

Abstract
Although Africa has played a central role in human evolutionary history, certain studies have suggested that not all contemporary human genetic diversity is of recent African origin. We investigated 35 simple polymorphic sites and one Tn microsatellite in an 8-kb segment of the dystrophin gene. We found 86 haplotypes in 1,343 chromosomes from around the world. Although a classical out-of-Africa topology was observed in trees based on the variant frequencies, the tree of haplotype sequences reveals three lineages accounting for present-day diversity. The proportion of new recombinants and the diversity of the Tn microsatellite were used to estimate the age of haplotype lineages and the time of colonization events. The lineage that underwent the great expansion originated in Africa prior to the Upper Paleolithic (27,000–56,000 years ago). A second group, of structurally distinct haplotypes that occupy a central position on the tree, has never left Africa. The third lineage is represented by the haplotype that lies closest to the root, is virtually absent in Africa, and appears older than the recent out-of-Africa expansion. We propose that this lineage could have left Africa before the expansion (as early as 160,000 years ago) and admixed, outside of Africa, with the expanding lineage. Contemporary human diversity, although dominated by the recently expanded African lineage, thus represents a mosaic of different contributions.
Introduction
Africa has played a central role in human evolutionary history. The oldest skeletal fossils with modern human characteristics are found at sites dated at 90–120 thousand years ago (kya) that extend from the Klasies River mouth in South Africa up to Qafzeh and Skhul in Israel (Stringer and Andrews 1988; Tattersall 1995; Lahr and Foley 1998; Tattersall and Schwartz 2000) and at sites dated as early as 160 kya from the Awash in Ethiopia (White et al. 2003). Modern human remains found outside Africa and the Levant are much younger and together support the notion that Southeast Asia and Australia were colonized by Homo sapiens ∼50–60 kya, Europe was colonized 30–40 kya, and the Americas were colonized 12–25 kya (Klein 1999).
The recent African ancestry of modern humans is generally supported by genetic studies, of contemporary populations, that use either classical protein markers (Cavalli-Sforza et al. 1994) or polymorphisms in mtDNA (Cann et al. 1987; Chen et al. 1995), the Y chromosome (Hammer 1995; Underhill et al. 2000), and the recombining portion of the genome (Batzer et al. 1994; Bowcock et al. 1994; Tishkoff et al. 1996; Labuda et al. 2000; Fan et al. 2002; Rosenberg et al. 2002). The majority of studies of human genetic history have been based on the analysis of mitochondrial and Y-chromosome DNA. In spite of the undisputable virtues of these two systems, their value as markers of population history is limited, because (i) each represents but a single locus, (ii) their effective population size is one-fourth that of autosomal segments, and (iii) they reflect the history of either the maternal or paternal lineage only (Seielstad et al. 1998).
In addition to the use of mitochondrial and Y-chromosome DNA, to understand the genetic structure of human populations and to retrace their genetic past, we also need to collect information from autosomal and/or X-chromosome loci. However, such information, which has been collected for different loci and different populations (Batzer et al. 1994; Tishkoff et al. 1996; Harding et al. 1997; Clark et al. 1998; Harris and Hey 1999; Jaruzelska et al. 1999; Jin et al. 1999; Kaessmann et al. 1999; Kidd et al. 2000; Labuda et al. 2000; Yu et al. 2001; Fan et al. 2002; Rosenberg et al. 2002), has never achieved either the population depth or the worldwide breadth of mtDNA or Y-chromosome studies.
We have proposed an 8-kb intronic segment of the dystrophin locus, dys44 on Xp21.3, as a model system to investigate variability in the nuclear non-Y-chromosome DNA (Ziętkiewicz et al. 1997, 1998). The X chromosome is conducive to such studies, since it facilitates analysis by allowing direct haplotype reading and direct assessment of linkage disequilibrium in male samples. Our previous analysis of worldwide dys44 haplotype diversity indicated the existence of at least two separate founder lineages of modern humans (Labuda et al. 2000). Sub-Saharan Africans, rather than making up a uniform genetic pool, were found to represent two chromosomal lineages with distinct genetic histories that may result from fragmentation of early human population(s) during periods of glaciation. Expansion of one of these lineages led to the global colonization of all presently inhabited continents, whereas the second lineage remained local to Africa. Here, we report an extended survey of dys44 diversity in a set of 1,343 X chromosomes. Our data, in addition to supporting a split African ancestry, reveal the contribution of another, third lineage, represented by an ancient haplotype found in Eurasia and the Americas that is virtually absent in sub-Saharan Africa. We address the question of whether it represents a lineage that had been lost in Africa following the Upper Paleolithic out-of-Africa expansion or a lineage that could have left Africa much earlier, at the dawn of modern humans, to subsequently contribute to northern human populations.
Material and Methods
Population Samples
All DNA samples were nonnominative, originating either from existing collections or peripheral blood samples donated by consenting informed adults, following the protocol approved by the institutional review boards of Ste.-Justine Hospital (Montréal), Victoria Hospital (Prince Albert), and their collaborating institutions. We analyzed 1,343 chromosomes from 33 human populations representing five continental groups, as detailed in table 1. In addition to the genotypes reported elsewhere (Ziętkiewicz et al. 1997, 1998; Labuda et al. 2000), new data have been obtained for Ashkenazim (n=72 chromosomes), Basques (n=20), North American Natives (NaDene speakers from Saskatchewan, n=40; and Ojibwa from Ontario, n=30), five populations of eastern Indonesians (n=91), and eight additional Mongolian populations (n=267). Supplementary chromosomes have been added to the following previously analyzed populations: Siberian (additional n=26), Polish (n=5), Mayan (n=2), African American (n=8), Mossi (n=6), Rimaibe (n=8), Biaka (n=4) and M’Buti (n=2). In contrast, 106 French Canadian chromosomes reported by Labuda et al. (2000) were not included in the present study. Eleven Nigerian chromosomes, previously assigned with the Burkina Faso populations, were included in the African American group in the present study.
Table 1.
