised. SLC26A6 may contribute to pancreatic duct HCO3− secretion at levels up to 140 mM (Lohi et al, 2000; Ko et al, 2002; Steward et al, 2005). Activation of PKC reduces anion secretion in pancreatic duct cells (Cheng et al, 1999), which could be explained by effects on Cl−/HCO3− exchange activity of SLC26A6. The recent report that substance P acts through PKC to reduce DIDS-inhibitable pancreatic duct bicarbonate secretion (Hegyi et al, 2005) further supports a possible role of PKC-mediated metabolon disruption in the regulation of pancreatic bicarbonate secretion. Our data suggest that reduction of pancreatic duct PKC activity and strategies aimed at increasing CAII interaction with SLC26A6 are targets for therapies to increase pancreatic bicarbonate secretion.
STAS domains have emerged as the centre for regulation of SLC26A6 family transporters (Ko et al, 2004). The STAS domain of the proteins SLC26A3 and SLC26A6 mediates reciprocal regulation with the CF gene product, CFTR (Ko et al, 2004). We found that the CAB site (546D–F549) and the PKC site responsible for reduction of SLC26A6 transport rate (S553) are both within the STAS domain of SLC26A6, which spans E530–A741 on the basis of sequence alignments (Ko et al, 2004) (Figure 8A). The NMR structure for the STAS domain of the bacterial antisigma-factor antagonists of Bacillus subtilis, SPOIIAA (Figure 8B and C) (Kovacs et al, 1998), reveals that the CAB site and the S553 PKC sites are in a position corresponding to a surface loop, which is highly variable between SLC26A6 transporters, suggesting a variable role of the loop in transport regulation (Figure 8A–C). The interaction between the AE1 Cl−/HCO3− exchanger and CAII occurs through electrostatic interaction of an acidic CAB motif in the AE1 C-terminus and the basic N-terminal tail of CAII (Vince et al, 2000). SLC26A6 likely interacts in a similar manner using CAB motif found in the surface loop. SLC26A6/CAII interaction is disrupted by PKC phosphorylation at S553, perhaps through electrostatic effect. The critical role played by the STAS domain of anion transporters is supported by disease-causing mutations identified in the STAS domain of SLC26A2 and SLC26A3 and SLC26A4 (Hastbacka et al, 1996; Everett et al, 1997; Rossi and Superti-Furga, 2001; Makela et al, 2002).
Figure 8.
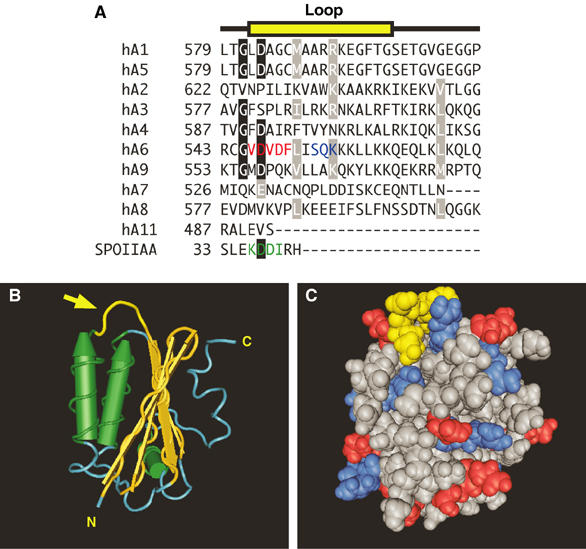
Structural basis for regulation of SLC26A6 by PKC: the STAS domain. (A) Multiple sequence alignment of the amino-acid sequence of a surface loop (yellow) in the STAS domain of human (h) SLC26A1–SLC26A11 (hA1–hA11) and the sporulation-specific sigma factor of B. subtilis, SPOIIAA. Conserved residues (black boxes) and conservative replacements (gray boxes) are indicated. The sequence for loop residues in SPOIIAA is highlighted (green). The CAII-binding site (red) and consensus PKC phosphorylation site (blue) are present only in SLC26A6. (B) Structural model of the STAS domain from SPOIIAA (PDB code 1BUZ) (Kovacs et al, 1998). Yellow structure highlighted with an arrow indicates the position of the variable loop between helix 1 and strand 3 (corresponding to the sequence highlighted in yellow in panel A). Cylinders represent α-helices and arrows represent β-sheets. N, amino-terminus; C, carboxyl-terminus. (C) Space-filling model of SPOIIAA oriented as in panel B. Loop region residues are yellow, while positive and negative residues are blue and red, respectively. Structures were rendered with Cn3D software.
