

Abstract
Background
HIV Associated Dementia (HAD) is a common complication of human immunodeficiency virus (HIV) infection that erodes the quality of life for patients and burdens health care providers. Intravenous drug use is a major route of HIV transmission, and drug use is associated with increased HAD. Specific proteins released as a consequence of HIV infection (e.g., gp120, the HIV envelope protein and Tat, the nuclear transactivating protein) have been implicated in the pathogenesis of HAD. In primary cultures of human fetal brain tissue, subtoxic doses of gp120 and Tat are capable of interacting with a physiologically relevant dose of cocaine, to produce a significant synergistic neurotoxicity. Using this model system, the neuroprotective potential of gonadal steroids was investigated.
Results
17β-Estradiol (17β-E2), but not 17α-estradiol (17α-E2), was protective against this combined neurotoxicity. Progesterone (PROG) afforded limited neuroprotection, as did dihydrotestosterone (DHT). The efficacy of 5α-testosterone (T)-mediated neuroprotection was robust, similar to that provided by 17β-E2. In the presence of the specific estrogen receptor (ER) antagonist, ICI-182,780, T's neuroprotection was completely blocked. Thus, T acts through the ER to provide neuroprotection against HIV proteins and cocaine. Interestingly, cholesterol also demonstrated concentration-dependent neuroprotection, possibly attributable to cholesterol's serving as a steroid hormone precursor in neurons.
Conclusion
Collectively, the present data indicate that cocaine has a robust interaction with the HIV proteins gp120 and Tat that produces severe neurotoxicity, and this toxicity can be blocked through pretreatment with ER agonists.
Background
A particularly devastating complication of HIV infection is a pervasive form of damage to the brain, HAD [1]. The overall incidence of severe HAD is estimated at about 30% of the HIV infected population [2]. However, HAD occurs more often in HIV-positive IV drug users than HIV-positive non-drug users [3-5]. Neuroimaging and autopsy studies demonstrate that the basal ganglia and frontal lobes are preferentially affected by HAD [5,6]. These structures may degenerate with chronic psychostimulant (methamphetamine, cocaine) abuse [7,8], eventually leading to a Parkinson type syndrome [9]. Injection of abused drugs, such as cocaine, has been noted to accelerate the progression of HIV infection to AIDS status and to HAD [8,10-12].
In the United States, cocaine use plays a larger role in HIV transmission to women than it does to men [13]. HIV infected women have lower initial viral loads than men, progress to AIDS status at the same rate as men, yet have higher mortality and lower life expectancy than men [14-16]. How sex differences contribute to the progression to HAD is largely unknown. However, female gender or estrogenic steroids are recognized as protective against several neurological insults, including animal models of ischemia, oxidative stress, and psychostimulant-induced neurotoxicity [17-22] and human neurodegenerative diseases such as Alzheimer's disease [23-26] and Parkinson's disease [27-29]. Tissue culture studies have found that the estrogenic steroid, 17β-E2, is protective against neurotoxic HIV proteins [30,31]. Collectively, these gender/hormonal effects suggest that gonadal hormones may play a differential role in effects of drug abuse, HIV infection, and HAD.
Neurotoxic interactions between the HIV proteins, Tat and gp120, and abused psychostimulant drugs have been previously reported [30,32]. More recently, 17β-E2 proved neuroprotective in vitro [30,32]; however, it is unknown whether this neuroprotection is specifically estrogenic, or also effected by PROG, and whether it contains an androgenic component. Therefore, the aim of the present study was to determine which gonadal steroids provide neuroprotection against the synergistic neurotoxicity of HIV proteins in the presence of cocaine. We investigated the potential for neuroprotection by T, whether this is mediated through an ER mechanism and the potential concentration-dependent neuroprotection by PROG, DHT and cholesterol.
We report here concentration dependent neuroprotection by T mediated through an ICI 182,780-sensitive mechanism. Incomplete neuroprotection at the concentrations (nM) tested was also provided by PROG, DHT, and cholesterol.
Results
17β-, not 17α-Estradiol, protects against HIV proteins plus cocaine synergistic neurotoxicity
Clear and robust synergistic neurotoxicity of the HIV proteins Tat and gp 120 was repeatedly observed when combined with a physiologically relevant dose of cocaine (Figs. 1C, 1D, 2 Left Panel, 3 Left Panel, 4 Top Panel, 5 Top Panel). False color visualization of the intensity of trypan blue within neurons verified the assay for cell death and demonstrated the synergistic toxicity of cocaine with the HIV proteins (Fig. 1). Furthermore, this neurotoxicity is precluded by pretreatment with 10 nM dose of 17β-E2, but not with 17α-E2. Neither the HIV proteins, Tat plus gp120, nor cocaine alone were more toxic than the Locke's buffer control; however, in combination they produced synergistic neurotoxicity (Fig. 2). The ANOVA confirmed the presence of a significant interaction of the HIV proteins with cocaine (F(1,24) = 15.38, p < 0.0008; n = 6 each point) (Fig. 2 Left Panel). The presence of this significant neurotoxic effect was demonstrated in every experiment at p < 0.005. The stereoisomers of estradiol demonstrated a significant treatment effect against this toxicity (F(2,15) = 6.95, p < 0.007). The 17β-stereoisomer of estradiol demonstrated significant neuroprotection (F(1,24) = 24.71, p < 0.0001; n = at least 3 each point). The percent neuronal death with 17α-E2 treatment was not significantly different from the synergistic toxicity control (Fig. 2 Right Panel).
Figure 1.
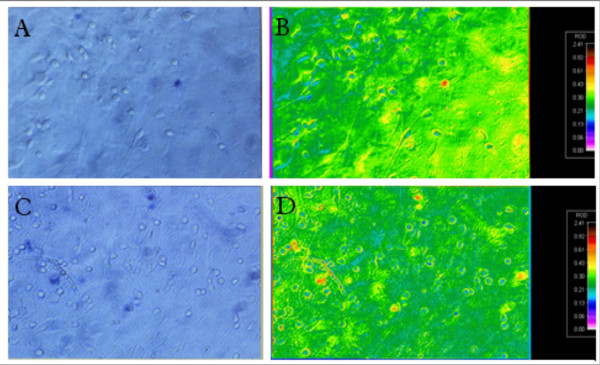
True and false color photomicrographs of human neurons (A-C). Control culture after trypan blue exclusion assay in true color (A) and false color (B). Cultures exposed to HIV proteins 40 nM Tat plus 32.5 pM gp120 and 1.6 μM cocaine show synergistic toxicity trypan blue exclusion assay in true color (C) and false color (D).
Figure 2.
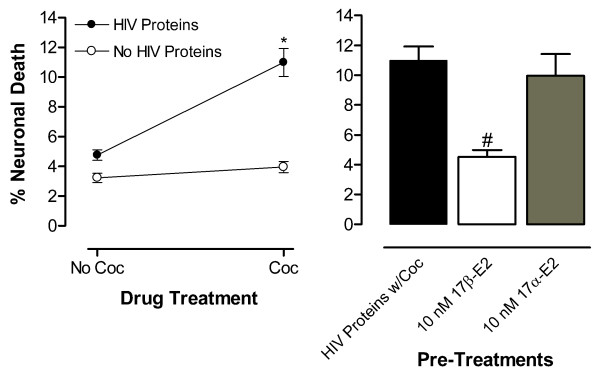
17β-, not 17α-E2, protects human fetal neurons against the synergistically toxic of incubation with Tat [40 nM] plus gp120 [32.5 pM] (HIV Proteins) plus cocaine (Coc) [1.6 μM]. Control conditions of incubation with Locke's Buffer vehicle or the HIV proteins or Coc only produced similar neuronal death. (Left Panel) Significant interaction of the HIV proteins with Coc, as illustrated by the lines diverging from parallel, produced toxic synergism (*p < 0.0008). (Right Panel) Significant neuroprotection by 17β-E2 against HIV proteins w/ Coc toxic synergism (#p < 0.0001).
DMSO at 10 nM (0.1%) served as the solvent for 17α-E2 and for ICI-182,780; therefore, it was tested for toxicity and was not significantly different from Locke's buffer incubation (data not shown). β-Cyclodextrin (100 nM) served as an encapsulating carrier to enhance solubility for the steroids 17β-E2, T, PROG and cholesterol and was therefore tested for neuroprotection. When coincubated with the toxic combination of HIV proteins plus cocaine, this carrier also had no significant effect on cell death (data not shown).
