

Abstract
Engineered macromolecules offer compelling means for therapy of conventionally undruggable interactions in human disease. However, their efficacy is limited by barriers to tissue and intracellular delivery. Inspired by recent advances in molecular barcoding and evolution, we developed BarcodeBabel, a generalized method for the design of libraries of peptide barcodes suitable for high-throughput mass spectrometry proteomics. Combined with PeptideBabel, a Monte Carlo sampling algorithm for the design of peptides with evolvable physicochemical properties and sequence complexity, we developed a barcoded library of cell penetrating peptides (CPP) with distinct physicochemical features. Using quantitative targeted mass spectrometry, we identified CPPs with improved nuclear and cytoplasmic delivery exceeding hundreds of millions of molecules per human cell, while maintaining minimal membrane disruption and negligible toxicity in vitro. These studies provide proof-of-concept for peptide barcoding as a homogeneous high-throughput method for macromolecular screening and delivery. BarcodeBabel and PeptideBabel are available open-source from https://github.com/kentsisresearchgroup/.
Keywords: Peptide barcoding, mass spectrometry proteomics, cell penetration, molecular screening, macromolecular drug delivery
Graphical Abstract
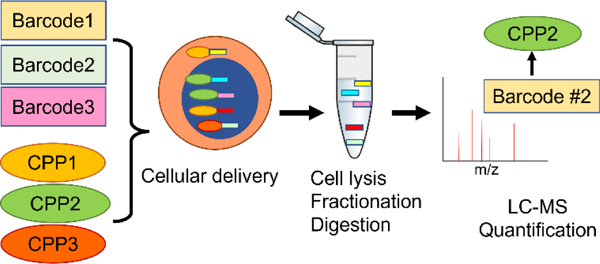
Introduction
Macromolecular drugs have emerged as a promising category of treatment for severe diseases including neurodegenerative disorders and cancers, both as a complement and an alternative to small molecules. Macromolecules in general, and polypeptides specifically, offer many advantages to small-molecule therapeutics due to greater selectivity and specificity, and are thus able to target otherwise undruggable factors such as protein-protein interactions.1–5
However, the relatively large size and polar features of many biological macromolecules render them impermeable to cellular lipid membranes. The recognition of naturally occurring moieties that allow for membrane penetration has led to the development of various cell penetrating peptides (CPPs) and protein transduction domains (PTDs) to assist with drug delivery, which are thought to work by membrane pore formation and other interfacial processes, endocytosis with or without involvement of transmembrane proteins, and translocation via inverted micelles followed by their intracellular dissolution.6–10 However, the diversity and complexity of these mechanisms have been a major barrier to the development of explicit structure-activity relationships, which are necessary for the ultimate design and development of efficient, selective and safe CPPs and PTDs for macromolecular drug delivery.11, 12 An efficient and robust method to quantify the subcellular localization of the CPPs is therefore highly desired.
Recent advances in molecular evolution and high-throughput screening approaches permit the identification of molecules with specific biological activities. In particular, Hoffmann et al. and Kauffmann et al. applied phage and RNA display and molecular evolution techniques, respectively, to develop improved CPPs.13, 14 Similarly, recent advances in mass spectrometry proteomics permit quantitative and sensitive measurements of diverse molecules in complex biological samples. Egloff et al. used genetically encoded peptide barcodes for measuring binding of libraries of engineered proteins.15 Peptide barcoding has also been implemented recently for diversity-oriented small molecule screening, demonstrating its increased stability and information coding capacity, as compared to nucleic acid barcoding.16 In principle, it is possible to track CPPs and other bioactive peptides directly without barcoding, but this is often challenging, and in some cases like the cationic polyarginine TAT CPP, is not possible due to the lack of specific signature ions.
Inspired by these distinct approaches, we sought to develop peptide barcoding as a homogeneous method for macromolecular screening and delivery. Implemented in open-source algorithms BarcodeBabel and PeptideBabel, this approach permits the design of libraries of peptide barcodes and novel CPPs optimized for screening and analysis by quantitative mass spectrometry. With this approach, we identified CPPs with improved nuclear and cytoplasmic delivery exceeding hundreds of millions of molecules per human cell, while maintaining minimal membrane disruption and negligible toxicity in vitro. These methods should be useful for a wide variety of molecular evolution and screening applications.
Materials and Methods
Reagents
The Fmoc amino acids, OxymaPure, and Rink Amide ProTide Resin used for solid phase peptide synthesis were purchased from CEM Corporation (Charlotte, NC, USA). N,N′-diisopropylcarbodiimide (DIC), 2,2’-(Ethylenedioxy) diethanethiol (DODT), triisopropylsilane (TIPS), sodium nitrite, sodium phosphate monobasic, 4-mercaptophenylacetic acid (MPAA), trifluoroacetic acid (TFA), piperidine, dimethylformamide (DMF), dichloromethane (DCM), diethyl ether, guanidine hydrochloride (Gn·HCl), 2-mercaptoethanol, and tris(2-carboxyethyl)phosphine hydrochloride (TCEP·HCl) were purchased from Sigma-Aldrich (St. Louis, MO, USA). 2,2’-Azobis[2-(2-imidazolin-2-yl)propane] dihydrochloride (Va-044) was purchased from Wako Chemicals (Richmond, VA, USA). HPLC-grade reagents including water, methanol, acetonitrile, and formic acid were purchased from Thermo Fisher Scientific (Waltham, MA, USA). Digitonin was purchased from MilliporeSigma (Burlington, MA, USA). cOmplete protease inhibitors were obtained from Roche Diagnostics GmbH (Mannheim, Germany).
Synthetic Chemistry
Peptide barcodes and CPPs were synthesized using the Liberty Blue HT12 microwave peptide synthesizer, according to the manufacturer’s instructions (CEM Corporation, Charlotte, NC).17, 18 0.2 M stock solutions of amino acids were made in DMF. 20% piperidine in DMF was used as the Fmoc-deprotecting solvent. 1 M OxymaPure ethyl 2-cyano-2-(hydroxyimino)acetate was used with 0.5 M N,N′-diisopropylcarbodiimide (DIC) in the carbodiimide approach to form peptide bonds, with inhibition of racemization and improved coupling efficiencies.19 All the barcode sequences were designed with N-terminal alanine (A) and C-terminal arginine (R), and one tryptophan (W) in their sequences to aid in UV absorbance detection, as listed in Table S1. In order to proceed with native chemical ligation, all the barcode peptides were initially synthesized with N-terminal cysteine (C), instead of alanine (A), using rink amide resin resulting in C-terminal amides. CPP peptides were synthesized using 2-chlorotrityl chloride (CTC) resin to prepare C-terminal hydrazide (R-NHNH2).20 The synthesized peptides were cleaved off the resin using the TFA:H2O:TIPS:DODT solution (92.5:2.5:2.5:2.5), and purified by adding 10-fold volume of cold diethyl ether to the TFA cleavage cocktail. Short barcode peptides (Table S1) used for generating signal-response standard curves were synthesized as SpikeTides (JPT Technologies, Berlin, Germany).
