

Abstract
Background
Genome-wide or application-targeted microarrays containing a subset of genes of interest have become widely used as a research tool with the prospect of diagnostic application. Intrinsic variability of microarray measurements poses a major problem in defining signal thresholds for absent/present or differentially expressed genes. Most strategies have used fold-change threshold values, but variability at low signal intensities may invalidate this approach and it does not provide information about false-positives and false negatives.
Results
We introduce a method to filter false-positives and false-negatives from DNA microarray experiments. This is achieved by evaluating a set of positive and negative controls by receiver operating characteristic (ROC) analysis. As an advantage of this approach, users may define thresholds on the basis of sensitivity and specificity considerations. The area under the ROC curve allows quality control of microarray hybridizations. This method has been applied to custom made microarrays developed for the analysis of invasive melanoma derived tumor cells. It demonstrated that ROC analysis yields a threshold with reduced missclassified genes in microarray experiments.
Conclusions
Provided that a set of appropriate positive and negative controls is included on the microarray, ROC analysis obviates the inherent problem of arbitrarily selecting threshold levels in microarray experiments. The proposed method is applicable to both custom made and commercially available DNA microarrays and will help to improve the reliability of predictions from DNA microarray experiments.
Background
Microarrays are a powerful tool to investigate differential gene expression of thousands of genes of a cell type, tissue, or organism [1,2]. While traditional microarray experiments strive to establish the 'global view' of the activity of all genes (i.e., the genome) in response to environmental conditions, they may also be used to characterize and quantitatively describe gene expression behavior of a selected set of genes as a true genotypic correlate of a particular phenotype. Application-targeted arrays and array reagents are already commercially available (Operon, Clontech, Incyte Pharmaceuticals, Affymetrix) for research in diverse areas such as cancer, stress and aging, toxicology, hematology, cell cycle, neurology and apoptosis. Contrary to 'genome-wide' chips, custom-fabricated microarrays are less expensive and more readily adapted to the economically sensitive environment of the molecular diagnostics laboratory, where relatively few interrogations are relevant for clinical investigation of a patient specimen.
Because typical microarray results are usually burdened with substantial amounts of noise [3], rigorous statistical methods must be applied to interpretation of data. Methods for systematically addressing noise in the analysis of the microarray data are only beginning to be described [4-10]. Such noise in microarray experiments may arise from non-specific hybridization of the labeled samples to elements printed on the microarray, print-tip effects, slide inhomogeneities, and variability in RNA isolation, purity, labeling and detection [6,9-12]. Among these, hybridization variance contributes most significantly to the overall variation [12].
Non-specific hybridization can be measured through the use of specificity controls on the microarray and addressed as a statistical problem [8,13]. The most common strategy in microarray experiments is to focus on fluorescent signal ratios in two-color competitive hybridization experiments. The problem with using ratio data alone is that it does not take into account the absolute signal intensity measurements used to calculate the ratios. While this approach may work adequately for ratios of moderate to highly expressed genes that yield bright fluorescent signals, weak signals arising from low transcript levels may be masked or biased by noise (non-specific hybridization). Non-specific hybridization is a characteristic of cDNA microarray hybridization and may be attributed to the uniform hybridization condition applied for all sequences on the chip [4,6,7]. The frequently used fold change threshold values of 2–3 to define a significant change are often arbitrarily chosen and do not take into account the statistical significance of absolute signal intensity. For example, microarray data showing a 4-fold change derived from low signal intensities may have no statistical significance whereas a 1.4 fold change derived from strong signal intensities may be highly significant in terms of reflecting actual changes in mRNA concentration within a biological sample. Thus, focusing on fold-changes alone is insufficient and confidence statements about differential expression must take into account absolute signal intensities [8]. In this study we have adapted a statistical method that utilizes absolute signal intensities from a reference set of positive controls and negative non-homologous control sequences to determine the absolute intensity range in each channel that may be used with a certain confidence level on a particular microarray to calculate expression ratios. The method of analysis proposed in this paper was originally developed along with radar and detailed results go back to the area of signal processing. ROC curves have been used for many years in numerous other areas, including psychology [14,15], and other areas of medical diagnostics [16]. For this purpose, diagnostic accuracy ROC curves have been used to depict the pattern of sensitivities and specificities observed when the performance of a diagnostic test is evaluated at several different thresholds. Here, receiver operating characteristic curves describe the ability of a particular parameter (e.g. fluorescence intensity) to discriminate between two conditions (e.g specific and non-specific hybridization). We illustrate our approach using gene expression data from a study comparing expression profiles of highly invasive and poorly invasive human melanoma cell lines [17]. The goal of these experiments is to identify candidate genes that may regulate the invasive behavior of melanoma cells. The area under the ROC curve may be used to assess the quality of individual microarray hybridizations which is particularly important because the quality of microarray hybridization can vary significantly [9,12]. The statistical evaluation of a reference set of genes to measure the sensitivity and specificity of each microarray hybridization reaction dramatically improves the ability to quality control the resulting data, a key requirement for the use of microarrays in diagnostic applications. Here, we propose the design of a prototype diagnostic microarray with respect to control sequences and we show how a ROC analysis using percentile ranks of specific and non-specific hybridization signals can be used to establish signal detection thresholds and hybridization quality assessments for each individual microarray experiment.
Results
The reliability of ratios measured to describe changes in gene expression depends on the absolute signal intensities. While ratios from highly abundant transcripts may be accurate, rare transcripts give absolute intensities that may be obscured by non-specific hybridization. Thus both ratio and absolute signal intensity are important to evaluate differential gene expression properly. Calibrating the appropriate signal and noise intensity thresholds for a given microarray hybridization requires the analysis of a set of positive and negative reference genes. At low signal intensities, both reference groups yield overlapping signal distributions (Figure 1a and 1b). Test signals from array 1 and 2 falling within the overlap region cannot easily be categorized as either present or absent and calculating ratios may lead to the identification of false positives (or false negatives) (Figure 1c and 1d).