Progesterone provides partial neuroprotection against HIV proteins plus cocaine synergistic neurotoxicity
To study the effect of PROG on HIV protein and cocaine synergistic toxicity we treated cultures with 1 nM, 10 nM, and 100 nM doses of PROG prior to adding the toxic combination of HIV proteins and cocaine (Fig. 3 Right Panel). PROG demonstrated significant neuroprotection (F(1,28) = 11.03, p < 0.0025; n = 5 each point). There was no concentration-dependent effect on percent neuronal death (F(1,28) < 1.0). Further, the neuroprotection was incomplete at these concentrations as the magnitude of cell death was significantly greater than that observed with Locke's Buffer control (ps < 0.01 for all concentrations tested). Fig. 3 Left Panel shows the significant interaction of the HIV proteins with cocaine confirming the presence of synergistic neurotoxicity (F(1,16) = 10.97, p < 0.0044; n = 5 each point;).
Figure 3.
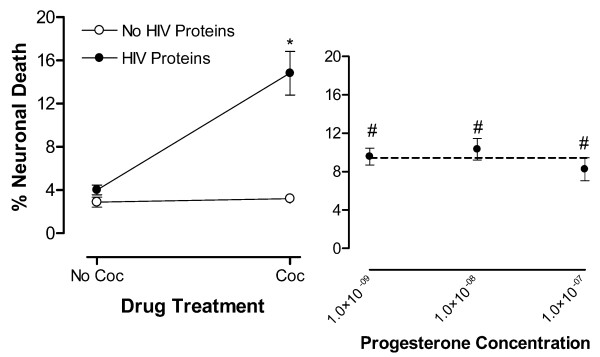
PROG [M] is partially neuroprotective against synergistic neurotoxicity of HIV proteins with cocaine (Coc). Control conditions of incubation with Locke's Buffer vehicle or the HIV proteins or Coc only produced similar neuronal death. (Left Panel) Significant interaction of the HIV proteins with Coc, as illustrated by the lines diverging from parallel, produced toxic synergism (*p < 0.0044). (Right Panel) PROG provided partial neuroprotection against HIV proteins w/ Coc toxic synergism at all concentrations tested (#p < 0.0025) however, the neuroprotection was not concentration dependent.
Testosterone completely protects against HIV proteins plus cocaine synergistic neurotoxicity
Concentrations of 1 pM, 100 pM, 1 nM, 10 nM, 100 nM and 10 μM of T were tested against the synergistically neurotoxic combination of HIV proteins and cocaine (Fig. 4). A significant interaction of the HIV proteins with cocaine (F(1,20) = 8.47, p < 0.0087; n = at least 6 each point) confirmed the presence of synergistic neurotoxicity (Fig. 4 Top Panel). T treatment provided significant neuroprotection (F(1,57) = 12.71, p < 0.0007; n = at least 4 each point). Specifically, there was a concentration-dependent effect of T with a significant linear decrease in percent neuronal death as a function of increasing dose (F(1,57) = 22.56, p < 0.0001) (Fig. 4 Middle Panel). The neuroprotective effect of T was characterized against the negative control of Locke's buffer vehicle and the positive control of the synergistically toxic combination of HIV proteins with cocaine. A ceiling effect of maximal neuroprotection at T concentrations of 10 nM or greater, was noted; magnitude of cell death was not significantly different from incubation with Locke's buffer vehicle. A floor effect was identified at the lowest concentrations tested, 100 pM or less, with the magnitude of cell death not significantly different from that observed by the toxic synergism of the HIV proteins with cocaine.
Figure 4.
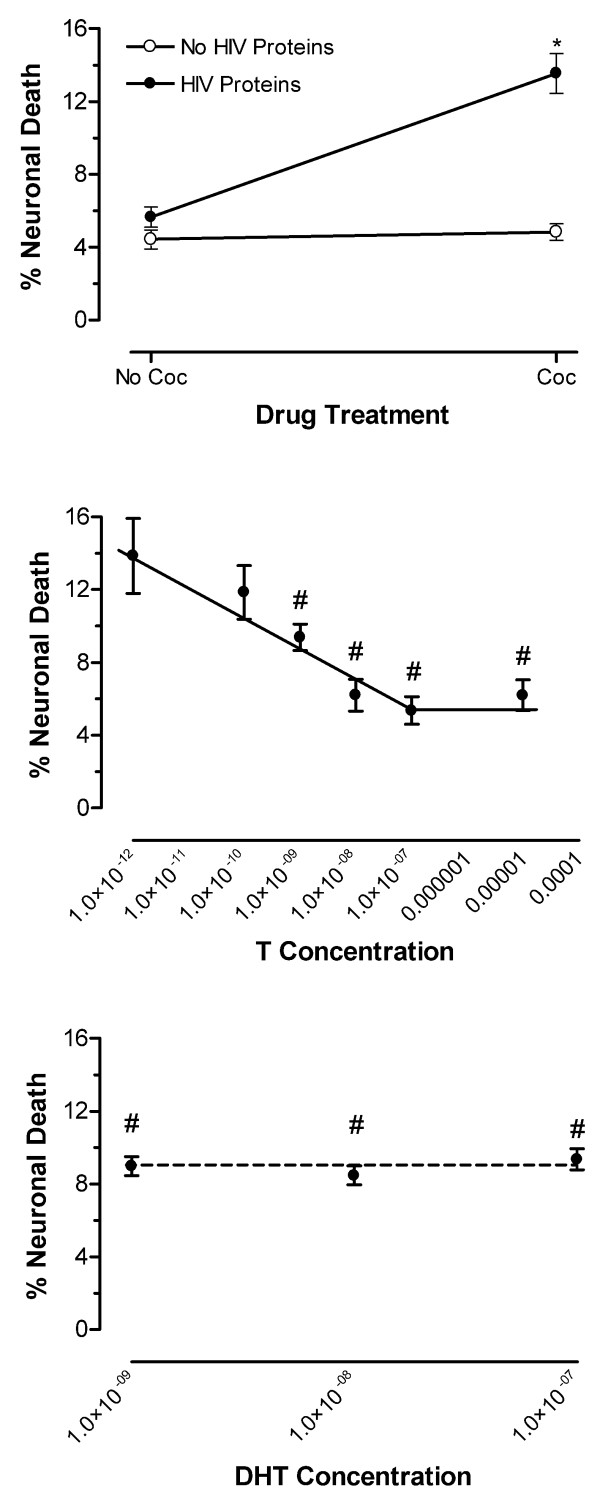
T [M] provides concentration-dependent neuroprotection against synergistic neurotoxicity of HIV proteins with cocaine (Coc); DHT's [M] neuroprotection is limited. Control conditions of incubation with Locke's Buffer vehicle or the HIV proteins or Coc only produced similar neuronal death. (Top Panel) Significant interaction of the HIV proteins with Coc, as illustrated by the lines diverging from parallel, produced toxic synergism (*p < 0.0087) (Middle Panel). T provides significant concentration-dependent neuroprotection against HIV proteins w/ Coc toxic synergism (#p < 0.0001). (Bottom Panel). DHT mediated partial protection against synergistic neurotoxicity of HIV proteins w/ Coc (#p < 0.023); however, the neuroprotection was not concentration dependent.
Dihydrotestosterone provides limited neuroprotection against HIV proteins plus cocaine synergistic neurotoxicity
DHT, the androgen receptor (AR) active metabolite of T, was tested for neuroprotection in the linear range of the T concentration-effect curve, 1 nM, 10 nM and 100 nM, against the toxic synergism of HIV proteins with cocaine (Fig. 4). DHT treatment was confirmed to provide significant neuroprotection (F(1,57) = 10.66, p < 0.002; n = at least 4 each point). However, there was no concentration-dependent effect on percent neuronal death (F(1,57) <1.0). Further, the neuroprotection was incomplete across these concentrations as the magnitude of cell death was significantly greater than that observed with Locke's Buffer control (F(1,57) = 6.72, p < 0.012) (Fig. 4 Bottom Panel). Because the DHT treatments were run in parallel with the T concentration effects, the control values are the same for both sets of treatments. That is, the presence of synergistic neurotoxicity was indicated by the same significant interaction of the HIV proteins with cocaine (F(1,20) = 8.47, p < 0.0087) as it was for the T treatments (Fig. 4 Top Panel). Ethanol at 10 nM (0.1%) served as the solvent for DHT; therefore, it was tested for toxicity and was found not to differ significantly from Locke's buffer incubation (data not shown).
