

Abstract
FKBP-type peptidyl prolyl cis/trans isomerases (PPIases) are folding helper enzymes involved in the control of functional regrowth of damaged sciatic, cortical cholinergic, dopaminergic and 5-HT neurones. Here, we show that the constitutively inactive human FK506-binding protein 38 (FKBP38) is capable of responding directly to intracellular Ca2+ rise through formation of a heterodimeric Ca2+/calmodulin/FKBP38 complex. Only complex formation creates an enzymatically active FKBP, displaying affinity for Bcl-2 mediated through the PPIase site. Association between Bcl-2 and the active site of Ca2+/calmodulin/FKBP38 regulates Bcl-2 function and thereby participates in the promotion of apoptosis in neuronal tissues. FKBP38 proapoptotic function mediated by this interaction is abolished by either potent inhibitors of the PPIase activity of the Ca2+/calmodulin/FKBP38 complex or RNA interference-mediated depletion of FKBP38, promoting neuronal cell survival.
Keywords: apoptosis, Bcl-2, CaM, FK506-binding protein, PPIase
Introduction
FK506-binding protein 38 (FKBP38), a multidomain protein, is characterized by pronounced expression of its corresponding mRNA in human brain tissue (Lam et al, 1995), and its specific antitumor effects in mice caused by regulation of the anti-invasive syndecan 1 gene expression and suppression of the proinvasive MMP9 gene (Fong et al, 2003). The protein belongs to the FKBP (FK506-binding protein) family of peptidyl prolyl cis/trans isomerases (PPIases) by sequence similarity, but neither enzymatic activity nor FK506 binding has been detected so far (Lam et al, 1995; Shirane and Nakayama, 2003). PPIases form a class of folding helper enzymes that assist biogenesis, assembly and intracellular trafficking of proteins by catalyzing the cis/trans isomerization between native-state prolyl bond isomers of different biological activity (Harrar et al, 2001; Fischer and Aumuller, 2003). Prolyl bond conformational dynamics were shown to be involved in the control of the cell cycle (Ryo et al, 2002), p53 transcriptional activity (Zheng et al, 2002) and the formation of neurofibrillary lesions in Alzheimer's disease (Lu et al, 2003). In addition, the FKBP38-related FKBP6 was described as a critical regulator in homologous chromosome pairing during male meiosis (Crackower et al, 2003).
Several studies highlight the importance of multidomain FKBPs in the control of signal transduction pathways. For example, FKBP52 was found in steroid hormone receptor complexes and is thought to play an important role in complex formation and translocation of receptor–ligand complexes from the cytosol to the nucleus (Silverstein et al, 1999). Notably, this multidomain FKBP shares structural characteristics as shown by an N-terminal PPIase domain followed by three tetratricopeptide repeats (TPR) and a putative calmodulin (CaM)-binding motif. A membrane anchor at the C-terminus, as previously described for its Arabidopsis thaliana homolog AtFKBP42 (Kamphausen et al, 2002), completes the FKBP38 polypeptide chain.
Using the yeast two-hybrid system, FKBP38 was found to bind Bcl-2. This interaction is thought to play a role in regulation of apoptosis (Shirane and Nakayama, 2003). The regulation of Bcl-2 function is a crucial event in various apoptotic pathways, because resistance to apoptosis can lead to cancer, whereas enhanced apoptosis contributes to acute neurodegenerative or neuromuscular diseases (Mattson, 2000). In this respect, it is noteworthy that various peptidomacrolides and their derivatives, such as FK506 and GPI1046, exhibit antiproliferative, neurotrophic, antineoplasmic and antimigratory effects via binding to members of the large FKBP family. Some of the observed effects of FKBP inhibitors are due to the formation of a binding platform by the immunophilin for FK506 presenting the bound drug to cellular interaction partners. In contrast, neuroregenerative properties of FKBP ligands are directly caused by drug-mediated inhibition of the PPIase activity of various FKBPs (Gold, 2000).
The nonimmunosuppressive GPI1046, for instance, has been reported to possess neuroprotective effects in the 6-hydroxydopamine toxicity model in rats (Zhang et al, 2001), to display neurotrophic effects on the penile innervation that preserve cavernous tissue structure and promote erectile function recovery in rats after extensive nerve injury (Sezen et al, 2001; Burnett and Becker, 2004) and is antiapoptotic in H2O2-stressed NG 108-15 cells (Tanaka et al, 2001). Currently, 16 FKBP genes have been identified in the human genome, of which FKBP12 and FKBP52 have been discussed to mediate neurotrophic actions of FKBP ligands (Gold et al, 1999; Christner et al, 2001).
However, neurotrophic properties of FKBP inhibitors and inhibition of residual PPIase activity measured under cellular conditions indicate the existence of at least one, still unrecognized FKBP involved in neuronal survival and regeneration being the cellular target of neurotrophic FKBP ligands (Christner et al, 2001).
Here, we report that FKBP38 exhibits a Ca2+/CaM-stimulated PPIase activity that is essential for FKBP38–Bcl-2 interaction to occur. Thus, FKBP38 represents the first example of a cofactor-regulated PPIase. This activity participates in Bcl-2-mediated apoptosis control, and might provide the cellular mediator of neurotrophic effects of neuroimmunophilin inhibitors.
Results
FKBP38 interacts with CaM in a Ca2+-dependent manner
Based on the prediction of a putative CaM-binding site, FKBP38 was investigated for its potential to interact with CaM. Therefore, we analyzed the interaction of both proteins in co-immunoprecipitation experiments using the endogenous protein of SH-SY5Y neuroblastoma cells. As shown in Figure 1A, endogenous FKBP38 interacted with CaM in the presence of Ca2+, whereas both proteins showed no interaction in the absence of Ca2+. In order to verify the Ca2+ dependence of this interaction, we incubated recombinant FKBP38 with CaM-Sepharose beads in the presence of Ca2+ or EGTA, indicating a Ca2+-dependent interaction of FKBP38 with the immobilized CaM, as well (Figure 1B). Experiments using isothermal titration calorimetry revealed a 1:1 stoichiometry for the proteins assembling the Ca2+/CaM/FKBP38 complex. The dissociation constant calculated from the titration curve was 1.8±0.4 μM.
Figure 1.
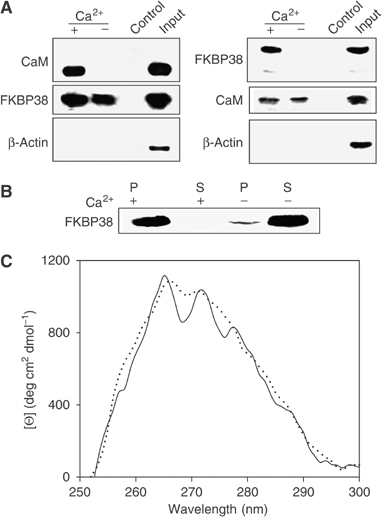
FKBP38 interacts with CaM in the presence of Ca2+. (A) Co-immunoprecipitation of endogenous FKBP38 and CaM. SH-SY5Y cell lysate preincubated with 500 μM EGTA was incubated with rabbit anti-FKBP38 antibody. Antibody/protein complexes were bound to protein G-Sepharose. Samples were washed. Precipitates and input were subjected to SDS–PAGE and analyzed by Western blot using mouse anti-CaM antibody. As a control, cell lysate preincubated with control rabbit immunoglobin was used. In an additional experiment, cell protein was incubated with mouse anti-CaM antibody and analyzed with rabbit anti-FKBP38 antibody. β-Actin and mouse immunoglobin were used as loading controls. In the presence of 1 mM Ca2+, FKBP38 bound to CaM. No interaction occurred in the absence of Ca2+. (B) Recombinant FKBP38 (30 μg) was subjected to CaM-Sepharose in the presence of 1 mM CaCl2 or 1 mM EGTA. Bound protein (P) and supernatant (S) were subsequently analyzed by SDS–PAGE with Coomassie blue staining. (C) Near-UV CD spectra of 1 μM FKBP38 and 5 μM CaM were measured in the presence of 2 mM CaCl2 either separated (dotted line) or mixed (solid line) in a tandem silica cell (Hellma, Germany).
Using tandem silica cells, near-UV circular dichroism (CD) spectra of a sample of FKBP38/CaM in buffer containing Ca2+ showed significant spectral changes in the range between 268 and 280 nm in comparison to the sum of the spectra of the separated proteins (Figure 1C). Under conditions of high calcium and CaM concentrations, FKBP38 is fully complexed and spectral contributions of the uncomplexed immunophilin can be neglected. The ellipticity of FKBP38 dominates the near-UV CD spectrum, because CaM had only a minor contribution in this spectral region. The appearance of fine structure of the spectrum points to aromatic side chains that rearrange during heterodimer formation. Spectral changes were not found either in the absence of Ca2+ or in the presence of EGTA, indicating strict requirement for calcium ions. Far-UV CD spectra did not reveal spectral changes upon complex formation, indicating lack of secondary structure rearrangements (data not shown). In conclusion, the CD spectrum of the Ca2+/CaM/FKBP38 complex exhibits a substantially increased level of tertiary structure, with only a minor change in the secondary structure.