Figure 1.
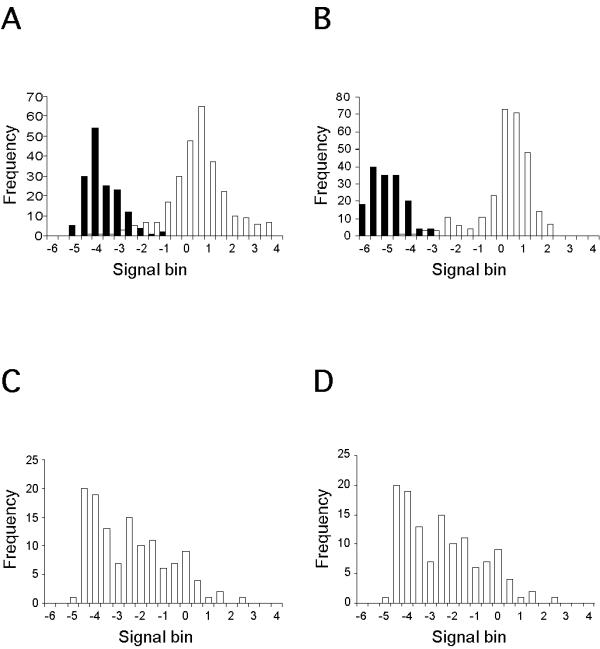
Signal distributions for specific and non-specific hybridizations overlap at low absolute intensities. The median intensity of 4 B.subtilis genes (n = 24 replicates per gene × 4 = 96) was used as a linear scaling factor to balance the Cy3 and Cy5 channels. Following this normalization step, normalized intensities were Log2 transformed for efficient graphical illustration. Positive control spots (open bars) and negative control spots (filled bars) from (A) array 1 and (B) array 2 microarray hybridizations. The positive control group includes seven housekeeping genes (n = 42) and four B.subtilis genes (24 repeats per sequence; n = 96) representing sequence-specific hybridization. The negative control sequences (six repeats per sequence) include three plant genes (n = 18), three E. coli genes (n = 18), and seven human cytomegalovirus (hCMV) genes (n = 42) representing non-specific hybridization events. Data for Cy3 and Cy5 signals were pooled. Signal distributions for test genes (n = 154) from (C) array 1 and (D) array 2.
Traditionally, cut-offs for microarrays have been calculated from negative controls. Here, we compare three widely used thresholds (TX = mean ; T0.5X = median; TX2SD = mean + 2 standard deviations) to the ROC- analyses derived threshold (TM = the threshold with maximum specificity and senitivity (TM) obtained as the point of intersection in figure 2) in terms of specificity (Sp) and sensitivity (Se) (Table 1). For TX and T0.5X the Se is ≅ 100%, however, the Sp would only be ≅ 50% indicating that approximately every second signal would be a false positive arising from non-specific hybridization. As a benefit, however, >99% of test genes would be included in data analyses (Table 1). If TX2SD or TM is the desired threshold, the Sp can be increased to >95% with only minor reductions in Se, however, the trade-off is an increase in the number of genes excluded from analyses (Table 1). If Sp and Se are plotted as a function of the thresholds, the intersection point of the two curves indicates maximum Sp and Se which can be directly read from the graph (Figure 2). As the criterion for a positive test becomes more stringent, the point on the curve corresponding to Sp and Se (point c, Figure 3a; point d, Figure 3b) moves down and to the left (lower Se, higher Sp); if less evidence is required for a positive test, the point on the curve corresponding to Sp and Se (point a, Figure 3a and 3b) moves up and to the right (lower Sp, higher Se). The area under the ROC curve (Figure 3, Table 2) is a measure of how well positive and negative signal can be distinguished in individual microarray experiments and indicates hybridization quality.
Figure 2.
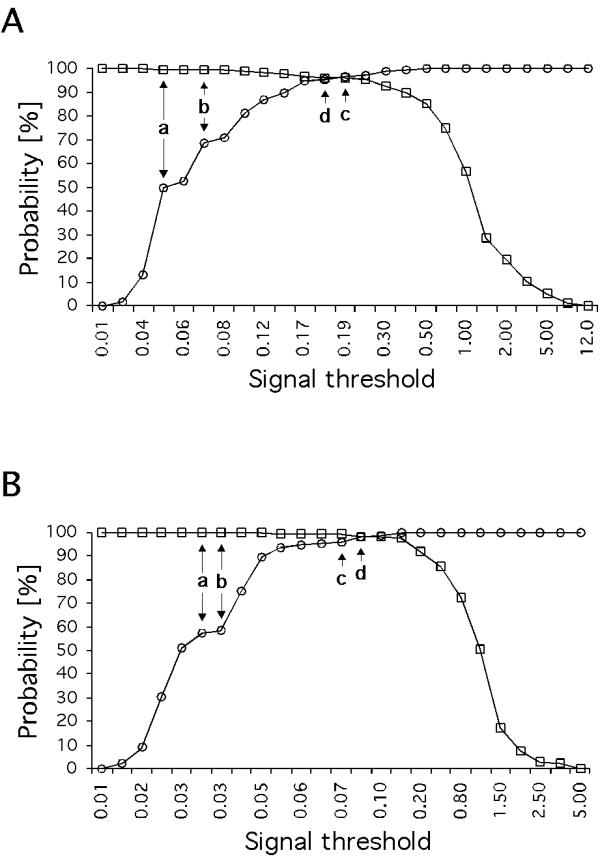
Specificity and sensitivity of select cut-offs for individual microarrays. Specific (spiked B. subtilis and housekeepers) and non-specific hybridization control groups (plant, bacterial and viral genes) represent sensitivity (squares) and specificity (circles), respectively. The intersection point of the two graphs indicates the threshold TM at which Sp equals Se. TM values were 0.18 (array1) and 0.09 (array 2). Indicated thresholds a-d are described in table 1. The Tj values presented in Table 1 were used to construct these curves. Note the different signal range (abcissa values) for array 1 (A) and array 2 (B).