ICI-182,780 blocks testosterone's neuroprotection against HIV proteins plus cocaine synergistic neurotoxicity
The cultures were pretreated with the specific ER antagonist, ICI-182,780 at 100 nM before addition of T (10 nM) then challenged with the HIV proteins plus cocaine synergistic toxicity (Fig. 5). A significant interaction of ICI-182,780 with T was observed (F(1,22) = 33.71, p < 0.0001; n = at least 6 each point). Importantly, the ICI compound was not more toxic to the cultures than was Locke's buffer; nor was it neuroprotective against the toxic challenge. T at 10 nM provided neuroprotection (F(1,65) = 121.26, p < 0.0001), which was not significantly different from incubation with Locke's buffer vehicle alone. This neuroprotective effect of T was fully blocked by pretreatment of the cultures with ICI-182,780 (100 nM) (Fig. 5 Middle Panel). A significant interaction of the HIV proteins with cocaine indicated the presence of synergistic neurotoxicity (F(1,28) = 61.04, p < 0.0001; n = 8 each point) (Fig. 5 Top Panel).
Figure 5.
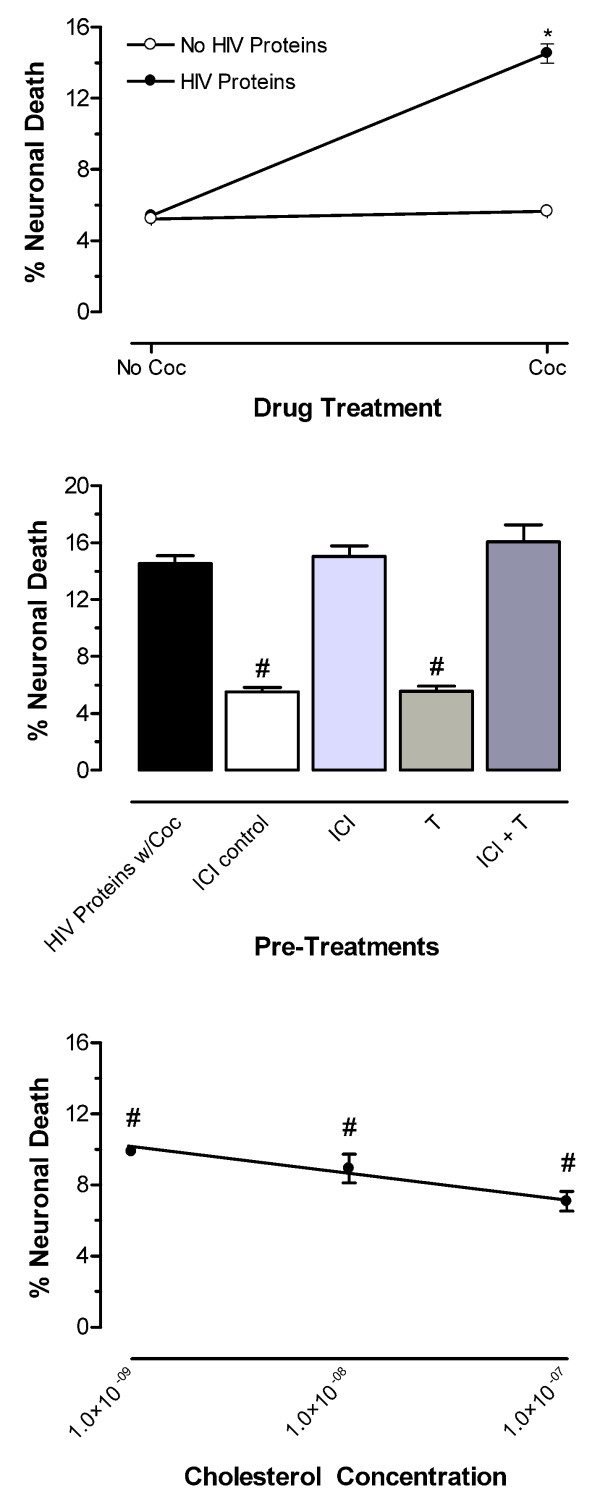
ER mediation of T's protection against synergistic neurotoxicity of HIV proteins with cocaine (Coc); cholesterol's [M] neuroprotection is incomplete. Control conditions of incubation with Locke's Buffer vehicle or the HIV proteins or Coc only produced similar neuronal death. (Top Panel) Significant interaction of the HIV proteins with Coc, as illustrated by the lines diverging from parallel, produces toxic synergism (*p < 0.0001. (Middle Panel) The ER specific antagonist, ICI-182,780 (ICI), completely blocked T-mediated neuroprotection. ICI's toxicity and T's neuroprotection did not differ from vehicle control (#p < 0.0001). (Bottom Panel) Cholesterol produced significant, concentration-dependent protection against synergistic neurotoxicity of HIV proteins w/ Coc (#p < 0.0001); however full protection was demonstrated only at 100 nM.
Cholesterol provides concentration dependent neuroprotection against HIV proteins plus cocaine synergistic neurotoxicity
Cholesterol was added to the cultures at concentrations of 1 nM, 10 nM, and 100 nM prior to treatment with the synergistic toxic combination of HIV proteins plus cocaine (Fig. 5). Significant neuroprotection by cholesterol against this synergistic toxicity was observed (F(1,65) = 55.74, p < 0.0001; n = at least 3 each point) (Fig. 5 Bottom Panel). There was a concentration-dependent effect of cholesterol with a significant linear decrease in percent neuronal death as concentration increased (F(1,65) = 4.4042.73, p < 0.0398). The neuroprotection afforded by the high dose (100 nM) of cholesterol was asymptotic to a ceiling effect; the magnitude of cell death was not significantly different from that observed by Locke's buffer incubation. Because the cholesterol treatments were performed concurrently with the ICI-182,780 treatments, the control values are the same for both sets of treatments. The presence of synergistic neurotoxicity was indicated by the same significant interaction of the HIV proteins with cocaine as it was for the ICI-182,780 treatments (F(1,28) = 61.04, p < 0.0001) (Fig. 5 Top Panel).
Discussion
Neuroprotection is clearly afforded by physiological concentrations of 17β-E2 in this human fetal neuronal model of gp120 and Tat HIV protein neurotoxic synergism with cocaine [30]. In the current studies, we have replicated and extended these findings to examine the ability of other gonadal steroids to provide neuroprotection against this combined insult. We now show robust neuroprotection mediated through the ER is provided by T while PROG, DHT and cholesterol provide only partial or incomplete neuroprotection in the same concentration range.
There are at least two mechanisms for the neuroprotective actions of estradiol: receptor mediated cellular events; and non-receptor mediated anti-oxidant effects. Stereo-specific neuroprotection by estradiol (Fig. 2) suggests a receptor-mediated mechanism rather than global scavenging of reactive oxygen species (ROS) by the phenolic ring. Both of the estradiol stereoisomers, but not PROG, T, DHT, nor cholesterol contain the phenolic moiety of estradiol. In contradiction to our results, neuroprotection by neutralization of ROS would be exhibited by both estradiol isomers but not the other steroid compounds tested. Moreover, the phenolic ring's efficacy in quenching ROS usually requires higher concentrations than needed for receptor-mediated activity [33-35]. Our data suggest that there is a specific ER modulation of the neurotoxic cascade preventing neural death, rather than nonspecific scavenging of ROS by estrogens.
In these studies, the neurons were exposed to estrogens for 15 hours. This time course (15-hour incubation) does not preclude estrogen-mediated genomic activity; however, estrogens are known to act through both genomic and non-genomic pathways [36,37]. Nevertheless, the lack of effect by the transcriptionally inactive stereoisomer of estradiol (17α-E2) provides evidence for a genomic neuroprotective mechanism. Neuroprotection by 17α-E2 has been reported in human neuronal cultures [38,39]. This neuroprotection, which was not seen in our model, was realized in some non-genomic manner such as activation of signal transduction. Research reports also support three other non-genomic actions of steroids that may protect neurons from toxic insults [40]: 1) an indirect effect due to structural and functional perturbation of membrane properties by the intercalation of the cholesterol moiety comprising the steroids; 2) a direct activation of membrane-bound steroid specific protein recognition sites; and 3) modulation of classical neurotransmitter receptor activity. Furthermore, neuroprotection conferred by an individual agent may not be attributable to a single mode of action. All steroid hormones, including 17α-E2, might be expected to modulate neurotransmission by altering the relative steroidal composition of the neuronal membrane. Since 17α-E2 failed to show neuroprotection, these nonspecific membrane effects are ruled out, but activity at a membrane receptor or modulation of neurotransmitter activity is not. We tested six steroids (the two estradiol stereoisomers, PROG, T, DHT and cholesterol) at physiologically relevant nanomolar (nM) concentrations against the synergistic neurotoxic challenge to delineate neuroprotection by steroids in this model of HAD, while considering that each steroid may operate by different as well as pleiotropic means.
