

Abstract
Heterochromatin is generally associated with gene silencing, yet in Drosophila melanogaster, heterochromatin harbors hundreds of functional protein-encoding genes, some of which depend on heterochromatin for expression. Here we document a recent evolutionary transition of a gene cluster from euchromatin to heterochromatin, which occurred <20 million years ago in the drosophilid lineage. This finding reveals evolutionary fluidity between these two genomic compartments and provides a powerful approach to identifying differences between euchromatic and heterochromatic genes. Promoter mapping of orthologous gene pairs led to the discovery of the “slippery promoter,” characterized by multiple transcriptional start sites predominately at adenines, as a common promoter type found in both heterochromatic and euchromatic genes of Drosophila. Promoter type is diverse within the gene cluster but largely conserved between heterochromatic and euchromatic genes, eliminating the hypothesis that adaptation to heterochromatin required major alterations in promoter structure. Transition to heterochromatin is consistently associated with gene expansion due to the accumulation of transposable elements and increased A-T content. We conclude that heterochromatin-dependent regulation requires specialized enhancers or higher-order interactions and propose a facilitating role for transposable elements.
Keywords: heterochromatin, promoter, transposable element
Heterochromatin was originally defined by its densely staining cytological appearance, but it is also typically associated with other biological features such as low recombination rate, late replication, and ability to modulate gene activity. The silencing effect of heterochromatin on gene expression is well documented in eukaryotes. The classical example is transcriptional silencing of euchromatic genes abnormally juxtaposed to heterochromatin by chromosome rearrangement, a phenomenon known as position effect variegation (PEV), which was first described for Drosophila. Studies of genetic suppressors of PEV [Su(var)s] have identified numerous chromatin proteins that are enriched in heterochromatin and mediate its silencing effect in a dosage-dependent manner (reviewed in ref. 1). Another defining characteristic of heterochromatin is the extreme abundance of the so-called “junk” repetitive DNAs such as transposable elements (TEs) and satellite sequences. Recent studies suggest that heterochromatin is also a natural platform for RNA interference-based phenomena (2-4). Small RNAs produced from repetitive DNAs have been implicated in heterochromatin formation and transcriptional silencing (4). It seems likely that these RNA interference phenomena are derived from naturally occurring mechanisms that defend the genome against the deleterious mobilization of TEs (2).
Given the prevailing views of heterochromatin, it is surprising that functional single-copy genes are embedded within heterochromatin in Drosophila (1, 5). These heterochromatic genes are immune to the silencing effect of heterochromatin and, in some cases, are known to depend on a heterochromatic location and Su(var) proteins for normal expression (6, 7). The notion that repetitive elements create heterochromatic repressive environments by recruiting silencing components is well accepted (4, 8), but it is unknown how this environment can facilitate expression of heterochromatic genes. In principle, there are two possibilities. First, these genes might be insulated from repressive heterochromatin, essentially maintaining a microenvironment that is compatible with transcriptional capacity. In this case, these genes might be considered euchromatic, with expression possible because of the establishment of specialized chromatin boundaries or other features that maintain separation of local domains of heterochromatin and euchromatin. Second, these genes might have adopted a specialized gene regulation mechanism tailored for heterochromatin. This proposed alteration could have involved acquisition of novel core promoters, enhancer elements, trans-acting factors, or a combination of these elements. An attractive possibility is an acquisition of TE-derived promoters given the predominance of TE-like sequences in heterochromatin and the finding that some TE promoters are transcribed in heterochromatin (9).
To distinguish among these possibilities, it is important to understand the evolutionary origin of heterochromatic genes and to identify their conserved and dynamic features. Here we show that a cluster of heterochromatic genes has evolved relatively recently from euchromatic ancestors in drosophilids. Structural comparisons of the heterochromatic and euchromatic orthologues have allowed us to infer a model to explain how these genes have adapted to heterochromatin to acquire heterochromatin-dependent expression.
Materials and Methods
Drosophila Strains and Germ-Line Transformation. Strains of Drosophila melanogaster used were wild-type Canton-S, yw, and the isochromosomal y; cn bw sp. These strains and other Drosophila species strains were obtained from the Bloomington Stock Center (Bloomington, IN), from the Tucson Stock Center (Tucson, AZ), or from R. Levis (Carnegie Institution, Baltimore). Germ-line transformation was carried out by using standard techniques.