Table 1.
Use of ROC analysis for the selection of cut-off values for individual microarray experiments.
| Array 1 | Array 2 | ||||||
| Threshold | Specificity [%] | Sensitivity [%] | % genes below threshold* | Threshold | Specificity [%] | Sensitivity [%] | % genes below threshold* |
| 0.010 | 0.0 | 100 | 0 | 0.005 | 0.0 | 100 | 0 |
| 0.030 | 1.8 | 100 | 0 | 0.010 | 2.5 | 100 | 0 |
| 0.040 | 13.4 | 100 | 0 | 0.015 | 9.3 | 100 | 0 |
| a0.057 | 49.7 | 99.7 | 0 | 0.020 | 30.5 | 100 | 0 |
| 0.060 | 52.7 | 99.6 | 1 | a0.027 | 51.0 | 100 | 0 |
| b0.077 | 68.6 | 99.4 | 7 | 0.030 | 57.7 | 100 | 1 |
| 0.080 | 71.1 | 99.4 | 7 | b0.031 | 58.6 | 100 | 1 |
| 0.100 | 81.3 | 99.0 | 17 | 0.040 | 75.4 | 99.9 | 12 |
| 0.120 | 86.8 | 98.2 | 26 | 0.050 | 89.9 | 99.8 | 20 |
| 0.140 | 90.0 | 97.7 | 36 | 0.060 | 93.9 | 99.6 | 29 |
| 0.170 | 94.9 | 96.5 | 44 | 0.062 | 94.9 | 99.6 | 31 |
| d0.180 | 95.6 | 96.1 | 44 | 0.064 | 95.6 | 99.9 | 33 |
| c0.192 | 96.6 | 95.8 | 47 | c0.066 | 96.0 | 99.5 | 36 |
| 0.200 | 97.4 | 95.7 | 48 | d0.090 | 98.1 | 98.5 | 43 |
| 0.300 | 98.6 | 92.8 | 65 | 0.100 | 99.1 | 98.3 | 45 |
| 0.400 | 99.5 | 89.9 | 77 | 0.110 | 100 | 97.6 | 48 |
| 0.500 | 100 | 85.0 | 79 | 0.200 | 100 | 92.0 | 65 |
| 0.700 | 100 | 74.7 | 83 | 0.500 | 100 | 85.7 | 81 |
| 1.000 | 100 | 56.6 | 89 | 0.800 | 100 | 72.4 | 90 |
| 1.500 | 100 | 28.7 | 95 | 1.000 | 100 | 50.8 | 94 |
| 2.000 | 100 | 19.4 | 98 | 1.500 | 100 | 17.1 | 97 |
| 3.000 | 100 | 10.2 | 98 | 2.000 | 100 | 7.7 | 98 |
| 5.000 | 100 | 5.4 | 99 | 2.500 | 100 | 2.9 | 98 |
| 10.000 | 100 | 1.2 | 100 | 3.000 | 100 | 2.1 | 99 |
| 12.000 | 100 | 0.0 | 100 | 5.000 | 100 | 0.0 | 100 |
Specificity and sensitivity for 25 signal cut-offs from 2 microarray experiments. Three thresholds (a-c) are calculated from the negative reference controls only and compared to the threshold with maximum specificity and senitivity (TM) obtained as the point of intersection in figure 2. a: mean (TX), b: median (T0.5X), c: mean + 2 standard deviations (TX2SD); d:TM Note the different signal range for array 1 and array 2. *normalized Cy3 and Cy5 pool.
Figure 3.
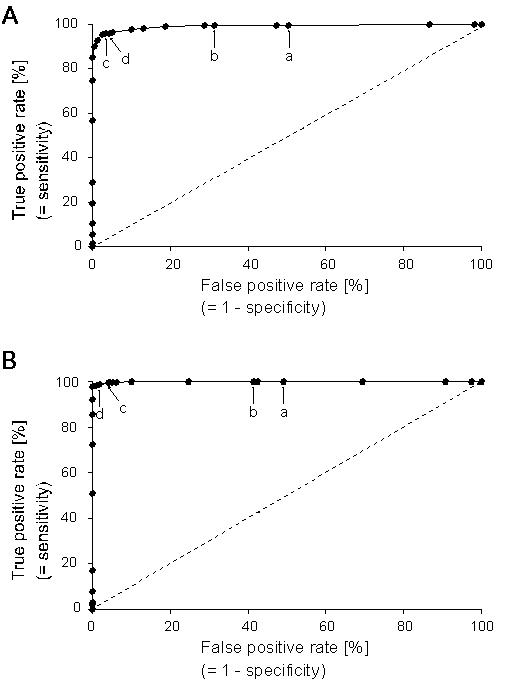
ROC analysis of selected signal cut-off values as a predictor for specific hybridization. ROC curves demonstrate the capacity to discriminate between the absence or presence of sequence-specific hybridization in individual microarray experiments. The closer an ROC curve is to the upper left hand corner of the graph, the more accurate it is because the true positive rate is 100% and the false positive rate is 0%. ROC plots based on percentile rank calculations for 25 cut-off signal thresholds (taken from table 1). The meaning of the position of thresholds a-d (table 1) are explained in the text. The area under the ROC curve was (A) 0.994 (array 1) and (B) 0.999 (array 2). Rising diagonal indicates no discrimination between positiv and negative control signals.
Table 2.