Barcoded CPP libraries were generated using native chemical ligation.21 All reactions were carried out in the 0.2 M phosphate buffer containing 6 M guanidine hydrochloride, pH 3 (NCL buffer). For each peptide, 3.6 mg of CPP peptide was dissolved in 0.4 ml NCL buffer, and stirred at −15°C for 15 min. Separately, 13.6 mg MPAA and 1.25 mg barcode peptide were dissolved in 0.4 ml NCL buffer, pH 6.5. The pre-cooled CPP peptide solution was oxidized with 40 µL 0.5 M NaNO2 by stirring at −15°C for 15 min. MPAA and barcode mixtures were added dropwise into the tube containing CPP peptide, and the tube was then warmed to room temperature, and its pH was adjusted to 6.8–7.0. After overnight incubation with stirring at room temperature, reaction mixtures were reduced by the addition of 0.4 ml of 0.1 M TCEP (NCL buffer, pH 6.0–7.0) and stirring for 20 minutes.22 Cysteine desulfurization was carried out as previously described.23 Specifically, 2 mg of NCL-ligated barcoded-CPP was dissolved in 300 µL of NCL buffer, into which 300 uL of aqueous 0.5 M TCEP·HCl, 20 uL of 2-mercaptoethanol (10% v/v), and 20 uL of 0.1 M VA-044 were added. The pH of reaction mixture was adjusted to neutral, and the reaction was stirred at 45°C for 45 min. All peptides were purified by high-performance liquid chromatography (HPLC) using with XBridge Peptide BEH C18 OBD Prep Column (#186008193, Waters, Milford, MA) and acetonitrile gradient 5–40% with 0.1% trifluoroacetic acid, to achieve at least 90% purity as measured by LC-MS.
Cell culture
Kasumi-1, HEK293T, and HUVEC cells were obtained from American Type Culture Collection (ATCC, Manassas, Virginia, USA). Huh-7 cells were kindly provided by Dr. Hao Zhu from UT Southwestern Medical Center. All cell lines were verified by short tandem repeats (STR) analysis (Integrated Genomics Operation, Memorial Sloan Kettering Cancer Center, New York, NY, USA). Cultures were confirmed to be free of mycoplasma contamination using Lonza MycoAlert (Lonza Walkersville, Inc., Walkersville, MD, USA). Cells were cultured at a concentration of 1×10^6 cells/mL in 5% CO2 in humidified atmosphere at 37 °C, in complete media supplemented with 10% fetal bovine serum, 100 U/mL penicillin, and 100 ug/mL streptomycin. RPMI-1640 medium (Corning Life Science, Corning, NY, USA) was used for Kasumi-1 cells; DMEM medium (Corning Life Science, Corning, NY, USA) was used for HEK293T and Huh-7 cells; the EGM-2 Endothelial Cell Growth Medium (Lonza Inc., Morristown, NJ, USA) was used for HUVEC cells.
Membrane permeabilization and cytotoxicity measurements
Membrane stability was evaluated using lactate dehydrogenase (LDH) release assay (#ab65393, AbCam, Cambridge, UK) and cell viabilty was measured using the CellTiter-Glo luminescent assay, according to manufacturer’s instructions (#G7571, Promega Corporation, Madison, WI, USA). Cells were counted with Countess II Automated Cell Counter (Thermo Fisher Scientific, Waltham, MA, USA) and 10K cells were aliquoted into each well of 96-well cell culture plates. Suspension Kasumi-1 cells were aliquoted immediately before treatment, while adherent HEK293T, Huh-7 and HUVEC cells were plated 17 hours before treatment. 10 µL barcoded CPP stock solutions (with varying concentrations) were added into 90 µL cell suspensions, resulting in final barcoded CPP concentrations ranging from tens of nanomolar to hundreds of micromolar. Then the cells were incubated in complete media in 5% CO2 at humidified atmosphere at 37 °C for 3 and 24 hour, respectively, and assayed using the TECAN Infinite M1000 Pro microplate reader (Tecan Group Ltd., Männedorf, Switzerland).
Cell internalization and fractionation
Cells were aliquoted into 12-well cell culture plates. Suspension Kasumi-1 cells (300K) were aliquoted immediately before treatment, while adherent HEK293T (175K), Huh-7 (240K) and HUVEC (280K) cells were plated 17 hours before treatment. 10 µL of 100 µM barcoded CPP stock solutions were added into 990 µL cell suspensions, resulting in 1 µM final barcoded CPP concentration. Then the cells were incubated in 5% CO2 at humidified atmosphere at 37 °C for 3 and 24 hour, respectively.Kasumi-1 cells were pelleted by centrifugation, while the other three adherent cell lines were harvested using cell scraper and then spun down. The cell pellets were washed once in PBS. For cell fractionation, a cell pellet of 2–3 million cells was resuspended in 90–150 µL of hypotonic buffer (10 mM HEPES, pH 7.9, 10 mM NaCl, 1 mM MgCl2, 0.5 mM DTT, cOmplete protease inhibitors) containing 0.1% digitonin, and was incubated at 25 °C for 5 min. The suspension was then centrifuged for 15 min at 3300 g at 4°C, and cytoplasmic supernatant was collected. The nuclei pellet was then resuspended in 500 µL of sucrose resuspension buffer containing 0.25 M sucrose and 10 mM MgCl2 (PBS buffer, pH 7.4). The suspension was layered onto a 500 µL sucrose cushion buffer (0.88 M sucrose and 0.05 mM MgCl2 in PBS, pH 7.4), and centrifuged at 1200 g at 4°C for 11 min. The nuclei pellet thus obtained was subjected to nuclear lysis and protein extraction in 70–110 µL of lysis buffer (6 M guanidine hydrochloride in PBS buffer, pH 7.4), sonicated by Covaris S220 ultrasonicator (Covaris, LLC., Woburn, MA, USA) at peak power 125W, duty factor of 10%, cycle/burst of 200, and duration of 360 sec. The protein content in both nuclear and cytoplasmic fractions was quantified using Pierce BCA Protein Assay Kit, according to the manufacturer’s instructions (Thermo Fisher Scientific, Waltham, MA, USA). Purified samples were stored at −80 °C.
Proteomics sample preparation
Purified protein extracts were digested with sequencing grade modified trypsin protease (Promega Corporation, Madison, WI, USA), with a protein-to-trypsin mass ratio of 10:1. The digestion mixture was incubated at 37 °C overnight. Digestion was halted by addition of formic acid to 3.36% v/v, and 650 fmol of the Pierce Retention Time Calibration Mixture (PRTC) was added into each sample as internal standards. Tryptic peptides were purified using solid phase extraction with BioPureSPN MIDI columns (#HEM S18V, Nest Group, Southborough, MA, USA) according to manufacturer’s protocol. Briefly, the spin column was washed with 200 µL methanol and activated with 200 µL acetonitrile, then equilibrated twice with 200 µL 0.1% formic acid in water. Samples were loaded onto equilibrated columns, and then washed twice with 200 µL 0.1% formic acid in water. Peptides were eluted with 60% acetonitrile containing 0.1% formic acid, and lyophilized by vacuum centrifugation. For analysis, purified peptides were resuspended in 0.1% formic acid in water. Calibration curves of barcoded CPPs were established by adding variable amounts of synthetic peptides into cell lysates, followed by their digestion and purification as described above.