Distribution of dys44 Haplotypes and Their Associated Tn Alleles in all 33 Populations
|
No. of Occurrences ina |
||||||||||||||||||||||||||||||||||
| Africa |
Europe |
Asia |
America |
Indonesia/PNG |
||||||||||||||||||||||||||||||
| Haplotype | Total | AAb | Mo | Ri | Bi | Mb | Eu | Po | It | Ba | As | Si | Kha | Kho | Ka | Za | Ur | Ol | My | De | Bt | Ch | Ja | Na | Oj | Ma | Kr | Hi | Te | Ro | Fl | Al | PNGc | PNGh |
| B001 (14) | 1 | 1 | ||||||||||||||||||||||||||||||||
| B001 (15) | 397 | 4 | 4 | 3 | 18 | 5 | 8 | 10 | 10 | 3 | 26 | 15 | 12 | 16 | 21 | 14 | 15 | 12 | 12 | 8 | 8 | 38 | 39 | 17 | 8 | 21 | 11 | 6 | 6 | 7 | 9 | 5 | 2 | 3 |
| B001 (16) | 3 | 2 | 1 | |||||||||||||||||||||||||||||||
| B002 (14) | 9 | 7 | 2 | |||||||||||||||||||||||||||||||
| B002 (15) | 250 | 9 | 1 | 3 | 12 | 2 | 2 | 2 | 1 | 10 | 10 | 7 | 7 | 12 | 8 | 8 | 5 | 7 | 14 | 9 | 25 | 18 | 1 | 9 | 6 | 8 | 8 | 8 | 11 | 27 | ||||
| B002 (16) | 19 | 5 | 2 | 11 | 1 | |||||||||||||||||||||||||||||
| B002 (17) | 15 | 6 | 2 | 3 | 1 | 3 | ||||||||||||||||||||||||||||
| B003 (14) | 5 | 3 | 1 | 1 | ||||||||||||||||||||||||||||||
| B003 (15) | 123 | 4 | 1 | 3 | 9 | 8 | 7 | 4 | 27 | 5 | 1 | 1 | 3 | 1 | 3 | 3 | 1 | 3 | 1 | 5 | 1 | 10 | 8 | 9 | 1 | 1 | 2 | 1 | ||||||
| B004 (15) | 104 | 1 | 1 | 1 | 1 | 3 | 1 | 1 | 2 | 3 | 1 | 1 | 1 | 3 | 1 | 25 | 55 | 2 | 1 | |||||||||||||||
| B005 (15) | 3 | 3 | ||||||||||||||||||||||||||||||||
| B005 (21) | 3 | 1 | 1 | 1 | ||||||||||||||||||||||||||||||
| B005 (22) | 53 | 4 | 2 | 4 | 2 | 1 | 1 | 4 | 2 | 1 | 5 | 3 | 2 | 4 | 2 | 3 | 6 | 2 | 1 | 4 | ||||||||||||||
| B005 (23) | 3 | 3 | ||||||||||||||||||||||||||||||||
| B006 (15) | 17 | 1 | 1 | 1 | 1 | 2 | 1 | 2 | 3 | 3 | 2 | |||||||||||||||||||||||
| B006 (16) | 74 | 3 | 3 | 3 | 1 | 4 | 3 | 6 | 1 | 1 | 2 | 8 | 8 | 16 | 15 | |||||||||||||||||||
| B008 (14) | 19 | 2 | 3 | 2 | 2 | 2 | 1 | 1 | 3 | 2 | 1 | |||||||||||||||||||||||
| B011 (15) | 1 | 1 | ||||||||||||||||||||||||||||||||
| B011 (22) | 6 | 1 | 1 | 2 | 1 | 1 | ||||||||||||||||||||||||||||
| B013 (22) | 5 | 1 | 1 | 1 | 1 | 1 | ||||||||||||||||||||||||||||
| B016 (15) | 7 | 3 | 1 | 1 | 2 | |||||||||||||||||||||||||||||
| B019 (15) | 3 | 1 | 2 | |||||||||||||||||||||||||||||||
| B019 (17) | 2 | 1 | 1 | |||||||||||||||||||||||||||||||
| B023 (15) | 5 | 1 | 1 | 3 | ||||||||||||||||||||||||||||||
| B024 (15) | 6 | 3 | 1 | 1 | 1 | |||||||||||||||||||||||||||||
| B025 (15) | 3 | 1 | 1 | 1 | ||||||||||||||||||||||||||||||
| B030 (15) | 7 | 2 | 1 | 2 | 2 | |||||||||||||||||||||||||||||
| B031 (15) | 2 | 1 | 1 | |||||||||||||||||||||||||||||||
| B034 (15) | 3 | 1 | 1 | 1 | ||||||||||||||||||||||||||||||
| B035 (15) | 2 | 1 | 1 | |||||||||||||||||||||||||||||||
| B039 (22) | 3 | 1 | 1 | 1 | ||||||||||||||||||||||||||||||
| b052 (16) | 3 | 1 | 2 | |||||||||||||||||||||||||||||||
| B043 (15) | 1 | 1 | ||||||||||||||||||||||||||||||||
| b068 (15) | 1 | 1 | ||||||||||||||||||||||||||||||||
| B082 (16) | 1 | 1 | ||||||||||||||||||||||||||||||||
| b084 (15) | 1 | 1 | ||||||||||||||||||||||||||||||||
| b089 (14) | 1 | 1 | ||||||||||||||||||||||||||||||||
| B104 (15) | 1 | 1 | ||||||||||||||||||||||||||||||||
| B014 (15) | 8 | 2 | 2 | 2 | 2 | |||||||||||||||||||||||||||||
| B015 (15) | 4 | 3 | 1 | |||||||||||||||||||||||||||||||
| B026 (15) | 2 | 2 | ||||||||||||||||||||||||||||||||
| B036 (15) | 2 | 2 | ||||||||||||||||||||||||||||||||
| B041 (15) | 1 | 1 | ||||||||||||||||||||||||||||||||
| B048 (15) | 1 | 1 | ||||||||||||||||||||||||||||||||
| B049 (15) | 1 | 1 | ||||||||||||||||||||||||||||||||
| b054 (15) | 1 | 1 | ||||||||||||||||||||||||||||||||
| b100 (15) | 1 | 1 | ||||||||||||||||||||||||||||||||
| b053 (15) | 2 | 2 | ||||||||||||||||||||||||||||||||
| b064 (16) | 1 | 1 | ||||||||||||||||||||||||||||||||
| b066 (15) | 1 | 1 | ||||||||||||||||||||||||||||||||
| B020 (15) | 3 | 3 | ||||||||||||||||||||||||||||||||
| B028 (15) | 2 | 2 | ||||||||||||||||||||||||||||||||
| B029 (15) | 5 | 1 | 4 | |||||||||||||||||||||||||||||||
| B042 (22) | 4 | 2 | 2 | |||||||||||||||||||||||||||||||
| B051 (22) | 1 | 1 | ||||||||||||||||||||||||||||||||
| B051 (23) | 1 | 1 | ||||||||||||||||||||||||||||||||
| b087 (15) | 1 | 1 | ||||||||||||||||||||||||||||||||
| B105 (15) | 1 | 1 | ||||||||||||||||||||||||||||||||
| B007 (17) | 1 | 1 | ||||||||||||||||||||||||||||||||
| B007 (19) | 1 | 1 | ||||||||||||||||||||||||||||||||