The heterodimeric Ca2+/CaM/FKBP38 complex exhibits enzymatic activity
To investigate whether the observed interaction influences the enzymatic properties of FKBP38, we examined recombinant FKBP38. This protein was completely inactive in PPIase assays in the absence of Ca2+/CaM. However, considerable rate acceleration of the peptidyl prolyl cis/trans isomerization of the standard PPIase tetrapeptide substrate succinyl-AFPF-4-nitroanilide was observed in the presence of Ca2+/CaM (Figure 2A). The PPIase activity that was found was strictly depending on the concentration of FKBP38 under conditions of Ca2+/CaM saturation (Figure 2A, inset). Excess FK506 competed with the substrate for binding to the PPIase site, and thus decreased the rate of the cis to trans isomerization of the substrate down to the rate of the spontaneous interconversion. Neither Ca2+ nor CaM alone promoted FKBP38 activity, indicating that only the heterodimeric Ca2+/CaM/FKBP38 complex is the active form of FKBP38. At Ca2+/CaM saturation, the limiting value for the bimolecular rate constant of FKBP38 catalysis was calculated to be kcat/KM=1.1 × 104 M−1 s−1 for succinyl-AFPF-4-nitroanilide as substrate (Figure 2B). To determine the required calcium concentrations activating FKBP38, we measured the FKBP38 PPIase activity in the presence of 5 μM CaM and various calcium concentrations. Figure 2B, inset, shows that about 70% of the fully activated FKBP38 was achieved at 500 nM calcium (about 50% active enzyme is present at 290 nM). Therefore, FKBP38 is activated by the second messenger calcium and its sensor protein CaM at concentration levels found intracellularly after Ca2+ bursts (Feske et al, 2001). In the presence of CaM inhibitory MLCK peptide (1 μM), the PPIase activity of the Ca2+/CaM/FKBP38 complex disappeared.
Figure 2.
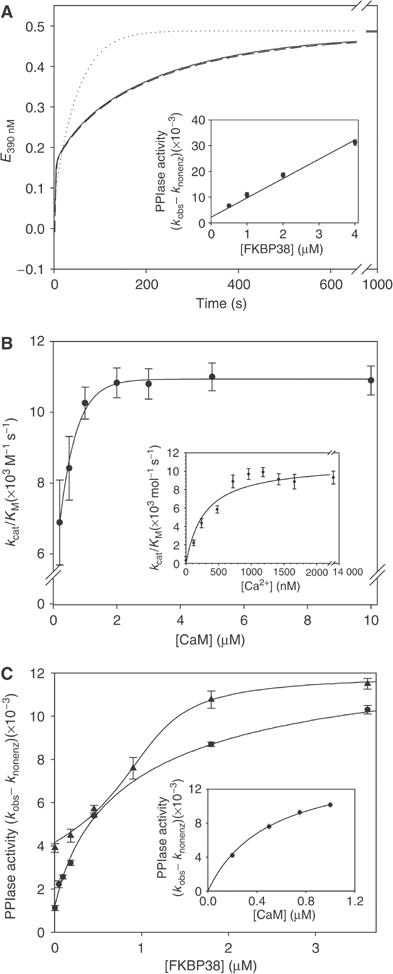
FKBP38 exhibits enzymatic activity after complex formation with Ca2+/CaM. (A) PPIase activity of 1 μM FKBP38 was measured in the presence (dotted line) and the absence (solid line) of 5 mM CaCl2 and 5 μM CaM in the protease-coupled standard PPIase assay. The dashed line represents the PPIase activity of FKBP38 in the presence of Ca2+/CaM and 2 μM FK506. The inset shows the dependence of PPIase activity on FKBP38 concentration at constant calcium (5 mM) and CaM (10 μM) concentrations. (B) CaM dependence of PPIase activity of 1 μM FKBP38. The Ca2+ dependence of the activation of FKBP38 by CaM was determined using 1 μM FKBP38, 5 μM CaM and various concentrations of calcium in the range of 50 nM–12 μM (inset). (C) Inhibition of Ca2+/CaM/FKBP38 PPIase activity by FK506 (•) and GPI1046 (▴) was studied in a competition assay with FKBP12. PPIase activity of 12 nM FKBP12 was inhibited by preincubation with 20 nM FK506 or 1140 nM GPI1046, respectively, 5 mM calcium and 5 μM CaM and subsequently recovered by addition of FKBP38. Because of competition of both FKBPs for inhibitor binding, inhibition constants can be determined. Calculation of inhibition constants was performed using Dynafit software. Regain of FKBP12 activity from the FKBP12/FK506 complex in the presence of 500 nM FKBP38 is strictly dependent on the CaM concentration (inset).
Now, the basis for the inhibitory action of FK506 is provided since only the Ca2+/CaM/FKBP38 assembly forms the binding pocket for FK506 (Figure 2C, inset). Usually, the inhibitory interaction of FKBP-type PPIases and FK506 is characterized to follow tight-binding kinetics. Micromolar concentrations of Ca2+/CaM/FKBP38 required in the PPIase assay render calculations of Ki values that are based on this inhibition type impossible. To determine inhibition constants for FK506, rapamycin and GPI1046, a novel assay was established on the basis of competition between Ca2+/CaM/FKBP38 and FKBP12 for inhibitor binding. Superior catalytic efficiency is achieved by FKBP12 (kcat/KM=1.2 × 106 M−1 s−1) compared with Ca2+/CaM/FKBP38 (kcat/KM=8 × 103 M−1 s−1) in a PPIase assay using the succinyl-ALPF-4-nitroanilide substrate. When both FKBPs exhibit comparable inhibitor affinities, a fraction of highly active FKBP12 emerged from a fully inhibited enzyme sample when the Ca2+/CaM/FKBP38 complex was allowed to sequester FKBP12-bound inhibitor molecules. Dose-dependent changes in emerging PPIase activity allowed the derivation of inhibition constants Ki for the drug–FKBP38 interaction (Figure 2C). In this assay, the nonimmunosuppressive, neurotrophic compound GPI1046 proved to be as potent as FK506 in inhibition of Ca2+/CaM/FKBP38 with a Ki of 48 nM. In contrast, rapamycin inhibited Ca2+/CaM/FKBP38 less efficiently with a Ki value of 499 nM.
In comparison, FKBP12 was found to be inhibited by FK506 and rapamycin with Ki values of about 1 nM, but GPI1046 was less efficient in inhibiting FKBP12 with a Ki value of 364, showing that GPI1046 will be preferentially bound by FKBP38 in neuronal cells.
The active site of Ca2+/CaM/FKBP38 interacts with Bcl-2
Based on suggestions of the physical interaction between FKBP38 and Bcl-2 (Shirane and Nakayama, 2003), we investigated whether Ca2+/CaM plays a role in this interaction. In co-immunoprecipitation experiments, endogenous FKBP38 and Bcl-2 associated only in the presence of Ca2+/CaM in SH-SY5Y cell lysate. In the absence of Ca2+, no interaction was observed. GPI1046 disrupted the Bcl-2–FKBP38 interaction, indicating that the active site of FKBP38 is important for Bcl-2 binding (Figures 3A and 7B).
Figure 3.