ROC parameters for different negative control groups.
| Threshold with maximum specificity and sensitivity* | β error | α error | Threshold value for α = 0.05 | Discriminatory power (Area under ROC plot) | |
| Array 1 | 0.40 (+SSC) | 0.101 | 0.096 | 0.50 | 0.969 |
| 0.18 (-SSC) | 0.039 | 0.044 | 0.17 | 0.994 | |
| Array 2 | 0.25 (+SSC) | 0.089 | 0.099 | 0.33 | 0.984 |
| 0.09 (-SSC) | 0.015 | 0.019 | 0.06 | 0.999 |
SCC spots or spots with deposited DNA perform differently in ROC-analysis yielding different areas under the ROC curve as well as different thresholds with maximum specificity and sensitivity. The area under the ROC curve may be used as an indicator for microarray hybridization quality. *characterized by smallest α and β errors. Note that in this case the α error can be >0.05.
The discriminatory power of a threshold based on the receiver operating characteristics depends on the proper controls included in the study and this choice will affect the interpretation of the data. Unlike spots containing printed control DNA (plant, bacterial, or viral), blank spots printed with 3X SSC often show strong signals indicating that unusually large amounts of label are inexplicably deposited on these manifestly blank areas. (The morphology of SSC spots giving relatively high signals are quite distinct from spots where DNA has been deposited, often showing small, highly concentrated areas of fluorescence. The cause of this fluorescence is not well understood. Their signal distribution is skewed towards large signals as compared to plant, bacterial, and viral genes; data not shown). Consequently, the inclusion of SSC spots results in a considerable overlap with positive controls resulting in increased thresholds and a lower overall discriminatory power (Table 2).
To illustrate the importance of signal threshold detection, we categorized differentially expressed genes of the melanoma study [17] according to confidence based on our predictive model (Table 3). For this purpose, we use TM as the threshold. Group A contains gene expression ratios derived from signals that exceed the selected threshold in both channels and are assigned high confidence ratios. In group B, either Cy3 or Cy5 signals were below that threshold, whereas in group C signals from both channels were below the defined detection limit. 'Risky' absolute intensities can be flagged and displayed when further processing the data (e.g. clustering analysis).
Table 3.
Analysis of fluorescence ratios of genes known to be involved in cancer invasion.
| Gene | Invasive signal | Non-invasive signal | Ratio | Confidence category | |||
| Array 1 | Array 2 | Array 1 | Array 2 | Array 1 | Array 2 | ||
| TIMP-1 | 1.70 | 0.92 | 0.25 | 0.30 | 6.9 | 3.1 | A |
| TIMP-2 | 0.63 | 0.25 | 0.24 | 0.70 | 2.5 | 0.4 | |
| MMP-9 | 4.16 | 2.22 | 1.70 | 1.01 | 2.5 | 2.2 | |
| Ln-5, γ2 | 1.27 | 1.22 | 0.06 | 0.03 | 22.5 | 35.5 | B |
| Integrin α3 | 0.24 | 0.16 | 0.12 | 0.06 | 2.0 | 2.8 | |
| Ln-5, β3 | 0.15 | 0.06 | 0.09 | 0.04 | 1.9 | 1.6 | C |
Ratios were qualitatively assigned to confidence categories according to their level of expression. All ratios met p < 0.05. TM was 0.18 for array 1 and 0.09 for array 2.
Discussion
Microarray data are generated from multi-step biochemical reactions, scanning/data collection, image analysis and data processing. This process is inherently prone to variability that affects the specificity and sensitivity of the assay, thus requiring evaluation of each microarray data set [9,10,12]. In order to calibrate the sensitivity and specificity of the output data, appropriate statistical tools applied to reference sequences composed of positive and negative controls may be used to quality control data from a given hybridization. We argue that any procedure which uses raw intensity ratios alone to infer differential expression may be inefficient and thus may lead to excessive errors.
Since ratios are simply the result of uneven signal distributions between Cy5 and Cy3 channels, analyzing these distributions will help interpret the biological relevance of an observed ratio. Signals are the result of specific and non-specific binding when a complex probe DNA mixture is incubated on the slide surface containing target DNA. The quality of DNA microarray data rests on the ability to measure non-specific components of a spot signal and eliminate them from ratio analysis. Such a component analysis on spotted DNA microarrays is not possible with today's technology and the proportion of non-specific binding will vary for each spot because of competitive binding in the presence of sequence specific hybridization (Note: GeneChip arrays from Affymetrix using perfect vs. mismatch oligonucleotide pairs do, to a certain extent, measure the non-specific binding component of every sequence; see technical note discussing probe length and performance, http://www.affymetrix.com/support/technical/technotes/25mer_technote.pdf). Since the influence of non-specific binding is more severe for probes where no or little specific hybridization occurs [8], we treat the problem as one of detecting a threshold value that is both determined by the highest signals attributable to spots representing non-specific hybridization and the lowest signals from spots where sequence specific hybridization must be assumed. Simply put, we determine a threshold separating specific from non-specific hybridization assuming that the former usually results in stronger signals than the latter [18]. A similar approach has been reported for Affymetrix GeneChip arrays, so called 'LUT based scoring system' [8] (tables to check noise level of particular chips or noise filtering look up tables).
Methods used to determine a signal threshold include the use of arbitrary fluorescence intensities [19], relative errors in Cy3/Cy5 ratios [9,20,21] or certain signal-to-background ratios [22]. However, these methods lack information about the specificity and sensitivity of the threshold, which are crucial parameters for estimation of the diagnostic accuracy of microarray hybridizations. To select a threshold, we have exploited a reference set of positive and negative control genes based on presence or absence of their cognate labeled cDNAs in the hybridization mix.
Positive controls may be spiked RNAs from non-homologous species or transcripts known to be expressed in the sample i.e. housekeepers [13,23]. Signals from positive controls should cover the range of test signals. This can be achieved by appropriate spiking and/or selection of housekeeping genes that fulfill this criterion.
The negative controls should be chosen to lack sequence homology to test genes, however, choosing appropriate control sequences for a ROC plot analysis is crucial: we conclude that SSC spots show a distinct signal pattern different from plant, bacterial and viral DNA deposits. Unlike spots containing control DNA, blank spots are not representative of non-specific hybridization to cellular probe DNA, do not behave well as control spots, and should be disregarded for threshold detection on custom arrays.