| B007 (20) | 33 | 3 | 1 | 1 | 2 | 26 | ||||||||||||||||||||||||||||
| B009 (15) | 15 | 2 | 2 | 7 | 4 | |||||||||||||||||||||||||||||
| B010 (21) | 4 | 2 | 2 | |||||||||||||||||||||||||||||||
| B010 (22) | 6 | 4 | 2 | |||||||||||||||||||||||||||||||
| B010 (24) | 2 | 1 | 1 | |||||||||||||||||||||||||||||||
| B012 (17) | 6 | 1 | 2 | 3 | ||||||||||||||||||||||||||||||
| B012 (20) | 3 | 2 | 1 | |||||||||||||||||||||||||||||||
| B017 (19) | 3 | 2 | 1 | |||||||||||||||||||||||||||||||
| B018 (19) | 5 | 2 | 2 | 1 | ||||||||||||||||||||||||||||||
| B021 (20) | 3 | 3 | ||||||||||||||||||||||||||||||||
| B022 (22) | 2 | 1 | 1 | |||||||||||||||||||||||||||||||
| B027 (14) | 3 | 2 | 1 | |||||||||||||||||||||||||||||||
| B032 (15) | 7 | 6 | 1 | |||||||||||||||||||||||||||||||
| B037 (16) | 4 | 4 | ||||||||||||||||||||||||||||||||
| B038 (20) | 1 | 1 | ||||||||||||||||||||||||||||||||
| B040 (19) | 1 | 1 | ||||||||||||||||||||||||||||||||
| B044 (15) | 1 | 1 | ||||||||||||||||||||||||||||||||
| B045 (14) | 1 | 1 | ||||||||||||||||||||||||||||||||
| B046 (19) | 1 | 1 | ||||||||||||||||||||||||||||||||
| B046 (20) | 4 | 4 | ||||||||||||||||||||||||||||||||
| B047 (22) | 1 | 1 | ||||||||||||||||||||||||||||||||
| B050 (17) | 1 | 1 | ||||||||||||||||||||||||||||||||
| b056 (20) | 2 | 2 | ||||||||||||||||||||||||||||||||
| b057 (14) | 1 | 1 | ||||||||||||||||||||||||||||||||
| b058 (15) | 1 | 1 | ||||||||||||||||||||||||||||||||
| b059 (20) | 1 | 1 | ||||||||||||||||||||||||||||||||
| b060 (21) | 1 | 1 | ||||||||||||||||||||||||||||||||
| b061 (19) | 1 | 1 | ||||||||||||||||||||||||||||||||
| b061 (20) | 4 | 4 | ||||||||||||||||||||||||||||||||
| b062 (19) | 1 | 1 | ||||||||||||||||||||||||||||||||
| b063 (16) | 1 | 1 | ||||||||||||||||||||||||||||||||
| b067 (21) | 1 | 1 | ||||||||||||||||||||||||||||||||
| b072 (20) | 1 | 1 | ||||||||||||||||||||||||||||||||
| b088 (21) | 1 | 1 | ||||||||||||||||||||||||||||||||
| b110 (21) | 2 | 1 | 1 | |||||||||||||||||||||||||||||||
| b115 (20) | 1 | 1 | ||||||||||||||||||||||||||||||||
| b116 (21) | 1 | 1 | ||||||||||||||||||||||||||||||||
| b117 (21) | 2 | 2 | ||||||||||||||||||||||||||||||||
| b118 (15) | 1 | 1 | ||||||||||||||||||||||||||||||||
| b119 (19) | 1 | 1 | ||||||||||||||||||||||||||||||||
| b120 (19) | 1 | 1 | ||||||||||||||||||||||||||||||||
| b121 (21) | 1 | 1 | ||||||||||||||||||||||||||||||||
| b122 (21) | 1 | 1 | ||||||||||||||||||||||||||||||||
| b123 (15) | 1 | 1 | ||||||||||||||||||||||||||||||||
| b124 (21) | 1 | 1 | ||||||||||||||||||||||||||||||||
| b125 (17) | 1 |
1 |
||||||||||||||||||||||||||||||||
| Total | 1,343 | 86 | 30 | 31 | 85 | 58 | 30 | 30 | 26 | 20 | 72 | 48 | 28 | 30 | 54 | 29 | 34 | 30 | 30 | 31 | 29 | 80 | 65 | 40 | 30 | 78 | 83 | 18 | 13 | 17 | 27 | 16 | 24 | 41 |
Population abbreviations are as follows: AA = African American; Mo = Mossi; Ri = Rimaibe; Bi = Biaka; Mb = Mbuti; Eu = mixed European; Po = Poland; It = Italian; Ba = Basque; As = Ashkenazim; Si = Siberian; Kha = Khalkha; Kho = Khoton; Ka = Kazakh; Za = Zakhchin; Ur = Uriankhai; Ol = Olet; My = Myangad; De = Derbet; Bt = Bait; Ch = Chinese; Ja = Japanese; Na = NaDene; Oj = Ojibwa; Ma = Maya; Kr = Karitiana; Hi = Hiri; Te = Ternate; Ro = Roti; Fl = Flores; Al = Alores; PNGc = PNG coastal; PNGh = PNG highland.
Sample includes 11 Nigerian chromosomes.
Genotyping
The genomic segment dys44 consists of exon 44 (148 bp) and its surrounding intronic sequence (positions −2782 to −1 of intron 43 and positions 1 to 4987 of intron 44) of the human dystrophin gene at Xp21 (GenBank accession number U94396) (Ziętkiewicz et al. 1998). Thirty-six simple intronic polymorphisms resulting from 33 nucleotide substitutions (including one three-allele site), two 3-nt deletions, and one 8-nt duplication were previously ascertained by SSCP/heteroduplex analysis of 7,622 bp within dys44 segment in 250 worldwide-distributed chromosomes (Ziętkiewicz et al. 1997). In addition, this segment contains a poly-T microsatellite, T14–24. A 7,917-bp distance between upstream and downstream flanking polymorphic sites determines the length of the haplotype. The newly examined genomic samples were typed by allele-specific oligonucleotide (ASO; see Ziętkiewicz et al. 1997) hybridization to determine alleles at the 36 segregating sites. The Tn polymorphism was typed by standard polyacrylamide gel electrophoresis.