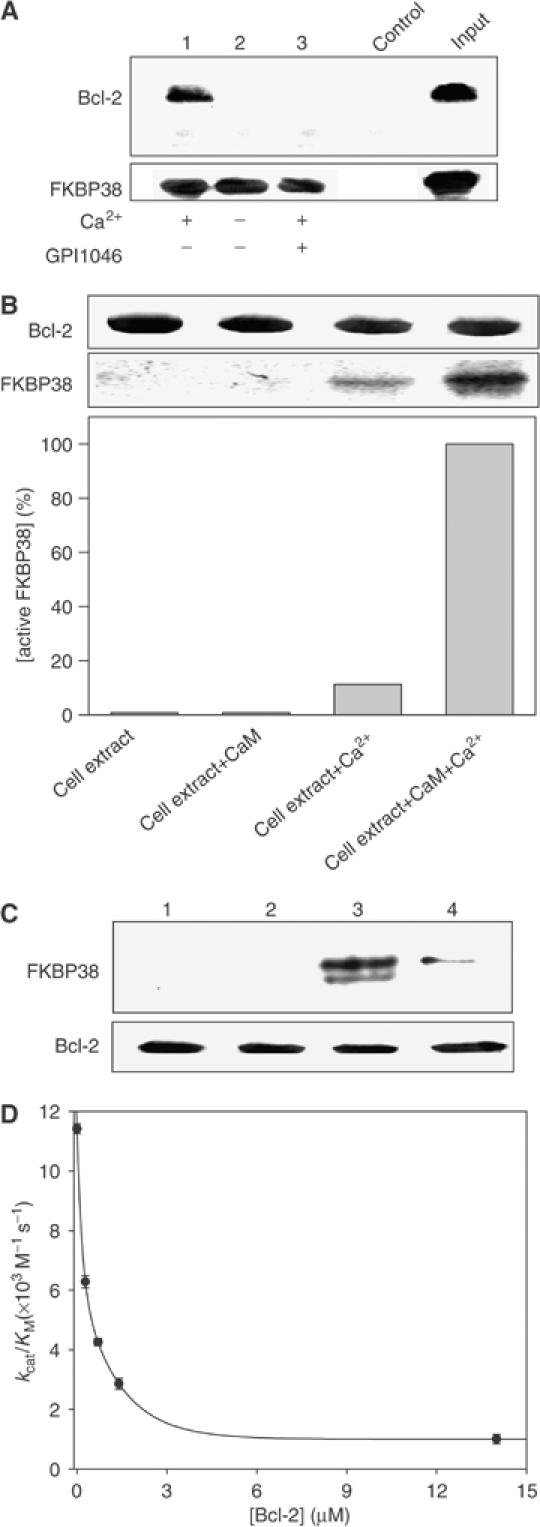
Only enzymatically active FKBP38 interacts with Bcl-2. (A) Co-immunoprecipitation of endogenous FKBP38 and Bcl-2. After preincubation with 500 μM EGTA, SH-SY5Y cell lysate was incubated with rabbit anti-FKBP38 antibody. Antibody/protein complexes were bound to protein G-Sepharose. Samples were washed. Precipitates (lanes 1–3) and input were subjected to SDS–PAGE and analyzed by Western blot using mouse anti-Bcl-2 antibody. Cell lysate preincubated with rabbit immunoglobin was used as a control. FKBP38 bound to Bcl-2 in the presence of 1 mM calcium. This interaction was disrupted by 1 μM GPI1046. No interaction was observed in the absence of Ca2+. (B) SH-SY5Y crude cell extract was applied in the absence and presence of 20 μM CaM and 2 mM Ca2+ to maltose-binding protein-Bcl-2 fusion protein (MBP-Bcl-2) immobilized on amylose resin. After three washing steps, protein was eluted by 200 mM maltose and analyzed by Western blot using polyclonal anti-FKBP38 antibody. The proportion of active endogenous FKBP38 was quantified by Biorad Multi-Analyst software. (C) MBP-Bcl-2 was immobilized on amylose resin and incubated with (1) FKBP38, (2) FKBP38 and CaM, (3) FKBP38 and Ca2+/CaM and (4) FKBP38, Ca2+/CaM and 200 nM GPI1046. After washing, protein was eluted and analyzed by Western blot using polyclonal anti-FKBP38 antibody. (D) Inhibition of PPIase activity of 1 μM FKBP38 by Bcl-2 was measured in the PPIase assay in the presence of 5 mM CaCl2 and 5 μM CaM. The calculated Ki value was 0.74 μM.
Figure 7.
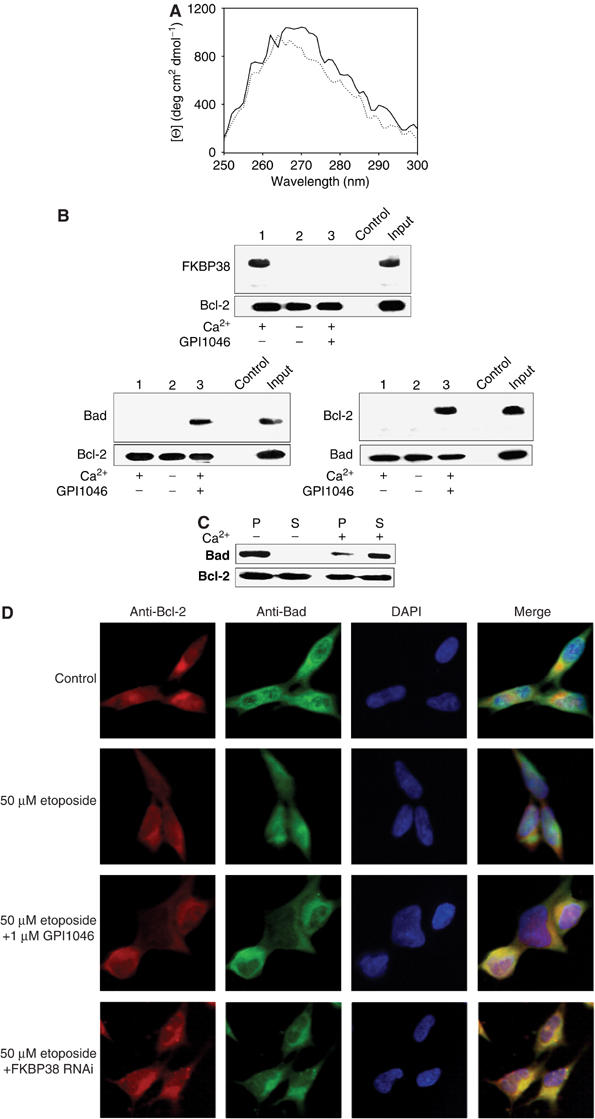
FKBP38 activity influences Bcl-2 function. (A) Near-UV CD spectra of 1 μM Ca2+/CaM/FKBP38 and 3 μM Bcl-2 were measured either separated (dotted line) or mixed (solid line) in a tandem cuvette. (B) Co-immunoprecipitation. SH-SY5Y cells were stimulated to apoptosis by 50 μM etoposide. Cell lysate preincubated with 500 μM EGTA was incubated with monoclonal mouse anti-Bcl-2 antibody. Antibody/protein complexes were bound to protein G-Sepharose. Samples were washed. Precipitates (lanes 1–3) and input were subjected to SDS–PAGE and analyzed by Western blot using rabbit anti-FKBP38 antibody. Cell lysate preincubated with control mouse immunoglobin was used as a control. FKBP38 bound to Bcl-2 in the presence of 1 mM Ca2+, whereas 1 μM GPI1046 abolished the interaction. Bcl-2/Bad interaction was investigated by incubation of SH-SY5Y cell lysate with hamster anti-Bcl-2 antibody and protein was detected using mouse anti-Bad antibody. Bad/Bcl-2 interaction was observed in the presence of 1 mM calcium and 1 μM GPI1046. In addition, SH-SY5Y cell lysate was incubated with monoclonal mouse anti-Bad antibody, and antibody/protein complexes formed were bound to protein G-Sepharose. Precipitates (lanes 1–3) and input were subjected to SDS–PAGE and analyzed by Western blot using hamster anti-Bcl-2 antibody. Cell lysate preincubated with mouse immunoglobin was used as a control. Bcl-2 bound to Bad in the presence of 1 mM Ca2+ and 1 μM GPI1046, indicating that GPI1046 interfered with the Bcl-2/FKBP38 interaction, allowing Bad/Bcl-2 complexes to form. (C) MBP-Bcl-2 fusion protein was immobilized on amylose beads and incubated with Bad (Sigma), FKBP38 and CaM either in the presence or absence of 2 mM Ca2+. Pellet (P) and supernatant (S) were subjected to 12.5% SDS–PAGE and analyzed by Western blotting using mouse anti-Bad antibody and hamster anti-Bcl-2 antibody. (D) Subcellular distribution of Bcl-2 and Bad in SH-SY5Y neuroblastoma cells was analyzed by immunostaining with FITC-conjugated goat anti-mouse antibody against mouse anti-Bad antibody and Cy5-conjugated goat anti-hamster antibody against hamster anti-Bcl-2 antibody. Nuclei were stained with DAPI. Cells were treated for 16 h with 50 μM etoposide either in the absence or presence of 1 μM GPI1046. Cells transfected with FKBP38 RNAi construct were analyzed with ImageBlue-conjugated goat anti-mouse antibody against mouse anti-Bad antibody and TRITC-conjugated goat anti-hamster antibody against hamster anti-Bcl-2. Nuclei were stained by 7-AAD. Because of better understanding, colors of this panel were adapted to other results presented in this figure. Again, inhibition of FKBP38 activity allows Bcl-2/Bad interaction after induction of apoptosis by 50 μM etoposide.
Furthermore, we analyzed the interaction of endogenous FKBP38 from SH-SY5Y cell lysate in a binding assay with Bcl-2. As shown in Figure 3B, Western blot immunodetection using anti-FKBP38 antibody revealed binding of maltose-binding protein-Bcl-2 fusion protein (MBP-Bcl-2) to endogenous FKBP38 present in the cell extract, but only to a small extent. The addition of Ca2+ and CaM (final concentrations were 2 mM and 20 μM, respectively) increased Bcl-2 binding about 120-fold. Ca2+ alone sufficed to get a 13-fold increase in FKBP38 molecules that were able to bind Bcl-2, indicating a limiting role of CaM under high cytosolic calcium concentrations.