DNA Probes and Hybridization. Genomic Southern blot and chromosomal FISH analyses were carried out with standard protocols, which are available on request. For these assays, we used D. melanogaster genomic or cDNA probes (10) for detecting light (lt) genes in the D. melanogaster species subgroup. A 600-bp Dp-lt(eu) probe was obtained by PCR from Drosophila pseudoobscura using primers matched to sequences within exons 9 and 11 of the D. melanogaster lt gene, because this showed the most consistent cross-species hybridization on genomic Southern blots. A 2.45-kb Dv-lt(eu) cDNA was isolated from a Drosophila virilis ovarian cDNA library and used for probing Drosophila ananassae and D. virilis. The Dv-lt cDNA and the corresponding genomic region were sequenced.
Mapping Transcriptional Start Sites. Poly(A)+ RNA was isolated from ovaries from D. virilis, D. pseudoobscura, and D. melanogaster (y; cn bw sp strain) adult females and used to map transcriptional start sites by using RNA ligase-mediated RACE (RLM-RACE) according to the manufacturer's specifications (Ambion, Austin, TX). RNA samples were first treated with alkaline phosphatase to remove 5′-phosphate group from all degraded or truncated RNAs. The sample was then treated with pyrophosphatase to remove the 5′ cap from full-length mRNAs, leaving a single 5′-phosphate group. The RNA adapter was ligated to this newly exposed 5′-phosphate group with T4 RNA ligase. The adapter-ligated RNAs, derived from the 5′-capped full-length mRNAs, were amplified by RT-PCR by using adapter-specific primers and gene-specific primers. The gene-specific primers hybridized near the predicted start codon of each gene. Primer sequences are available on request. A parallel experiment in which pyrophosphatase treatment was omitted served as a negative control. The PCR products were ligated to pCR2.1 vector (Invitrogen) transformed into Escherichia coli. A minimum of six randomly selected clones were sequenced for each gene. We repeated the experiment with an alternative RNA adapter (5′-GCUGAUGGCGAUGAAUGAACACUGCGUUUGCUGGCUUUGAUGAAC-3′) that was custom-synthesized by Ambion. We also repeated the analysis for D. melanogaster using testis mRNA.
Results
Evolutionary Transition of the lt Gene from Euchromatin to Heterochromatin. To identify the distinguishing features of heterochromatic genes, we focused on the lt gene, an autosomal heterochromatic gene of D. melanogaster. It is an essential gene located in chromosome 2L heterochromatin, and its dependence on heterochromatin and Su(var) proteins for expression is well established (6, 7, 11). We examined the copy number and chromosomal location of the orthologous gene in nine additional Drosophila species. Genomic Southern blots established that each species has a single lt gene. We used FISH on salivary gland polytene chromosomes to distinguish heterochromatic from euchromatic chromosomal location. As shown for D. melanogaster, the hallmark of a heterochromatic gene is a dispersed FISH signal in the chromocenter due to irregular alignment of chromatids in regions enriched in repetitive DNA (Fig. 1A) (12). Similar hybridization patterns were observed in six species most closely related to D. melanogaster (e.g., Fig. 1 B and C). However, a tight FISH signal aligned with a proximal euchromatic band in D. ananassae and D. pseudoobscura and in more distal euchromatin D. virilis (Fig. 1 D-F). In each case, the signal was located in the chromosome arm corresponding to the D. melanogaster 2L. The most parsimonious explanation based on the phylogeny of Drosophila (Fig. 1G) (13, 14) is that the ancestral lt gene was euchromatic but was heterochromatized in the lineage that gave rise to the melanogaster subgroup species, an event estimated to have occurred <20 million years ago (13, 14).
Fig. 1.

Chromosomal location of the lt gene in drosophilids. Location of the gene was mapped by FISH on polytene salivary gland chromosomes in D. melanogaster (A), D. yakuba (B), Drosophila erecta (C), D. ananassae (D), D. pseudoobscura (E), and D. virilis (F). The arrow indicates the FISH signal (red); chromosomes counterstained with DAPI (blue) and heterochromatic chromocenter (cc) are indicated. (Scale bar, 20 μm.) (Insets) Higher-magnification views of the FISH signals. (G) The phylogenetic tree of drosophilids (13, 14). FISH analysis was performed on 10 species (red), and those with heterochromatic lt genes are shown in the blue box.
The discovery of euchromatic and heterochromatic orthologues allowed us to examine lt gene features in two different chromatin contexts. The D. virilis and D. pseudoobscura lt [denoted Dv-lt(eu) and Dp-lt(eu), respectively] were compared with the heterochromatic D. melanogaster gene [Dm-lt(het)]. Predicted amino acid sequences are highly similar (72-83% identity and 84-89% similarity among three species). Based on cDNAs for the Dm and Dv genes and the predicted gene structure for Dp, we conclude that the exon-intron structures are essentially identical (Fig. 2A). Vicinities of the euchromatic lt genes are entirely single-copy, whereas the Dm-lt(het) is characterized by a high density of repetitive DNAs in flanking regions and in the two largest introns (Fig. 2 A) (10). The repetitive DNAs result in a substantial expansion of the transcription unit and are heterogeneous. Most are scrambled TE-like sequences (Fig. 2 A) (5).