Derivation of Haplotypes
Converting dys44 genotype data into haplotypes was straightforward, by direct observation, in hemizygous males as well as in homozygous and single-heterozygous females; these unambiguous haplotypes were assigned names starting with an uppercase letter “B.” Multiple-heterozygous female genotypes were initially resolved into haplotypes, as described elsewhere (Labuda et al. 2000). In brief, if a genotype represented a unique sum of two previously reported haplotypes, these were accepted. Where more than one solution was possible, the most likely haplotype combination was chosen, taking into account the candidate haplotype frequencies in the respective populations (the most frequent ones being preferred). If only one known haplotype could be recognized within the individual genotype, the novel haplotype was inferred indirectly by subtracting the known one from the genotypes. Such a new haplotype was accepted if it could be derived from the already known haplotypes by a single or double recombination or by a simple mutation (the simple “genealogy” rule) and was assigned a name beginning with a lowercase “b.” The only case in which a genotype was solved into two lowercase “b” haplotypes was in the case of a heterozygous individual carrying a singleton variant allele at position 86. According to the above rules, this genotype could have been resolved into one known (B002) and one novel haplotype, made up of the mutated “86” allele assigned to the rare haplotype b124; instead, we chose to assign the singleton variant to the frequent haplotype B002, resulting in the new haplotype b125. The Tn polymorphism was not considered when naming haplotypes, such that a given “B” haplotype may be accompanied by different Tn alleles (e.g., B002 is present as B002_14, B002_15, B002_16, and B002_17). However, because of their linkage with the remaining portion of the haplotype, Tn alleles aided in resolving some of the genotypes. In two cases in which several solutions seemed equally likely, as well as in one case in which no simple sequence of recombination or mutation could be inferred that linked the novel haplotype to the known ones, solutions were accepted on arbitrary bases. A few genotypes that could not be resolved into known haplotypes remained unsolved and were excluded from further analyses; the number of the reported haplotypes is thus conservative. Our inference of haplotype pairs from genotypes was independently assessed by PHASE software (Stephens et al. 2001), which essentially arrived at the same solution. In a few cases, we “disagreed” with PHASE, using the criteria discussed above. However, this does not affect the emerging pattern of dys44 diversity and bears no consequence for the conclusions of this study.
Distance Trees
Haplotype clustering was performed by the NEIGHBOR program (which implements a neighbor-joining [NJ] algorithm), through use of a distance matrix of haplotype sequences (excluding the Tn polymorphism) obtained by applying equal weighting to all allelic sites. Population trees were obtained by NEIGHBOR, KITCH, and FITCH, using distance matrices obtained by GENDIST. One set of these trees was based on the population frequencies of alleles at individual polymorphic sites (the frequencies of non-new alleles in the hypothetical ancestral population were set to 1.0). A second set of trees was based on the population frequencies of haplotypes (i.e., regarding dys44 as a single multiallelic site); the haplotype frequencies in the outgroup were set at 0. All programs were from the Phylip package 3.57 (Felsenstein 1993).
Haplotype Diversity Analysis
To test for selective neutrality and population equilibrium, the observed frequency distribution of the haplotypes was compared with that expected under the infinite-allele model (Ewens 1972; Watterson 1978), through use of the Arlequin package, version 1.1 (Schneider et al. 1997). For a detailed description of the test used, please refer to the Arlequin help manual (Arlequin's Home on the Web).
Provided that mutations, occurring at the rate μ, are rare, such that recurrent events can be neglected, the proportion of intact (i.e., nonmutated) chromosomes after one generation is P1=(1-μ) and after g generations is Pg=(1-μ)g, such that
Similarly, when we consider recombinations rather than mutations in creating haplotype diversity,
where rapp corresponds to the apparent recombination rate—that is, the rate of nonsilent recombinations (those that result in new, observable recombinants). On the basis of the mapping data of Kong et al. (2002), the ratio of genetic to physical distance in the dystrophin exon 44 region is 2 cM/Mb, from which we obtain the estimate of the rate of recombination as r=2×10-8 per bp. If we assume an equal contribution of both sexes to the effective population size, one-third of the X-chromosome population (males) is not involved in meiotic recombinations. Correcting for this, one obtains an estimate of r=2/3×2×10-8 per bp per generation, or 1.06×10-4 per 7,917 bp of dys44 haplotype. Since only ∼30% of dys44 recombinations are informative, leading to newly observable recombinants (data not shown; also see Stephens 1986), the apparent average recombination rate should be lowered to rapp=0.32×10-4 per segment per generation.
To estimate Tn microsatellite diversity, we calculated the variance in the allele length (number of repeats) in a population sample (Di Rienzo et al. 1998):
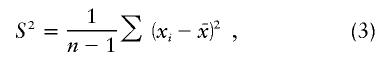 |
where x1, x2, …, xn denotes the number of repeats in each of the n chromosomes sampled and 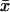 denotes their average repeat number. Assuming a simple stepwise mutation model (Pritchard and Feldman 1996), we expect, for a constant population size scenario,
denotes their average repeat number. Assuming a simple stepwise mutation model (Pritchard and Feldman 1996), we expect, for a constant population size scenario,
where N denotes the constant effective population size expressed in number of chromosomes and μ denotes the mutation rate per generation. Under a rapid population growth scenario, we expect
where ge is the number of generations since the rapid expansion started. S2 was estimated using MSA (microsatellite analyzer) software by Dieringer and Schlötterer (2003).
In a population that experiences a founder effect, a rare mutation (or a rare haplotype) that goes through the bottleneck and spreads rapidly because of the subsequent demographic growth appears younger by
generations, where d is the rate of population growth, m is the rate of genetic events (mutations, recombinations) used to time the founder effect (Luria and Delbrück 1943; Labuda et al. 1997), and fd=ed/(ed-1); if d is small, then fd∼d-1.