Treatments with PROG may be neuroprotective [41-43]. The neuroprotective capability of PROG in the physiological range [44] was also exhibited against our neurotoxic challenge (Fig. 3). Although PROG's neuroprotection was significant, it was incomplete in the nM concentration range tested, as PROG was significantly different from the negative control (i.e., relative to incubation in Locke's buffer). The linear regression analysis confirmed the significance of the neuroprotection. However, PROG as well as some of its metabolites are known to act both as agonists at the intracellular and membrane PROG receptors and as allosteric modulators of some neurotransmission [45].
In contrast, T (10 nM) produced neuroprotection of a similar magnitude to that achieved by 17β-E2 (Fig. 3). A log plot of the data (Fig. 4 Middle Panel) describes a linear function between 100 pM and 10 nM. No protection is seen at 1 pM, whereas concentrations above 100 nM show maximal protection, thus establishing the EC50 for the neuroprotective effect of T between 1 pM and 100 nM. The pharmacokinetics of a concentration-dependent effect are consistent with a receptor-mediated mechanism of neuroprotection. Because T can be aromatized to estradiol by P450 aromatase expressed by astrocytes [46] and by neurons [47] in rat brain, it is not entirely surprising to see a robust neuroprotective effect of T, as was seen with 17β-E2. Such neuroprotection could be produced through activation of the ER. Therefore, the question remained whether an androgenic component to the presumptive steroid receptor-mediated neuroprotection is operating.
T is not specific in identifying the receptor mediating its effects because it is the prohormone for both DHT, the principal ligand at the AR, and 17β-E2, the principal ligand at the ER. DHT is a useful androgen for evaluating the relative contributions of AR and ER-mediated neuroprotective effects because it does not serve as an agonist at the ER nor can it be converted to estradiol. Our toxic challenge test results with DHT suggest that the AR may mediate neuroprotection, but do not exclude the possibility of effects by other means (Fig. 4 Bottom Panel). To clarify whether T was acting at the AR or the ER, we used the specific ER antagonist, ICI-182,780, while treating the cultures with T and the toxic combination of HIV proteins and cocaine. The ER antagonist completely blocked T's neuroprotection thus demonstrating that the effect is mediated through the ER (Fig. 5 Middle Panel) at physiological concentrations [48,49]. If an AR-mediated component were active in T's neuroprotection it would have been revealed when T was tested while the ER was pharmacologically blocked by ICI-182,780. Therefore, we conclude that T's neuroprotection is mediated through the ER with no portion attributable to AR activation or to non-genomic pathways.
As a control for general steroidal structural and receptor activation we tested cholesterol, which is known to modulate membrane fluidity but is neurally inactive. We saw that cholesterol's concentration-dependent neuroprotection against synergistic toxicity of the HIV proteins with cocaine is more efficacious than that of PROG and DHT (Fig. 5 Bottom Panel) in that 100 nM cholesterol was fully neuroprotective whereas the same dose of PROG or DHT was not. However, cholesterol is less potent than the gonadal steroids, 17β-E2 and T in that 10 nM cholesterol was not fully neuroprotective, whereas the same dose of the steroids was. The mechanism of this protection is interesting because the cholesterol moiety is common to all steroids, but 17α-E2 was not neuroprotective. Furthermore, the ICI-182,780-sensitive neuroprotection provided by T does not accommodate a nonspecific cholesterol-mediated component. In this model, cholesterol may not promote neuronal survival by any intrinsic activity of its own, but may be serving as a synthesis precursor for an explicitly protective steroid, e.g., 17β-E2. Others have also shown increased bioavailability of cholesterol to interact with effects mediated by steroids in the brain [50,51]. Evidence for all the necessary enzymes of synthesis in human brain has been reported [52,53] except for one, which has been identified in rat brain [54]. Because we use primary cell cultures derived from human fetal brain, it is likely that the metabolic machinery is present, operational and facilitating cholesterol's neuroprotection through an estrogenic end product. In support of our findings, neuroprotection by locally synthesized estrogen has recently been demonstrated [55], which when considered with the current findings, suggests possibilities for steroid prodrugs serving as neuroprotective therapeutics. However, others have demonstrated that cholesterol significantly reduces the neurotoxicity of gp120 by modulating membrane fluidity. In human neuroblastoma cells enriching cholesterol significantly reduced gp120 binding and consequently, the biochemical events triggered by the viral protein leading to necrotic death, while depleting cholesterol had the opposite effect [56]. Therefore the physiochemical properties of the membrane in mediating neurotoxicity/protection may be as significant as some pharmacological agents.
Synergistic neurotoxicity of the HIV proteins gp120 and Tat with cocaine was demonstrated in each experiment. Although neither the HIV protein combination nor cocaine alone was more neurotoxic than the Locke's buffer media, the simultaneous incubation of cells with both the HIV proteins and cocaine produced significant neuronal death. The mechanism by which cocaine facilitates the HIV protein toxicity is not well characterized. Recombinant variants of Tat protein (Tat 1–72 and Tat 1–86) are known to induce neuronal degeneration by mechanisms that involve triggering of apoptotic cascades [57,58]. Cocaine was recently shown to enhance gp120 induced neuronal apoptosis in vivo via a mechanism implicating glutamate excitotoxicity and iNOS expression with abnormal NO production in the neocortex of rat [59]. In vitro work in our lab with rat hippocampal cells supports a similar mechanism for cocaine's enhancement of Tat mediated toxicity by augmentation of ROS production and mitochondrial depolarization [60]. Likewise cocaine at very high concentrations in vitro may induce neuronal apoptosis [61]. Accordingly, estrogen neuroprotection could be via stimulation of the expression of anti-apoptotic proteins [62,63], or alternatively suppression of pro-apoptotic gene transcripts [21,64], although both of these interesting possibilities await further experimentation.
Conclusion
In conclusion, we demonstrated that T provides neuroprotection through an ICI-182,708-sensitive mechanism against the synergistic toxicity of gp120 and Tat HIV proteins with cocaine. Additionally, we show that PROG, DHT and cholesterol provide some less efficacious neuroprotection than do the steroids, 17β-E2 and T. It is conceivable that gonadal steroids may provide biologically based gender-specific prophylaxis and/or amelioration either in the time of onset or symptomology of HAD and drugs of abuse. These results have implications for the innovation of therapeutic strategies, including steroid based neuroprotective prodrugs. Moreover, given that the neuropathology of HAD may have commonalities with dementias of other etiologies, the development of successful pharmacology for HAD would surely have applications for other equally devastating and costly neurodegenerative conditions.
Methods
Culture of human brain cells
Neuronal cultures were prepared as described previously [65-67]. Human fetal brain specimens of gestational age 12–14 weeks were obtained with the consent of women opting for elective termination of pregnancy. Protocols were carried out with strict adherence to the guidelines of the National Institutes of Health (NIH) and with approval from the University of Kentucky Institutional Review Board. Briefly, after mechanical dissociation, the cells were suspended in Opti-MEM with 5% heat-inactivated fetal bovine serum, 0.2% N2 supplement and 1% antibiotic solution (penicillin G sodium 104 units/ml, streptomycin sulfate10 μg/ml and amphotericin-B 25 μg/ml) (GIBCO). The cells were maintained in culture flasks at 37°C in a humidified atmosphere of 5% CO2 and 95% air for at least a month. At least 3 days before the experiment was performed the cells were plated in flat bottom 96 well plates coated with poly-d-lysine. Sample cultures were stained for the neuron-specific enolase, microtubule-associated protein-2, synaptophysin, and glial fibrillary astrocytic protein (GFAP). The cultures were thus characterized as more than 80% homogenous for neurons. The remaining cells were predominantly astrocytes (GFAP positive) with less than 1% microglia/macrophages, as determined by RCA-1 lectin and CD68 staining [67-69]-. Sample cultures were further characterized for the presence of ER-α and ER-β and dopaminergic neurons. As described earlier, ERs were visualized in 5–10% of cultured astrocytes and neurons in the cytoplasm and nuclei of both by immunostaining. Antigenicity for dopamine and dopamine transporter were co-localized in nearly 60% of neurons. Dopamine receptor D1A was seen in 50% of cells while receptor D2 was seen in 40% of cells. ER co-localized with cells staining for dopamine as well as D1A and D2 receptor containing neurons [30].