Fig. 2.

Comparisons of the heterochromatic and euchromatic lt genes. (A) Structure of the lt genes, showing exons (black blocks) and TE-related sequence content. Orientation and type of TE-like repetitive DNAs are shown by the colored arrows (blue, LTR retroposons; green, non-LTR retroposons; red, inverted-repeat transposons). (B) RACE-PCR products assayed on ethidium bromide-stained gels. The - lanes show negative controls (see Materials and Methods). (C) Promoter structures of the lt genes determined by RLM-RACE. Each arrow corresponds to a randomly selected clone of the RACE-PCR product. The number of arrows at each position is therefore an indication of relative frequency of usage as a transcription start site. For Dm-lt(het), the start sites detected by using the alternative RNA adapter are indicated by dashed arrows, and the start sites found in testis are shown underneath the sequence. Circles indicate conserved sequence motifs. Note that the TATA motif in Dm-lt(het) is replaced by GATA in the corresponding region of Dp-lt(eu), but this noncanonical TATA-box has been observed for other genes (37).
Promoter Comparison of the Heterochromatic and Euchromatic lt Genes. To test the possibility that Dm-lt(het) has adapted to heterochromatin by acquiring a new type of core promoter, we mapped the transcriptional start site of the lt gene from three species, then searched for potential promoter motifs. We used RLM-RACE, which involves ligation of a defined RNA adapter specifically to the 5′ ends of full-length mRNAs (15), followed by RT-PCR and sequencing of cloned products. We used ovarian mRNA because lt is abundantly expressed in this tissue (10).
We observed two promoter types. The Dv-lt(eu) promoter has the simplest structure, with a single transcription start site (Fig. 2 B and C). Two sequence motifs that correspond to promoter motifs 1 and 5 of Ohler et al. (16) and are commonly found in Drosophila reside within 30 nt of the Dv-lt(eu) start site. These features indicate that Dv-lt(eu) has a “typical” promoter. We showed that this Dv-lt(eu) promoter is recognized in D. melanogaster. A transgene containing 710 bp upstream of the start site and the entire coding sequence of Dv-lt(eu) fully rescued lt- mutation in D. melanogaster when inserted into euchromatic locations. This result also shows that the promoter and the encoded protein are compatible in both species.
We obtained strikingly different results for the Dm-lt(het) gene. The RLM-RACE products were heterogeneous in size (Fig. 2 B and C), and sequencing of randomly selected clones revealed numerous start sites (seven different sites identified among 11 sequenced clones). A TATA-box motif is found at ≈30 bp upstream of the 5′-most start site identified. Curiously, the choice of transcription start sites is nonrandom; there is strong preference for adenine (A) as the start site. We repeated the mapping using testis mRNA and found that testis start sites largely overlapped with those in ovary (Fig. 2C).
To verify that the observed preference for A start sites was not due to a technical artifact of the ligation step in the RLM-RACE procedure, we repeated the ovarian mRNA start-site mapping using an alternative RNA adapter. The adapter serves as a ligation acceptor, and the decapped mRNA serves as ligation donor. Because it is known that 3′ sequence of ligation acceptor may affect the choice of the ligation donor (17, 18), we tested an alternative RNA adapter whose sequence was identical to the standard adapter provided by the manufacturer except that the 3′ end was AAC-3′ instead of AAA-3′. The predominance of A start sites in Dm-lt(het) was reproduced by using this alternative RNA adapter (Fig. 2C). Specifically, we found that six of seven sequenced clones, representing five different start sites, started with A. One exception, a C start site, was found much further downstream (data not shown). These results demonstrate that the A preference reflects the in vivo transcription start sites rather than biased ligation efficiency in the RNA ligation step. Moreover, in the assay conducted by England and Uhlenbeck (17), A was rather less favored a donor when AAAC-3′ oligo was used as acceptor, further strengthening our conclusion.
We refer to the promoter documented here that has multiple start sites with predominance of A as the “slippery promoter.” The discovery of this newly described promoter type in association with the Dm-lt(het) gene was intriguing and led us to hypothesize that it may be a distinguishing feature of a heterochromatic gene. However, mapping of the Dp-lt(eu) transcription start sites showed a similar pattern of multiple start sites (Fig. 2 B and C), allowing us to conclude that this promoter type is not heterochromatin-specific. Interestingly, the Dp-lt(eu) promoter bears sequence similarity to both the Dv-lt(eu) start-site region (indicated by open circles in Fig. 2C) and the Dm-lt(het) TATA-box region (filled circles), although the first T is replaced by G (see Fig. 2 legend). The promoter sequence similarities observed for the Dp-lt(eu) promoter are accompanied by the corresponding transcription start sites (Fig. 2C). Thus, it is tempting to view the Dp-lt(eu) promoter as an “intermediate” between Dv-lt(eu) and Dm-lt(het) promoters given the phylogenetic relationships.