In order to substitute endogenous FKBP38 in in vitro binding experiments, recombinant FKBP38 was tested for Bcl-2 interaction. Again, only the Ca2+/CaM/FKBP38 complex was able to interact with Bcl-2, whereas FKBP38 did not show affinity for Bcl-2 in the absence of Ca2+ or CaM (Figure 3C). GPI1046 and Bcl-2 competed for binding to FKBP38, as observed with endogenous FKBP38 (Figure 3C, lane 4). Based on these results, the PPIase activity of the Ca2+/CaM/FKBP38 complex was tested in the presence of recombinant Bcl-2. As shown in Figure 3D, Bcl-2 reversibly and competitively inhibited enzymatic activity of FKBP38 with a Ki value of 415±63 nM, supporting the notion of FKBP38 active site involvement in Bcl-2 binding.
Inhibition of FKBP38 prevents apoptosis
Because the Ca2+/CaM/FKBP38 complex interacts with Bcl-2, we were interested in the effect of FKBP38 activity on apoptosis of neuroblastoma cells. Therefore, the influence of FKBP38 inhibition by FKBP38 RNA interference (RNAi) and the FKBP ligands GPI1046, FK506 and rapamycin on apoptotic SH-SY5Y cells was tested. Rapamycin and GPI1046 were found to be especially discriminating among FKBP inhibitors tested.
As shown in Figures 4A–F, GPI1046 prevented apoptosis in neuroblastoma cells and increased the cell survival rate up to 75% in a concentration-dependent manner when cells were subjected to the apoptosis-inducing compound etoposide (Figures 4A and G). Similar antiapoptotic effects of GPI1046 on cells were observed when apoptosis was induced by daunorubicin, ionomycin and camptothecin (Figures 4C, D and F). However, neither FK506 nor rapamycin increased cell survival rates (Figures 4A–F).
Figure 4.
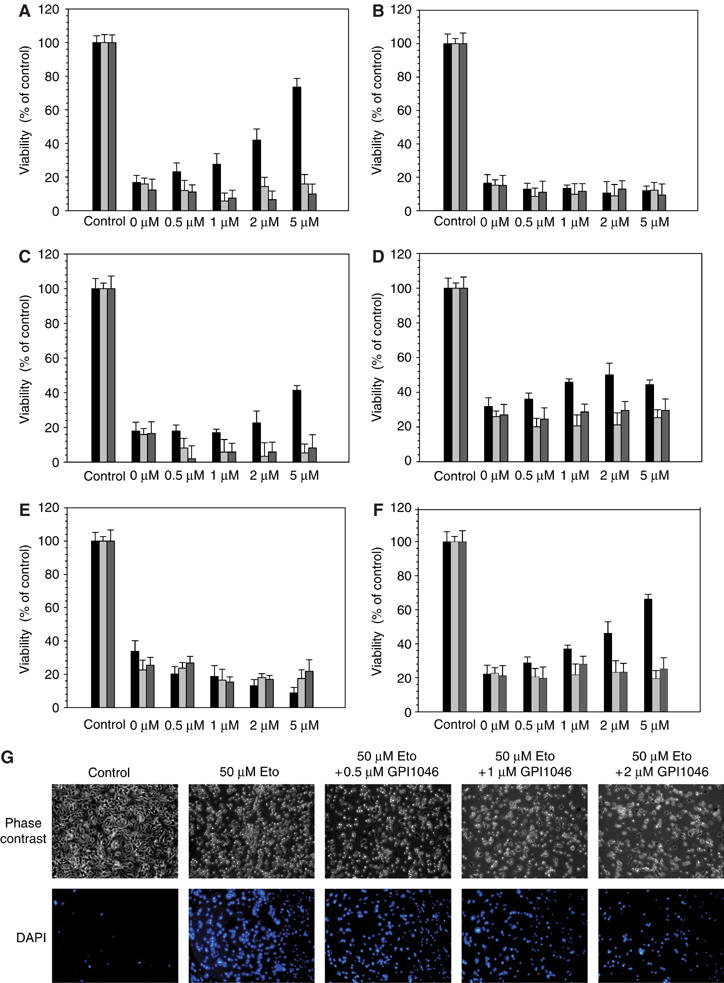
Inhibition of FKBP38 activity protects neuroblastoma cells from apoptosis. Survival rates of SH-SY5Y cells treated with 50 μM etoposide (A), 150 μM antimycin A2 (B), 50 nM daunorubicin (C), 10 μM ionomycin (D), 100 nM staurosporin (E) and 5 μM camptothecin (F) and various concentrations of GPI1046 (black bar), FK506 (gray bar) and rapamycin (dark gray bar) were determined by using Guava Viability Assay with a Guava personal cell analyzer. (G) SH-SY5Y cells treated with 50 μM etoposide (Eto) and various concentrations of GPI1046 were analyzed by phase-contrast microscopy. Apoptotic cells were visualized with DAPI staining using a fluorescence microscope.
To analyze whether the neuroprotective effect of GPI1046 is due to the inhibition of FKBP38, we performed RNAi experiments. FKBP38 RNAi expression resulted in a significant reduction of endogenous FKBP38 protein in neuroblastoma cells (Figure 5A). A concomitant enhancement of cell survival (Figure 5B) was observed. The antiapoptotic effect of FKBP38 RNAi was exclusively obtained with GPI1046-sensitive apoptotic stimuli of wild-type cells (etoposide, daunorubicin, camptothecin), but not with GPI1046-insensitive stimuli (antimycin A2, staurosporin) (Figures 4A–F). Thus, the antiapoptotic effect of FKBP38 depletion by RNAi paralleled the effect of blocking the PPIase site of FKBP38 by GPI1046. In further support of the role of FKBP38 inhibition by GPI1046 for neuronal cell survival, the drug inhibited the residual FKBP38 activity in etoposide-treated cells containing FKBP38 RNAi, completely abolishing the proapoptotic effect of FKBP38. This compound proved to be completely inactive in both staurosporin- and antimycin A2-treated wild-type cells and FKBP38-depleted cells. These findings are in contrast to previous observations of FKBP38 depletion by RNAi in HeLa cells (Shirane and Nakayama, 2003). To analyze whether the contradicting data were due to the different cell lines used, we investigated the influence of our FKBP38 RNAi construct on HeLa cells. Indeed, HeLa cells transfected with FKBP38 RNAi showed increased cell survival rates compared to cells transfected with the control vector (Supplementary Figure 1). The differences in the effects of RNAi between the neuronal SH-SY5Y cell line and the epithelial-like HeLa cell line are perhaps cell type specific due to different apoptotic pathways (Bursztajn et al, 2001; Fulda et al, 2001), various Bcl-2 interaction partners and their expression pattern (Mackrill et al, 1997; Makin et al, 2001). For example, depletion of endoplasmic reticulum (ER) calcium that leads to an increased cytosolic calcium concentration appeared to have a protective function for HeLa cells, whereas depletion of ER calcium was toxic for SH-SY5Y cells (Pinton et al, 2001; Nguyen et al, 2002).
Figure 5.
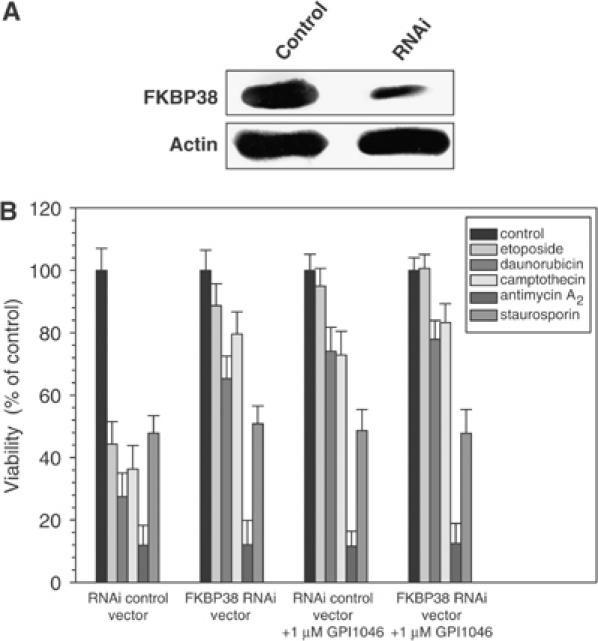
FKBP38 depletion by RNAi inhibits apoptosis in neuroblastoma cells. (A) SH-SY5Y neuroblastoma cells were transfected with a vector producing specific FKBP38 RNAi or the empty vector (control). Further controls using an unspecific oligonucleotide and a vector construct containing the unspecific sequence were performed showing no effect of the vector control on SH-SY5Y cell survival rates (data not shown). Expression of FKBP38 was analyzed by Western blot using rabbit polyclonal anti-FKBP38 antibody. β-Actin was used as a loading control. (B) Cells transfected with FKBP38 RNAi vector or empty vector were stimulated to apoptosis with 50 μM etoposide, 50 nM daunorubicin, 5 μM camptothecin, 150 μM antimycin A2 and 150 nM staurosporin in the absence and presence of 5 μM GPI1046. Survival rates of SH-SY5Y cells were determined by using Guava Viability Assay with a Guava personal analyzer.