Recombinant Tat and gp120 proteins
Recombinant Tat was prepared as described previously [67,70] with minor modifications. The tat gene encoding the first 72 amino acids was amplified from HIVBRU obtained from Dr. Richard Gaynor through the AIDS repository at the NIH and inserted into an E coli vector PinPoint Xa-2 (Promega). This construct allowed the expression the Tat1-72 protein as a fusion protein naturally biotinylated at the N-terminus. The biotinylated Tat protein was purified on a column of soft release avidin resin, cleaved from the fusion protein using factor Xa, eluted from the column, then desalted with a PD10 column. The Tat protein was further purified of endotoxin by passage through a polymycin B, cyanogen bromide, immobilized on cross-linked 4% beaded agarose column (Sigma). All purifications steps contained dithiothreitol to prevent oxidation of the proteins. Tat Protein was more than 95% pure as determined by SDS-PAGE followed by silver staining. Analysis by HPLC using a C4 column showed a single symmetrical peak. Western blot analysis showed that this preparation contained both monomeric and dimeric forms of Tat1-72. The functional activity of Tat1-72 was confirmed using a transactivation assay in HL3T1 cells containing an HIV-1 LTR-CAT construct. Chiron Corporation made a gift of gp120 from HIVSF2 which was described previously [30]. Briefly, recombinant gp120 was made in a Chinese Hamster Ovary cell line. Purification yielded 95% pure gp120 as determined by Western blot analysis. The Tat and gp120 preparations contained less than 1 pg/ml endotoxin as determined using a Pyrochrome Chromogenic test kit (Associates of Cape Cod, Inc., Falmouth, MA). The Tat proteins were stored in a lyophilized form and gp120 as a stock solution in water at -80°C in endotoxin-free siliconized microfuge tubes until taken for experimentation. Tat and gp120 are highly susceptible to degradation and loss of biological activity with each freeze-thaw cycle. Therefore, single aliquots were used for each experiment. Any remaining solution was discarded. Prior work has shown that no significant toxicity is seen with Tat less than 80 nM, gp120 less than 40 pM, or Tat (40 nM) plus gp120 (30 pM) [30]. For these studies, we used subtoxic dosage levels of Tat1-72 at 40 nM and gp120 at 32.5 pM.
Drug levels
Cocaine hydrochloride obtained from the NIDA Drug Supply Program was solvated in Locke's buffer (pH 7.2) immediately prior to use. The dosage of cocaine (500 ng/ml) in culture translates to 1.6 μM, which is several orders of magnitude less than studies demonstrating a direct cytotoxic effect in culture [71-73]-. Fetal brain levels of cocaine of 1750 +/- 250 ng/ml were associated with fetal plasma levels of 500 ng/ml following repeated daily dosing in rat [74]. The plasma levels of cocaine from fetuses of mothers exposed to cocaine using the rat IV model [75] was 500 ng/ml as this represents 1/5 the peak brain levels in the fetus. This is also a concentration within the physiological levels experienced by human IV cocaine drug users [76]. Thus 500 ng/ml (1.6 μM) represents a physiologically relevant dose of cocaine to the neurons. Cocaine levels as high as 16 μM are not neurotoxic in human fetal neuronal cultures [30]. In these studies the cocaine solution was diluted such that addition of 1:10 (v/v) to the culture dishes resulted in a final concentration of 1.6 μM. This dose of cocaine was added concurrently with the Tat and gp120 proteins to the cultures.
Neurotoxicity assay
At the time of the experimental treatment, the culture media was replaced with Locke's buffer containing (in mM) 154 NaCl, 5.6 KCl, 2.3 CaCl2, 1 MgCl2, 3.6 NaHCO3, 5 glucose, 5 N-2-hydroxyethylpiperazine-N'2-ethanesulfonic acid (HEPES) and 1% antibiotic solution (penicillin G sodium 104 units/ml, streptomycin sulfate10 μg/ml and amphotericin-B 25 μg/ml) (pH 7.2). Cells were incubated for 15 hours in Locke's buffer or with subtoxic concentrations of the HIV proteins, Tat1-72 (40 nM) plus gp120 (32.5 pM) [30], or cocaine (1.6 μM), or the HIV proteins plus cocaine to demonstrate the synergistic effect on cell death of HIV proteins with cocaine. To replicate the stereoisomer specific neuroprotective properties of estrogen, the cultures were treated with either 17β-E2 (10 nM), or 17α-E2 (10 nM) immediately preceding addition of the HIV proteins plus cocaine. To investigate the neuroprotective properties of other gonadal steroids, the cells were treated with various concentrations of PROG or T, immediately followed by exposure to the HIV proteins plus cocaine. To determine the mechanism of T's neuroprotection, the cultures were treated with various concentrations of DHT (5α-Androstan-17β-ol-3-one; 17β-Hydroxy-5α-androstan-3-one), or ICI-182,780 (100 nM) plus T (10 nM) before the HIV proteins plus cocaine were added. Various concentrations of cholesterol were used as treatment prior to incubation with the HIV proteins plus cocaine to further characterize the nature of gonadal steroid neuroprotection. ICI-182,780, DMSO (10 nM), β-cyclodextrin (100 nM) and ethanol (10 nM) were also tested for neurotoxic/protective effects. ICI-182,780 and 17α-E2 were solvated in DMSO. 17β-E2, T, PROG and cholesterol were β-cyclodextrin encapsulated to provide water solubility. Ethanol was used to solvate DHT. Tat1-72 was produced as indicated above, whereas gp120 was a gift from Chiron Corporation. The ICI-182,780 compound was obtained from Tocris Cookson, Inc. (Ellisville, MO). All other chemical agents were supplied by Sigma Chemicals (St. Louis, MO). At the end of the incubation period neuronal death was assessed by trypan blue (Sigma) exclusion assay as described previously [42,65,66]. In brief, each experiment was conducted in triplicate and at least three independent squads were treated with each pharmacological agent. Five randomly selected fields from each well were photographed using an inverted microscope (Nikon Diphot, 40X) and coded by a technician blinded to the well treatments. Viable and dead neurons in each photomicrograph were counted before decoding for statistical analysis. In representative fields the neurotoxicity assay was verified by false color visualization using the MCID digital camera-based system interfaced with a computer (Imaging Research, Ontario, Canada).
The neuronal and glial phenotypes seen in these cultures have been well characterized immunohistochemically and morphologically in our hands [65,66] as well as by others [77-79]-. Furthermore, the glia readily attach to the surface of the culture dish, whereas neurons do not [80]. In our hands, astrocytic attachment to the culture dish is requisite for the survival of the neurons, which in turn, attach to the substrate of glia. In this way, the glia are seen in a specific plane of microscopy that differs from the microscopic plane in which the neurons are seen. Therefore, we are confident of our ability to visually recognize and enumerate neurons as distinct from glia in these cultures.
Analysis
Data are expressed as percent neuronal cell death/total viable neurons (means ± SEM). A 2 (presence or absence of the viral proteins gp120 and Tat) × 2 (presence or absence of cocaine) analysis of variance (ANOVA) design was employed to ascertain the potential for synergistic viral protein/cocaine-induced neurotoxicity. A significant interaction of the HIV proteins with Coc, as graphically illustrated by the lines diverging from parallel, provided evidence of toxic synergism. One-way ANOVAs were used to assess the neuroprotective potential of the various gonadal steroid pre-treatments. Regression analysis was used to determine concentration-effect relationships [81]. Planned Tukey-Kramer comparisons were used to determine specific treatment effects. An α level of p < 0.05 was considered significant for all statistical tests employed. Computer assisted analyses utilized BMDP Statistical Software, Release 7, Los Angeles, CA (1993) and SPSS Statistical Software release 11.5.0, SPSS, Inc., Chicago, IL (2004).
Authors' contributions
SLK participated in the conception and design of the study, carried out the cell culture studies, quantified and analyzed the data, and drafted the manuscript. CFA participated in the design and coordination of the study. AN participated in the conception and design of the study and manuscript preparation. JT-C characterized the cell cultures and participated in manuscript preparation. CLL helped interpret the data and contributed to the preparation of the manuscript. CFM performed statistical and graphical analyses and helped draft the manuscript. RMB participated in the conception and design of the study and manuscript preparation and was the primary mentor of SLK. All authors read and approved the final manuscript.
Acknowledgments
Acknowledgements
This research was supported by NIH research grants to Rosemarie M. Booze (DA014401, DA13137), Avindra Nath (NS039253, P20RR015592, R01NS039253), and Charles F. Mactutus (HD043680, DA09160). Sherie L. Kendall was supported by training grants from the NHLB (K30 HL04163) and NIA (T32 AG00242). We also thank Philip D. Ray for technical assistance in producing the Tat protein used in these studies. Dr. Michael Aksenov and Dr. Marina Aksenova provided technical assistance with imaging of the cultures and very helpful discussions for which we are grateful.