Nanoscale liquid chromatography and nanoelectrospray ionization mass spectrometry
All nanoscale liquid chromatography was performed using the Eksigent nanoLC425 chromatography system (Sciex, Framingham, MA, USA). The 50 cm uPAC pillar array reverse phase column (#COL-NANO050G1B, ThermoFisher Scientific, San Jose, CA, USA) was used for the LC-MS signal-response study of the pilot 96 peptide barcodes. Capillary reverse phase columns were used for cellular delivery quantitation. These capillary columns were fabricated by pressure filling the stationary phase into silica capillaries fritted with K-silicate, as previously described.24 Samples were resolved with constant flowrate of 300 nL/min using a 5–40% linear gradient of acetonitrile in water (both with 0.1% v/v/ formic acid) over 90 minutes. Ionization was accomplished using laser-fabricated emitters with terminal opening diameter of 2–3 µm, made from 50 µm silica capillaries, and connected to the outlet of the reversed phase column using a metal union that also served as the electrospray current electrode, as described.25 Eluting peptides were transferred to an Orbitrap Fusion mass spectrometer (ThermoFisher Scientific, San Jose, CA, USA) via the DPV-566 PicoView nano-electrospray ion source (New Objective, Woburn, MA, USA).
For data dependent acquisition (DDA), the full-scan spectra were acquired in positive ion mode at a fixed resolution of 120,000 and a mass range of 300–1200 m/z. Fragmentation spectra were acquired at a resolution of 7,500, and the precursor ions were isolated using 1.6 m/z width and fragmented with a fixed higher-energy collisional dissociation (HCD) collision energy of 30%.
For parallel reaction monitoring (PRM) scans, full-scan spectra were acquired in positive ion mode at a mass range of 300–1200 m/z and 120,000 resolution. Precursor ions were isolated in the quadrupole using 1.2 m/z windows and fragmented by HCD with normalized collision energy 30% (stepped collision energy +/−5%), before analysis of fragment ions in the Orbitrap at 7,500 resolution. The PRM method was scheduled using 10-min acquisition windows set for each barcode based on the DDA retention time results.26
For selected ion monitoring (SIM) scans, the precursor ions were isolated in the quadrupole using 0.7 m/z window and were detected in the Orbitrap at 240,000 resolution. The maximum injection time mode was set as dynamic, with six minimum points across the peak. The SIM method was scheduled using 10-min acquisition windows set for each barcode based on the DDA retention time results.
The mass spectra obtained from all methods were analyzed using Skyline27 (version 21.2.0.425). The target list was first established in Skyline, then the MS raw data files were imported to extract the target chromatographs. DDA MS1 spectra were filtered with a resolving power of 120,000 at m/z 400. SIM MS1 spectra were filtered with a resolving power of 240,000 at m/z 200. PRM MS2 spectra were filtered using 60,000 resolving power at m/z 200, and at least 3 fragments per precursor are required for valid quantitation. For the LC-MS signal-response study, the MS1 and MS2 ion intensities of each barcode precursor in all serially diluted samples were extracted and were linearly fitted to obtain a signal-response function for each barcode. For the absolute quantitation of digested cell fractions, MS2 ion intensities of all barcodes were exported and normalized to the ion intensity of PRTC ionization standards. A baseline sample (untreated cell lysate) was measured, and any ion intensities of the cell fractions that were lower or comparable to the baseline signal were considered non-detectable. Only valid signals from the cell fractions were calculated using the calibration curves to obtain the final delivery quantities.
Numerical and statistical analyses were performed using Origin Pro (OriginLab Corporation, Northampton, MA).
BarcodeBabel
BarcodeBabel is a Python algorithm to generate libraries of peptide barcodes with user-defined features for optimal detectability by nanoscale liquid chromatography tandem mass spectrometry. User-selected rules include m/z range, absence of homopolymers, specific hydrophobicity range, enzyme cleavage sites, residue frequencies, and library size. In addition, users can specify reference proteome to remove any naturally occurring interfering sequences. The default enzyme for barcode cleavage is trypsin. Specific residues are programmed to be avoided: lysine (K), arginine (R), and histidine (H) were omitted to prevent trypsin cleavage within the barcode sequences; methionine (M) and cysteine (C) were omitted to avoid oxidation and crosslinking events; proline (P) was omitted as it may skew fragmentation (though could be included to promote fragmentation at the proline residue); glutamine (Q) and asparagine (N) were avoided due to their propensity for deamidation; isoleucine (I) was omitted because it cannot be distinguished from leucine (L) by conventional mass spectrometry. Peptide properties were calculated at default pH of 3 (for conventional positive ion mode electrospray). Default m/z range was set to 550–850 to fall within the optimal detection window of high-field Orbitrap mass analyzers. Default hydrophobicity range (−0.5 to 2.5) is specified to allow for consistent peptide elution in the middle of conventional reverse phase chromatography gradients. Users can also list specific reference proteomes, as well as common contaminants to avoid sequence overlap. BarcodeBabel is implemented open-source via https://github.com/kentsisresearchgroup/BarcodeBabel.
PeptideBabel
PeptideBabel is Python algorithm for the generation of novel bioactive peptide sequences using Metropolis-Hastings sampling. This algorithm generates novel peptides by exploring the sequence space around a set of seed sequences using Markov chain Monte Carlo sampling (Metropolis-Hastings) with permutation of seed sequences mapped to a density function based on physicochemical features or k-mer sequence complexity, as specified by users. This allows efficient generation of hundreds to millions of peptide sequences for subsequent empiric validation, tailored to application based on input peptides. Briefly, after the user uploads a list of references peptides (seed library), PeptideBabel identifies key properties of the seed library, including sequence length, hydrophobicity (windowed Kyte-Doolittle scale), isoelectric point (Bjellqvist), secondary structure propensity (fraction helical, turn-like, and sheet-like based on residue composition using the Garnier-Osguthorpe-Robson method). The sampling algorithm permutes peptide sequences (substitutions, insertions, deletions) at user defined sequence length constraints and sampling density, and implements a random-walk Metropolis-Hastings, favoring steps moving up the density function at user-defined probability to generate new sequences or down the density function to sample similar sequences. PeptideBabel is implemented open-source via https://github.com/kentsisresearchgroup/PeptideBabel.
Results
Mass spectrometry proteomics enables the detection and quantitation of specific macromolecules based on tandem fragmentation and high-accuracy measurements of their mass and charge. This enables the resolution of unique polypeptide sequences, differing by as little as a single amino acid. To enable the generation of libraries of unique peptide barcodes for studies of engineered macromolecules in complex biological samples, we developed BarcodeBabel, a python script and corresponding Jupyter notebook, which compute arbitrary numbers of unique amino acid peptide sequences, flanked by specific enzyme cleavage sites for their release in complex biological samples, e.g. trypsin (Figure 1A). The script can exclude user-defined internal amino acids, which may interfere with correct enzymatic cleavage and consistent mass spectrometric detection or fragmentation, i.e. lysine, arginine, histidine, cysteine, methionine, proline, and isoleucine. Users may also specify frequencies of amino acids to generate libraries with specific features, such as inclusion of tryptophan residue for optical measurements, as well as specific ranges of m/z values and amino acid lengths. The script also implements a hydrophobicity estimator based on the Kyte-Doolittle scale for optimal reverse phase separations that accompany high-resolution mass spectrometry measurements. To calibrate this parameter, we analyzed 175,579 unique tryptic peptides of length from 5 to 15 residues,28 and found that peptides with hydrophobicity scores of −0.5 to 2.5 exhibit monotonically variable retention times in reverse phase chromatography most often coupled with modern high-resolution mass spectrometry instruments, which we implement as the default values for BarcodeBabel (Figure S1). Finally, BarcodeBabel is configured with a user-specified reference proteome to ensure that the designed barcode sequences do not match any naturally occurring or common contaminant proteins.
