

Abstract
Hydrops-ectopic calcification-“moth-eaten” (HEM) or Greenberg skeletal dysplasia is an autosomal recessive chondrodystrophy with a lethal course, characterized by fetal hydrops, short limbs, and abnormal chondro-osseous calcification. We found elevated levels of cholesta-8,14-dien-3β-ol in cultured skin fibroblasts of an 18-wk-old fetus with HEM, compatible with a deficiency of the cholesterol biosynthetic enzyme 3β-hydroxysterol Δ14-reductase. Sequence analysis of two candidate genes encoding putative human sterol Δ14-reductases (TM7SF2 and LBR) identified a homozygous 1599–1605TCTTCTA→CTAGAAG substitution in exon 13 of the LBR gene encoding the lamin B receptor, which results in a truncated protein. Functional complementation of the HEM cells by transfection with control LBR cDNA confirmed that LBR encoded the defective sterol Δ14-reductase. Mutations in LBR recently have been reported also to cause Pelger-Huët anomaly, an autosomal dominant trait characterized by hypolobulated nuclei and abnormal chromatin structure in granulocytes. The fact that the healthy mother of the fetus showed hypolobulated nuclei in 60% of her granulocytes confirms that classic Pelger-Huët anomaly represents the heterozygous state of 3β-hydroxysterol Δ14-reductase deficiency.
Hydrops-ectopic calcification-“moth-eaten” (HEM) skeletal dysplasia (MIM 215140), also known as “Greenberg dysplasia,” is a rare autosomal recessive chondrodystrophy characterized by early in utero lethality and, therefore, considered to be nonviable (Greenberg et al. 1988; Chitayat et al. 1993; Horn et al. 2000). Affected fetuses typically present with fetal hydrops, short-limbed dwarfism, and a marked disorganization of chondro-osseous calcification and may present with polydactyly (Chitayat et al. 1993) and additional nonskeletal malformations (Horn et al. 2000).
After obtaining informed consent, we conducted biochemical and molecular investigations in cells from a fetus with HEM—detected by fetal ultrasound performed on a 24-year-old woman of Turkish descent—who presented with intrauterine growth retardation at gestational age 17 wk. The fetus showed severe hydrops and short-limb skeletal dysplasia. Amniotic fluid examination showed a 46,XY karyotype, and a tentative diagnosis of thanatophoric dysplasia was made. Intrauterine death occurred at 18 wk, and delivery was induced. Fetal examination showed severe hydrops, extremely shortened edematous limbs, and postaxial polydactyly on both hands. Radiographic examination showed severe platyspondyly, short irregular ribs, a “moth-eaten” aspect of scapular and pelvic bones, and very short tubular bones with angulated diaphyses. Histopathology showed almost complete absence of enchondral ossification, severe disorganization of cartilage (with nodular calcification deposits), and defective or absent joint formation. On the basis of these findings, the fetus was diagnosed with HEM/Greenberg skeletal dysplasia. The consanguineous parents (F⩾1/16) were both healthy, and the family history was noncontributory.
Since sterol analysis elsewhere of tissues and cells of six fetuses with HEM obtained from a skeletal dysplasia repository suggested a deficiency of 3β-hydroxysterol Δ14-reductase (R. I. Kelley, unpublished data; Kelley and Herman 2001), we analyzed the sterol content in cultured primary skin fibroblasts obtained from the fetus with HEM. To this end, the cells were cultured in lipoprotein-depleted medium for 4 d, after which the sterols were extracted and analyzed by gas chromatography/mass spectrometry (GC/MS), as described elsewhere (Waterham et al. 2001). This revealed elevated levels of cholesta-8,14-dien-3β-ol (fig. 1A), a cholesterol precursor that was not detected in fibroblasts from healthy control subjects or in chorionic villi and amniotic fluid cells from fetuses of similar age. In analogy with sterol Δ14-reductase-deficient erg24 mutants of yeast, which accumulate ergosta-8,14-dien-3β-ol (Crowley et al. 1996; Silve et al. 1998), this finding pointed to a deficiency of the enzyme 3β-hydroxysterol Δ14-reductase, which catalyzes the reduction of the Δ14double bond in early sterol intermediates (fig. 1B).
Figure 1.