Comparative Analysis of the Heterochromatic Genes and the Euchromatic Orthologues. To determine whether our conclusions from studies of the lt gene could be more generally applied to other genes, we extended the analysis to additional D. melanogaster-D. pseudoobscura orthologous gene pairs. This comparison was possible because of the recent publication of a draft sequence that includes >96% of the euchromatic genome of D. pseudoobscura (19). Eleven protein-encoding genes, including Dm-lt(het), are located within a 594-kb heterochromatic contig analyzed by the Drosophila Heterochromatin Project (Fig. 3A) (5). As shown in Fig. 3B, we found orthologues for seven genes, including lt, within a 224-kb D. pseudoobscura contig assembled by the Baylor College of Medicine Human Genome Sequencing Center (19) and localized to euchromatin of chromosome 4 (Fig. 1E). These data indicate that the evolutionary transition of these seven genes from euchromatin to heterochromatin occurred at the level of a large genomic segment rather than on a gene-by-gene basis. Of the remaining four genes found in the D. melanogaster heterochromatic contig, the closest homologues for three genes (Cht3, CG17715, and CG40016) are found elsewhere in the D. pseudoobscura euchromatin. Cht3 and CG17715 are closely linked, being separated by ≈10 kb. Interestingly, the closest homologue for CG40439 is found in the chromosome U in D. pseudoobscura, which is a collection of unassigned short sequence fragments that likely represents heterochromatic DNA. Thus, CG40439 may be an example of a more ancient heterochromatic gene.
Fig. 3.

Comparisons of the heterochromatic and euchromatic gene clusters. (A) Gene content of the 594-kb D. melanogaster heterochromatic lt contig (5). Arrows indicate the direction of transcription. (B) Gene content of the 224-kb D. pseudoobscura euchromatic lt contig. Gene annotation was based on blastx hits (E < 10-3) against melanogaster genes. The genes are named according to the closest homologues in D. melanogaster to facilitate comparisons for this study. The cytogenetic locations of the closest homologues in melanogaster are noted in the parentheses. For gene names, see Table 1, which is published as supporting information on the PNAS web site. Note that A and B are not drawn to scale. Relative gene sizes and distances between the genes were also neglected. (C) Comparison of A-T content of the ORFs. Filled diamonds, D. melanogaster heterochromatic genes; open diamonds, D. pseudoobscura euchromatic genes. (D) Comparison of A-T content of the individual non-TE-containing introns. The symbols are the same as in C.
Repetitive DNAs occupy >50% of the Dm-lt(het) contig (5) but <5% of the Dp-lt(eu) contig. Manual annotation of the Dp-lt(eu) contig based on similarity to D. melanogaster genes predicts ≈35 protein-encoding genes. Thus, the Dp-lt(eu) contig has ≈8.4-fold greater gene density than the Dm-lt(het) contig (Fig. 3 A and B). Comparisons for the seven orthologous gene pairs show that the heterochromatic genes are consistently larger in size because of TE accumulation and are 5-10% more AT-rich in the coding sequences (Fig. 3C) as well as in the introns (Fig. 3D) than their euchromatic counterparts.
We mapped the transcription start sites for all seven orthologous gene pairs (Figs. 2C and 4). Three gene pairs have slippery promoters. The promoters of orthologous gene pairs do not often share extensive sequence identity, but there is a striking correlation in the “slipperiness” that they exhibit. In summary, not all heterochromatic genes use slippery promoter, slippery promoter is not restricted to the heterochromatic genes, and the promoter structure seems to be largely conserved between each heterochromatic gene and its euchromatic orthologue.
Fig. 4.

Comparisons of promoter structures, as in Fig. 2C. Circles indicate conserved motifs, and underlining indicates similar sequences. For C and E, some sequences are omitted (slashed lines) to show the similarity in the upstream.
Discussion
Euchromatic genes are repressed when experimentally juxtaposed to heterochromatin, and few, if any, exceptions are known to this rule. Here we provide evidence for a relatively recent evolutionary transition of euchromatic genes to heterochromatic genes that occurred in the melanogaster lineage <20 million years ago. The discovery that 7 of 11 genes found in D. melanogaster 2L heterochromatin have orthologues in D. pseudoobscura that are clustered on the corresponding chromosome arm suggests that an infiltration of heterochromatin occurred in the melanogaster lineage.