Figure 1.

BarcodeBabel algorithm generates unique peptide barcodes that are readily detectable by high-resolution mass spectrometry. A. Schematic of BarcodeBabel to generate barcode libraries. B. Signal-response functions for the library of 96 designed barcodes using PRM. The MS2 XIC signal is used as y axis. C. Signal response (MS signal intensity/amol of barcodes) of the signal-response functions of each barcode obtained from SIM, DDA, and PRM.
To test BarcodeBabel, we designed a library of 96 unique barcode sequences, synthesized them using solid phase synthesis, and serially diluted purified peptides in whole-cell extracts of human OCI-AML2 cells. The abundance of peptides in whole-cell proteomes was quantified using selected ion monitoring (SIM), data-dependent acquisition (DDA), and parallel reaction monitoring (PRM), with limits of quantitation determined using Skyline. We found that PRM exhibited superior limits of quantitation (LOQ), as compared to SIM and DDA (mean LOQ of 75 versus 550 and 280 amol, respectively; Figure 1B). We also found that PRM exhibited more uniform signal-response for the quantitation of barcode peptide abundance, as compared to SIM and DDA (mean 0.67 ± 0.22 versus 0.60 ± 0.34 and 0.80 ± 0.30 intensity/amol, respectively; Figure 1C). Thus, BarcodeBabel permits the construction of libraries of specific peptide barcodes, which can be quantitatively deconvoluted using high-resolution mass spectrometry.
Pioneering studies of first-generation of CPPs and PTDs for macromolecular delivery used naturally inspired peptides derived from TAT and penetratin.29, 30 Since then, a variety of cationic and amphipathic CPPs have been identified experimentally, as most recently catalogued in the CPPsite 2.0 database.31, 32 Comparative studies of specific CPPs have identified several key propensities, such as the optimal number of eight guanidine side chains for cationic polyarginine CPPs.33 However, the development of explicit structure-activity relationships for efficient, selective and safe CPPs and PTDs has been challenging, at least in part due to the diversity and complexity of CPPs and their membrane penetration and cellular internalization mechanisms.
We reasoned that the mechanisms of CPP membrane penetration and cellular internalization ultimately can be learned from large-scale structure-function studies. To generate libraries of candidate CPPs for high-throughput studies, we implemented a Monte Carlo sampling algorithm, PeptideBabel. PeptideBabel uses the Metropolis-Hastings algorithm to introduce random changes in the amino acid composition of seed peptide sequences, followed by their acceptance or rejection based on user-specified density functions of either the physicochemical properties of peptides or their k-mer sequence complexity (Figure 2). Current version of PeptideBabel includes 1552 seed sequences of putative membrane penetration domains from diverse viral pathogens,34 as well as curated members from CPPsite 2.0 (Figure S2 and Table S4). PeptideBabel samples the chemical space according to the physicochemical scoring function of the seed sequences: is based on the linear combination of peptide length, estimated isoelectric point, hydrophobicity (Kyte-Doolittle), and secondary structure propensity (Garnier-Osguthorpe-Robson). Alternatively, PeptideBabel can sample peptide sequences based on their sequence complexity, as measured using k-mer scoring.35 The Monte Carlo sampling and design can be executed to generate unique sequences with physicochemical or complexity properties that are either similar to the seed sequences, or alternatively, those that are of increasing diversity from the seed sequences. Convergence of sampling can be assessed using multiple independent calculations, starting from different initial conditions.36 Thus, PeptideBabel is expected to permit the construction of diverse libraries of candidate bioactive peptides.
Figure 2. Schematic of the PeptideBabel algorithm, which generates bioactive peptide sequences using Markov chain Monte Carlo sampling with permutation of seed sequences (sequence 0) mapped to a density function.

PeptideBabel implements a random-walk Metropolis-Hastings algorithm, accepting steps moving up and rejecting steps moving down the density function using user-defined probability functions. The model parameters (θ) and proposed parameters (z) are based on k-mer space that indicates sequence complexity or the physiochemical space (e.g., sequence length, isoelectric point, secondary structure propensity, and hydrophobicity). The figure is based on Jin et al.36
As proof-of-concept of this strategy, we designed a library of seven barcoded CPPs, including representative cationic and amphipathic CPPs, as well as novel chimeric CPPs, and their anionic negative controls that do not transit across anionic mammalian cell membranes (Table 1). We chose unique barcodes with diverse physicochemical properties, as estimated by their m/z, net charge, and hydrophobicity values (Figure S3). Barcode peptides and CPPs were synthesized using solid phase synthesis, and assembled using native chemical ligation.22 Because native chemical ligation involves cysteines that can undergo chemical reactions in cells, we used desulfurization to convert them into alanines.23 This allowed us to generate barcoded CPPs up to 42-amino acids in length with >90% purity, as confirmed using LC-MS (Figure S4).
Table 1.
The CPP candidates chosen for chemical synthesis and cellular delivery assay.
| ID | CPP | Sequence | CPP type | Sequence source | Barcode |
|---|---|---|---|---|---|
| 1 | TAT | YGRKKRRQRRR | Cationic | Prototypic CPP, NLS from HIV TAT protein | B64 |
| 2 | P14 | RKKRWFRRRRPKWKK | Cationic | TAT/penetratin hybrid | B91 |
| 3 | TAT-P-Ebola | GAAIGLAWIPYFGPAAYPRKKRRQRRR | Hydrophobic+cationic (moderate hydrophobicity) | Good penetrator in pilot study, chimera design of TAT + Ebola coat | B121 |
| 4 | TAT-G-EBV | IYNGWYAYGRKKRRQRRR | Hydrophobic+cationic (moderate hydrophobicity) | TAT+Epstein Barr Virus coat | B107 |
| 5 | KLAL-TAT | KLALKLALKALKAALKLAGCYGRKKRRQRRR | Amphipathic | Model amphipathic peptide + TAT | B82 |
| 6 | RLAL-TAT | RLALRLALRALRAALRLAGCYGRKKRRQRRR | Amphipathic | Model amphipathic peptide + TAT | B108 |
| 7 | badTAT | YGEKKEEQRRR | Negative ctrl | Negative control | B55 |
NLS: nuclear localization sequence
CPPs can disrupt cellular membranes, causing both cytotoxicity and artifactual internalization due to cell death. Therefore, we measured the effects of individual barcoded CPPs on cell viability, as assessed with cellular ATP content, and membrane stability with assays of LDH release using a panel of four biologically diverse cell lines: hematopoietic Kasumi-1 cells, mesenchymal HEK293T cells, endothelial HUVEC cells, and epithelial Huh-7 cells (Figure 3 and S5, S6). We found that TAT-[barcode64] CPP showed no measurable cytotoxicity up to 15 µM either at 3 or 24 hours of exposure on Kasumi-1 cells, and up to 100 µM across the other three cell lines tested. In contrast, 3-hour incubation of chimeric KLAL-TAT-[barcode82] CPP promoted LDH release and impaired cell viability with an IC50 of 1.4 and 2.3 µM, respectively in HUVEC cells, and 34 and 10 µM, respectively in HEK293T cells, with the other two cell lines having intermediate values (Figure 3 and S5–6). We found that Kasumi-1 cells were most sensitive to various CPPs, whereas Huh-7 and HEK293T cells were largely insensitive. Importantly, all barcoded CPPs had negligible cytotoxicity and membrane disruption at 1 µM, thereby establishing this dose for cell delivery studies in vitro (Figure 6–7 and S9–11). Though many prior studies have used 10–100 µM CPP cell treatments,37 we chose to use this lower concentration in order to avoid any potential confounding effects from membrane disruption and cytotoxicity.