Given the GPI1046-induced protection of neuroblastoma cells and the fact that GPI1046 interferes with Bcl-2/Ca2+/CaM/FKBP38 complex formation, the GPI1046-dependent subcellular distribution of Bcl-2 and FKBP38 was studied using various apoptotic stimuli. In our experiments, FKBP38 and Bcl-2 colocalize in the presence and absence of GPI1046 (Figure 6A). These results indicate that PPIase activity of FKBP38 does not influence the subcellular distribution of Bcl-2 in SH-SY5Y cells. The subcellular localization of FKBP38 was investigated comparing the immunostaining of mitochondria, ER and actin filaments (Figure 6B). FKBP38 colocalized with the staining of mitochondria and ER of the neuroblastoma cells, but not with actin staining. The mitochondrial and endoplasmic localization of FKBP38 is similar to the localization of Bcl-2, as could be inferred from published data (Kim et al, 2004). However, the Ca2+/CaM-stimulated PPIase activity of FKBP38 does not mediate delocalization of its substrate Bcl-2 (Figure 6A).
Figure 6ab.
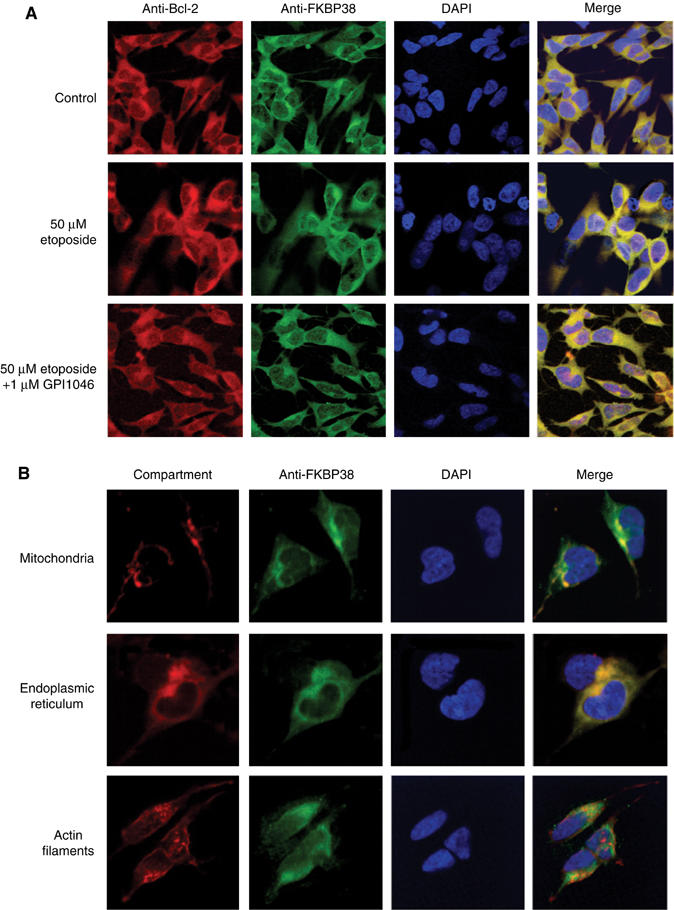
FKBP38 and Bcl-2 colocalize in neuroblastoma cells. (A) Localization of Bcl-2 and FKBP38 in SH-SY5Y neuroblastoma cells was analyzed by immunostaining with Cy5-conjugated goat anti-hamster antibody against hamster anti-Bcl-2 antibody and FITC-conjugated goat anti-rabbit IgG against rabbit anti-FKBP38 antibody. Cells were treated for 16 h with 50 μM etoposide and 2 μM GPI1046. Nuclei were stained with DAPI. (B) Subcellular distribution of FKBP38 in SH-SY5Y cells was studied by immunostaining with FITC-conjugated goat anti-rabbit IgG against rabbit anti-FKBP38 antibody and a subcellular structure localization kit (Chemicon).
The mitochondrial and ER localization of FKBP38 and Bcl-2 in neuroblastoma cells was confirmed by analyzing isolated fractions of mitochondria, ER, nuclei and cytosol of SH-SY5Y cells by Western blotting (Figure 6C). FKBP38 is found in the ER and mitochondrial fractions. Bcl-2 can be detected in these fractions as well. Only a small portion of this protein was additionally detected in the nuclear fraction of neuroblastoma cells.
Figure 6c.
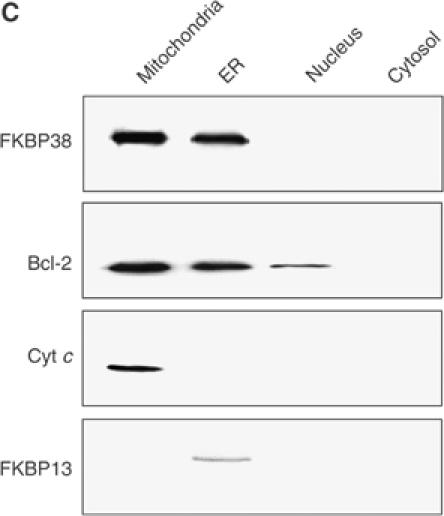
(C) Subcellular localization of FKBP38 and Bcl-2 was studied by preparing mitochondria, ER, nucleus and cytosol from SH-SY5Y cells and analyzing these fractions by Western blotting using anti-FKBP38 and anti-Bcl-2 antibodies. Antibodies detecting cytochrome c (Cyt c) and FKBP13 were used as controls for mitochondrial and ER localization, respectively.
Bcl-2 function is regulated by its interaction with FKBP38
We have shown by co-immunoprecipitation and PPIase activity assays that Bcl-2 binds to the active site of FKBP38. Therefore, we examined the tertiary structure of Bcl-2 complexed with Ca2+/CaM/FKBP38 by near-UV CD spectroscopy. As shown in Figure 7A, complex formation of Ca2+/CaM/FKBP38 with Bcl-2 changes significantly the near-UV CD spectrum realizing a blue shift of about 3 nm, and a concomitant amplitude reduction in the range of 270–290 nm indicating the formation of a ternary complex. Altogether, the structural and kinetic data showed features consistent with the idea that Bcl-2 turned out to be a substrate for FKBP38 that undergoes bioactivity relevant changes that might be related to a prolyl cis/trans isomerization. We next tested whether the observed Bcl-2/FKBP38 interaction affects the antiapoptotic function of Bcl-2 in neuroblastoma cells. The cellular function of Bcl-2 is determined by the ability to bind to several proapoptotic proteins preventing apoptosis initiation. Therefore, we investigated whether FKBP38 influences the interaction of Bcl-2 with a binding partner, such as Bad. In a co-immunoprecipitation experiment, FKBP38 was able to block Bad interaction with Bcl-2 in the presence of Ca2+, whereas in samples containing a specific inhibitor of the FKBP38 active site, both proteins interact with each other (Figure 7B). Furthermore, we tested whether FKBP38 interferes with Bad/Bcl-2 complex formation in a binding assay, using Bcl-2 immobilized on maltose beads. As shown in Figure 7C, FKBP38 is only able to disrupt Bad/Bcl-2 complexes in the presence of calcium and CaM.
Based on the interaction assays, the influence of the FKBP38 inhibitor GPI1046 and FKBP38 RNAi on the subcellular localization of Bad was studied. In untreated cells, Bad did not colocalize with Bcl-2. Similarly, colocalization did not occur when cells were stimulated to apoptosis (Figure 7D). However, in the presence of the PPIase inhibitor GPI1046 or in neuroblastoma cells transfected with FKBP38 RNAi, Bad and Bcl-2 colocalize when cells are stimulated to apoptosis. These data suggest that the active form of FKBP38 might change the tertiary structure of Bcl-2 and influence thereby the ability of Bcl-2 to interact with its partner proteins. However, at this point, a steric hindrance of Bad/Bcl-2 interaction by FKBP38 cannot be excluded.
Discussion
Here, we demonstrated that the activity of FKBP38 is controlled by its association with Ca2+/CaM. Enzymatic activity was observed at calcium concentrations below 1 μM. Simultaneously, appearance of an FK506-binding site in the heterodimeric complex was observed. In the absence of Ca2+/CaM, the enzyme remained completely inactive, and commonly known FKBP ligands, such as immunosuppressive and nonimmunosuppressive peptidomacrolides and their derivatives, failed to bind.
Our study provides the first example for a cofactor-regulated folding helper enzyme. In order to verify enzyme activation by intracellular Ca2+ rise, the active site concentration of endogenous FKBP38 was determined by co-immunoprecipitation and affinity absorption on MBP-Bcl-2 amylose beads. The inactive form of FKBP38 dominates in unstimulated SH-SY5Y cells.