The robustness of ROC analysis to yield TM and ROC area values to discriminate 'good' from 'poor' microarray hybridizations relies on the relative positions of signal ranges from positive and negative controls as well as from target genes. We can imagine two szenarios making ROC analysis inappropriate for determination of threshold and/or microarray hybridization quality: (i) If the set of positive controls is in the high signal range, ROC analysis will yield a higher TM and ROC areas close to 1.0 (indicating good microarray hybridization) because positive and negative signals are well separated, irrespective of the distance between the greatest observation in the negative and the lowest observation in the positive sample. Consequently, a large portion of target genes will be discarded because of the relatively high TM. (ii) Alternatively, if the positive controls are spiked below the detection limit of microarrays (i.e. typically 1:500,000 wt/wt), their signal range may resemble the one from negative controls. This scenario will produce a low TM and ROC areas close to 0.5 falsley indicating 'poor' hybridization.
The overall performance of individual microarray hybridizations can be assessed by the position of the receiver operating characteristic line (Figure 3) using one single parameter: the area under the ROC curve (Table 2). Poor microarray hybridizations have lines close to the rising diagonal (or values ~0.5), whereas the lines for 'perfect' hybridizations would rise steeply and pass closely to the top left hand corner (or values ~1.0), where both, the specificity and sensitivity are 100%. In high-throughput applications such as routine diagnostic examinations, where a large number of hybridizations may be performed using a standard microarray-design, the ROC-plot area may be used as a 'hybridization quality checkpoint' to either accept or discard individual microarray hybridizations (for example 0.990 for array 1 and 2, Table 2). The area under the ROC curve represents a summary statistic of the overall performance of individual microarray hybridizations. A modification of the Wilcoxon rank-sum procedure may then be used as a statistical test to determine whether two ROC curves are significantly different [15].
Among 25 thresholds calculated here we have compared Sp and Se of 3 commonly used cut-offs with the ROC analysis-derived threshold TM (Table 1). The median or mean of a negative control group is regarded an adequate measure for non-specific hybridization [13], however, due to low specificity (~50%) we conclude that neither thresholds should be used if maximum specificity is required. In such a case, we find that the widely used cut-off value defined as the mean plus two standard deviations of the negative reference sample may be used adequately for DNA microarrays. The underlying rationale of using this threshold is to establish a cut-off value providing a specificity of 97.5% [24,25]. In our own example (Figure 1) skewness to the left makes the TX2SD overly conservative, which will sacrifice sensitivity unnecessarily. Hence, TX2SD may be inadequate and to adjust the sensitivity one should use TM. Most importantly, the TX2SD procedure does not account for the sensitivity of the threshold. Although the ROC-analysis derived cut-off resembles closely the cut-off defined as the mean + 2 SD, it is entirely possible that choosing a finer-grained partition of the signal space would alter the relative positions of theses two points. Likewise, this may be a characteristic for the 2 exemplary microarrays. Usually, the cut-off selection procedure is an informed decision based on the motivation of the individual to accept false positives (high Sp) or false negatives (high Se) that takes into account whether it is crucial to exclude any false positives (high Sp) or to cover the broadest signal range possible (high Se). Which cutoff to use depends on the objective of the experiment: If one needs to make sure that the 'present' or 'absent' call for a particular gene is correct, a cut-off with high Sp should be chosen, whereas if one is willing to accept false-positives where signals are low, high Se will be the driving force.
The signal intensity is the most critical parameter that influences the informative value of ratio estimates [26]. Therefore, ratios should be judged based on the absolute signal intensity of each gene. To diagnose the metastatic potential of highly versus poorly invasive melanoma cells we compared their gene expression profile with our metastasis chip, which contains genes critical for aspects of the metastatic process, including tumor cell motility and the ability to form primitive tubular networks [17,27]. Each ratio was tested for the Null hypothesis that there is no difference between the means of the ranks of the Cy5 and Cy3 signals over 6 replicate spots representing a unique sequence. At signal intensities below the threshold with maximum specificity and sensitivity some genes gave a ratio greater than 1.6-fold (at a confidence level of p < 0.05). In such a case, however, ratios are not optimal estimators because the low denominator value introduces large artifacts [8]. Therefore we sought to determine ratio confidence categories based on the absolute signal intensities [13]. Assuming that the ROC-analysis derived threshold is an 'appropriate' cut-off for distinguishing absent/present genes, the proposed 'confidence categories' may be interpreted as follows:
(A) The gene is present in both samples, and this is the best estimate of the true ratio, while further statistical evaluation should be applied to take into account the variability of the measurements.
(B) The gene is present in one sample and absent in the other. Ratios are meaningless, but this is still an extremely significant biological result!
(C) The gene is absent in both samples. Not only are the ratios meaningless, but so are the intensity estimates.
As a result of threshold setting, certain genes may be falsely included (=false positives) or, less frequently [5], falsely discarded (=false negatives) from further analysis (i.e. ratio calculations, clustering analysis, etc.) as exemplified here with the microarray experiment for investigating invasion in cancer. The ROC-derived threshold correctly classifies the signal for Laminin-5, γ2 as 'absent', whereas the mean- or median -derived thresholds would produce a false-positive result. Since this gene product plays a significant role in vasculogenic mimicry [17]. Classifying the expression level is biologically crucial. ROC analysis leads to a result (Type B, above) that is in line with data obtained otherwise, whereas both mean- or median -derived threshold would have resulted in accepting falsely a change (type A, above) in Laminin-5, γ2 expression.
Collectively, the present study demonstrated that microarray-derived signals from positive and negative controls may be used to compute accurately type I and type II errors for a series of signal thresholds. We have introduced a new model for signal threshold determination for gene expression microarray experiments that greatly eases the interpretation and comparison of these data. This model is based on analysis of signal intensities and distributions of a reference set of positive and negative controls included on each microarray. It provides a framework for determination of detection limits, confidence about fluorescent ratios and for pre-processing data for subsequent data analyses, such as cluster analyses [23,28].