Figure 3. Membrane destabilization and cytotoxicity measurements of CPPs.
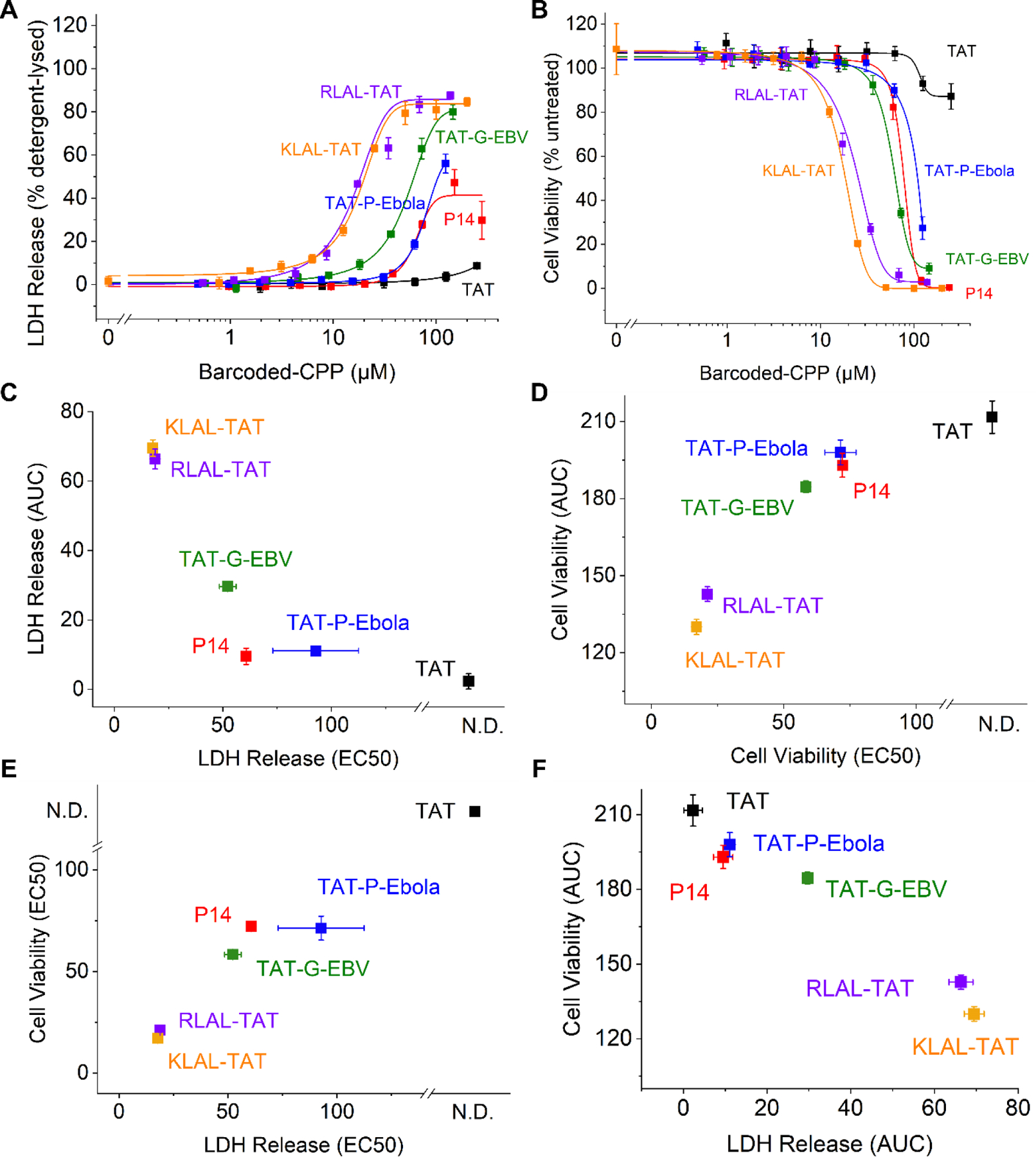
A. LDH release of Huh-7 cells after 3-hour incubation of barcoded CPPs. B. Cell viability of Huh-7 cells after 3-hour incubation of barcoded CPPs. C. EC50 values as a function of AUC of the LDH release measurements. D. EC50 values as a function of AUC of the cell viability. E. EC50 values of LDH release as a function of EC50 values of cell viability. F. AUC of LDH release as a function of the AUC of cell viability. Symbols and whiskers represent mean and standard deviation values of three biologic replicates, respectively.
Figure 6. Barcoded TAT-P-Ebola peptides exhibit efficient cellular penetration without apparent membrane disruption and cytotoxicity in vitro.

A. LDH release measurements of Kasumi-1 cells upon 3-hour incubation with barcoded TAT-P-Ebola peptides with various barcodes, as indicated. B. Cell viability of Kasumi-1 cells upon 3-hour incubation with barcoded TAT-P-Ebola peptides with various barcodes, as indicated. C. Abundance of barcoded TAT-P-Ebola CPPs in nuclear and cytoplasmic fractions upon 1 mM treatment for 3 hours. Symbols and whiskers indicate mean and standard deviation values of three biologic replicates, respectively. The specific sequence of TAT-P-Ebola and the barcodes are listed in supplemental Table 1 and S1.
Figure 7. Chimeric CPPs exhibit improved cell type-specific subcellular penetration.

A. Abundance of barcoded CPPs in nuclear and cytoplasmic fractions of Huh-7 cells upon 3-hour treatment. TAT (black) serves as conventional cationic CPP. badTAT (pink) serves as non-penetrating negative control. B. Abundance of barcoded CPPs in nuclear and cytoplasmic fractions of Huh-7 cells upon 24-hour treatment. C-D. Abundance of barcoded KLAL-TAT CPP (C) and TAT-P-Ebola CPP (D) in nuclear and cytoplasmic fractions of HUVEC, Kasumi-1, Huh-7, and HEK293T cells, as indicated, upon 3-hour treatment. Symbols and whiskers represent mean and standard deviation values for three biologic replicates, respectively. The specific sequence of the CPP candidates and the barcodes are listed in supplemental Table 1 and S1.
To assess membrane penetration and subcellular delivery of various CPPs, we first sought to establish a robust cellular fractionation method to measure nuclear versus cytoplasmic accumulation of barcoded CPPs. We found that plasma membrane extraction using 0.1% digitonin, followed by sucrose density sedimentation produced specific separation of nuclear versus cytoplasmic compartments, as validated by Western immunoblotting against Lamin B1, Histone H3 and GAPDH, respectively (Figure 4 and S7; Table S2–3). We then established a quantitative procedure for measuring the absolute abundance of specific barcoded CPPs using cell fractionation, combined with targeted PRM mass spectrometry, with variation in ionization efficiency controlled by normalization to synthetic PRTC peptides (Figure 5). Figure 5b shows signal-response function for representative barcode B91.