Contributor Information
Sherie L Kendall, Email: skend2@uky.edu.
Caroline F Anderson, Email: cander24@jhmi.edu.
Avindra Nath, Email: anath@jhmi.edu.
Jadwiga Turchan-Cholewo, Email: turchan@uky.edu.
Cantey L Land, Email: ratfantastic@yahoo.com.
Charles F Mactutus, Email: mactutus@gwm.sc.edu.
Rosemarie M Booze, Email: Booze@gwm.sc.edu.
References
- Buchanan RJ, Wang S, Huang C. Analyses of nursing home residents with HIV and dementia using the minimum data set. JAIDS. 2001;26:246–255. doi: 10.1097/00042560-200103010-00006. [DOI] [PubMed] [Google Scholar]
- Dougherty RH, Skolasky RLJ, McArthur JC. Progression of HIV-associated dementia treated with HAART. AIDS Read. 2002;12:69–74. [PubMed] [Google Scholar]
- Chiesi A, Vella S, Dally LG, Pedersen C, Danner S, Johnson AM, Schwander S, Goebel FD, Glauser M, Antunes F, Lundgren JD, X Epidemiology of AIDS dementia complex in Europe by the AIDS in Europe study group. J Acquir Immune Defic Syndr Hum Retrovirol. 1996;11:39–44. doi: 10.1097/00042560-199601010-00005. [DOI] [PubMed] [Google Scholar]
- Davies J, Everall IP, Weich S, McLaughlin J, Scaravilli F, Lantos PL. HIV-associated brain pathology in the United Kingdom: an epidemiological study. AIDS. 1997;11:1145–1150. doi: 10.1097/00002030-199709000-00010. [DOI] [PubMed] [Google Scholar]
- Bell JE, Brettle RP, Chiswick A, Simmonds P. HIV encephalitis, proviral load and dementia in drug users and homosexuals with AIDS. Effect of neocortical involvement. Brain. 1998;121:2043–2052. doi: 10.1093/brain/121.11.2043. [DOI] [PubMed] [Google Scholar]
- Berger JR, Nath A, Greenberg RN, Andersen AH, Greene RA, Bognar A, Avison MJ. Cerebrovascular changes in the basal ganglia with HIV dementia. Neurology. 2000;54:921–926. doi: 10.1212/wnl.54.4.921. [DOI] [PubMed] [Google Scholar]
- Holman BL, Garada B, Johnson KA, Mendleson J, Hallgring E, Teoh SK, Worth J, Navia B. A comparison of brain perfusion SPECT in cocaine abuse and AIDS dementia complex. J Nucl Med. 1992;33:1312–1315. [PubMed] [Google Scholar]
- Nath A, Maragos WF, Avison MJ, Schmitt FA, Berger JR. Acceleration of HIV dementia with methamphetamine and cocaine. J Neurovirol. 2001;7:66–71. doi: 10.1080/135502801300069737. [DOI] [PubMed] [Google Scholar]
- Mirsattari SM, Power C, Nath A. Parkinsonism with HIV infection. Movement Disorders. 1998;13:684–689. doi: 10.1002/mds.870130413. [DOI] [PubMed] [Google Scholar]
- Webber MR, Shoenbaum EE, Gourevitch MN, Buono D, Klein RS. A prospective study of HIV disease progression in female and male drug users. AIDS. 1999;13:257–262. doi: 10.1097/00002030-199902040-00014. [DOI] [PubMed] [Google Scholar]
- Bouwman FH, Skolasky RL, Hes D, Selnes OA, Glass JD, Nance-Sproson TE, Royal W, Dal Pan GJ, McArthur JC. Variable progression of HIV-associated dementia. Neurology. 1998;50:1814–1820. doi: 10.1212/wnl.50.6.1814. [DOI] [PubMed] [Google Scholar]
- Margolin A, Avants SK, Warburton LA, Hawkins KA. Factors affecting cognitive functioning in a sample of human immunodeficiency virus-positive injection drug users. AIDS Patient Care and STDs. 2002;16:255–267. doi: 10.1089/10872910260066697. [DOI] [PubMed] [Google Scholar]
- Holmberg SD. The estimated prevalence and incidence of HIV in 96 large US metropolitan areas. Am J Public Health. 1996;86:642–654. doi: 10.2105/ajph.86.5.642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farzadegan JH, Hoover DR, Astemborski J, Lyles CM, Margolick JB, Markham RB, Quinn TC, Vlahov D. Sex differences in HIV-1 viral load and progression to AIDS. Lancet. 1998;352:1510–1514. doi: 10.1016/S0140-6736(98)02372-1. [DOI] [PubMed] [Google Scholar]
- Sterling TA, Vlahov D, Astermborski J, Hoover DR, Margolick JB, Quinn TC. Initial plasma HIV-1 RNA levels and progression to AIDS in women and men. N Engl J Med. 2001;344:720–725. doi: 10.1056/NEJM200103083441003. [DOI] [PubMed] [Google Scholar]
- Squires KE. Treating HIV infection and AIDS in women. AIDS Read. 2003;13:228–240. [PubMed] [Google Scholar]
- Jover T, Tanaka H, Calderone A, Oguro K, Bennett MVL, Etgen AM, Zukin RS. Estrogen protects against global ischemia-induced neuronal death and prevents activation of apoptotic signaling cascades in the hippocampal CA1. J Neurosci. 2002;22:2115–2124. doi: 10.1523/JNEUROSCI.22-06-02115.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang SH, Liu R, Wu SS, Simpkins JW. The use of estrogens and related compounds in the treatment of damage from cerebral ischemia. Ann N Y Acad Sci. 2003;1007:101–107. doi: 10.1196/annals.1286.010. [DOI] [PubMed] [Google Scholar]
- Merchenthaler I, Dellovade TL, Shugrue PJ. Neuroprotection by estrogen in animal models of global and focal ischemia. Ann N Y Acad Sci. 2003;1007:89–100. doi: 10.1196/annals.1286.009. [DOI] [PubMed] [Google Scholar]
- Rau SW, Dubal DB, Böttner M, Gerhold LM, Wise PM. Estradiol attenuates programmed cell death after stroke-like injury. J Neurosci. 2003;23:11420–11426. doi: 10.1523/JNEUROSCI.23-36-11420.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawada H, Ibi M, Kihara T, Urushitani M, Honda K, Nakanishi M, Akaike A, Shimohama S. Mechanisms of antiapoptotic effects of estrogens in nigral dopaminergic neurons. FASEB J. 2000;14:1202–1214. doi: 10.1096/fasebj.14.9.1202. [DOI] [PubMed] [Google Scholar]
- Gajjar TM, Anderson LI, Dluzen DE. Acute effects of estrogen upon methamphetamine induced neurotoxicity of the nigrostriatal dopaminergic system. J Neural Transm. 2003;110:1215–1224. doi: 10.1007/s00702-003-0045-3. [DOI] [PubMed] [Google Scholar]
- Tang MT, Jacobs D, Stern Y, Marder K, Schofield P, Gurland B, Andrews H, Mayeux R. Effect of oestrogen during menopause on risk and age at onset of Alzheimer's disease. Lancet. 1996;348:429–432. doi: 10.1016/S0140-6736(96)03356-9. [DOI] [PubMed] [Google Scholar]
- Kawas C, Resnick S, Morrison A, Brookmeyer R, Corrada M, Zonderman A, Bacal C, Lingle DD, Metter E. A prospective study of estrogen replacement therapy and the risk of developing Alzheimer's disease: the Baltimore Longitudinal Study of Aging. Neurology. 1997;48:1517–1521. doi: 10.1212/wnl.48.6.1517. [DOI] [PubMed] [Google Scholar]
- Manly JJ, Merchant CA, Jacobs DM, Small SA, Bell K, Ferin M, Mayeux R. Endogenous estrogen levels and Alzheimer's disease among postmenopausal women. Neurology. 2000;54:833–837. doi: 10.1212/wnl.54.4.833. [DOI] [PubMed] [Google Scholar]
- Asthana S, Baker LD, Craft S, Stanczyk FZ, Veith RC, Raskind MA, Plymate SR. High-dose estradiol improves cognition for women with AD. Neurology. 2001;57:605–612. doi: 10.1212/wnl.57.4.605. [DOI] [PubMed] [Google Scholar]
- Saunders-Pullman R, Gordon-Elliott J, Parides M, Fahn S, Saunders HR, Bressman S. The effect of estrogen replacement on early Parkinson's disease. Neurology. 1999;52:1417–1421. doi: 10.1212/wnl.52.7.1417. [DOI] [PubMed] [Google Scholar]