Materials & Methods
Synthesis of PCR products
IMAGE cDNA clones representing 72 human genes involved in cell migration were purchased from Research Genetics (Huntsville, AL) and sequence verified in-house. Control cDNA clones (GenBank Accession numbers are in brackets) from Arabidopsis thaliana (T76518, T45394, H37673), human cytomegalovirus (NP_039949, NP_039950, NP_039952, NP_039955, NP_039971, NP_039974, NP_039976) E. coli (J04423: BioB, BioC, BioD) and B. subtilis (X04603, M24537, X17013, L38424) were provided by the Scripps Research Institute's microarray facility and were chosen to lack homology to human sequences. Plasmids were prepared from IMAGE clones using Qiagen's (Santa Clara, CA) plasmid mini prep kits following manufacturer's protocols. All plasmid inserts were amplified in 100 μl reactions using the following primer sequences:
M13 Forward 5'-GTTTTCCCAGTCACGACGTTG-3'
M13 Reverse 5'-TGAGCGGATAACAATTTCACACAG
PCR reactions were cycled 35 times under the following conditions: 94 degrees for 30 seconds, 55 degrees for 1 minute and 72 degrees for 2 minutes. All PCR products were analyzed by gel electrophoresis to confirm the presence of a single band. PCR products showing weak and/or multiple bands were reamplified using variable MgCl2 concentrations (1.5–2.5 mM) until single bands were generated. The PCR products were purified using Qiagen's 96-well PCR purification kit according to manufacturer's protocols. Purified PCR products were dried down and resuspended in 80 ul 3X SSC (standard saline citrate- 1X SSC = 150 mM NaCl, 15 mM sodium citrate) with 0.01% sodium dodecyl sulfate (SDS).
Microarray production
Each gene was spotted 6 times at various locations across one of the four subarrays printed on each glass slide. DNA was printed on poly-lysine coated slides using a custom built robotic arrayer in the TSRI's DNA Microarray Core Facility. This facility houses a custom made spotter built by Robotic Labware Designs (Carlsbad, CA). The arraying instrument was conceptually modeled after that of Dr. Patrick Brown and colleagues at Stanford University (details can be obtained from: http://cmgm.stanford.edu/pbrown/array.html). Spotting tips (CMPB2) are purchased from Telechem International (San Jose, CA). Polylysine coated slides were prepared as described in the Brown lab website (see above).
Preparation of Cy3 and Cy5 labeled cDNA probes
Cy3 and Cy5 -labeled cDNA probes were synthesized as described in (14) Briefly, 2 μg mRNA spiked with B. subtilis RNA cocktail (described below) at indicated amounts. mRNA was added to 4 μg of oligo (dT21) primer (Life Technologies, Rockville, MD), DEPC water was used to bring the volume to 27 μl and the mixture was incubated at 65°C for 10 min and placed on ice. A reaction mixture consisting of mRNA, first-strand buffer (Life Technologies), DTT (0.1 M), d(GAT)TP (25 mM), dCTP (1 mM), Cy3-dCTP (2 mM)(Amersham, Piscataway, NJ) and Superscript RT II (200 units) were added and incubated at room temperature for 10 minutes and then at 42°C for 2 hours. The cDNA:mRNA hybrid was isolated on a Qiaquick PCR purification column (Qiagen) and degraded after addition of 2.5 μl 1 N NaOH for 10 minutes at 37°C. After addition of 2.5 μl 1 N Tris/HCl (pH 7.5), labeled cDNA was concentrated by ETOH precipitation and resuspended in 2 μl water.
Generation of B. subtilis RNA cocktail
Four B. subtilis clones containing engineered polyA tails were purchased as plasmids from ATCC (Manassas, VA; ATCC No. pglbs-lys, pglbs-phe, pglbs-thr, pglbs-dap). Polyadenylated control RNA was prepared from these clones by in-vitro transcription of linearized plasmids using the AmpliScribe transcription kit (Epicenter Technologies, Madison, WI) following the manufacturer's instructions. The B. subtilis control cocktail contained equal proportions (wt/wt) of each RNA species and was spiked into cDNA reactions at 125 pg/2 ug mRNA in order to normalize Cy3 and Cy5 signals after scanning.
Hybridization, image processing and normalization
Resuspended probes were mixed with 4 μl of 5 × hybridization buffer (20 × SSX, 1%SDS), 1 μl human Cot-1 DNA (10 μg/μl), 1 μl polyA DNA (2 μg/μl) (Amersham) and 8 μl formamide (Sigma, St. Louis, MO), heated to 65°C for 2 min and then centrifuged briefly. Samples were hybridized to a previously prepared microarray overnight at 42°C in a humid chamber in the dark. Following hybridization, slides were washed serially in 2× SSC, 0.2% SDS, once in 2× SSC and once in 0.2× SSC for 5 min at room temperature each. The slides were then air dried and scanned on a ScanArray 5000 (GSI Lumonics, Watertown, MA) using the appropriate excitation and emission filter wavelengths for Cy3 and Cy5. Image files were analyzed using ImaGene 4.0 (Biodiscovery). Background subtracted fluorescence intensities were normalized by dividing all signals on the microarray by the median signal generated by the four B. subtilis genes dap, lys, phe and thr (n = 96 = 4 × 24) [29-31].; for reviews about current normalization strategies see [10,23].