Figure 4. Efficient cell fractionation achieved using digitonin and sucrose density sedimentation.

A. Representative Western blot with cytoplasmic (Cyt) marker GAPDH, and nuclear (Nu) markers Lamin B1 and Histone H3, as a function of various numbers of freeze-thaw cycles (left), time of treatment with 0.1% digitonin (DGT), and time of treatment with 0.03% DGT, as indicated. B. Fluorescence densitometry quantitation of 0.1% DGT-treated cells of Histone H3 abundance (grey) and GAPDH abundance (pink) in nuclear (Nuc) and cytoplasmic (Cyto) fractions. The x-axis corresponds with the Western blot columns in panel A. Histone H3 (% Cyto/Nuc) is the % ratio calculated by band intensity of Histone H3 in cytoplasmic fraction divided by band intensity of Histone H3 in nuclear fraction. Lower ratios (%) correspond to more efficient cell fractionation. Bars and whiskers represent mean and standard deviation values of three biologic replicates, respectively.
Figure 5. Barcoding and screening strategy results for absolute quantitation of CPP penetration.

A. Schematic of the cell treatment, fractionation, and LC-MS analysis. B. Signal-response function of representative barcode 91, showing MS2 ion current intensity as a function of peptide abundance. Solid line indicates linear fit; dashed line indicates baseline noise value from untreated cell lysate. C. Schematic for data analysis to calculate extracted ion chromatograms, normalized for variation in ionization efficiency using PRTC reference standards and background noise from untreated control samples.
Using this approach, we first sought to determine the potential contribution of barcode peptides on the cellular penetration activity of chimeric TAT-P-Ebola CPP, chosen because it contains both cationic and amphipathic CPP components (Figure 6). Library of TAT-P-Ebola GAAIGLAWIPYFGPAAYPRKKRRQRRR-[barcode] CPPs containing 8 diverse peptide barcodes showed no significant variation in their membrane destabilization or cytotoxicity, as measured by LDH release and ATP content, respectively (mean IC50 = 22 ± 6.0 and 12 ± 2.2 mM, respectively; Figure 6a–b). In contrast, TAT-P-Ebola variants with AFSVDAETLWR [Barcode82] and AGLDELAAFGWR [Barcode91] barcodes tested at non-toxic 1 µM concentrations exhibited significantly enhanced cytoplasmic accumulation, as compared to B55, B64, B81, B107, B108, and B121 barcodes, which were nearly exclusively nuclear (mean cytoplasmic abundance = 22 and 9.9 millions of molecules/cell, two-tailed unpaired Student’s t-test p = 0.018 and 0.022, respectively; Figure 6c). These results indicate that peptide barcoding can be used both to quantitatively measure and to modulate biologic properties of CPPs.
We then investigated the nuclear and cytoplasmic delivery of barcoded CPPs in our panel of cell lines, tested at 1 µM concentrations with negligible cytotoxicity and membrane disruption (as shown in Figure 3, S5 and S6). As a negative control, we used the anionic version of TAT, termed badTAT, in which key arginine residues have been replaced with glutamates (YGEKKEEQRRR-[barcode55]), preventing its membrane translocation which consistently led to lack of measurable accumulation of badTAT either in cytoplasmic or nuclear fractions (Figure 7a). We detected nuclear accumulation of TAT-[barcode64] and P14-[barcode91] CPPs, which is consistent with prior studies but at relatively low levels, due to the low 1 µM concentration of treatment, in order to be more relevant for future therapy development, and to minimize the confounding effects of cytotoxicity; TAT and P14 CPPs are used at >10 µM concentrations in many prior studies.14, 38
Notably, the novel chimeric CPPs TAT-P-Ebola-[barcode121], TAT-G-EBV-[barcode107], and RLAL-TAT-[barcode108] exhibited significantly higher nuclear accumulation (mean nuclear abundance = 403, 251 and 196 millions of molecules/cell, two-tailed unpaired Student’s t-test p < 0.0001 versus TAT-[barcode64]; Figure 7a). In addition, TAT-P-Ebola-[barcode121] and RLAL-TAT-[barcode108], but not TAT-G-EBV-[barcode107] exhibited time-dependent increase in nuclear accumulation after 24 hours of cell exposure (Figure 7b). Interestingly, KLAL-TAT-[barcode82] also exhibited significant time-dependent cytoplasmic accumulation, as compared to other barcoded CPPs (mean cytoplasmic abundance = 27 and 42 millions of molecules/cell at 3 and 24 hours, two-tailed unpaired Student’s t=test p = 0.0001 and 0.0004 versus TAT-[barcode64], respectively; Figure 7a–b). This effect may be potentiated by the specific contribution of the AFSVDAETLWR B82 barcode to apparent CPP activity (Figure 6c).
We also found that the activity of novel barcoded CPPs was cell type specific. For example, KLAL-TAT-[barcode82] showed increased cytoplasmic accumulation in mesenchymal HEK293T, epithelial Huh-7, and hematopoietic Kasumi-1 cells, but not in endothelial HUVEC cells (mean cytoplasmic abundance = 31, 27 and 15 versus 0 millions of molecules/cell, respectively; Figure 7c). In contrast, TAT-P-Ebola-[barcode121] exhibited increased nuclear accumulation in Huh-7 and HUVEC cells, as compared to HEK293T and Kasumi-1 cells (mean nuclear abundance = 420 and 291 versus 112 and 30.5 millions of molecules/cell, respectively; Figure 7d). Thus, the combination of peptide barcoding and de novo CPP design can be used to discover CPPs with improved cellular penetration activities and reduced toxicities (Figure 8). In addition, comparative studies can reveal time-dependent and cell type-specific differences in activity, thereby identifying potential targets for mechanistic studies.
Figure 8. Schematic for peptide barcoding and screening for profiling of bioactive macromolecules using BarcodeBabel and PeptideBabel.

The subcellular delivery of specific CPPs is deconvoluted using quantitative mass spectrometry of peptide barcodes.
Discussion
Nucleic acid barcoding has become a highly enabling technology for diverse high-throughput biological studies, such as binding studies using diversity-oriented synthesis and DNA barcoding, peptide and protein engineering using RNA display and RNA barcoding, among others.39 Peptide barcoding is particularly compelling for the engineering, screening, and other studies of biological molecules, because of their homogeneous biochemical properties, superior stability and information content, in contrast to mixed macromolecules with nucleic acid barcodes.16
Here, we developed an open-source algorithm BarcodeBabel, designed to construct libraries of unique peptide barcodes with optimal properties for high-throughput mass spectrometry proteomics studies. This enables the design of unique sequences not present in canonical biological proteomes, e.g., human tissues, with optimal ionization, separation, and fragmentation properties, as empirically validated using a library of 96 designed peptide barcodes.
BarcodeBabel is accompanied by an open-source algorithm PeptideBabel, which implements Monte Carlo sampling of user-defined seed sequences to generate novel libraries of peptides with diverse physicochemical and sequence complexity properties and to create bioactive macromolecules that can be uniquely identified and quantified in complex biological environments. We tested this approach empirically using a pilot library of barcoded cell penetration peptides (CPPs). We found that peptide barcoding can be used to quantitatively measure cell penetration and subcellular distribution of CPPs, as validated using known CPPs and their inactive negative controls. This work also implemented a targeted mass spectrometry method, suitable for quantitative studies of absolute molecular abundance of peptide barcodes in complex biological samples.