Sterol synthesis in cells from a control and the fetus with HEM. A, GC/MS sterol analysis in primary skin fibroblasts cultured for 4 d in lipoprotein-depleted medium revealed elevated levels of cholesta-8,14-dien-3β-ol (peak 2) in cells of the fetus with HEM, whereas, in control cells, only cholesterol (peak 1) was detected. B, The accumulation of cholesta-8,14-dien-3β-ol in HEM cells points to a deficiency of the enzyme 3β-hydroxysterol Δ14-reductase, which catalyses the reduction of the C14–15double bond of cholesta-8,14-dien-3β-ol to produce cholesta-8-en-3β-ol.
Two different genes have been reported to encode human sterol Δ14-reductases: LBR (GenBank accession number L25932–L25941) at 1q42 and TM7SF2 (GenBank accession number AF096303) at 11q13. LBR encodes the lamin B receptor, a 70.4-kDa protein of the inner nuclear membrane that contains an N-terminal lamin B/DNA-binding domain of ∼200 amino acids followed by a C-terminal sterol reductase-like domain of ∼450 amino acids (Holmer et al. 1998), which exhibits sterol Δ14-reductase activity when expressed in yeast (Silve et al. 1998). TM7SF2 encodes a sterol reductase of 46.4 kDa located in the endoplasmic reticulum (ER) membrane (Holmer et al. 1998), which exhibits sterol Δ14-reductase activity when overexpressed in COS-7 cells (Roberti et al. 2002). On the basis of the highest sequence similarity of the encoded protein with fungal sterol Δ14-reductases and its localization in the ER membrane (as are most other sterol reductases known to date), the TM7SF2 gene was considered the best candidate to encode the defective 3β-hydroxysterol Δ14-reductase. However, sequencing, by means of −21M13 and M13rev fluorescent primers (Waterham et al. 2001), of all exons and flanking intron sequences of TM7SF2 amplified by PCR (TM7SF2-specific primer sequences available upon request) did not identify mutations. Subsequent sequencing of PCR-amplified exons and flanking intron sequences of the LBR gene (table I), by means of −21M13 and M13rev fluorescent primers (Waterham et al. 2001), identified a homozygous substitution of seven nucleotides in exon 13 (fig. 2). Homozygosity was confirmed by finding the same mutation in heterozygous form in genomic DNA of the mother; no DNA from the father was available. The same homozygous substitution was also observed in LBR cDNA (GenBank accession number L25931) prepared from total RNA isolated from the fetus’s cells, which indicates that the mutation neither affects transcription nor is part of a nontranscribed pseudogene. The 1599–1605TCTTCTA→CTAGAAG substitution introduces a stop codon (underlined) that leads to the production of a truncated protein lacking the C-terminal 82 amino acids. Since the C-terminus is highly conserved among the various sterol reductases (Holmer et al. 1998; Waterham et al. 1998), it is very likely that no functional sterol Δ14-reductase is produced.
Table 1.
Primers and PCR Conditions Used for Mutation Analysis of LBR Gene
| Exon(s) | Forward Primer (5′→3′)a | Reverse Primer (5′→3′)b | PCR programc |
| 1 and 2 | [−21M13]TGATCAGCCTGTGGAAAAAGAC | [M13rev]TAATATTATTAACTGCAAGTAACTTG | Standard |
| 3 and 4 | [−21M13]CCAAAACAGTAAGTTTTTACCATC | [M13rev]ACCAGGCTCATTACCTTGCC | Touchdown |
| 5 | [−21M13]AAATAAATAAACCAGATTGAAATTTG | [M13rev]CCTCATCTTATTAAAACAAAACAG | Standard |
| 6 | [−21M13]ATATTACTAAATTTCCATGTTACAAG | [M13rev]TAAACAGTTGCTCTTCCAAC | Touchdown |
| 7 | [−21M13]AGTGGCGTAAGTTTCATGTGAG | [M13rev]AATCTATTCAAATCTGGAAATGGC | Standard |
| 8 | [−21M13]AGTGGCGTAAGTTTCATGTGAG | [M13rev]TTTATGGAAAGCCATAATTATCCC | Touchdown |
| 9 | [−21M13]TCTGCCAGGTGTCTCAGCTG | [M13rev]CGTCCTCCTCTCAAGCAGCC | Touchdown |
| 10 and 11 | [−21M13]AATGTTGATTGTGGACCCGCCC | [M13rev]AGTACATTTTTAATGATGTCGAGAC | Touchdown |
| 12 and 13 | [−21M13]GGAAAGCTTGCACGTAAGTATTG | [M13rev]TGTTTTTGCAAATGGCAGCTGG | Standard |
−21M13: tgt aaa acg acg gcc agt.