The origin of the autosomal heterochromatic genes we studied differs from the mechanism proposed for the D. melanogaster Y-linked genes, which involved the duplication and interchromosomal transposition of individual genes onto the Y chromosome (20). It is also distinct from the origin of gene cluster located within the heterochromatic knob of Arabidopsis thaliana (21). These genes arose from an undated segmental duplication of a euchromatic region, and the euchromatic and heterochromatic paralogues coexist in the genome. As in the case of the D. melanogaster Y-linked genes, the Arabidopsis gene pairs are evolving under conditions of functional redundancy. A comparison of two Arabidopsis gene clusters shows that TEs have invaded intergenic regions in the heterochromatic gene cluster, but the genes themselves are devoid of TE insertions, histone H3 methylated at lysine-9, and DNA methylation, which are markers considered typical of heterochromatin. Hence, expression of these genes is possible, because they have essentially remained as isolated islands of euchromatin. The situation may be transitional. According to conventional views of heterochromatin, the expectation is that, with continued invasion of TEs, the genes will lose their ability to be expressed. Arabidopsis genes apparently have the additional constraint on intron size, because only one intron is predicted to exceed 5 kb based on whole-genome analysis (www.arabidopsis.org). Hence, in Arabidopsis, TE or non-TE insertions of any type that substantially increase intron size will be detrimental to gene expression.
Unlike Arabidopsis, Drosophila and mammals tolerate large introns. Indeed, Drosophila autosomal heterochromatic genes have accumulated TEs within their introns as well as flanking regions (Fig. 2 A) (10). In addition, they take advantage of heterochromatin-enriched proteins such as Su(var) proteins and have become dependent on them for normal levels of expression (7). Moreover, the Drosophila heterochromatic genes are single-copy in all species we have examined, and some (e.g., lt) are essential. Thus, their essential function may have required that they adopt a more robust strategy to adapt to heterochromatin, one that ensures tolerance to ever-accumulating TEs.
Carvalho and Clark (22) recently described another interesting case that permits intra- and interspecific comparisons of heterochromatic and euchromatic gene counterparts. Their data suggest that chromosomal translocations occurred between the sex chromosomes and autosomes in the D. pseudoobscura lineage and led to genomic reorganizations such that formerly Y-linked genes are now autosomal. These genes have apparently shrunk in size after the translocation, because they lack the megabase-size introns and extraordinarily large intergenic regions characteristic of more ancestral Y-linked orthologues present in other Drosophila species. Carvalho and Clark (22) suggest that these once heterochromatic genes of D. pseudoobscura are evolving toward a more euchromatic nature, a reverse transition of heterochromatin to euchromatin. Taken together, these Arabidopsis and Drosophila studies demonstrate surprising evolutionary fluidity between heterochromatic and euchromatic domains.
Our discovery of euchromatic orthologues of the heterochromatic genes of D. melanogaster permits a comparative analysis of genes in two genomic contexts. In addition to substantial gene expansion associated with massive TE accumulation, we found elevated A-T content in the coding sequences and the non-TE-containing introns of heterochromatic genes relative to their euchromatic counterparts (Fig. 3 C and D). This observation is consistent with the idea that there is regional mutation bias and/or fixation bias depending on the chromosomal location. Features that may contribute to this bias include replication timing, subnuclear compartmentalization, and, perhaps most critically, recombination rate (23, 24), all of which are known to differ between heterochromatin and euchromatin.
During this study, we fortuitously discovered a newly described promoter type, which we refer to as the “slippery promoter.” It is defined by the occurrence of multiple start sites with predominance of A and is seen in TATA-box-containing and TATA-less genes (Figs. 2C and 4). This promoter bears superficial resemblance to the multiple start-site promoters described in mammals (25, 26) and yeast (27). In mammals, the TATA-box mediates recruitment and positioning of the RNA polymerase active site, so transcription starts ≈30 bp downstream (28). Start-site multiplicity in mammalian cells is associated with a lack of the TATA-box (25, 26, 29). However, in yeast, transcription typically starts further downstream of the TATA-box (up to 120 bp) and often at multiple sites. The positions are defined by start-site sequences and not by distance from TATA-box (27). Studies of Drosophila have shown that TATA-box function is more similar to that of mammals than yeast (16, 30). Although our analysis of start sites is not exhaustive, current data suggest that the Dm-lt(het) and Dp-lt(eu) TATA-boxes are involved in RNA polymerase positioning, but the choice of the exact start site is permissive. In this regard, the slippery promoter represents a new paradigm. We note that start-site multiplicity has been previously noted for GC-rich rather than AT-rich promoters in mammals (26), and predominance of A in the multiple start-site promoters has not been noted in mammals or yeast.