Here we provided evidence for metabolon disruption as a novel mechanism to regulate membrane transport. The human SLC26A6 Cl−/HCO3− exchanger binds CAII, the enzyme responsible for HCO3− production. This binding maximizes SLC26A6-mediated transmembrane HCO3− flux in a manner analogous to that seen for other BTs. PKC reduced SLC26A6 activity and also reduced SLC26A6/CAII interaction. The proximity of the CAII-binding site to the identified PKC phosphorylation immediately suggests a regulatory mechanism: PKC-mediated displacement of CAII reduces SLC26A6 transport activity. This is the first example of regulatory metabolon disruption, displacement of the enzyme that produces transport substrate from the surface of the transporter.
Materials and methods
Molecular biology
Expression constructs for human SLC26A6 (accession number AAF81911, the SLC26A6a, or S+Q variant in the nomenclature used by Alper (Chernova et al, 2005)) and human CAII have been described previously (Lohi et al, 2000; Sterling et al, 2001). Bacterial expression constructs encoding GST-fusion proteins consisting of the cDNA for GST fused to either cDNA corresponding to amino acids 497–633, amino acids 531–633, amino acids 570–633 or 602–633 of SLC26A6 C-terminal tail were constructed.
Protein expression
SLC26A6, SLC26A6 mutants, AT1a receptor, α1a-adrenergic receptor, and V143Y CAII (Khandoudi et al, 2001; Sterling et al, 2001; Stanasila et al, 2003) were expressed by transient transfection of HEK293 cells, using the calcium phosphate method (Alvarez et al, 2001a). Cells were grown at 37°C in an air/CO2 (19:1) environment in Dulbecco's Modified Eagles Medium (DMEM) medium, supplemented with 5% (v/v) fetal bovine serum and 5% (v/v) calf serum. GST fusion proteins were expressed and purified as described previously (Sterling et al, 2002a).
Cl−/HCO3− exchange assays
HEK293 cells, grown on 6 × 11 mm2 glass, were transfected with cDNAs. At 2 days post transfection, coverslips were incubated in serum-free DMEM, containing 2 μM 2′,7′-bis (2-carboxyethyl)-5 (and -6)-carboxyfluorescein, acetoxymethyl ester (BCECF-AM) (Molecular Probes), 37°C, for 20 min. Coverslips in a fluorescence cuvette were perfused at 3.5 ml/min alternately with Ringer's buffer (5 mM glucose, 5 mM potassium gluconate, 1 mM calcium gluconate, 1 mM MgSO4, 2.5 mM, NaH2PO4, 25 mM NaHCO3, 10 mM Hepes, pH 7.40) containing either 140 mM NaCl (Cl− containing) or 140 mM sodium gluconate (Cl−-free). Both buffers were continuously bubbled with air/5% CO2. Fluorescence changes were monitored in a Photon Technologies International RCR fluorimeter at excitation wavelengths 440 and 502 nm and emission wavelength 528 nm. All transport data were corrected for background activity of HEK293 cells transfected with pcDNA3 vector alone. The intrinsic buffer capacity (βi) was negligible at pHi values above 7.10 (Alvarez et al, 2004), so that 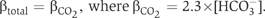 The total flux of proton equivalents was calculated as: JH+=βtotal × ΔpHi (Alvarez et al, 2004), where ΔpHi was determined by linear regression of the first 100 s of alkalinization or acidification, using Kaleidagraph software.
The total flux of proton equivalents was calculated as: JH+=βtotal × ΔpHi (Alvarez et al, 2004), where ΔpHi was determined by linear regression of the first 100 s of alkalinization or acidification, using Kaleidagraph software.