M13rev: cag gaa aca gct atg acc.
The amplification by PCR of genomic DNA, by means of a “standard” or a “touchdown” PCR amplification program, was performed as described elsewhere (Houten et al. 2001), using the indicated LBR-specific primers.
Figure 2.

Sequence analysis of the entire LBR gene from the fetus with HEM revealed a homozygous 1599–1605TCTTCTA→CTAGAAG substitution in exon 13. The mother was heterozygous for the same mutation.
To confirm that LBR indeed encodes the defective sterol Δ14-reductase, the patient’s fibroblasts were transfected transiently with control LBR cDNA by means of retroviral transduction, using the Phoenix retroviral system (Kinsella and Nolan 1996; Michiels et al. 2000). To this end, the coding region of LBR cDNA was amplified by PCR from control cDNA using a BamHI-tagged LBR cDNA–specific forward primer (5′-TTAAAGGATCCTCGTGTCACCGCCGGAACC-3′) and an LBR cDNA–specific reverse primer (5′-AAGTACAGACCCTGTCAGTGC-3′). The amplified LBR cDNA was cloned first in the pGEM-T vector (Clontech) and sequenced to exclude PCR-introduced errors. Subsequently, the LBR cDNA was released from pGEM-T with BamHI and NotI and ligated into the BamHI and NotI sites of the pLZRS-IRES-ZEO/pBR plasmid (Michiels et al. 2000). The resulting pLZRS-LBR-IRES-ZEO/pBR plasmid and, as a control, empty pLZRS-IRES-ZEO/pBR plasmid were transfected into amphotrophic Phoenix ΦNX-A packaging cells (gift from Dr. G. P. Nolan) using lipofectamine-plus (Invitrogen), and helper-free recombinant retrovirus was produced as described elsewhere (Michiels et al. 2000). LBR-containing and, as a control, non-LBR–containing viral supernatants (7 ml) were added to patient’s fibroblasts, seeded 1 d prior to the experiment at 40% confluence in T75 culture dishes. After 24 h of incubation, the viral supernatants were replaced by lipoprotein-depleted Dulbecco modified Eagle medium. After 4 d of culturing in the lipoprotein-depleted medium, cells were harvested, and sterols were extracted and analyzed by GC/MS, as described elsewhere (Waterham et al. 2001). The levels of cholesta-8,14-dien-3β-ol were corrected for total protein and recovery of the internal sterol standard epicoprostanol, which was added prior to the sterol extractions. In two independent experiments, we observed a marked reduction of 41% and 49.5% of cholesta-8,14-dien-3β-ol in the patient’s cells that were transduced with the LBR-containing retroviral supernatant, when compared with the identically treated cells transduced with the non-LBR–containing retroviral supernatant.
When this work was near completion, Hoffmann et al. (2002) reported that heterozygous mutations in LBR are responsible for Pelger-Huët anomaly (PHA [MIM 169400]). This is a benign autosomal dominant disorder of leukocyte development characterized by hypolobulated nuclei and abnormal chromatin structure in granulocytes of heterozygotes but without evident clinical symptoms (Pelger 1928; Huët 1932). Presumed PHA homozygotes present with ovoid granulocyte nuclei; may have developmental delay; display variable skeletal abnormalities, such as polydactyly and metacarpal shortening (Aznar and Vaya 1981; Von Siegert et al. 1983; Hoffmann et al. 2002); and may reach adult age (Hoffmann et al. 2002). Moreover, PHA homozygosity leads to short-limbed chondrodysplasia in rabbits (Nachtsheim 1950) and cats (Latimer et al. 1988). These findings combined with ours imply that HEM/Greenberg skeletal dysplasia and homozygous PHA are allelic disorders that display a wide clinical spectrum, with nonviable fetuses with HEM representing the severe end and minor limb defects representing the mild end of the spectrum. It is unfortunate that no sterol analysis to further substantiate this was performed in the single individual with homozygous PHA reported by Hoffmann et al. (2002). However, the mother of the fetus showed hypolobulated nuclei in 60% of her granulocytes (fig. 3), which demonstrates that classic PHA represents the heterozygous state of 3β-hydroxysterol Δ14-reductase deficiency.
Figure 3.