Results
Allelic Structure and Continental Distribution of dys44 Haplotypes
Eighty-six dys44 haplotypes, composed of the previously ascertained 35 variant sites with 36 derived alleles, were found in the sample of 1,343 X chromosomes from 33 populations (table 1). These haplotypes were either directly observed or inferred by genotype solving, as described in the “Material and Methods” section. Haplotypes are presented as strings of alleles at polymorphic positions (fig. 1), with the new alleles indicated and the ancestral (i.e., not new) ones left blank. Assuming an absence of recurrent mutations, we considered an allele to be ancestral if it was identical by state (IBS) to that found at the orthologous position in at least two ape species (Ziętkiewicz et al. 1998; Labuda et al. 2000). The observed high number of haplotypes and their allelic composition indicate that, in addition to mutations, recombinations (and, presumably, gene conversion events) largely contributed to their evolution.
Figure 1.

Allelic structure of dys44 haplotypes. Position IDs and haplotype names are arbitrary (consistent with Ziętkiewicz et al. 1997, 1998; Labuda et al. 2000). For clarity, only new (derived) alleles are shown, and an empty space implies identity with the ancestral allele (the same as that found in nonhuman primates), as shown at the top of the figure. The new alleles of the worldwide polymorphic loci (21 sites) are highlighted in color; those of the continentally restricted polymorphisms (15 sites) are indicated by bold letters, whereas the numbers in the column separating sites 72 and 85 indicate the associated length alleles of the Tn microsatellite. Geographic affiliations are indicated on the left (Af = African; Am = American; As = Asiatic; Eu = European; In = Indonesian/PNG; continentally shared haplotypes are left blank); haplotype names are color coded to indicate their structural clustering into haplogroups (see text and fig. 2), reflected by similarity to the dominant most frequent haplotype; and boxes indicate the shortest putative recombination/conversion sites relating the rare recombinant to the dominant haplotype of the group (only for non-African haplotypes).
Haplotype sequence differences were recorded in a simple distance matrix, with each allelic difference counted as one, and haplotypes were subsequently clustered using the NJ algorithm (Saitou and Nei 1987). Although the detailed topology of the resulting tree depended on the number of haplotypes, on their order in the input file, or on the seed number used, we found that the overall clustering pattern remained constant. A representative tree topology is shown in figure 2. Structural similarities within the haplotype clusters can be appreciated in figure 1, where the haplotypes are ordered in accordance with the output of the NJ clustering, with only small modifications to highlight the clustering. Histograms of continental (or regional) frequencies of the corresponding haplotypes, shown next to the NJ tree in figure 2, demonstrate that almost every cluster is dominated by a relatively frequent haplotype. Figure 1 shows that haplotypes within the clusters can be related to the dominating frequent haplotype through a point mutation, recombination, or gene conversion event. For example, a C→T transition at site 87 of a B001 chromosome explains the appearance of haplotype B026. A double recombination or gene conversion event could be inferred to have given rise to haplotype B031, in which allele G at site 70 becomes A on the otherwise intact B001 haplotype background. In haplotype b054, a simple recombination explains the substitution of the rightmost segment of B001 (sites 90–95) from, for example, a B002 haplotype. The same recombination, exchanging the rightmost segments of B002 and B001, could have reciprocally created haplotype B004. However, whereas b054 in our sample was observed only once, in Asia, B004 was seen as >100 copies on different continents. Interestingly, the geographical maxima of B004 and B002, which is the essential ingredient for recreation of B004 by recombination, do not overlap (fig. 2; table 1); in the Americas, where B004 occurs at its highest frequency, haplotype B002 is virtually absent. This strongly suggests that the multiple copies of B004 chromosomes in American populations are identical by descent (IBD), having spread as a result of a founder effect rather than being due to recurrent recombinations. More-detailed analysis of the outcomes of recombination events is presented in appendix A and suggests that most, if not all, of the haplotypes we report are likely to have originated only once. Hence, the present-day population frequencies of dys44 haplotypes are likely to reflect their times of appearance in a population and the demographic events that followed, such as migrations or founder effects. Thus, to learn about the genetic history of this locus, we have to analyze the evolution of haplotype structure as well as the frequencies of variant haplotypes across populations. In the following, we will refer to a haplotype cluster by the name of its dominating haplotype and to its structurally associated haplotypes as a “haplogroup.” The term “haplogroup” was originally coined to denote a monophyletic cluster of mitochondrial sequences (Torroni et al. 1993); the same term has been adopted for the Y-chromosome haplotype lineages (Bianchi et al. 1998). However, in those nonrecombining systems, a haplogroup consists of chromosomes with new polymorphic sites that arose on the ancestral haplotype background. Here, mutation as well as recombination/conversion events participated in the process; assignment of the new recombinant haplotype to a haplogroup is based on the proportion of the parental background in the recombinant’s structure.
Figure 2.

NJ tree (A) of haplotype sequences and the corresponding haplotype frequencies (counts) (B) in five continental regions. Structural affiliations of haplotypes use the same color code as in figure 1. Asterisks (*) indicate haplotypes that occur in a single continental group.
Clustering of haplotypes on the basis of their sequence similarity (figs. 1 and 2) reflects their “immediate” genealogical relations. Recombinants tend to fall into the haplogroup of their recipient haplotype (i.e., the one of the two recombining haplotypes that constitutes the major portion of the recombinant haplotype). The majority of haplotypes found outside of Africa can easily be connected to the most frequent haplotype of its haplogroup, if we assume simple mutation and/or recombination events (the putative sites of the latter are indicated by boxes in fig. 1). In contrast, the structural/genealogical relations of the haplotypes endemic to sub-Saharan populations are less straightforward and therefore more difficult to trace. Most of the endemic African haplotypes not only form separate clusters but often require multiple genetic events to be mutually reconnected; some intermediate haplotypes may be missing (fig. 1). Taken together, these observations suggest a longer history of the African-only haplogroups. On the other hand, some of the African haplotypes fall within the worldwide-distributed haplogroups, such as B001/B003 or B002/B004, and seem to have simpler genealogical relations within these groups, which might suggest a recent origin.
Sequence Trees versus Population Trees
The NJ tree obtained from the distance matrix of haplotype sequences in figure 2 can be compared with the NJ tree of populations presented in figure 3. The matrices of interpopulation distances used to obtain population trees were based on either allele frequencies or haplotype frequencies. Whereas the sequence tree was rooted with the hypothetical haplotype featuring ancestral (i.e., nonhuman primate) alleles at all sites, the outgroup in population trees was obtained by setting the ancestral allele frequencies at all sites to one or by setting the frequencies of all present-day haplotypes to zero. The topologies of both population trees were very similar; the one shown in figure 3 was obtained using haplotype frequencies. The use of different options of the GENDIST program did not significantly influence tree topology, and the trees preserved all geographical groupings with the exception of the Maya and Karitiana, which were swapped between European and Asian clusters (fig. 3). African populations were always at the root of the tree, separated from non-Africans. The population and regional subdivisions among non-Africans were, almost without exception, very shallow.