We suggest that, in certain genes, the transcriptional machinery binds slightly promiscuously to DNA or slides in cis along DNA, resulting in multiple start sites. The reason the slippery promoter may have escaped attention in previous in vivo studies of Drosophila transcription may the use of assays less sensitive than RLM-RACE or the general inclination of researchers to regard only cDNAs with the longest 5′ extension as “complete.”
Three major conclusions can be drawn from our promoter analyses. First, the slippery promoter is a distinct and common promoter type among Drosophila genes regardless of chromosomal locations and can be maintained throughout evolution. Second, promoter structures of the heterochromatic genes are quite diverse (Fig. 4). For the genes reported here, we have observed slippery and nonslippery promoters, TATA- and TATA-less promoters, and a pyrimidine-rich promoter, which is characteristic of the yip6 gene and other ribosomal protein genes (31). Therefore, usage of any particular promoter type is not a prerequisite for adaptation to heterochromatin. Third, and most importantly, evolutionary adaptation to heterochromatin has been achieved without changing the basic promoter type. Occasional alteration of promoter type occurs, as we observed for the Dv-lt(eu) and Dm-lt(het) genes, but is not correlated with the major change of chromosomal environment.
From our data, we suggest a model for evolution of the heterochromatic lt gene in the melanogaster lineage. Ancestrally, lt was a typical euchromatic gene (Fig. 5A). The lt and neighboring genes may have resided quite proximally in the euchromatin on the chromosome arm, or they may have been relocated closer to the centromere by chromosome rearrangement. In either case, gradual accumulation of TEs is proposed to play a major role in their evolution, leading to expansion of the genes and separation of essential enhancer elements from the promoter (Fig. 5B). The increasing distance between enhancers and promoter could be a threat for maintenance of transcriptional capacity, which could be maintained only if long-distance interactions could bring cis-regulatory regions into proximity. Su(var) proteins, actively recruited by the repetitive DNAs, perhaps through RNA interference-related mechanisms, could facilitate interactions between interspersed repetitive DNAs and bring remote enhancer elements in close proximity to the promoter (Fig. 5C). Comparisons among Drosophila simulans, Drosophila yakuba, and D. melanogaster, three closely related species that share ≈90% DNA sequence identity genome-wide, show that the lt gene TATA-box region is particularly well conserved. However, there is no recognizable sequence similarity at least in the next several kb upstream because of differences in TE insertions (data not shown). Moreover, our attempts to drive expression from Dm-lt(het) transgene constructs that contain up to 7 kb of 5′-flanking region have been unsuccessful. These observations are consistent with the idea that essential enhancer elements have been displaced from the promoter by the TE insertions.
Fig. 5.
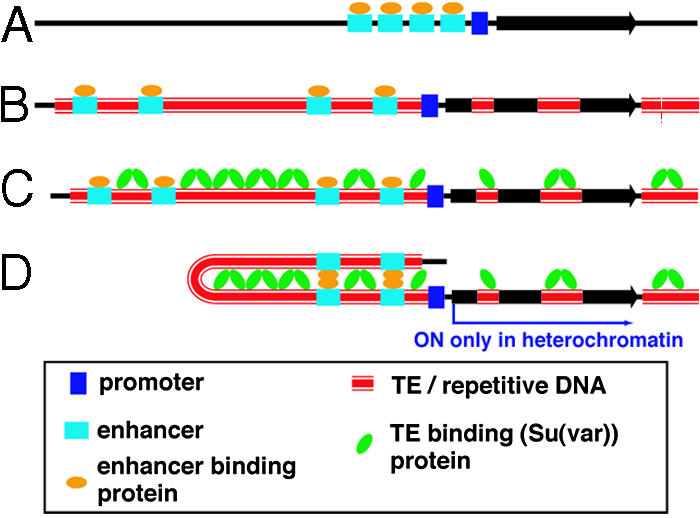
A model for the evolutionary transition from a euchromatic gene to a heterochromatic gene. (A) A compact euchromatic gene is expanded by the insertions of TEs. Expansion is proposed to occur gradually by the accumulation of TEs, resulting in the separation of enhancers from the promoter (B). (C) The Su(var) chromatin proteins bind repetitive sequences, including TEs, and facilitate enhancer-promoter interactions by DNA looping. (D) These interactions account for the requirement of heterochromatic genes for a heterochromatic environment and the Su(var) proteins for transcription.