Immunoblotting
Samples (10 μg protein) were resolved by SDS–PAGE on 10% acrylamide gels. Proteins were transferred to PVDF membranes, and then incubated with either rabbit anti-human SLC26A6 (Lohi et al, 2003), or rabbit anti-rat AT1a-AngII receptor antibody (Santa Cruz Biotechnology), with sheep anti-human CAII antibody (Serotec), or anti-GST rabbit polyclonal antibody (Z-5, Santa Cruz Biotechnology), as appropriate. Immunoblots were incubated with either donkey anti-rabbit IgG conjugated to horseradish peroxidase or donkey anti-sheep IgG conjugated to horseradish peroxidase, or rabbit anti-goat IgG conjugated to horseradish peroxidase (Sterling et al, 2002a). Blots were visualized and quantified using enhanced chemiluminescence (ECL) reagent and a Kodak Image Station.
Gel overlay assays
Gel overlay assays to detect SLC26A6 interactions with CAII were performed as described previously (Sterling et al, 2001, 2002a).
CAII-binding assays
The ability of CAII to bind the C-terminal tail of human SLC26A6 was investigated using a microtiter assay, described previously (Vince and Reithmeier, 1998, 2000). After washing, bound fusion proteins were detected by sequential incubation with rabbit anti-GST antibody (Santa-Cruz), biotinylated anti-rabbit IgG (Amersham), and peroxidase-labeled biotin/streptavidin (Amersham). Plates were then incubated with the peroxidase substrate o-phenylenediamine dihydrochloride (Sigma) and product formation detected at 450 nm in a Labsystems Mutiskan MCC microplate reader.
Immunoprecipitation
HEK293 cells transiently transfected with SLC26A6 cDNA, or sham transfected, were grown in 100 mm tissue culture plates, for 48 h. Cells were washed with PBS (140 mM NaCl, 3 mM KCl, 6.5 mM Na2HPO4, 1.5 mM KH2PO4, pH 7.5) and harvested by lysis in 500 μl of lysis buffer (PBS buffer, containing 1% (v/v) Triton X-100, 5 mM EDTA, and protease inhibitor cocktail (MiniComplete Tablet, Roche). In experiments to examine the effect of PMA on CAII binding by SLC26A6, cells were serum-starved for 1 h, and incubated for 1 h with either 200 nM of αPMA or PMA, at 37°C. Cells were washed with PBS, and harvested in 500 μl of lysis buffer, containing 1 mM Na3VO4 and 10 mM NaF. Lysates were clarified by centrifugation at 16 300 g for 15 min at 4°C. Samples were immunoprecipitated with 2 μl of anti-N-terminal SLC26A6 antibody (Lohi et al, 2003), using a protocol described previously (Alvarez et al, 2003). Immunoprecipitates were analyzed on immunoblots, probed with anti-C-terminal SLC26A6 antibody (Lohi et al, 2003), or anti-CAII antibody.
Confocal microscopy
Confocal microscopy was performed following standard procedures (see Supplementary Materials and methods).
Numerical analysis
Statistical significance was evaluated using unpaired t-test, paired t-test, or one-way ANOVA (followed by Bonferroni test) as indicated, with P<0.05 considered significant. Error bars show standard error of the mean. Kd values were determined by curve fitting using GraphPad Prism® Software.
Supplementary Material
Supplementary Information
Acknowledgments
We thank Dr H Lohi for the SLC26A6 antibody and cDNA, and Drs Marek Michalak and James Young for helpful comments on the manuscript. JRC is a Senior Scholar of the Alberta Heritage Foundation for Medical Research. BVA holds a fellowship from the Canadian Cystic Fibrosis Foundation. This work was supported by a grant from the Heart and Stroke Foundation of Alberta.