Figure 3.

NJ tree of populations from the distance matrix calculated from haplotype frequencies. The bootstrapping values show how many times in 100 runs the cluster to the right was observed.
It is interesting that, in the haplotype sequence tree in figure 2, the dys44 haplogroups endemic to sub-Saharan Africa, such as B010, B007, and B012, appeared in the middle of the tree. The haplotype that emerged closest to the root was B006, which differed from the ancestral haplotype by only four new (derived) alleles. Two of these new alleles (sites 10 and 85; see fig. 1) were found only in B006 and related recombinants, which, together with the scarcity of new alleles on the haplotype, made this haplotype structurally distinct from the remaining haplogroups. It is most interesting that B006 was virtually absent in sub-Saharan Africa. Among the African populations studied, it could be inferred only in the Rimaibe from Burkina Faso, in which a single B006 was found in a female sample whose genotype had been resolved into B006 and a new haplotype, b118. This singular occurrence of B006 in the Rimaibe might not, in fact, be exceptional. A partial haplotype (which is thus not reported in table 1) in another Rimaibe sample represented a mosaic of an African-specific haplotype (from site 58 to 72) and B006 (from site 85 to 95). B006 was also found in three African American chromosomes, but we considered this finding as representing the result of Amerindian admixture. What points towards an Amerindian rather than an African origin of B006 among African Americans is the fact that B006 is preponderant in Amerindians and that the B006 chromosomes in our African American samples were found associated with an Amerindian variant of a very informative compound-microsatellite marker located 5 kb downstream of dys44 (V.Y., T.W., and D.L, unpublished data). Thus, if the presence of B006 in Rimaibe is not accidental (e.g., if it is not due to recent gene flow), they remain unique as the sole carriers of B006 in our sub-Saharan African sample.
Distribution of dys44 Haplotypes across Continents and Population Expansion
The occurrence of the 86 dys44 haplotypes across populations is reported in table 1. Nineteen of these haplotypes were shared by at least two continental groups, whereas 67 had a limited distribution: 6 were restricted to Europe, 9 to Asia, 8 to the Indonesian Islands and Papua New Guinea (PNG), 3 to the Americas, and as many as 41 to sub-Saharan Africa. However, it was not only the number of continent-specific haplotypes that distinguished Africa from the other continents. Continentally restricted haplotypes were carried by only 5% of all non-African chromosomes (n=50 out of 1,053), whereas, among African chromosomes, the African-specific haplotypes represented 48% of the sample (n=139 out of 290) (see table 1). Thus, the majority (95%) of chromosomes outside of Africa carried frequent, continentally shared haplotypes, as shown in figure 4. Such a frequency distribution, dominated by a few frequent haplotypes followed by a tail of rare ones, is suggestive of a population expansion. Indeed, both in the total sample and in both groups when subdivided into African and non-African samples, the distribution deviated significantly from neutral expectations under the infinite allele model (Ewens 1972).
Figure 4.

Population frequencies of the frequent, continentally shared haplotypes. Different colors, if present, indicate different Tn alleles shared by the same B haplotype. Note that B006 is not shown in the African American sample, because of the strong evidence that it is a result of recent admixture with the Amerindian population (see text).
Assuming a bottleneck scenario (Tishkoff et al. 1996), we may consider continentally shared haplotypes to represent the ancestral population before expansion and consider rare, region-specific recombinant haplotypes (table 1) to represent the diversity acquired after the range expansion. We used equation (2) (with Pg corresponding to the proportion of continentally shared haplotypes in a given population cluster) to estimate the time elapsed since these proposed colonization events occurred (table 2). Thus, the time of separation between non-Africans and sub-Saharan Africans is estimated at ∼55 kya, the colonization of Americas at ∼13 kya, and the colonization of Europe at ∼27 kya. The time estimate for the colonization of the Indonesian Islands and PNG combined is much greater (∼145 kya), whereas the Indonesian samples alone yield estimates comparable to those of the Asiatic sample (∼33–35 kya). The latter difference reflects the greater proportion of recombinants in the PNG sample. This proportion, augmented by the presence of multiple, presumably IBD copies of the recombinants (table 1), reflects a greater departure from the starlike phylogeny model in PNG than in the other population groups studied and may also account for the much greater variance associated with the corresponding time estimate (Di Rienzo et al. 1998; Kimmel et al. 1998).
Table 2.
Estimates of the Time Since Expansion for Each Continent Based on Recombination within the dys44 Segment
| Area/Group | No. ofChromosomes | No. ofRegion-SpecificRecombinants | -lnP | Putative TimeSince Expansiona(g×25)(years) |
| Americas | 231 | 4 | .017 | 13,300 |
| Asia | 488 | 20 | .042 | 32,700 |
| Europe | 178 | 6 | .034 | 27,300 |
| Indonesia/PNG | 156 | 15 | .101 | 79,000 |
| Indonesia | 91 | 4 | .045 | 35,100 |
| PNG | 65 | 11 | .185 | 144,800 |
| Non-Africans | 1,053 | 45 | .044 | 34,100 |
| Non-Africans versus Africans | 1,053 | 72 | .073 | 55,300 |
The putative expansion time was calculated from equation (5), using rapp=.32×10-4 (see the “Material and Methods” section) and an arbitrary 25 years as generation length. Note that, given the possible variation in local recombination rates and the sampling error and simplifying assumptions of the underlying demographic model, the variance associated with these estimates may be very large.
Although, given the large uncertainty associated with the obtained values, the results have to be treated with caution, they do provide a reasonable insight into past events. An alternative method of obtaining independent estimates of the timing of past events is the assessment of the dys44 Tn allele distribution, as considered in the next section.
Tn—Microsatellite Variance in Populations
Frequency distributions of the Tn alleles (with length ranging from T14 to T24) are shown in figure 5. In the world sample (fig. 5A), the distribution is bimodal, with the larger peak at T15 and a smaller peak at T22; T18 was not observed. Outside of Africa (fig. 5B), the picture is even simpler: the major peak at T15 is flanked by a few chromosomes carrying alleles T14 and T16; the smaller peak, T22, is likewise accompanied by its neighboring alleles, T21 and T23. In the African sample (fig. 5C), both length modes are present, but the second peak is at T20 rather than T22. Furthermore, the T15 mode in Africa is preferentially associated with the chromosomes carrying continentally shared haplotypes (fig. 5E), whereas the T20 mode is found with the subset of exclusively African chromosomes (fig. 5D).