Near-UV CD spectroscopy revealed activation of FKBP38 in the Ca2+/CaM/FKBP38 complex by changes of the tertiary structure-related signals of the enzyme. As observed for most Ca2+/CaM-regulated enzymes, the mechanism of cofactor activation might involve an altered conformation of a regulatory segment of FKBP38 that complements FKBP38 in the FKBP-like catalytic core. In calcineurin (CaN), the Ca2+/CaM-mediated displacement of an autoinhibitory segment caused an increased accessibility of the catalytic site (Klee et al, 1998). The identification of the putative autoinhibitory domain of FKBP38 requires further experiments.
Previous investigations using yeast two-hybrid system revealed physical interaction of FKBP38 and Bcl-2 (Shirane and Nakayama, 2003). Our data demonstrate that this interaction occurs only with the active form of FKBP38. Therefore, it can be assumed that the identified interaction of both proteins in yeast depended on the calcium concentration as well. A highly homologous form of CaM present in yeast likely mediated FKBP38 activation (Cyert, 2001). In addition, it was shown that the interaction of Bcl-2 and FKBP38 is involved in apoptosis. In contrast to these findings in HeLa cells, we observed a proapoptotic role of FKBP38 in SH-SY5Y cells that is strictly dependent on the formation of PPIase active form of FKBP38. It is noteworthy that protein–protein interaction experiments, confocal imaging and apoptosis assays using specific FKBP38 inhibition demonstrate that the cellular functions of FKBP38 investigated here are not mediated by the membrane anchor, as previously suggested. Our results show that the cellular function of FKBP38 depends on the enzymatic activity of the protein.
Furthermore, in the same report, FKBP38 and Bcl-2 were exclusively identified in mitochondrial membranes (Shirane and Nakayama, 2003). However, the mitochondrial and ER localization of Bcl-2 and FKBP38 in our assay agrees well with the colocalization of FKBP38 and Bcl-2 and published data about the subcellular distribution of Bcl-2 (Vander Heiden and Thompson, 1999; Demaurex and Distelhorst, 2003; Kim et al, 2004). The colocalization of FKBP38 and Bcl-2 is most likely because both proteins contain a C-terminal membrane anchor and are found in the same cellular compartments. However, interaction between both proteins occurs only upon calcium influx and subsequent FKBP38 activation. The proposed dual function of FKBP38, mitochondrial targeting of Bcl-2 and direct inhibition of the protein phosphatase CaN (Shirane and Nakayama, 2003) do not play a part in the apoptotic pathway of neuroblastoma cells, because CaN is inert against FKBP38 (Weiwad et al, 2005). More specifically, our data indicate that FKBP38 neither in the presence nor absence of Ca2+ inhibits or interacts with CaN. Only the FK506/Ca2+/CaM/FKBP38 complex has affinity to CaN, inactivating its protein phosphatase activity (Weiwad et al, 2005).
Competition experiments revealed participation of the active site of Ca2+/CaM/FKBP38 in Bcl-2 binding. These findings suggest that Bcl-2 belongs to the cellular substrates of FKBP38. The Ca2+/CaM-mediated structural changes of FKBP38-bound Bcl-2 might indicate proline-directed conformational changes, thereby altering the biological activity of the protein. It has been demonstrated that PPIases are able to display cis/trans isomer ratios of substrates in the Michaelis complexes that are rather different from those obtained for the unbound substrates (Howard et al, 2003). Such an isomer shift may be important in isomer-specific reactions if the concentration of the Michaelis complex is high (Fischer and Aumuller, 2003). In addition, we have shown that formation of the Ca2+/CaM/FKBP38 complex subsequently led to disruption of the Bcl-2 interaction with physiological interaction partners in vitro and in vivo, implying that the homeostasis of Bcl-2 complexes is disturbed in cells with high FKBP38 level after calcium rise.
The neuroimmunophilin inhibitor GPI1046 efficiently blocks progression of apoptosis induced by various stimuli in neuroblastoma cells. Using FKBP38 RNAi, the concentration of endogenous FKBP38 was significantly reduced in neuroblastoma cells. The RNAi experiments demonstrated that GPI1046 targets the Ca2+/CaM/FKBP38 complex and blocks the proapoptotic function of endogenous FKBP38 in SH-SY5Y cells, because an antiapoptotic effect by RNAi was only observed with GPI1046-sensitive apoptotic stimuli. Antimycin A2- and staurosporin-induced apoptosis that is not affected by FKBP38 RNAi was not attenuated by GPI1046 as well. Remarkably, most of the GPI1046-sensitive stimuli have been described to be antagonized by Bcl-2 overexpression (Eliseev et al, 2003; Kim et al, 2003). The effect of RNAi-mediated FKBP38 depletion on apoptosis correlated well with the antiapoptotic effect of the FKBP38 active site inhibition by GPI1046, showing a relation between FKBP38 activity and apoptosis in neuroblastoma cells.
The antiapoptotic effect of FKBP inhibitors is probably mediated by preventing the interaction of Bcl-2 with the Ca2+/CaM/FKBP38 complex. Subsequent to stimulation, GPI1046 administration led to changed subcellular localization of the Bcl-2 interaction partner Bad in SH-SY5Y cells. These data are consistent with a recent report showing that FKBP38 reverses the antiapoptotic effect of Bcl-2 (Massaad et al, 2004).
Rapamycin, which inhibits constitutively active FKBPs preferably, did not increase cell survival rates. The lack of a pronounced antiapoptotic effect for FK506 is, in part, at variance to its relatively high affinity for FKBP38. The manifold of biochemical processes affected by FK506 renders cellular effects of FK506 difficult to interpret (Fischer and Aumuller, 2003). In this respect, it is noteworthy that constitutively active FKBPs will not respond to Ca2+ rise in their FK506 affinity. However, they collectively form an efficient sink for FK506, reducing its availability to the active site of the Ca2+/CaM/FKBP38 complex.
Our study indicates that the proapoptotic function of FKBP38 can be explained partly by the regulation of Bcl-2 interaction with Bad. In this respect, it may be interesting to see whether FKBP38 affects binding of Bcl-2 to other interaction partners. It would be of special interest to elucidate the functional mechanism of the Ca2+/CaM-mediated activation of the FKBP38 PPIase site and the mechanism by which this activity regulates Bcl-2. Interestingly, a proline-directed conformational change was observed in the Bcl-2-related Bax protein that is functionally important (Schinzel et al, 2004). In addition, it is important to clarify the inhibition of different FKBP activities by the ligands available to gain an insight into the physiological processes that these proteins are involved in. The identification of the first FKBP functionally controlled by a second messenger raises the question of whether the physiological processes involving similarly structured multidomain FKBPs might also respond to calcium signals.
Materials and methods
Sources of enzymes used in the experiments are as follows: recombinant human FKBP38, lacking the membrane anchor (FKBP381–336), recombinant human FKBP12, recombinant human CaM and human Bcl-2 were recombinantly expressed by using a pET28a-vector in Escherichia coli Rosetta™ cells. MBP-Bcl-2 fusion protein and His-tagged Bad were purchased from Sigma (Deisenhofen, Germany). The FKBP38 antibody was an affinity-purified section 4 polyclonal antibody from rabbit against purified FKBP38 domain (amino acids 1–165). Additional antibodies used were polyclonal rabbit anti-actin (Sigma), monoclonal hamster anti-Bcl-2 (Pharmingen), monoclonal mouse anti-Bcl-2 (Santa Cruz), polyclonal rabbit anti-CaM (Santa Cruz), monoclonal mouse anti-CaM (Sigma), monoclonal mouse anti-Bad (Pharmingen) and polyclonal goat anti-mouse ImageBlue labeled (Leinco). The subcellular structure localization kit (Chemicon) was used.
Peptide substrates used were obtained from Bachem (Heidelberg, Germany). FK506 and rapamycin were purchased from Calbiochem (La Jolla, CA). Low-molecular-weight substances were synthesized according to standard procedures (Christner et al, 1999) and characterized by ESI mass spectrometry.
Measurement of PPIase activity
PPIase activity was determined using protease-coupled assays (Fischer et al, 1989). Typically, FKBP38 PPIase activity was measured in a reaction mixture containing 1 μM FKBP38, 5 μM recombinant human CaM and 5 mM CaCl2. Inhibition constants for the PPIase activity of the Ca2+/CaM/FKBP38 complex by low-molecular-weight inhibitors were determined by a competition assay using recombinant FKBP12 and succinyl-ALPF-4-nitroanilide substrate. Based on different acceleration rates for FKBP12 and FKBP38 (kcat/KM=0.9 × 106 and 1 × 104 M−1 s−1, respectively), it is possible to detect exclusively FKBP12 activity. Because of competition of both FKBPs for inhibitor binding, PPIase activity of FKBP12 increases after addition of FKBP38. PPIase activity of 12 nM FKBP12 was inhibited either by 20 nM FK506, 23 nM rapamycin or 1140 nM GPI1046 and subsequently recovered by addition of FKBP38. Assay mixtures were allowed to equilibrate for 20 min. Because of competition of both FKBPs for inhibitor binding, inhibition constants could be determined by using Dynafit software (Kuzmic, 1999) according to the following reaction mechanism:
![]()
where E and F represent the concentrations of FKBP12 and FKBP38, respectively, and I the concentration of the FKBP inhibitors.