References
- Alvarez BV, Fujinaga J, Casey JR (2001a) Molecular basis for angiotensin II-induced increase of chloride/bicarbonate exchange in the myocardium. Circ Res 89: 1246–1253 [DOI] [PubMed] [Google Scholar]
- Alvarez BV, Kieller DM, Quon AL, Markovich D, Casey JR (2004) Slc26a6: a cardiac chloride–hydroxyl exchanger and predominant chloride–bicarbonate exchanger of the mouse heart. J Physiol 561: 721–734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez BV, Loiselle FB, Supuran CT, Schwartz GJ, Casey JR (2003) Direct extracellular interaction between carbonic anhydrase IV and the human NBC1 sodium/bicarbonate co-transporter. Biochemistry 42: 12321–12329 [DOI] [PubMed] [Google Scholar]
- Alvarez L, Fanjul M, Carter N, Hollande E (2001b) Carbonic anhydrase II associated with plasma membrane in a human pancreatic duct cell line (CAPAN-1). J Histochem Cytochem 49: 1045–1053 [DOI] [PubMed] [Google Scholar]
- Aravind L, Koonin EV (2000) The STAS domain—a link between anion transporters and antisigma-factor antagonists. Curr Biol 10: R53–R55 [DOI] [PubMed] [Google Scholar]
- Cheng HS, Wong WS, Chan KT, Wang XF, Wang ZD, Chan HC (1999) Modulation of Ca2+-dependent anion secretion by protein kinase C in normal and cystic fibrosis pancreatic duct cells. Biochim Biophys Acta 1418: 31–38 [DOI] [PubMed] [Google Scholar]
- Chernova MN, Jiang L, Friedman DJ, Darman RB, Lohi H, Kere J, Vandorpe DH, Alper SL (2005) Functional comparison of mouse slc26a6 anion exchanger with human SLC26A6 polypeptide variants: differences in anion selectivity, regulation, and electrogenicity. J Biol Chem 280: 8564–8580 [DOI] [PubMed] [Google Scholar]
- Cousin JL, Motais R (1976) The role of carbonic anhydrase inhibitors on anion permeability into ox red blood cells. J Physiol 256: 61–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahl NK, Jiang L, Chernova MN, Stuart-Tilley AK, Shmukler BE, Alper SL (2003) Deficient HCO3− transport in an AE1 mutant with normal Cl− transport can be rescued by carbonic anhydrase II presented on an adjacent AE1 protomer. J Biol Chem 278: 44949–44958 [DOI] [PubMed] [Google Scholar]
- Everett LA, Glaser B, Beck JC, Idol JR, Buchs A, Heyman M, Adawi F, Hazani E, Nassir E, Baxevanis AD, Sheffield VC, Green ED (1997) Pendred syndrome is caused by mutations in a putative sulphate transporter gene (PDS). Nat Genet 17: 411–422 [DOI] [PubMed] [Google Scholar]
- Everett LA, Green ED (1999) A family of mammalian anion transporters and their involvement in human genetic diseases. Hum Mol Genet 8: 1883–1891 [DOI] [PubMed] [Google Scholar]
- Gross E, Pushkin A, Abuladze N, Fedotoff O, Kurtz I (2002) Regulation of the sodium bicarbonate cotransporter kNBC1 function: role of Asp(986), Asp(988) and kNBC1-carbonic anhydrase II binding. J Physiol 544: 679–685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastbacka J, Superti-Furga A, Wilcox WR, Rimoin DL, Cohn DH, Lander ES (1996) Atelosteogenesis type II is caused by mutations in the diastrophic dysplasia sulfate-transporter gene (DTDST): evidence for a phenotypic series involving three chondrodysplasias. Am J Hum Genet 58: 255–262 [PMC free article] [PubMed] [Google Scholar]
- Hegyi P, Rakonczay Z Jr, Tiszlavicz L, Varro A, Toth A, Racz G, Varga G, Gray MA, Argent BE (2005) Protein kinase C mediates the inhibitory effect of substance P on HCO3− secretion from guinea pig pancreatic ducts. Am J Physiol Cell Physiol 288: C1030–C1041 [DOI] [PubMed] [Google Scholar]
- Khandoudi N, Albadine J, Robert P, Krief S, Berrebi-Bertrand I, Martin X, Bevensee MO, Boron WF, Bril A (2001) Inhibition of the cardiac electrogenic sodium bicarbonate cotransporter reduces ischemic injury. Cardiovasc Res 52: 387–396 [DOI] [PubMed] [Google Scholar]