Figure 5.

Frequency distribution of length alleles of the Tn microsatellite worldwide (A), in non-Africans (B), and in sub-Saharan Africans (C), subdivided further into chromosomes carrying African-specific haplotypes (D) and those shared with other continents (E).
In the Tn distribution in figure 5B (non-African chromosomes), the flanking alleles differ from the major alleles (T15 or T22 ) by one length unit only and thus appear to have been derived from the major allele by a simple addition or deletion of one T unit at a time. The distribution of the Tn alleles considered in association with individual B haplotypes (table 1) is also consistent with such a mechanism; there is no evidence of mutations occurring in jumps greater than one (three chromosomes carrying haplotype B005 associated with T15 are most likely explained by a recombination rather than Tn mutation). Therefore, it appears that this system conforms to a simple version of the stepwise mutation model (Ohta and Kimura 1973). Accordingly, Tn diversity can be described by the variance S2 in the number of repeats (eq. [3]). The results obtained for our population samples are summarized in table 3. When all samples are considered together, the estimated S2 value of 4.3 is lower than the estimated value of 7.2 for sub-Saharan Africa and slightly higher than the estimated values of 3.3–3.9 for the other continents (disregarding the exceptionally low S2 value of 0.2 in the Americas).
Table 3.
Variance in Allele Length of the Tn Microsatellite across Continents for the Entire Range of Allele Lengths (S2), for Modal Length 15, Comprising Alleles 14–17 (S2(15)), and for Modal Length 22, Comprising Alleles 19–24 (S2(22))
| Area/Group | N | S2 | S2(15) | S2(22) |
| Americas | 231 | .173 | .173 | … |
| Asia | 488 | 3.870 | .047 | .097 |
| Europe | 178 | 3.403 | .170 | .077 |
| Indonesia/PNG | 156 | 3.323 | 0 | .091 |
| Non-Africans | 1,053 | 2.913a | .098 | .093 |
| Sub-Saharan Africa | 290 | 7.161 | .727 | 1.168 |
| Shared | 151 | 4.323 | .674 | .066 |
| Specific | 139 | 6.139 | .862 | .966 |
| Overall | 1,343 | 4.338 | .204 | 1.316 |
When the Americas were excluded, the total variance (n=822) was S2=3.658.
Under the constant-population-size model, S2 is expected to correspond to the product of mutation rate μ and population size N (eq. [4]), whereas, in the case of rapid population growth, it is expected to correspond to the product of the mutation rate and the time since expansion, ge (eq. [5]). Using the Tn world variance estimated above and assuming the constant effective population size of 11,200—that is, 33,600 X chromosomes (Ziętkiewicz et al. 1998)—one would estimate the Tn mutation rate, from equation (4), at μ=1.3×10-4 per generation. This estimate, although falling within the usual range of microsatellite mutation rates, does not fit the rest of our data. In particular, the S2 values estimated separately within the abundantly represented haplotype lineages, such as B001, B002, B003, or B004, do not exceed 0.01 in non-Africans (table 4). Substituting these S2 values and the mutation rate above into equation (2) would lead to the unrealistically small population size of ⩽100 chromosomes carrying each of these frequent haplotypes. Using a more appropriate scenario of rapid population growth (i.e., using eq. [5] rather than eq. [4]) would lead, in turn, to a time estimate of ⩽100 generations (2,500–3,000 years) since the expansion of these lineages, which is equally untenable. The source of the incongruity seems to lie in the above estimate of the Tn mutation rate. Given the bimodal distributions of allele length (fig. 5), it is likely that the overall estimates of S2 are inflated and that we should rather obtain S2 separately for the short (T15) and long (T20/22) modes, which are naturally demarcated by the nonexistent allele T18 . From the resulting values of S2(15) and S2(20/22) (table 3) and using equation (4), we obtained mutation rate estimates of 0.6 and 3.9×10-5 (average μ=2.3×10-5) for the worldwide sample, and 2.2 and 3.5×10-5 (average μ=2.8×10-5) for the African sample alone. In the above, equation (4) was used because we considered that the impact of the stationary phase, extended over a long evolutionary time period and described by a constant-population-size model, would prevail in these two samples, in spite of any subsequent population expansion. However, if population growth was preceded by a bottleneck (Tishkoff et al. 1996), one would expect to observe a substantial loss of diversity. Indeed, the estimates of S2(15) and S2(20/22) (0.098 and 0.093, respectively) in non-Africans were smaller by one order of magnitude (table 3), consistent with a bottleneck scenario and suggesting that equation (5) may be more appropriate for non-Africans. Accordingly, assuming the mutation rate μ=2.3×10-5, we estimated the time of the non-African lineage expansion (ge from eq. [5]) to be 4,040–4,260 generations, or 101–107 kya.
Table 4.
Variance in Allele Length of the Tn Microsatellite for the World, Non-Africans, and Africans, by Haplotype[Note]
|
World |
Non-Africans |
Africans |
|||||||
| Haplotype | N | S2 | -lnP | n | S2 | -lnP | n | S2 | -lnP |
| B001 | 401 | .010 | .010 | 366 | .011 | .011 | 34 | .0 | .0 |
| B002 | 292 | .284 | .159 | 222 | .005 | .004 | 69 | .955 | .938 |
| B003 | 128 | .038 | .040 | 116 | .009 | .009 | 12 | .242 | .410 |
| B004 | 104 | .0 | .0 | 102 | .0 | .0 | 2 | .0 | .0 |
| B005a | 59 | .103 | .107 | 48 | .106 | .110 | 11 | .091 | .095 |
| B006b | 91 | .154 | .207 | 87 | .152 | .203 | 1 | .0 | .0 |
| B019 | 5 | 1.200 | .511 | 3 | .0 | .0 | 2 | .0 | .0 |
| B051 | 2 | .500 | NA | 2 | .500 | NA | … | … | … |
| B007 | 35 | .281 | .06 | … | … | … | 35 | .281 | .59 |
| B010 | 12 | 1.091 | .69 | … | … | … | 12 | 1.091 | .693 |
| B012 | 9 | 2.250 | .401 | … | … | … | 9 | 2.250 | .401 |
| B046 | 5 | .200 | .223 | … | … | … | 5 | .200 | .223 |
| b061 | 5 | .200 | .223 | … | … | … | 5 | .200 | .223 |
Note.— Only haplotypes showing variable Tn are shown. In calculating the proportion of intact ancestral alleles, P, the major allele was assumed to be ancestral, which is a fragile assumption when made with a low number of chromosomes. The -lnP values were nevertheless calculated, for comparison with S2. NA = not applicable.