- Tsang KL, Ho SL, Lo SK. Estrogen improves motor disability in parkinsonian postmenopausal women with motor fluctuations. Neurology. 2000;54:2292–2298. doi: 10.1212/wnl.54.12.2292. [DOI] [PubMed] [Google Scholar]
- Wooten GF, Curie LJ, Bovbjerg VE, Lee JK, Patrie J. Are men at greater risk for Parkinson's disease than women? J Neurosurg Psychiatry. 2004;75:637–639. doi: 10.1136/jnnp.2003.020982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turchan J, Anderson C, Hauser KF, Sun Q, Zhang J, Liu Y, Wise PM, Kruman I, Maragos W, Mattson MP, Booze R, Nath A. Estrogen protects against the synergistic toxicity by HIV proteins, methamphetamine and cocaine. BMC Neurosci. 2001;2:3. doi: 10.1186/1471-2202-2-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zemlyak I, Brooke SM, Sapolsky RM. Protection against gp120-induced neurotoxicity by an array of estrogenic steroids. Brain Res. 2002;958:272–276. doi: 10.1016/S0006-8993(02)03558-8. [DOI] [PubMed] [Google Scholar]
- Nath A, Haughey NJ, Jones M, Anderson C, Bell JE, Geiger JD. Synergistic neurotoxicity by human immunodeficiency virus proteins Tat and gp120: protection by memantine. Ann Neurol. 2000;47:186–194. doi: 10.1002/1531-8249(200002)47:2<186::AID-ANA8>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Behl C, Skutella T, Lezoualc'h F, Post A, Widman M, Newton C, Newton C, Holsboer F. Neuroprotection against oxidative stress by estrogens: structure-activity relationship. Mol Pharmacol. 1997;51:535–541. [PubMed] [Google Scholar]
- Culmsee C, Vedder H, Ravati A, Junker V, Otto D, Ahlemeyer B, Krieg JC, Krieglstein J. Neuroprotection by estrogens in a mouse model of focal cerebral ischemia and in cultured neurons: Evidence for a receptor-independent antioxidative mechanism. J Cereb Blood Flow Metab. 1999;19:1263–1269. doi: 10.1097/00004647-199911000-00011. [DOI] [PubMed] [Google Scholar]
- Moosmann B, Behl C. The antioxidant neuroprotective effects of estrogens and phenolic compounds are independent from their estrogenic properties. Proc Natl Acad Sci U S A. 1999;96:8867–8872. doi: 10.1073/pnas.96.16.8867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss RL, Gu J, Wong M. Recent Progress in Hormone Research. The Endocrine Society; 1997. Estrogen: Nontranscriptional signaling pathway; pp. 33–69. [PubMed] [Google Scholar]
- Woolley CS. Effects of estrogen in the CNS. Curr Opin Neurbiol. 1999;9:349–354. doi: 10.1016/S0959-4388(99)80051-8. [DOI] [PubMed] [Google Scholar]
- Green PS, Bishop J, Simpkins JW. 17a-Estradiol exerts neuroprotective effects on SK-N-SH cells. J Neurosci. 1997;17:511–515. doi: 10.1523/JNEUROSCI.17-02-00511.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond J, Quynh L, Goodyer C, Gelfand M, Trifiro M, LeBlanc A. Testosterone-mediated neuroprotection through the androgen receptor in human primary neurons. J Neurochem. 2001;77:1319–1326. doi: 10.1046/j.1471-4159.2001.00345.x. [DOI] [PubMed] [Google Scholar]
- Golden GA, Mason PE, Rubin RT, Mason RP. Biophysical membrane interactions of steroid hormones: A potential complementary mechanism of steroid action. Clinical Neuropharmacology. 1998;21:181–189. [PubMed] [Google Scholar]
- Jiang N, Chopp M, Stein D, Feit H. Progesterone is neuroprotective after transient middle cerebral artery occlusion in male rats. Brain Res. 1996;735:101–107. doi: 10.1016/0006-8993(96)00605-1. [DOI] [PubMed] [Google Scholar]
- Vongher JM, Frye CA. Progesterone in conjunction with estradiol has neuroprotective effects in an animal model of neurodegeneration. Pharmacol Biochem Behav. 1999;64:777–785. doi: 10.1016/S0091-3057(99)00140-9. [DOI] [PubMed] [Google Scholar]
- Ciriza I, Axcoitia I, Garcia-Segura LM. Reduced progesterone metabolites protect rat hippocampal neurones from kainic acid excitotoxicity in vivo. J Neuroendocrin. 2004;16:58–63. doi: 10.1111/j.1365-2826.2004.01121.x. [DOI] [PubMed] [Google Scholar]
- Chernecky CC, Berger BJ. In: Laboratory Tests and Diagnostic Procedures. 3. Chernecky CC and Berger BJ, editor. Philadelphia, WB Saunders; 2001. [Google Scholar]
- Rupprecht R, Friess E, Holsboer F. The Neuropsychopharmacological Potential of Neurosteroids. In: Baulieu EE, Robel P and Schumacher M, editor. Neurosteroids: A new regulatory function in the nervous system. 1. Vol. 20. Totowa, NJ, Humana Press; 1999. pp. 349–364. (Contemporary Endocrinology). Conn PM. [Google Scholar]
- Zwain IH, Yen SSC, Cheng CY. Astrocytes cultured in vitro produce estradiol-17b and express aromatase cytochrome P-450 (P-450 AROM) mRNA. Biochim Biophys Acta. 1997;1334:338–348. doi: 10.1016/s0304-4165(96)00115-8. [DOI] [PubMed] [Google Scholar]
- Hojo Y, Hattori T, Enami T, Furukawa A, Suzuki K, Ishii H, Mukai H, Morrison JH, Janssen WGM, Kominami S, Harada N, Kimoto T, Kawato S. Adult male rat hippocampus synthesizes estradiol from pregnenolone by cytochromes P45017a and P450 aromatase localized in neurons. Proc Natl Acad Sci U S A. 2003;PNAS Early Edition:1–6. doi: 10.1073/pnas.2630225100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daly RC, Su TP, Schmidt PJ, Pagliaro M, Pickar D, Ruiz-Lozano P. Neuroendocrine and behavioral effects of high-dose anabolic steroid administration in male normal volunteers. Psychoneuroendocrinology. 2003;28:317–331. doi: 10.1016/S0306-4530(02)00025-2. [DOI] [PubMed] [Google Scholar]
- Labrie F, Bélanger A, Cusan L, Candas B. Physiological changes in dehydroepiandrosterone are not reflected by serum levels of active androgens and estrogens but of their metabolites: Intracrinology. J Clin Endocrinol Metab. 1997;82:2403–2409. doi: 10.1210/jc.82.8.2403. [DOI] [PubMed] [Google Scholar]
- Papadopoulos V, Guarneri P, Krueger KE, Guidotti A. Pregnenolone biosynthesis in C6-2B glioma cell mitochondria: Regulation by a mitochondrial diazepam binding inhibitor receptor. Proc Natl Acad Sci U S A. 1992;89:5113–5117. doi: 10.1073/pnas.89.11.5113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korneyev A, Pan BS, Plol A, Romeo E, Guidotti A, Costa E. Stimulation of brain pregnenolone synthesis by mitochondrial diazepam binding inhibitor receptor ligands in vivo. J Neurochem. 1993;61:1515–1524. doi: 10.1111/j.1471-4159.1993.tb13647.x. [DOI] [PubMed] [Google Scholar]
- Stoffel-Wagner B. Neurosteroid metabolism in the human brain. Eur J Endocrinology. 2001;145:669–679. doi: 10.1530/eje.0.1450669. [DOI] [PubMed] [Google Scholar]
- Weill-Engerer S, David JP, Sazdovitch V, Liere P, Schumacher M, Delacourte A, Baulieu EE, Akwa Y. In vitro metabolism of dehydroepiandrosterone (DHEA) to 7a-hydroxy-DHEA and D5-androstene-3b,17b-diol in specific regions of the aging brain from Alzheimer's and non-demented patients. Brain Res. 2003;969:117–125. doi: 10.1016/S0006-8993(03)02288-1. [DOI] [PubMed] [Google Scholar]
- Zwain IH, Yen SSC. Dehydroepiandrosterone: Biosynthesis and metabolism in the brain. Endocrinology. 1999;140:880–887. doi: 10.1210/en.140.2.880. [DOI] [PubMed] [Google Scholar]