Using this proof-of-concept study, we identified novel CPPs with improved nuclear and cytoplasmic delivery exceeding hundreds of millions of molecules per human cell, with distinct cell type specific activities, while maintaining minimal membrane disruption and negligible toxicity in vitro. While we observed improved nuclear and cytoplasmic delivery of novel chimeric CPPs such as TAT-P-Ebola and KLAL-TAT, future studies will be required to define specific structure-activity relationships for CPPs, their precise mechanisms of membrane penetration and subcellular distribution, potential cellular receptors, and mechanisms of cell type-specific internalization. Interestingly, we found that in some cases, the specific barcode modulated the apparent CPP activity, suggesting that peptide barcodes may themselves be incorporated into the design of bioactive molecules. This also indicates that high-throughput screens should utilize multiple independent barcodes in order to discern specific biological activities, as is practiced with nucleic acid barcoding and other high-throughput technologies.
We anticipate BarcodeBabel and PeptideBabel should be useful for diverse screening, design, and analytical studies. For example, this approach may be used to design novel protein binders and quantify their binding affinities and kinetics using libraries of purified barcoded proteins in vitro.15, 40 Given the high sensitivity and resolving power of modern mass spectrometers, similar screens may also be performed with libraries of barcoded CPPs injected intravenously, and quantified using proteomics of specific tissues and organs in vivo. BarcodeBabel and PeptideBabel enable the construction of barcoded libraries of peptidic macromolecules with varied biological activities, and thus should be useful for a wide variety of molecular evolution and screening applications.
Supplementary Material
Acknowledgements
We acknowledge Markus Seeger and Pascal Egloff for helpful discussions, and members of our lab for critical advice.
Funding
This work was supported by NIH R01 CA214812, U54 CA243124, P30 CA08748, Starr Cancer Consortium, Doris Duke Charitable Foundation, and Mr. William H and Mrs. Alice Goodwin and the Commonwealth Foundation for Cancer Research the Center for Experimental Therapeutics at MSKCC. AK is a Scholar of the Leukemia & Lymphoma Society.
Footnotes
Associated Content
SUPPORTING INFORMATION:
The following supporting information is available free of charge at ACS website http://pubs.acs.org
Captions for Supplementary Figures 1–13 and Tables 1–3 (PDF), and Supplementary Table S4 (XLSX):
Figure S1. The design strategy and design parameters of BarcodeBabel.
Table S1. The pilot 96 barcode sequences designed by BarcodeBabel.
Figure S2. The workflow to identify candidate CPPs by cataloguing viral peptide sequences.
Figure S3. Physiochemical properties of the 96 barcode library.
Figure S4. Representative LC chromatograph and MS spectrum of the synthetic barcoded CPPs.
Figure S5. The LDH release and cell viability profiles of the barcoded CPPs using four different cell lines after incubation for 3 hours.
Figure S6. The LDH release and cell viability profiles of the barcoded CPPs using four different cell lines after incubation for 24 hours.
Table S2. Physiochemical methods tested for cell fractionation efficiency.
Figure S7. Evaluating the cell fractionation efficiency of various physical and chemical methods.
Table S3. Experimental conditions tested for freeze-thaw and digitonin-based cell fractionation methods.
Figure S8. Calibration curves of all the barcodes used in the cellular delivery assays.
Figure S9. The cytotoxicity profiles and cellular delivery of barcoded TAT-P-Ebola peptides.
Figure S10. The extracted MS2 ion current signal for each barcode in each cell fraction across four different cell lines.
Figure S11. Quantitative cellular delivery of the barcoded CPPs in four different cell lines.
Figure S12. Image of the entire western blot membrane shown in Figure 4.
Figure S13. Image of the entire western blot membrane shown in Figure S7.
Table S4. The list of seed sequences used in the current version of PeptideBabel (XLSX).
Conflict of Interest
Alex Kentsis is a consultant for Novartis, Rgenta, Blueprint Medicines, and Syndax.
Data Availability Statement
All raw and processed mass spectrometry data as well as Skyline chromatogram documents are available via ProteomeXchange with the identifier PXD048412.
References
- 1.Wang L; Wang N; Zhang W; Cheng X; Yan Z; Shao G; Wang X; Wang R; Fu C, Therapeutic peptides: Current applications and future directions. Signal Transduction and Targeted Therapy 2022, 7 (1), 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lu H; Zhou Q; He J; Jiang Z; Peng C; Tong R; Shi J, Recent advances in the development of protein–protein interactions modulators: mechanisms and clinical trials. Signal transduction and targeted therapy 2020, 5 (1), 213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Muttenthaler M; King GF; Adams DJ; Alewood PF, Trends in peptide drug discovery. Nature reviews Drug discovery 2021, 20 (4), 309–325. [DOI] [PubMed] [Google Scholar]
- 4.Ebrahimi SB; Samanta D, Engineering protein-based therapeutics through structural and chemical design. Nature Communications 2023, 14 (1), 2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cao L; Coventry B; Goreshnik I; Huang B; Sheffler W; Park JS; Jude KM; Marković I; Kadam RU; Verschueren KH, Design of protein-binding proteins from the target structure alone. Nature 2022, 605 (7910), 551–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guidotti G; Brambilla L; Rossi D, Cell-penetrating peptides: from basic research to clinics. Trends in pharmacological sciences 2017, 38 (4), 406–424. [DOI] [PubMed] [Google Scholar]
- 7.Frankel AD; Pabo CO, Cellular uptake of the tat protein from human immunodeficiency virus. Cell 1988, 55 (6), 1189–93. [DOI] [PubMed] [Google Scholar]
- 8.Green M; Loewenstein PM, Autonomous functional domains of chemically synthesized human immunodeficiency virus tat trans-activator protein. Cell 1988, 55 (6), 1179–88. [DOI] [PubMed] [Google Scholar]
- 9.Agrawal P; Bhalla S; Usmani SS; Singh S; Chaudhary K; Raghava GP; Gautam A, CPPsite 2.0: a repository of experimentally validated cell-penetrating peptides. Nucleic Acids Res 2016, 44 (D1), D1098–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kondo E; Saito K; Tashiro Y; Kamide K; Uno S; Furuya T; Mashita M; Nakajima K; Tsumuraya T; Kobayashi N; Nishibori M; Tanimoto M; Matsushita M, Tumour lineage-homing cell-penetrating peptides as anticancer molecular delivery systems. Nat Commun 2012, 3, 951. [DOI] [PubMed] [Google Scholar]