- Knauf F, Yang CL, Thomson RB, Mentone SA, Giebisch G, Aronson PS (2001) Identification of a chloride–formate exchanger expressed on the brush border membrane of renal proximal tubule cells. Proc Natl Acad Sci USA 98: 9425–9430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko SB, Shcheynikov N, Choi JY, Luo X, Ishibashi K, Thomas PJ, Kim JY, Kim KH, Lee MG, Naruse S, Muallem S (2002) A molecular mechanism for aberrant CFTR-dependent HCO3− transport in cystic fibrosis. EMBO J 21: 5662–5672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko SB, Zeng W, Dorwart MR, Luo X, Kim KH, Millen L, Goto H, Naruse S, Soyombo A, Thomas PJ, Muallem S (2004) Gating of CFTR by the STAS domain of SLC26 transporters. Nat Cell Biol 6: 343–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacs H, Comfort D, Lord M, Campbell ID, Yudkin MD (1998) Solution structure of SpoIIAA, a phosphorylatable component of the system that regulates transcription factor sigmaF of Bacillus subtilis. Proc Natl Acad Sci USA 95: 5067–5071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Alvarez B, Casey JR, Reithmeier RA, Fliegel L (2002) Carbonic anhydrase II binds to and enhances activity of the Na+/H+ exchanger. J Biol Chem 277: 36085–36091 [DOI] [PubMed] [Google Scholar]
- Lohi H, Kujala M, Kerkela E, Saarialho-Kere U, Kestila M, Kere J (2000) Mapping of five new putative anion transporter genes in human and characterization of SLC26A6, a candidate gene for pancreatic anion exchanger. Genomics 70: 102–112 [DOI] [PubMed] [Google Scholar]
- Lohi H, Lamprecht G, Markovich D, Heil A, Kujala M, Seidler U, Kere J (2003) Isoforms of SLC26A6 mediate anion transport and have functional PDZ interaction domains. Am J Physiol Cell Physiol 284: C769–C779 [DOI] [PubMed] [Google Scholar]
- Loiselle FB, Morgan PE, Alvarez BV, Casey JR (2004) Regulation of the human NBC3 Na+/HCO3− cotransporter by carbonic anhydrase II and PKA. Am J Physiol Cell Physiol 286: C1423–C1433 [DOI] [PubMed] [Google Scholar]
- Mahieu I, Becq F, Wolfensberger T, Gola M, Carter N, Hollande E (1994) The expression of carbonic anhydrases II and IV in the human pancreatic cancer cell line (Capan 1) is associated with bicarbonate ion channels. Biol Cell 81: 131–141 [DOI] [PubMed] [Google Scholar]
- Makela S, Kere J, Holmberg C, Hoglund P (2002) SLC26A3 mutations in congenital chloride diarrhea. Hum Mutat 20: 425–438 [DOI] [PubMed] [Google Scholar]
- McMurtrie HL, Cleary HJ, Alvarez BV, Loiselle FB, Sterling D, Morgan PE, Johnson DE, Casey JR (2004) The bicarbonate transport metabolon. J Enzyme Inhib Med Chem 19: 231–236 [DOI] [PubMed] [Google Scholar]
- Mount DB, Romero MF (2004) The SLC26 gene family of multifunctional anion exchangers. Pflugers Arch 447: 710–721 [DOI] [PubMed] [Google Scholar]
- Petrovic S, Wang Z, Ma L, Seidler U, Forte JG, Shull GE, Soleimani M (2002) Colocalization of the apical Cl−/HCO3− exchanger PAT1 and gastric H–K–ATPase in stomach parietal cells. Am J Physiol Gastrointest Liver Physiol 283: G1207–G1216 [DOI] [PubMed] [Google Scholar]
- Pushkin A, Abuladze N, Gross E, Newman D, Tatishchev S, Lee I, Fedotoff O, Bondar G, Azimov R, Ngyuen M, Kurtz I (2004) Molecular mechanism of kNBC1–carbonic anhydrase II interaction in proximal tubule cells. J Physiol 559: 55–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi A, Superti-Furga A (2001) Mutations in the diastrophic dysplasia sulfate transporter (DTDST) gene (SLC26A2): 22 novel mutations, mutation review, associated skeletal phenotypes, and diagnostic relevance. Hum Mutat 17: 159–171 [DOI] [PubMed] [Google Scholar]
- Schwartz GJ (2002) Physiology and molecular biology of renal carbonic anhydrase. J Nephrol 15 (Suppl 5): S61–S74 [PubMed] [Google Scholar]