- McCullough LD, Blizzard K, Simpson ER, Ohran K, Hurn PD. Aromatase cytochrome P450 and extragonadal estrogen play a role in ischemic neuroprotection. J Neurosci. 2003;23:8701–8705. doi: 10.1523/JNEUROSCI.23-25-08701.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maccarrone M, Navarra M, Catani V, Corasaniti MT, Bagetta G, Finazazi-Agrò A. Cholesterol-dependent modulation of the toxicity of HIV-1 coat protein gp120 in human neuroblastoma cells. J Neurochem. 2002;82:1444–1452. doi: 10.1046/j.1471-4159.2002.01072.x. [DOI] [PubMed] [Google Scholar]
- Bonavia R, Bajetto A, Barbero S, Albini A, Noonan DM, Schettini G. HIV-1 Tat causes apoptotic death and calcium homeostasis alterations in rat neurons. Biochem Res Commun. 2001;288:301–308. doi: 10.1006/bbrc.2001.5743. [DOI] [PubMed] [Google Scholar]
- Song L, Nath A, Geiger JD, Moore A, Hochman S. Human immunodeficiency virus type 1 Tat protein directly activates neuronal N-methyl-D-aspartate receptors at an allosteric zinc-sensitive site. J Neurovirol. 2003;9:399–403. doi: 10.1080/13550280390201704. [DOI] [PubMed] [Google Scholar]
- Bagetta G, Piccirilli S, Del Duca C, Morrone LA, Rombolà L, Nappi G, De Alba J, Knowles RG, Corasaniti MT. Inducible nitric oxide synthase is involved in the mechanisms of cocaine enhanced neuronal apoptosis induced by HIV-1 gp120 in the neocortex of rat. Neurosci Lett. 2004;356:183–186. doi: 10.1016/j.neulet.2003.11.065. [DOI] [PubMed] [Google Scholar]
- Aksenova M, Aksenov MY, Nath A, Mactutus C, Booze R. Cocaine enhances oxidative stress-dependent toxicity of HIV-1 Tat in rat hippocampal cell cultures. Society for Neuroscience. 2004;573.8 [Google Scholar]
- Nassogne MC, Louahed J, Evrard P, Courtoy PJ. Cocaine induced apoptosis in cortical neurons in fetal mice. J Neurochem. 1997;68:2442–2450. doi: 10.1046/j.1471-4159.1997.68062442.x. [DOI] [PubMed] [Google Scholar]
- Singer C, Rogers K, Dorsa D. Modulation of Bcl-2 expression: A potential component of estrogen protection in NT2 neurons. Neuroreport. 1998;9:2565–2568. doi: 10.1097/00001756-199808030-00025. [DOI] [PubMed] [Google Scholar]
- Pike DJ. Estrogen modulates neuronal Bcl-xl expression and beta-amyloid induced apoptosis relevance to Alzheimer's disease. J Neurochem. 1999;72:1552–1563. doi: 10.1046/j.1471-4159.1999.721552.x. [DOI] [PubMed] [Google Scholar]
- Garnier M, DiLorenzo D, Albertini A, Maggi A. Identification of estrogen-responsive genes in neuroblastoma SK-ER3 cells. J Neurosci. 1997;17:4591–4599. doi: 10.1523/JNEUROSCI.17-12-04591.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnuson DS, Knudsen BE, Geiger JD, Brownstone RM, Nath A. Human immunodeficiency virus type 1 tat activates non-N-methyl-D-aspartate excitatory amino acid receptors and causes neurotoxicity. Ann Neurol. 1995;37:373–380. doi: 10.1002/ana.410370314. [DOI] [PubMed] [Google Scholar]
- Nath A, Psooy K, Martin C, Knudsen B, Magnuson DS, Haughey N, Geiger JD. Identification of a human immunodeficiency virus type 1 Tat epitope that is neuroexcitatory and neurotoxic. J Virol. 1996;70:1475–1480. doi: 10.1128/jvi.70.3.1475-1480.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- New DR, Ma M, Epstein LG, Nath A, Gelbard HA. Human immunodeficiency virus type 1 Tat protein induces death by apoptosis in primary human neuron cultures. J Neurovirol. 1997;3:168–173. doi: 10.3109/13550289709015806. [DOI] [PubMed] [Google Scholar]
- Gelbard HA, Nottet HS, Swindells S, Jett M, Dzenko KA, Genis P, White R, Wang L, Choi YB, Zhang D, Lipton SA, Tourtellotte WW, Epstein LG, Gendelman HE. Platelet-activating factor: a candidate human immunodeficiency virus type 1-induced neurotoxin. J Virol. 1994;68:4628–4635. doi: 10.1128/jvi.68.7.4628-4635.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- New DR, Maggirwar SB, Epstein LG, Dewhurst S, Gelbard HA. HIV-1 Tat induces neuronal death via tumor necrosis factor-a and activation of non-N-methyl-D-aspartate receptors by a NFkB-independent mechanism. The Journal of Biological Chemistry. 1998;273:17852–17858. doi: 10.1074/jbc.273.28.17852. [DOI] [PubMed] [Google Scholar]
- Ma M, Nath A. Molecular determinants for cellular uptake of Tat protein of human immunodeficiency virus type 1 in brain cells. J Virol. 1997;71:2495–2499. doi: 10.1128/jvi.71.3.2495-2499.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu J, Yassini P, Goldberg G, Shu W, Konat GW, Wiggins RC. Cocaine cytotoxicity in serum-free environment: C6 glioma cell culture. Neurotoxicology. 1993;14:19–22. [PubMed] [Google Scholar]
- Nassogne MC, Evrard P, Courtoy PJ. Selective neuronal toxicity of cocaine in embryonic mouse brain cocultures. Proc Natl Acad Sci U S A. 1995;92:11029–11033. doi: 10.1073/pnas.92.24.11029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennet BA, Hyde CE, Pecora JR, Clodfelter JE. Long-term cocaine administration is not neurotoxic to cultured fetal mesencephalic dopamine neurons. Neurosci Lett. 2003;153:210–214. doi: 10.1016/0304-3940(93)90324-E. [DOI] [PubMed] [Google Scholar]
- Robinson SE, Enters EK, Jackson GF, Chinchilli VM, Maher JR, McDowell KP, Allen HM, Guo H. Maternal and fetal brain and plasma levels of cocaine and benzoylecgonine after acute or chronic maternal intravenous administration of cocaine. J Pharmacol Exp Ther. 1994;271:1234–1239. [PubMed] [Google Scholar]
- Mactutus CF, Herman AS, Booze RM. Chronic intravenous model for studies of drug (Ab)use in the pregnant and/or group-housed rat: an initial study with cocaine. Neurotoxicology and Teratology. 1994;16:183–191. doi: 10.1016/0892-0362(94)90116-3. [DOI] [PubMed] [Google Scholar]
- Evans SM, Cone EJ, Henningfield JE. Arterial and venous cocaine plasma concentration in humans: Relationship to route of administration, cardiovascular effects and subjective effects. J Pharmacol Exp Ther. 1996;279:1345–1356. [PubMed] [Google Scholar]
- Chiu FC, Rozental R, Bassallo C, Lyman WD, Spray DC. Human fetal neurons in culture: intercellular communication and voltage- and ligand-gated responses. J Neurosci Res. 1994;38:687–697. doi: 10.1002/jnr.490380611. [DOI] [PubMed] [Google Scholar]
- Sanfeliu C, Sebastiá J, Ki SU. Methylmercury neurotoxicity in cultures of human neurons, astrocytes, neuroblastoma cells. Neurotoxicology. 2001;22:317–327. doi: 10.1016/S0161-813X(01)00015-8. [DOI] [PubMed] [Google Scholar]
- Messam CA, Hou J, Berman JW, Major EO. Analysis of the temporal expression of nestin in human fetal brain derived neuronal and glial progenitor cells. Developmental Brain Research. 2002;134:87–92. doi: 10.1016/S0165-3806(01)00325-X. [DOI] [PubMed] [Google Scholar]
- Fried G, Andersson E, Csöregh L, Enmark E, Gustafsson JA, Aanesen A, Österlund C. Estrogen receptor beta is expressed in human embryonic brain cells and is regulated by 17b-estradiol. Eur J Neurosci. 2004;20:2345–2354. doi: 10.1111/j.1460-9568.2004.03693.x. [DOI] [PubMed] [Google Scholar]
- Winter BJ. Statistical Principles in Experimental Design. 2. New York, McGraw Hill; 1971. [Google Scholar]