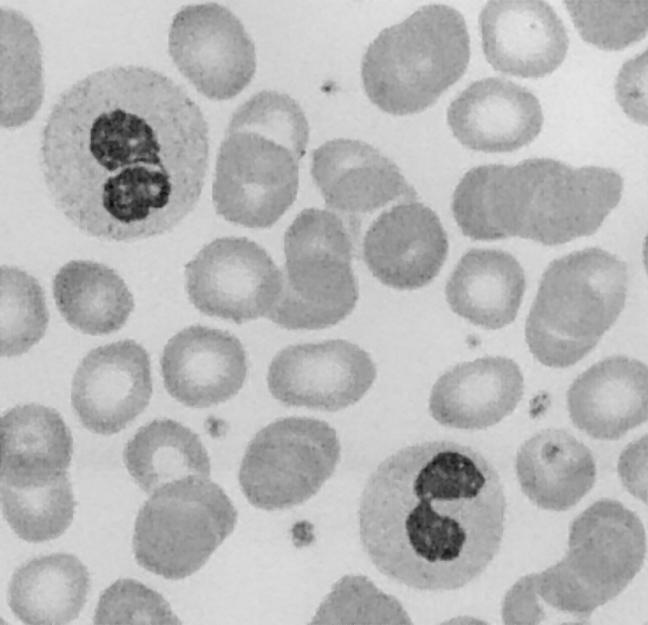
Blood smear of the mother of the present fetus with HEM showing the two characteristics of PHA: granulocytes with the typical pince-nez appearance of the bilobed nuclei and coarse clumping of chromatin.
HEM/Greenberg skeletal dysplasia is the sixth inherited disorder of cholesterol biosynthesis for which the molecular basis now has been resolved. Most of these disorders represent previously reported clinical entities of unknown etiology that could be attributed to different enzyme defects in the postsqualene segment of the cholesterol biosynthetic pathway after the finding of abnormally increased levels of intermediate sterol metabolites in patients, followed by the demonstration of disease-causing mutations in genes encoding the implicated enzymes (Kelley and Herman 2001; Porter 2002; Waterham 2002). Apart from autosomal recessive HEM/Greenberg skeletal dysplasia, these include the three autosomal recessive disorders Smith-Lemli-Opitz syndrome (MIM 270400), which is caused by 3β-hydroxysterol Δ7-reductase deficiency (Fitzky et al. 1998; Wassif et al. 1998; Waterham et al. 1998); desmosterolosis (MIM 602398), which is caused by 3β-hydroxysterol Δ24-reductase deficiency (Waterham et al. 2001); and lathosterolosis (MIM 607330), which is caused by 3β-hydroxysterol Δ5-desaturase deficiency (Brunetti-Pierri et al. 2002). In addition, the molecular basis of two X-linked dominant inherited disorders of cholesterol biosynthesis has been resolved: Conradi-Hünermann-Happle syndrome (MIM 302960) and CHILD syndrome (MIM 308050) are caused by deficiencies of sterol Δ8–Δ7isomerase (Braverman et al. 1999; Derry et al. 1999) and a sterol C-4 demethylase (Konig et al. 2000), respectively. Patients afflicted with these inborn errors of cholesterol biosynthesis are characterized, in general, by multiple morphogenic and congenital anomalies, including skeletal, internal organ, and/or skin abnormalities (Kelley and Herman 2001; Porter 2002; Waterham 2002).
The occurrence of an abnormal granulocyte chromatine structure in the hetero- and homozygous individuals with PHA and the presence of (severe) skeletal abnormalities in individuals with homozygous HEM/Greenberg skeletal dysplasia indicate that the lamin B receptor has two different physiological functions, namely, preserving chromatin structure by promoting heterochromatin binding to the inner nuclear membrane (Ye and Worman 1994) and functioning as the primary sterol Δ14-reductase in human cholesterol biosynthesis. The latter was somewhat unexpected, since all enzymes involved in the postsqualene cholesterol biosynthesis pathway have been localized to the ER membrane, whereas the lamin B receptor is present (predominantly) in the inner nuclear membrane. In this respect, the gene product of TM7SF2 a priori seemed a better candidate, since it is localized in the ER membrane (Holmer et al. 1998) and also exhibits sterol Δ14-reductase activity (Roberti et al. 2002). Now that TM7SF2 is not the primary sterol Δ14-reductase involved in human cholesterol synthesis, its physiological function remains to be discovered.