Our model (Fig. 5) can explain why heterochromatic genes can be transcribed in the usually repressive chromosomal environment and depend on the high concentration of Su(var) proteins found in heterochromatin (Fig. 5D) (7). We propose that this activating effect of Su(var)s is possible only where repetitive DNAs have accumulated in a way that promotes a chromatin configuration that is compatible with gene expression. This state can be achieved only through gradual accumulation of repetitive DNA and evolutionary selection for gene expression. Therefore, heterochromatic genes do not need to overcome the “silencing components”; components act to silence genes only if the insertions of TEs occur in “wrong” places relative to genes. The model proposes a role for repetitive DNA, including TEs, in positively regulating transcription of essential genes. In this regard, TEs are participating in a normal cellular function (32, 33). Our model is general in that it can apply to genes that have diverse types of promoters, gene-specific enhancers, and trans-acting factors. In this sense, it may be applicable to a large number of ≈400 heterochromatic genes in D. melanogaster (5).
Heterochromatin is a common entity in multicellular organisms. The paradoxical nature of heterochromatic genes was recognized decades ago by Drosophila geneticists, but it is only beginning to be recognized as such in other organisms (21, 34-36). Remaining questions include the following: Are there common evolutionary pathways and strategies to the adaptation of genes to heterochromatin? How diverse are effects of heterochromatin on gene activation? The repetitive nature of heterochromatin has presented more than the usual number of challenges for genome projects. However, as described here, comparative genomics can offer new tools and insights for exploring the content and evolution of heterochromatin.
Supplementary Material
Acknowledgments
We thank D. Slade and H. Kobayashi for technical assistance; J. Davison for assistance in Arabidopsis sequence analysis; and M. Fisher, K. Wilson, H. Malik, W. Swanson, and L. Comai for helpful comments. This work was supported by National Institutes of Health Training Grant Fellowship T32HD007183-26 (to J.C.Y.) and a National Science Foundation grant (to B.T.W.).
Author contributions: J.C.Y. and B.T.W. designed research; J.C.Y., C.H.D., and B.T.W. performed research; J.C.Y., C.H.D., and B.T.W. contributed new reagents/analytic tools; J.C.Y., C.H.D., and B.T.W. analyzed data; and J.C.Y. and B.T.W. wrote the paper.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: TE, transposable element; Dm-lt(het), Drosophila melanogaster heterochromatic light gene; Dp-lt(eu), Drosophila pseudoobscura euchromatic light gene; Dv-lt(eu), Drosophila virilis euchromatic light gene; RLM-RACE, RNA ligase-mediated RACE.
Data deposition: The sequences reported in this paper have been deposited in the GenBank database (accession nos. DQ118645 and DQ118646).
References
- 1.Weiler, K. S. & Wakimoto, B. T. (1995) Annu. Rev. Genet. 29, 577-605. [DOI] [PubMed] [Google Scholar]
- 2.Volpe, T. A., Kidner, C., Hall, I. M., Teng, G., Grewal, S. I. & Martienssen, R. A. (2002) Science 297, 1833-1837. [DOI] [PubMed] [Google Scholar]
- 3.Pal-Bhadra, M., Leibovitch, B. A., Gandhi, S. G., Rao, M., Bhadra, U., Birchler, J. A. & Elgin, S. C. (2004) Science 303, 669-672. [DOI] [PubMed] [Google Scholar]
- 4.Verdel, A., Jia, S., Gerber, S., Sugiyama, T., Gygi, S., Grewal, S. I. & Moazed, D. (2004) Science 303, 672-676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hoskins, R. A., Smith, C. D., Carlson, J. W., Carvalho, A. B., Halpern, A., Kaminker, J. S., Kennedy, C., Mungall, C. J., Sullivan, B. A., Sutton, G. G., et al. (2002) Genome Biol. 3, RESEARCH0085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wakimoto, B. T. & Hearn, M. G. (1990) Genetics 125, 141-154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hearn, M. G., Hedrick, A., Grigliatti, T. A. & Wakimoto, B. T. (1991) Genetics 128, 785-797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dorer, D. R. & Henikoff, S. (1994) Cell 77, 993-1002. [DOI] [PubMed] [Google Scholar]