ROC analysis
For signal threshold determination we used normalized Cy3 and Cy5 signal intensities from a study carried out to compare highly invasive vs. poorly invasive uveal (MUM2B vs. MUM2C; array 1) and cutaneous (C8161 vs. C81–61; array 2) melanoma cells [17]. Although thresholds may be determined for each channel individually, normalizing fluorescence intensities allows the pooling of Cy3 and Cy5 data thus simplifying the process. A negative control group consisting of cDNAs from three A. thaliana, three E. coli and 7 human cytomegalovirus genes were printed on the microarrays and used to measure non-specific hybridization. cDNAs derived from 7 housekeeping genes (ribosomal proteins L13A, L30, S9; α-tubulin; β-actin; ubiquitin c and hypoxanthine guanine phosphoribosyl transferase) and four B. subtilis clones (designed to hybridize to the B. subtilis RNA cocktail spiked into the cDNA reactions) were printed on the microarrays to serve as specific hybridization controls (positive controls).
Steps for construction of the ROC curves
1: Divide the positive and negative control signals arithmetically into an appropriate number of intervals (e.g. 20–30) with the resulting limits termed 'thresholds' (Tj). Alternatively, each observed measurement point (i.e. signal from positive and negative controls) may be used as Tj. (Scott, DW; 1976. Nonparametric Probability Density Estimation by Optimization Theoretic Techniques, Technical report TR476-131-1, http://www.stat.rice.edu/stat/techr.html)
2: Specificity Spj and Sensitivity Sej obtained with each threshold value Tj are calculated as the proportion of positive results in the positive and negative results in the negative reference group, respectively. Note that these calculations are estimates of type I and type II errors (equivalent to α and β errors) for each threshold.
3: Draw a ROC curve by plotting the false positive rate or (100 – specificity) on the x-axis. The y-axis shows the true positive rate or sensitivity. The area under the ROC plot is an estimator of the discriminatory power of selecting a cut-off and varies from array to array. The area of the ROC plot was calculated as the sum of
Area = Σ {[αt(n) – (αt(n+1))]/ [1-βt(n)]}
where t(n) denotes the threshold n = 1, 2,...22, α = alpha error, and β= beta error. (For a detailed description of ROC-area calculation: [15,16]). A sum of 0.5 indicates that negative and positive test groups cannot be distinguished. A ROC curve representing a parameter with no discrimination at all is a 45° diagonal line from the left lower corner (0% true positive rate and 0% false positive rate) to the upper right corner (100% true positive rate and 100% false positive rate) with an area under the curve of 0.5. A parameter with no overlap between the two conditions will discriminate perfectly and has an ROC curve passing along the y-axis to the upper left corner (100% true positive rate and 0% false positive rate) to end again in the upper right corner with an area under the curve of 1.0. Software for ROC analysis is available commercially or as spreadsheet calculation macros [24].
Acknowledgments
Acknowledgements
This work was supported by NIH grants GM46902 and CA47858 to Vito Quaranta and grants 13321-MED (Austrian Science Foundation), 8339 (Austrian National Bank Vienna) both to Gernot Desoye. We thank Jim Koziol for critical reading of the manuscript.
Contributor Information
M Bilban, Email: mbilban@scripps.edu.
LK Buehler, Email: l.buehler@ucsd.edu.
S Head, Email: shead@scripps.edu.
G Desoye, Email: gernot.desoye@kfunigraz.ac.at.
V Quaranta, Email: quaranta@scripps.edu.
References
- Young RA. Biomedical discovery with DNA arrays. Cell. 2000;102:9–15. doi: 10.1016/s0092-8674(00)00005-2. [DOI] [PubMed] [Google Scholar]
- Bilban M, Head S, Desoye G, Quaranta V. DNA microarrays: a novel approach to investigate genomics in trophoblast invasion – a review. Placenta. 2000;21:S99–105. doi: 10.1053/plac.1999.0517. [DOI] [PubMed] [Google Scholar]
- Claverie JM. Computational methods for the identification of differential and coordinated gene expression. Hum Mol Genet. 1999;8:1821–1832. doi: 10.1093/hmg/8.10.1821. [DOI] [PubMed] [Google Scholar]
- Kane MD, Jatkoe TA, Stumpf CR, Lu J, Thomas JD, Madore SJ. Assessment of the sensitivity and specificity of oligonucleotide (50 mer) microarrays. Nucleic Acids Res. 2000;28:4552–4557. doi: 10.1093/nar/28.22.4552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee ML, Kuo FC, Whitmore GA, Sklar J. Importance of replication in microarray gene expression studies: statistical methods and evidence from repetitive cDNA hybridizations. Proc Natl Acad Sci U S A. 2000;97:9834–9839. doi: 10.1073/pnas.97.18.9834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuchhardt J, Beule D, Malik A, Wolski E, Eickhoff H, Lehrach H, Herzel H. Normalization strategies for cDNA microarrays. Nucleic Acids Res. 2000;28:E47. doi: 10.1093/nar/28.10.e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes TR, Mao M, Jones AR, Burchard J, Marton MJ, Shannon KW, Lefkowitz SM, Ziman M, Schelter JM, Meyer MR, et al. Expression profiling using microarrays fabricated by an ink-jet oligonucleotide synthesizer. Nat Biotechnol. 2001;19:342–347. doi: 10.1038/86730. [DOI] [PubMed] [Google Scholar]