Acknowledgments
We thank Gerrit-Jan Romeijn for technical assistance in the initial stages of this study. H.R.W. is supported by a fellowship of the Royal Netherlands Academy of Arts and Sciences.
Electronic-Database Information
Accession numbers and URLs for data presented herein are as follows:
- GenBank, http://www.ncbi.nlm.nih.gov/Genbank/ (for TM7SF2 gene [accession number AF096303], LBR gene [accession numbers L25932–L25941]), and LBR cDNA [accession number L25931])
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for HEM/Greenberg skeletal dysplasia, Pelger-Huët anomaly, Smith-Lemli-Opitz syndrome, desmosterolosis, lathosterolosis, Conradi-Hünermann-Happle syndrome, and CHILD syndrome)
References
- Aznar J, Vaya A (1981) Homozygous form of the Pelger-Huët leukocyte anomaly in man. Acta Haematol 66:59–62 [DOI] [PubMed] [Google Scholar]
- Braverman N, Lin P, Moebius FF, Obie C, Moser A, Glossmann H, Wilcox WR, Rimoin DL, Smith M, Kratz L, Kelley RI, Valle D (1999) Mutations in the gene encoding 3 β-hydroxysteroid-Δ8, Δ7- isomerase cause X-linked dominant Conradi-Hunermann syndrome. Nat Genet 22:291–294 [DOI] [PubMed] [Google Scholar]
- Brunetti-Pierri N, Corso G, Rossi M, Ferrari P, Balli F, Rivasi F, Annunziata I, Ballabio A, Dello Russo A, Andria G, Parenti G (2002) Lathosterolosis, a novel multiple-malformation/mental retardation syndrome due to deficiency of 3β-hydroxysteroid-Δ5-desaturase. Am J Hum Genet 71:952–958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chitayat D, Gruber H, Mullen BJ, Pauzner D, Costa T, Lachman R, Rimoin DL (1993) Hydrops-ectopic calcification-moth-eaten skeletal dysplasia (Greenberg dysplasia): prenatal diagnosis and further delineation of a rare genetic disorder. Am J Med Genet 47:272–277 [DOI] [PubMed] [Google Scholar]
- Crowley JH, Smith SJ, Leak FW, Parks L (1996) Aerobic isolation of an ERG24 null mutant of Saccharomyces cerevisiae. J Bacteriol 178:2991–2993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derry JM, Gormally E, Means GD, Zhao W, Meindl A, Kelley RI, Boyd Y, Herman GE (1999) Mutations in a Δ8–Δ7 sterol isomerase in the tattered mouse and X-linked dominant chondrodysplasia punctata. Nat Genet 22:286–290 [DOI] [PubMed] [Google Scholar]
- Fitzky BU Witsch-Baumgartner M, Erdel M, Lee JN, Paik YK, Glossmann H, Utermann G, Moebius FF (1998) Mutations in the Δ7-sterol reductase gene in patients with the Smith-Lemli-Opitz syndrome. Proc Natl Acad Sci USA 95:8181–8186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg CR, Rimoin DL, Gruber HE, DeSa DJ, Reed M, Lachman RS (1988) A new autosomal recessive lethal chondrodystrophy with congenital hydrops. Am J Med Genet 29:623–632 [DOI] [PubMed] [Google Scholar]
- Hoffmann K, Dreger CK, Olins AL, Olins DE, Shultz LD, Lucke B, Karl H, Kaps R, Muller D, Vaya A, Aznar J, Ware RE, Sotelo Cruz N, Lindner TH, Herrmann H, Reis A, Sperling K (2002) Mutations in the gene encoding the lamin B receptor produce an altered nuclear morphology in granulocytes (Pelger-Huet anomaly). Nat Genet 31:410–414 [DOI] [PubMed] [Google Scholar]
- Holmer L, Pezhman A, Worman HJ (1998) The human lamin B receptor/sterol reductase multigene family. Genomics 54:469–476 [DOI] [PubMed] [Google Scholar]
- Horn LC, Faber R, Meiner A, Piskazeck U, Spranger J (2000) Greenberg dysplasia: first reported case with additional non-skeletal malformations and without consanguinity. Prenat Diagn 20:1008–1011 [DOI] [PubMed] [Google Scholar]