- 11.Guha S; Ghimire J; Wu E; Wimley WC, Mechanistic landscape of membrane-permeabilizing peptides. Chemical reviews 2019, 119 (9), 6040–6085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kauffman WB; Fuselier T; He J; Wimley WC, Mechanism matters: a taxonomy of cell penetrating peptides. Trends in biochemical sciences 2015, 40 (12), 749–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hoffmann K; Milech N; Juraja SM; Cunningham PT; Stone SR; Francis RW; Anastasas M; Hall CM; Heinrich T; Bogdawa HM, A platform for discovery of functional cell-penetrating peptides for efficient multi-cargo intracellular delivery. Scientific reports 2018, 8 (1), 12538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kauffman WB; Guha S; Wimley WC, Synthetic molecular evolution of hybrid cell penetrating peptides. Nat Commun 2018, 9 (1), 2568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Egloff P; Zimmermann I; Arnold FM; Hutter CAJ; Morger D; Opitz L; Poveda L; Keserue HA; Panse C; Roschitzki B; Seeger MA, Engineered peptide barcodes for in-depth analyses of binding protein libraries. Nat Methods 2019, 16 (5), 421–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rössler SL; Grob NM; Buchwald SL; Pentelute BL, Abiotic peptides as carriers of information for the encoding of small-molecule library synthesis. Science 2023, 379 (6635), 939–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chang CD; Meienhofer J, Solid-Phase Peptide Synthesis Using Mild Base Cleavage of Nαfluorenylmethyloxycarbonylamino Acids, Exemplified By a Synthesis of Dihydrosomatostatin. International journal of peptide and protein research 1978, 11 (3), 246–249. [DOI] [PubMed] [Google Scholar]
- 18.Behrendt R; White P; Offer J, Advances in Fmoc solid-phase peptide synthesis. Journal of Peptide Science 2016, 22 (1), 4–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Subirós-Funosas R; Prohens R; Barbas R; El-Faham A; Albericio F, Oxyma: An Efficient Additive for Peptide Synthesis to Replace the Benzotriazole-Based HOBt and HOAt with a Lower Risk of Explosion [1]. Chemistry–A European Journal 2009, 15 (37), 9394–9403. [DOI] [PubMed] [Google Scholar]
- 20.Huang Y-C; Chen C-C; Li S-J; Gao S; Shi J; Li Y-M, Facile synthesis of C-terminal peptide hydrazide and thioester of NY-ESO-1 (A39-A68) from an Fmoc-hydrazine 2-chlorotrityl chloride resin. Tetrahedron 2014, 70 (18), 2951–2955. [Google Scholar]
- 21.Dawson PE; Muir TW; Clark-Lewis I; Kent SB, Synthesis of proteins by native chemical ligation. Science 1994, 266 (5186), 776–779. [DOI] [PubMed] [Google Scholar]
- 22.Zheng J-S; Tang S; Qi Y-K; Wang Z-P; Liu L, Chemical synthesis of proteins using peptide hydrazides as thioester surrogates. Nature Protocols 2013, 8 (12), 2483–2495. [DOI] [PubMed] [Google Scholar]
- 23.Wan Q; Danishefsky SJ, Free-radical-based, specific desulfurization of cysteine: a powerful advance in the synthesis of polypeptides and glycopolypeptides. Angewandte Chemie 2007, 119 (48), 9408–9412. [DOI] [PubMed] [Google Scholar]
- 24.Cifani P; Kentsis A, High sensitivity quantitative proteomics using automated multidimensional nano-flow chromatography and accumulated ion monitoring on quadrupole-Orbitrap-linear ion trap mass spectrometer. Molecular & Cellular Proteomics 2017, 16 (11), 2006–2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cifani P; Dhabaria A; Kentsis A, Fabrication of nanoelectrospray emitters for LC-MS. Protocol Exchange 2015. [Google Scholar]
- 26.Schilling B; MacLean B; Held JM; Sahu AK; Rardin MJ; Sorensen DJ; Peters T; Wolfe AJ; Hunter CL; MacCoss MJ, Multiplexed, scheduled, high-resolution parallel reaction monitoring on a full scan QqTOF instrument with integrated data-dependent and targeted mass spectrometric workflows. Analytical chemistry 2015, 87 (20), 10222–10229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pino LK; Searle BC; Bollinger JG; Nunn B; MacLean B; MacCoss MJ, The Skyline ecosystem: Informatics for quantitative mass spectrometry proteomics. Mass spectrometry reviews 2020, 39 (3), 229–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zolg DP; Wilhelm M; Schnatbaum K; Zerweck J; Knaute T; Delanghe B; Bailey DJ; Gessulat S; Ehrlich HC; Weininger M; Yu P; Schlegl J; Kramer K; Schmidt T; Kusebauch U; Deutsch EW; Aebersold R; Moritz RL; Wenschuh H; Moehring T; Aiche S; Huhmer A; Reimer U; Kuster B, Building ProteomeTools based on a complete synthetic human proteome. Nat Methods 2017, 14 (3), 259–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dupont E; Prochiantz A; Joliot A, Penetratin story: an overview. Cell-Penetrating Peptides: Methods and Protocols 2011, 21–29. [DOI] [PubMed] [Google Scholar]
- 30.Rizzuti M; Nizzardo M; Zanetta C; Ramirez A; Corti S, Therapeutic applications of the cell-penetrating HIV-1 Tat peptide. Drug discovery today 2015, 20 (1), 76–85. [DOI] [PubMed] [Google Scholar]
- 31.Agrawal P; Bhalla S; Usmani SS; Singh S; Chaudhary K; Raghava GP; Gautam A, CPPsite 2.0: a repository of experimentally validated cell-penetrating peptides. Nucleic acids research 2016, 44 (D1), D1098–D1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kardani K; Bolhassani A, CPPsite 2.0: An available database of experimentally validated cell-penetrating peptides predicting their secondary and tertiary structures. Journal of molecular biology 2021, 433 (11), 166703. [DOI] [PubMed] [Google Scholar]
- 33.Futaki S; Suzuki T; Ohashi W; Yagami T; Tanaka S; Ueda K; Sugiura Y, Arginine-rich peptides: an abundant source of membrane-permeable peptides having potential as carriers for intracellular protein delivery. Journal of Biological Chemistry 2001, 276 (8), 5836–5840. [DOI] [PubMed] [Google Scholar]
- 34.Hulo C; De Castro E; Masson P; Bougueleret L; Bairoch A; Xenarios I; Le Mercier P, ViralZone: a knowledge resource to understand virus diversity. Nucleic acids research 2011, 39 (suppl_1), D576–D582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Du Z; He Y; Li J; Uversky VN, Deepadd: protein function prediction from k-mer embedding and additional features. Computational Biology and Chemistry 2020, 89, 107379. [DOI] [PubMed] [Google Scholar]
- 36.Jin S-S; Ju H; Jung H-J, Adaptive Markov chain Monte Carlo algorithms for Bayesian inference: recent advances and comparative study. Structure and Infrastructure Engineering 2019, 15 (11), 1548–1565. [Google Scholar]
- 37.Ramaker K; Henkel M; Krause T; Rockendorf N; Frey A, Cell penetrating peptides: a comparative transport analysis for 474 sequence motifs. Drug Deliv 2018, 25 (1), 928–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Takao S; Forbes L; Uni M; Cheng S; Pineda JMB; Tarumoto Y; Cifani P; Minuesa G; Chen C; Kharas MG, Convergent organization of aberrant MYB complex controls oncogenic gene expression in acute myeloid leukemia. Elife 2021, 10, e65905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liszczak G; Muir TW, Nucleic Acid-Barcoding Technologies: Converting DNA Sequencing into a Broad-Spectrum Molecular Counter. Angewandte Chemie International Edition 2019, 58 (13), 4144–4162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Matsuzaki Y; Aoki W; Miyazaki T; Aburaya S; Ohtani Y; Kajiwara K; Koike N; Minakuchi H; Miura N; Kadonosono T, Peptide barcoding for one-pot evaluation of sequence–function relationships of nanobodies. Scientific Reports 2021, 11 (1), 21516. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All raw and processed mass spectrometry data as well as Skyline chromatogram documents are available via ProteomeXchange with the identifier PXD048412.