- Mills JC, Gordon JI. A new approach for filtering noise from high-density oligonucleotide microarray datasets. Nucleic Acids Res. 2001;29:E72. doi: 10.1093/nar/29.15.e72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng GC, Oh MK, Rohlin L, Liao JC, Wong WH. Issues in cDNA microarray analysis: quality filtering, channel normalization, models of variations and assessment of gene effects. Nucleic Acids Res. 2001;29:2549–2557. doi: 10.1093/nar/29.12.2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilban M, Buehler LK, Head S, Desoye G, Quaranta V. Normalizing DNA microarray data. Curr Issues Mol Biol. 2002. [PubMed]
- Bartosiewicz M, Trounstine M, Barker D, Johnston R, Buckpitt A. Development of a toxicological gene array and quantitative assessment of this technology. Arch Biochem Biophys. 2000;376:66–73. doi: 10.1006/abbi.2000.1700. [DOI] [PubMed] [Google Scholar]
- Yue H, Eastman PS, Wang BB, Minor J, Doctolero MH, Nuttall RL, Stack R, Becker JW, Montgomery JR, Vainer M, Johnston R. An evaluation of the performance of cDNA microarrays for detecting changes in global mRNA expression. Nucleic Acids Res. 2001;29:E41. doi: 10.1093/nar/29.8.e41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou XJ, Schena M, Horrigan FT, Lawn RM, Davis RW. Expression monitoring using cDNA microarrays. A general protocol. Methods Mol Biol. 2001;175:323–340. doi: 10.1385/1-59259-235-X:323. [DOI] [PubMed] [Google Scholar]
- Dorfman DD, Alf E., Jr Maximum likelihood estimation of parameters of signal detection theory – a direct solution. Psychometrika. 1968;33:117–124. doi: 10.1007/BF02289677. [DOI] [PubMed] [Google Scholar]
- Hanley JA, McNeil BJ. The meaning and use of the area under a receiver operating characteristic (ROC) curve. Radiology. 1982;143:29–36. doi: 10.1148/radiology.143.1.7063747. [DOI] [PubMed] [Google Scholar]
- Metz CE. Basic principles of ROC analysis. Semin Nucl Med. 1978;8:283–298. doi: 10.1016/s0001-2998(78)80014-2. [DOI] [PubMed] [Google Scholar]
- Seftor RE, Seftor EA, Koshikawa N, Meltzer PS, Gardner LM, Bilban M, Stetler-Stevenson WG, Quaranta V, Hendrix MJ. Cooperative interactions of laminin 5 gamma2 chain, matrix metalloproteinase-2, and membrane type-1-matrix/metalloproteinase are required for mimicry of embryonic vasculogenesis by aggressive melanoma. Cancer Res. 2001;61:6322–6327. [PubMed] [Google Scholar]
- Schmitt AO, Herwig R, Meier-Ewert S, Lehrach H. High-density cDNA grids for hybridization fingerprinting experiments. In: Innis MA, Gelfand DH, Sninsky JJ, editor. In PCR applications. Academic Press; 1999. pp. 457–472. [Google Scholar]
- Sakai K, Higuchi H, Matsubara K, Kato K. Microarray hybridization with fractionated cDNA: enhanced identification of differentially expressed genes. Anal Biochem. 2000;287:32–37. doi: 10.1006/abio.2000.4831. [DOI] [PubMed] [Google Scholar]
- Chen Y, Dougherty ER, Bittner ML. Ratio-based decisions and the quantitative analysis of cDNA microarray images. J Biomed optics. 1997;2:364–374. doi: 10.1117/1.429838. [DOI] [PubMed] [Google Scholar]
- Geiss GK, Bumgarner RE, An MC, Agy MB, 't Wout AB, Hammersmark E, Carter VS, Upchurch D, Mullins JI, Katze MG. Large-scale monitoring of host cell gene expression during HIV-1 infection using cDNA microarrays. Virology. 2000;266:8–16. doi: 10.1006/viro.1999.0044. [DOI] [PubMed] [Google Scholar]
- Cunningham MJ, Liang S, Fuhrman S, Seilhamer JJ, Somogyi R. Gene expression microarray data analysis for toxicology profiling. Ann N Y Acad Sci. 2000;919:52–67. doi: 10.1111/j.1749-6632.2000.tb06867.x. [DOI] [PubMed] [Google Scholar]
- Quackenbush J. Computational analysis of microarray data. Nat Rev Genet. 2001;2:418–427. doi: 10.1038/35076576. [DOI] [PubMed] [Google Scholar]
- Greiner M, Pfeiffer D, Smith RD. Principles and practical application of the receiver-operating characteristic analysis for diagnostic tests. Prev Vet Med. 2000;45:23–41. doi: 10.1016/S0167-5877(00)00115-X. [DOI] [PubMed] [Google Scholar]
- Rocke DM, Durbin B. A model for measurement error for gene expression arrays. J Comput Biol. 2001;8:557–569. doi: 10.1089/106652701753307485. [DOI] [PubMed] [Google Scholar]
- Yang MC, Ruan QG, Yang JJ, Eckenrode S, Wu S, McIndoe RA, She JX. A statistical method for flagging weak spots improves normalization and ratio estimates in microarrays. Physiol Genomics. 2001;7:45–53. doi: 10.1152/physiolgenomics.00020.2001. [DOI] [PubMed] [Google Scholar]
- Bittner M, Meltzer P, Chen Y, Jiang Y, Seftor E, Hendrix M, Radmacher M, Simon R, Yakhini Z, Ben Dor A, Sampas N, Dougherty E, Wang E, Marincola F, Gooden C, Lueders J, Glatfelter A, Pollock P, Carpten J, Gillanders E, Leja D, Dietrich K, Beaudry C, Berens M, Alberts D, Sondak V. Molecular classification of cutaneous malignant melanoma by gene expression profiling. Nature. 2000;406:536–540. doi: 10.1038/35020115. [DOI] [PubMed] [Google Scholar]
- Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci U S A. 1998;95:14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeRisi J, Penland L, Brown PO, Bittner ML, Meltzer PS, Ray M, Chen Y, Su YA, Trent JM. Use of a cDNA microarray to analyse gene expression patterns in human cancer. Nat Genet. 1996;14:457–460. doi: 10.1038/ng1296-457. [DOI] [PubMed] [Google Scholar]
- Schena M, Shalon D, Heller R, Chai A, Brown PO, Davis RW. Parallel human genome analysis: microarray-based expression monitoring of 1000 genes. Proc Natl Acad Sci U S A. 1996;93:10614–10619. doi: 10.1073/pnas.93.20.10614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eickhoff B, Korn B, Schick M, Poustka A, van der BJ. Normalization of array hybridization experiments in differential gene expression analysis. Nucleic Acids Res. 1999;27:e33. doi: 10.1093/nar/27.22.e33. [DOI] [PMC free article] [PubMed] [Google Scholar]