- Simpson JE, Gawenis LR, Walker NM, Boyle KT, Clarke LL (2005) Chloride conductance of CFTR facilitates basal Cl−/HCO3− exchange in the villous epithelium of intact murine duodenum. Am J Physiol Gastrointest Liver Physiol 288: G1241–G1251 [DOI] [PubMed] [Google Scholar]
- Spicer SS, Ge ZH, Tashian RE, Hazen-Martin DJ, Schulte BA (1990) Comparative distribution of carbonic anhydrase isozymes III and II in rodent tissues. Am J Anat 187: 55–64 [DOI] [PubMed] [Google Scholar]
- Stanasila L, Perez JB, Vogel H, Cotecchia S (2003) Oligomerization of the alpha 1a- and alpha 1b-adrenergic receptor subtypes. Potential implications in receptor internalization. J Biol Chem 278: 40239–40251 [DOI] [PubMed] [Google Scholar]
- Sterling D, Alvarez BV, Casey JR (2002a) The extracellular component of a transport metabolon. Extracellular loop 4 of the human AE1 Cl−/HCO3− exchanger binds carbonic anhydrase IV. J Biol Chem 277: 25239–25246 [DOI] [PubMed] [Google Scholar]
- Sterling D, Brown NJ, Supuran CT, Casey JR (2002b) The functional and physical relationship between the DRA bicarbonate transporter and carbonic anhydrase II. Am J Physiol Cell Physiol 283: C1522–C1529 [DOI] [PubMed] [Google Scholar]
- Sterling D, Casey JR (2002) Bicarbonate transport proteins. Biochem Cell Biol 80: 483–497 [DOI] [PubMed] [Google Scholar]
- Sterling D, Reithmeier RA, Casey JR (2001) A transport metabolon. Functional interaction of carbonic anhydrase II and chloride/bicarbonate exchangers. J Biol Chem 276: 47886–47894 [DOI] [PubMed] [Google Scholar]
- Steward MC, Ishiguro H, Case RM (2005) Mechanisms of bicarbonate secretion in the pancreatic duct. Annu Rev Physiol 67: 377–409 [DOI] [PubMed] [Google Scholar]
- Thomas WG, Thekkumkara TJ, Baker KM (1996) Cardiac effects of AII. AT1A receptor signaling, desensitization, and internalization. Adv Exp Med Biol 396: 59–69 [PubMed] [Google Scholar]
- Tsang SW, Cheng CH, Leung PS (2004) The role of the pancreatic renin–angiotensin system in acinar digestive enzyme secretion and in acute pancreatitis. Regul Pept 119: 213–219 [DOI] [PubMed] [Google Scholar]
- Vince JW, Carlsson U, Reithmeier RA (2000) Localization of the Cl−/HCO3− anion exchanger binding site to the amino-terminal region of carbonic anhydrase II. Biochemistry 39: 13344–13349 [DOI] [PubMed] [Google Scholar]
- Vince JW, Reithmeier RA (2000) Identification of the carbonic anhydrase II binding site in the Cl−/HCO3− anion exchanger AE1. Biochemistry 39: 5527–5533 [DOI] [PubMed] [Google Scholar]
- Vince JW, Reithmeier RAF (1998) Carbonic anhydrase II binds to the carboxyl-terminus of human band 3, the erythrocyte Cl−/HCO3− exchanger. J Biol Chem 273: 28430–28437 [DOI] [PubMed] [Google Scholar]
- Waldegger S, Moschen I, Ramirez A, Smith RJ, Ayadi H, Lang F, Kubisch C (2001) Cloning and characterization of SLC26A6, a novel member of the solute carrier 26 gene family. Genomics 72: 43–50 [DOI] [PubMed] [Google Scholar]
- Wang Z, Petrovic S, Mann E, Soleimani M (2002) Identification of an apical Cl−/HCO3− exchanger in the small intestine. Am J Physiol Gastrointest Liver Physiol 282: G573–G579 [DOI] [PubMed] [Google Scholar]
- Wang Z, Wang T, Petrovic S, Tuo B, Riederer B, Barone S, Lorenz JN, Seidler U, Aronson PS, Soleimani M (2005) Renal and intestinal transport defects in Slc26a6-null mice. Am J Physiol Cell Physiol 288: C957–C965 [DOI] [PubMed] [Google Scholar]
- Xie Q, Welch R, Mercado A, Romero MF, Mount DB (2002) Molecular characterization of the murine Slc26a6 anion exchanger: functional comparison with Slc26a1. Am J Physiol Renal Physiol 283: F826–F838 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information