- Houten SM, Koster J, Romeijn GJ, Frenkel J, Di Rocco M, Caruso U, Landrieu P, Kelley RI, Kuis W, Poll-The BT, Gibson KM, Wanders RJ, Waterham HR (2001) Organization of the mevalonate kinase (MVK) gene and identification of novel mutations causing mevalonic aciduria and hyperimmunoglobulinaemia D and periodic fever syndrome. Eur J Hum Genet 9:253–259 [DOI] [PubMed] [Google Scholar]
- Huët GJ (1932) Ueber eine bisher unbekannte familiaere Anomalie der Leukocyten. Klin Wschr 11:1264–1266 [Google Scholar]
- Kelley RI, Herman GE (2001) Inborn errors of sterol biosynthesis. Annu Rev Genomics Hum Genet 2:299–341 [DOI] [PubMed] [Google Scholar]
- Kinsella TM, Nolan GP (1996) Episomal vectors rapidly and stably produce high-titer recombinant retrovirus. Hum Gene Ther 7:1405–1413 [DOI] [PubMed] [Google Scholar]
- Konig A, Happle R, Bornholdt D, Engel H, Grzeschik KH (2000) Mutations in the NSDHL gene, encoding a 3β-hydroxysteroid dehydrogenase, cause CHILD syndrome. Am J Med Genet 90:339–346 [PubMed] [Google Scholar]
- Latimer KS, Rowland GN, Mahaffey MB (1988) Homozygous Pelder Huet anomaly and chondrodysplasia in a stillborn kitten. Vet Pathol 25:325–328 [DOI] [PubMed] [Google Scholar]
- Michiels F, Van der Kammen RA, Janssen L, Nolan G, Collard JG (2000) Expression of Rho GTPases using retroviral vectors. Meth Enzymol 325:295–302 [DOI] [PubMed] [Google Scholar]
- Nachtsheim H (1950) The Pelger-anomaly in man and rabbit. J Hered 41:131–137 [DOI] [PubMed] [Google Scholar]
- Pelger K (1928) Demonstratie van een paar zeldzaam voorkomende typen van bloedlichaampjes en bespreking der patienten. Ned Tijdschr Geneesk 72:1178 [Google Scholar]
- Porter FD (2002) Malformation syndromes due to inborn errors of cholesterol synthesis. J Clin Invest 110:715–724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberti R, Bennati AM, Galli G, Caruso D, Maras B, Aisa C, Beccari T, Della Fazia MA, Servillo G (2002) Cloning and expression of sterol Δ14-reductase from bovine liver. Eur J Biochem 269:283–290 [DOI] [PubMed] [Google Scholar]
- Silve S, Dupuy PH, Ferrara, P, Loison, G. (1998) Human lamin B receptor exhibits sterol C14-reductase activity in Saccharomyces cerevisiae. Biochim Biophys Acta 1392:233–244 [DOI] [PubMed] [Google Scholar]
- Von Siegert E, Beier L, Graebner H (1983) Ein Beitrag zur homozygoten Form der Pelger-Huetschen Kernanomalie. Kinderartzliche Praxis 51:164–169 [PubMed] [Google Scholar]
- Wassif CA, Maslen C, Kachilele-Linjewile S, Lin D, Linck LM, Connor WE, Steiner RD, Porter FD (1998) Mutations in the human sterol Δ7-reductase gene at 11q12–13 cause Smith-Lemli-Opitz syndrome. Am J Hum Genet 63:55–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterham HR (2002) Inherited disorders of cholesterol biosynthesis. Clin Genet 61:393–403 [DOI] [PubMed] [Google Scholar]
- Waterham HR, Koster J, Romeijn GJ, Hennekam RCM, Vreken P, Andersson HC, FitzPatrick DR, Kelley, RI, Wanders RJA (2001) Mutations in the 3β-hydroxysteroid Δ24-reductase gene cause desmosterolosis, an autosomal recessive disorder of cholesterol biosynthesis. Am J Hum Genet 69:685–694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterham HR, Wijburg FA, Hennekam RCM, Vreken P, Poll-The BT, Dorland L, Duran M, Jira PE, Smeitink JA, Wevers RA, Wanders RJA (1998) Smith-Lemli-Opitz syndrome is caused by mutations in the 7- dehydrocholesterol reductase gene. Am J Hum Genet 63:329–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Q, Worman HJ (1994) Primary structure analysis and lamin B and DNA binding of human LBR, an integral protein of the nuclear envelope inner membrane. J Biol Chem 269:11306–11311 [PubMed] [Google Scholar]