In the case of Bcl-2 and CaM, insensitivity to proteolytic digestion by α-chymotrypsin in the time range of the kinetic experiments was verified.
Protein–protein interaction
Co-immunoprecipitation: Cells were grown in flasks and incubated with 50 μM etoposide for 16 h and harvested. Cell lysis and co-immunoprecipitation experiments were performed according to the manufacturer's protocols of the immunoprecipitation starter kit (Amersham Bioscience). Prior to incubation, 0.5 mM EGTA was added to the samples. A 1 mM portion of CaCl2 was added to those samples incubated in the presence of Ca2+.
CaM-binding assay: CaM-Sepharose (Amersham-Pharmacia Biotech) was pre-equilibrated in 25 mM Tris–HCl buffer (pH 7.5, 200 mM NaCl, 1 mM DTT) in the presence of either 2 mM CaCl2 or 2 mM EGTA. Subsequently, 30 μg recombinant FKBP38 was incubated with CaM-Sepharose. Sepharose was washed and bound proteins were analyzed by 12.5% SDS–PAGE and Coomassie blue staining.
Bcl-2-binding assay: A 40 μl volume of 6 μM MBP-Bcl-2 fusion protein (MBP-Bcl-2) was subjected to 40 μl amylose resin (New England Biolabs, Beverly, MA) and incubated for 30 min. Thereafter, beads were washed twice with buffer B (25 mM Tris–HCl, pH 7.5, 200 mM NaCl, 1 mM DTT) and subsequently incubated for 1 h with 40 μl reaction mixture containing either FKBP38, FKBP38 (50 μM) and CaM (200 μM), FKBP38 and Ca2+/CaM, or FKBP38/Ca2+/CaM and 200 nM GPI1046. In some experiments, SH-SY5Y crude cell extract was applied in the absence and presence of 20 μM CaM and 2 mM Ca2+ to maltose-binding protein-Bcl-2 fusion protein (MBP-Bcl-2) immobilized on amylose resin. To prepare SH-SY5Y crude cell extract, cells were harvested, centrifuged for 10 min at 2000g and cell pellets were resuspended in the same volume of hypotonic lysis buffer, followed by sonication. Beads were pelleted, washed three times and mixed with an equal volume of Laemmli sample buffer. Then, samples were subjected to 12.5% SDS–PAGE and analyzed by Western blotting using a polyclonal rabbit anti-FKBP38 antibody.
Circular dichroism spectroscopy: Near-UV CD spectra were recorded at 25°C on a Jasco J-710 CD spectrometer in 10 mM HEPES pH 7.8, 2 mM CaCl2 containing 1 μM FKBP38 and 5 μM CaM either separated or mixed in a 10 mm tandem cuvette. Interaction of 1 μM FKBP38 and 3 μM Bcl-2 was measured in the presence of 1 mM CaCl2 and 5 μM CaM in a tandem cuvette as well.
Cell culture
To test the antiapoptotic effect of FKBP38 inhibitors, the neuroblastoma cell line SH-SY5Y (DSMZ Braunschweig, Germany) was used. Cells were cultured in DMEM (Biochrom, Berlin, Germany) supplemented with 2 mM L-glutamine and 10% (v/v) heat-inactivated FCS in a humidified incubator at 37°C in 10% (v/v) CO2. SH-SY5Y cells were seeded at 5 × 106 cells/ml in six-well plates.
Induction of apoptosis
The cells were preincubated with various concentrations of GPI1046, rapamycin or FK506 for 10 min at 37°C before apoptotic stimuli were applied. Stock solutions of the substances in DMSO (Sigma, Deisenhofen, Germany) were added to a final concentration of 0.5% (v/v) DMSO in each sample. After stimulation, cells were grown for 16 h at 37°C and 10% CO2.
Apoptosis assay
Cells were analyzed after treatment for 16 h by phase-contrast microscopy at 400-fold magnification. Survival rates of SH-SY5Y cells treated with apoptotic stimuli and various concentrations of GPI1046, FK506 and rapamycin were determined by using the ViaCount assay (Guava Technologies, Hayward, CA). The Guava ViaCount assay distinguishes viable and nonviable cells based on the differential cell permeability of a combination of DNA-binding dyes in the Guava ViaCount reagent. The assay was performed according to the manufacturer. The data presented are means±s.d. of three independent experiments.
Depletion of FKBP38 by RNAi
For depletion of FKBP38 by RNAi, a pair of 64 nt oligonucleotides containing the sequence 5′-AAGAGTGGCTGGACATTCT-3′ was constructed. The fragment of annealed oligonucleotides was cloned into the RNAi producing vector pSUPER.neo−GFP (Oligoengine, Seattle, WA) according to the manufacturer's instructions. The pSUPER construct was introduced into SH-SY5Y and HeLa cells by electroporation (Amaxa, Cologne, Germany). After 16 h, the transfection efficiencies between 75 and 85% were determined by FACS. The reduction of the cellular FKBP38 by RNAi was tested by Western blotting, and compared to cells containing the RNAi control vector.
Confocal imaging
SH-SY5Y and HeLa cells were cultured on 10 mm coverslips and transfected or incubated with 50 μM etoposide and 1 μM GPI1046 for 16 h. Cells were fixed in 3.7% paraformaldehyde in phosphate-buffered saline (PBS) for 30 min at room temperature. After washing in PBS, the cells were permeabilized for 30 min on ice with 0.5% saponine in PBS containing 10% FCS, blocked with PBS containing 10% FCS for 1 h, incubated with PBS containing primary antibody (1:100 dilution for hamster anti-human Bcl-2 antibody (BD Biosciences Pharmingen), 1:100 dilution for mouse anti-human Bad antibody (Pharmingen) and 1:50 dilution for rabbit anti-human FKBP38 antibody) and 10% FCS for 1 h and incubated with PBS containing secondary antibody (1:100 dilution for TRITC-conjugated goat anti-mouse antibody IgG and 1:100 dilution for FITC-conjugated goat anti-rabbit IgG, 1:50 dilution for Cy5-conjugated goat anti-hamster antibody (Biomol, Hamburg, Germany)) and 10% FCS for 30 min. Nuclei were stained with DAPI (BD Biosciences Pharmingen). Coverslips were mounted onto glass slides with Vectashield mounting medium H-1000 (Vector Laboratories Inc.). Cells were analyzed by confocal laser scanning microscopy (Carl Zeiss LSM 410) at 750-fold magnification.
Cellular fractions of SH-SY5Y cells were isolated using the Sigma ER isolation kit, mitochondrial isolation kit and Nuclei isolation kit PURE prep and analyzed by Western blot using rabbit anti-FKBP38, mouse anti-Bcl-2, mouse anti-Cyt c and rabbit anti-FKBP13 antibodies.
Supplementary Material
Supplementary Figure 1
Acknowledgments
We are grateful to Beate Rappsilber and Martina Heidler for technical assistance. Furthermore, we thank Professor Gunther Reuter and Anja Ebert for providing confocal microscope and support.