- 9.Danilevskaya, O. N., Arkhipova, I. R., Traverse, K. L. & Pardue, M. L. (1997) Cell 88, 647-655. [DOI] [PubMed] [Google Scholar]
- 10.Devlin, R. H., Bingham, B. & Wakimoto, B. T. (1990) Genetics 125, 129-140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Howe, M., Dimitri, P., Berloco, M. & Wakimoto, B. T. (1995) Genetics 140, 1033-1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Devlin, R. H., Holm, D. G., Morin, K. R. & Honda, B. M. (1990) Genome 33, 405-415. [DOI] [PubMed] [Google Scholar]
- 13.Powell, J. R. & DeSalle, R. (1995) in Evolutionary Biology, ed. Hecht, M. K. (Plenum, New York), Vol. 28, pp. 87-138. [Google Scholar]
- 14.Russo, C. A., Takezaki, N. & Nei, M. (1995) Mol. Biol. Evol. 12, 391-404. [DOI] [PubMed] [Google Scholar]
- 15.Schaefer, B. C. (1995) Anal. Biochem. 227, 255-273. [DOI] [PubMed] [Google Scholar]
- 16.Ohler, U., Liao, G. C., Niemann, H. & Rubin, G. M. (2002) Genome Biol. 3, RESEARCH0087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.England, T. E. & Uhlenbeck, O. C. (1978) Biochemistry 17, 2069-2076. [DOI] [PubMed] [Google Scholar]
- 18.Romaniuk, E., McLaughlin, L. W., Neilson, T. & Romaniuk, P. J. (1982) Eur. J. Biochem. 125, 639-643. [DOI] [PubMed] [Google Scholar]
- 19.Richards, S., Liu, Y., Bettencourt, B. R., Hradecky, P., Letovsky, S., Nielsen, R., Thornton, K., Hubisz, M. J., Chen, R., Meisel, R. P., et al. (2005) Genome Res. 15, 1-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Carvalho, A. B. (2002) Curr. Opin. Genet. Dev. 12, 664-668. [DOI] [PubMed] [Google Scholar]
- 21.Lippman, Z., Gendrel, A. V., Black, M., Vaughn, M. W., Dedhia, N., McCombie, W. R., Lavine, K., Mittal, V., May, B., Kasschau, K. D., et al. (2004) Nature 430, 471-476. [DOI] [PubMed] [Google Scholar]
- 22.Carvalho, A. B. & Clark, A. G. (2005) Science 307, 108-110. [DOI] [PubMed] [Google Scholar]
- 23.Meunier, J. & Duret, L. (2004) Mol. Biol. Evol. 21, 984-990. [DOI] [PubMed] [Google Scholar]
- 24.Singh, N. D., Arndt, P. F. & Petrov, D. A. (2005) Genetics 169, 709-722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ince, T. A. & Scotto, K. W. (1995) J. Biol. Chem. 270, 30249-30252. [DOI] [PubMed] [Google Scholar]
- 26.Reynolds, G. A., Basu, S. K., Osborne, T. F., Chin, D. J., Gil, G., Brown, M. S., Goldstein, J. L. & Luskey, K. L. (1984) Cell 38, 275-285. [DOI] [PubMed] [Google Scholar]
- 27.Struhl, K. (1989) Annu. Rev. Biochem. 58, 1051-1077. [DOI] [PubMed] [Google Scholar]
- 28.Kollmar, R. & Farnham, P. J. (1993) Proc. Soc. Exp. Biol. Med. 203, 127-139. [DOI] [PubMed] [Google Scholar]
- 29.Benoist, C. & Chambon, P. (1981) Nature 290, 304-310. [DOI] [PubMed] [Google Scholar]
- 30.Smale, S. T. & Kadonaga, J. T. (2003) Annu. Rev. Biochem. 72, 449-479. [DOI] [PubMed] [Google Scholar]
- 31.Dudov, K. P. & Perry, R. P. (1986) Proc. Natl. Acad. Sci. USA 83, 8545-8549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pardue, M. L. & DeBaryshe, P. G. (1999) Genetica 107, 189-196. [PubMed] [Google Scholar]
- 33.Miller, W. J., McDonald, J. F., Nouaud, D. & Anxolabehere, D. (1999) Genetica 107, 197-207. [PubMed] [Google Scholar]
- 34.Copenhaver, G. P., Nickel, K., Kuromori, T., Benito, M. I., Kaul, S., Lin, X., Bevan, M., Murphy, G., Harris, B., Parnell, L. D., et al. (1999) Science 286, 2468-2474. [DOI] [PubMed] [Google Scholar]
- 35.Nagaki, K., Cheng, Z., Ouyang, S., Talbert, P. B., Kim, M., Jones, K. M., Henikoff, S., Buell, C. R. & Jiang, J. (2004) Nat. Genet. 36, 138-145. [DOI] [PubMed] [Google Scholar]
- 36.She, X., Horvath, J. E., Jiang, Z., Liu, G., Furey, T. S., Christ, L., Clark, R., Graves, T., Gulden, C. L., Alkan, C., et al. (2004) Nature 430, 857-864. [DOI] [PubMed] [Google Scholar]
- 37.Fong, T. C. & Emerson, B. M. (1992) Genes Dev. 6, 521-532. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.