References
- Burnett AL, Becker RE (2004) Immunophilin ligands promote penile neurogenesis and erection recovery after cavernous nerve injury. J Urol 171: 495–500 [DOI] [PubMed] [Google Scholar]
- Bursztajn S, Feng JJ, Nanda A, Berman SA (2001) Differential responses of human neuroblastoma and glioblastoma to apoptosis. Brain Res Mol Brain Res 91: 57–72 [DOI] [PubMed] [Google Scholar]
- Christner C, Herdegen T, Fischer G (2001) FKBP ligands as novel therapeutics for neurological disorders. Mini Rev Med Chem 1: 377–397 [DOI] [PubMed] [Google Scholar]
- Christner C, Wyrwa R, Marsch S, Kullertz G, Thiericke R, Grabley S, Schumann D, Fischer G (1999) Synthesis and cytotoxic evaluation of cycloheximide derivatives as potential inhibitors of FKBP12 with neuroregenerative properties. J Med Chem 42: 3615–3622 [DOI] [PubMed] [Google Scholar]
- Crackower MA, Kolas NK, Noguchi J, Sarao R, Kikuchi K, Kaneko H, Kobayashi E, Kawai Y, Kozieradzki I, Landers R, Mo R, Hui CC, Nieves E, Cohen PE, Osborne LR, Wada T, Kunieda T, Moens PB, Penninger JM (2003) Essential role of Fkbp6 in male fertility and homologous chromosome pairing in meiosis. Science 300: 1291–1295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cyert MS (2001) Genetic analysis of calmodulin and its targets in Saccharomyces cerevisiae. Annu Rev Genet 35: 647–672 [DOI] [PubMed] [Google Scholar]
- Demaurex N, Distelhorst C (2003) Apoptosis—the calcium connection. Science 300: 65–67 [DOI] [PubMed] [Google Scholar]
- Eliseev RA, Gunter KK, Gunter TE (2003) Bcl-2 prevents abnormal mitochondrial proliferation during etoposide-induced apoptosis. Exp Cell Res 289: 275–281 [DOI] [PubMed] [Google Scholar]
- Feske S, Giltnane J, Dolmetsch R, Staudt LM, Rao A (2001) Gene regulation mediated by calcium signals in T lymphocytes. Nature Immunol 2: 316–324 [DOI] [PubMed] [Google Scholar]
- Fischer G, Aumuller T (2003) Regulation of peptide bond cis/trans isomerization by enzyme catalysis and its implication in physiological processes. Rev Physiol Biochem Pharmacol 148: 105–150 [DOI] [PubMed] [Google Scholar]
- Fischer G, Wittmann-Liebold B, Lang K, Kiefhaber T, Schmid FX (1989) Cyclophilin and peptidyl-prolyl cis–trans isomerase are probably identical proteins. Nature 337: 476–478 [DOI] [PubMed] [Google Scholar]
- Fong S, Mounkes L, Liu Y, Maibaum M, Alonzo E, Desprez PY, Thor AD, Kashani-Sabet M, Debs RJ (2003) Functional identification of distinct sets of antitumor activities mediated by the FKBP gene family. Proc Natl Acad Sci USA 100: 14253–14258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulda S, Meyer E, Friesen C, Susin SA, Kroemer G, Debatin KM (2001) Cell type specific involvement of death receptor and mitochondrial pathways in drug-induced apoptosis. Oncogene 20: 1063–1075 [DOI] [PubMed] [Google Scholar]
- Gold BG (2000) Neuroimmunophilin ligands: evaluation of their therapeutic potential for the treatment of neurological disorders. Expert Opin Investig Drugs 9: 2331–2342 [DOI] [PubMed] [Google Scholar]
- Gold BG, Densmore V, Shou W, Matzuk MM, Gordon HS (1999) Immunophilin FK506-binding protein 52 (Not FK506-binding protein 12) mediates the neurotrophic action of FK506. J Pharmacol Exp Ther 289: 1202–1210 [PubMed] [Google Scholar]
- Harrar Y, Bellini C, Faure JD (2001) FKBPs: at the crossroads of folding and transduction. Trends Plant Sci 6: 426–431 [DOI] [PubMed] [Google Scholar]
- Howard BR, Vajdos FF, Li S, Sundquist WI, Hill CP (2003) Structural insights into the catalytic mechanism of cyclophilin A. Nat Struct Biol 10: 475–481 [DOI] [PubMed] [Google Scholar]
- Kamphausen T, Fanghanel J, Neumann D, Schulz B, Rahfeld JU (2002) Characterization of Arabidopsis thaliana AtFKBP42 that is membrane-bound and interacts with Hsp90. Plant J 32: 263–276 [DOI] [PubMed] [Google Scholar]
- Kim PK, Annis MG, Dlugosz PJ, Leber B, Andrews DW (2004) During apoptosis bcl-2 changes membrane topology at both the endoplasmic reticulum and mitochondria. Mol Cell 14: 523–529 [DOI] [PubMed] [Google Scholar]
- Kim YH, Park JW, Lee JY, Surh YJ, Kwon TK (2003) Bcl-2 overexpression prevents daunorubicin-induced apoptosis through inhibition of XIAP and Akt degradation. Biochem Pharmacol 66: 1779–1786 [DOI] [PubMed] [Google Scholar]
- Klee CB, Ren H, Wang X (1998) Regulation of the calmodulin-stimulated protein phosphatase calcineurin. J Biol Chem 273: 13367–13370 [DOI] [PubMed] [Google Scholar]
- Kuzmic P (1999) General numerical treatment of competitive binding kinetics: application to thrombin–dehydrothrombin–hirudin. Anal Biochem 267: 17–23 [DOI] [PubMed] [Google Scholar]
- Lam E, Martin MM, Timerman AP, Sabers C, Fleischer S, Lukas T, Abraham RT, O'Keefe SJ, O'Neill EA, Wiederrecht GJ (1995) A novel FK506 binding protein can mediate the immunosuppressive effects of FK506 and is associated with the cardiac ryanodine receptor. J Biol Chem 270: 26511–26522 [DOI] [PubMed] [Google Scholar]
- Lu KP, Liou YC, Vincent I (2003) Proline-directed phosphorylation and isomerization in mitotic regulation and in Alzheimer's disease. BioEssays 25: 174–181 [DOI] [PubMed] [Google Scholar]
- Mackrill JJ, Challiss RA, O'Connell DA, Lai FA, Nahorski SR (1997) Differential expression and regulation of ryanodine receptor and myo-inositol 1,4,5-trisphosphate receptor Ca2+ release channels in mammalian tissues and cell lines. Biochem J 327: 251–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makin GW, Corfe BM, Griffiths GJ, Thistlethwaite A, Hickman JA, Dive C (2001) Damage-induced Bax N-terminal change, translocation to mitochondria and formation of Bax dimers/complexes occur regardless of cell fate. EMBO J 20: 6306–6315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massaad CA, Portier BP, Taglialatela G (2004) Inhibition of transcription factor activity by nuclear compartment-associated Bcl-2. J Biol Chem 279: 54470–54478 [DOI] [PubMed] [Google Scholar]
- Mattson MP (2000) Apoptosis in neurodegenerative disorders. Nat Rev Mol Cell Biol 1: 120–129 [DOI] [PubMed] [Google Scholar]
- Nguyen HN, Wang C, Perry DC (2002) Depletion of intracellular calcium stores is toxic to SH-SY5Y neuronal cells. Brain Res 924: 159–166 [DOI] [PubMed] [Google Scholar]
- Pinton P, Ferrari D, Rapizzi E, Di Virgilio F, Pozzan T, Rizzuto R (2001) The Ca2+ concentration of the endoplasmic reticulum is a key determinant of ceramide-induced apoptosis: significance for the molecular mechanism of Bcl-2 action. EMBO J 20: 2690–2701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryo A, Liou YC, Wulf G, Nakamura M, Lee SW, Lu KP (2002) PIN1 is an E2F target gene essential for Neu/Ras-induced transformation of mammary epithelial cells. Mol Cell Biol 22: 5281–5295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schinzel A, Kaufmann T, Schuler M, Martinalbo J, Grubb D, Borner C (2004) Conformational control of Bax localization and apoptotic activity by Pro168. J Cell Biol 164: 1021–1032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sezen SF, Hoke A, Burnett AL, Snyder SH (2001) Immunophilin ligand FK506 is neuroprotective for penile innervation. Nat Med 7: 1073–1074 [DOI] [PubMed] [Google Scholar]
- Shirane M, Nakayama KI (2003) Inherent calcineurin inhibitor FKBP38 targets Bcl-2 to mitochondria and inhibits apoptosis. Nat Cell Biol 5: 28–37 [DOI] [PubMed] [Google Scholar]
- Silverstein AM, Galigniana MD, Kanelakis KC, Radanyi C, Renoir JM, Pratt WB (1999) Different regions of the immunophilin FKBP52 determine its association with the glucocorticoid receptor, hsp90, and cytoplasmic dynein. J Biol Chem 274: 36980–36986 [DOI] [PubMed] [Google Scholar]
- Tanaka K, Fujita N, Yoshioka M, Ogawa N (2001) Immunosuppressive and non-immunosuppressive immunophilin ligands improve H2O2-induced cell damage by increasing glutathione levels in NG108-15 cells. Brain Res 889: 225–228 [DOI] [PubMed] [Google Scholar]
- Vander Heiden MG, Thompson CB (1999) Bcl-2 proteins: regulators of apoptosis or of mitochondrial homeostasis? Nat Cell Biol 1: 209–216 [DOI] [PubMed] [Google Scholar]
- Weiwad M, Edlich F, Erdmann F, Jarczowski F, Kilka S, Dorn M, Pechstein A, Fischer G (2005) A reassessment of the inhibitory capacity of human FKBP38 on calcineurin. FEBS Lett 579: 1591–1596 [DOI] [PubMed] [Google Scholar]
- Zhang CY, Steiner JP, Hamilton GS, Hicks TP, Poulter MO (2001) Regeneration of dopaminergic function in 6-hydroxydopamine-lesioned rats by neuroimmunophilin ligand treatment. J Neurosci 21: 156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H, You H, Zhou XZ, Murray SA, Uchida T, Wulf G, Gu L, Tang X, Lu KP, Xiao ZX (2002) The prolyl isomerase Pin1 is a regulator of p53 in genotoxic response. Nature 419: 849–853 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1