Three B005 chromosomes with Tn allele 15 were not considered in the calculation. We assumed that this length allele was acquired in B005 by recombination rather than by mutation (see text).
Because of the likely origin of three B006 haplotypes in African Americans by admixture (see text), only B006 found in the Rimaibe was retained in the African sample.
Chromosomal Lineages as Markers of Population Expansion
The expansion time estimation can be performed separately for each of the distinct B haplotype lineages, for which the issue of distinct modes of allele lengths no longer exists. The S2-based age estimates can be obtained from equation (5), as discussed above. Under the plausible assumption that the most frequent Tn allele is the ancestral one, one can apply equation (1) to assess the age of a haplotype lineage from -lnP (where P is the proportion of haplotypes carrying the ancestral Tn allele; also note that g from equation [1] is equivalent to ge from equation [5]). The results for all B haplotypes associated with more than one Tn allele are presented in table 4; haplotype B004 is included as well, to emphasize its lack of Tn allele diversity. The values of S2 and -lnP are generally very similar; at low variance, the results are identical. Their differences at higher values can be ascribed to the fact that the legitimacy of equation (1) was stretched when used for data with a level of divergence from the ancestral state of >10%. Table 4 demonstrates that, outside of Africa, there is little or no variance in the Tn allele length associated with either haplotype B002 and its recombinant product B004 or with B003. In contrast, among the African chromosomes, there is a large variance associated with B002 and B003. This discrepancy suggests that the founding event of the B002/B004 and B003 haplotypes outside Africa was relatively recent and was accompanied by a severe bottleneck. Such a bottleneck presumably reduced the originally high repeat-length variance, as observed today only in African B002/B004 and B003 chromosomes. In Africa, a large variance also accumulated in a number of other, continent-specific haplotypes, which never left the continent (table 4), either because they did not pass through the bottleneck or because they belonged to the local, nonexpanding lineage(s) (Labuda et al. 2000). The ubiquitous, young B001 haplotype, which is monomorphic at the Tn site on African chromosomes and is associated with a small variance in Tn length in non-Africans, appears to be a marker of the expanding lineage that spread both outside and within Africa (fig. 4).
Substituting the non-African S2 or -lnP values for the above haplotypes (B001, B002/B004, and B003; table 4) in equation (5) or equation (2), we arrived at out-of-Africa bottleneck time estimates of ∼5 and 12 kya. These dates appear too recent when compared with the historically documented Upper Paleolithic expansions dated at 35–50 kya. However, under the model of a bottleneck followed by population expansion (i.e., a founder effect), the time of the founding event estimated from genetic data is expected to appear younger than it actually is (Luria and Delbrück 1943; Hastbacka et al. 1992; Labuda et al. 1996, 1997). This can be corrected using the heuristic approach of Luria and Delbrück (1943), as described elsewhere (Labuda et al. 1997; Colombo 2000; Slatkin and Rannala 2000). Such a correction of the age of the founder effect is based on demographic growth and the rate of either recombination or mutation, whichever causes the decay of the ancestral haplotype/allele with time. Given a demographic growth rate of 0.002–0.005 per generation and given the above mutation rate, the correction (from eq. [6]) is very substantial (in the range of 27–56 ky), placing the age estimates for these out-of-Africa lineages within a plausible time range.
The non-African ancient haplotype B006 displays a relatively large Tn variance, an order of magnitude greater than in B001, B003, and B002/B004, although smaller than that of the African-only haplotypes (table 4). A similarly moderate level of Tn length variance characterizes haplotype B005, found in both Eurasia and Africa. B006 apparently did not go through the out-of-Africa bottleneck, since it is found only outside Africa; the same may be true for B005, for which the associated Tn variance in Eurasia is equal to if not higher than that in Africa. The estimated times of expansion of the B006 and B005 lineages—165 kya and 115 kya, respectively (obtained using eq. [5])—are older than the time of the out-of-Africa bottleneck estimated above (100 kya), suggesting that B006 and, possibly, B005 might have originated outside Africa.
Discussion
Human genetic history and the partitioning of genetic diversity across populations have been extensively studied through the analysis of mitochondrial and Y-chromosome DNA. The worldwide breadth and population depth of such studies have never been matched by investigations employing nuclear, non-Y DNA polymorphisms. However, to truly understand the genetic structure of human populations, we have to collect information from loci other than those representing the maternal or paternal lineages only. Towards this goal, we analyzed genetic diversity in the X-chromosome segment dys44 (Ziętkiewicz et al. 1997, 1998; Labuda et al. 2000), in 33 populations worldwide. This system provides a genomic record of a variety of mutations, including nucleotide substitutions, small insertions, deletions, and changes in a Tn repeat-length polymorphism, as well as a record of informative recombinations, as reflected in the structural diversity of dys44 haplotypes. The interpretation of this genomic record should take into account the temporal frame within which the underlying genetic events have occurred, the demographic histories of the analyzed populations, and the sampling scheme applied by the investigator.
The initial ascertainment of polymorphisms within the dys44 segment was limited to a sample of 250 chromosomes (Ziętkiewicz et al. 1997). Genotyping of the enlarged population sample in this study was performed using the ASO-hybridization technique, aimed at the already detected polymorphisms. Therefore, no new nucleotide variants were expected to show up. This limitation did not extend, however, to the ascertainment of new dys44 haplotypes or to the Tn alleles reported here. The proportion of non-African chromosomes with non-African continental-specific haplotypes was 5%, whereas 48% of African chromosomes carried African-specific haplotypes. Among the 35 polymorphic sites of the dys44 segment, there were 14 new SNP-like alleles endemic to Africa, versus 2 private alleles found outside Africa (fig. 1). The variance in Tn allele repeat length for both length modes (S2(15) and S2(20/22)) was ∼1 in Africa versus ∼0.1 in non-Africans. The comparisons above indicate a ratio of ∼10:1 for the different measures of dys44 variability between Africans and non-Africans. This observation is not new: the greater diversity in sub-Saharan Africans can be expected on the basis of previous studies (Cann et al. 1987; Batzer et al. 1994; Bowcock et al. 1994; Tishkoff et al. 1996; Jorde et al. 1997; Stoneking et al. 1997; Clark et al. 1998; Hammer et al. 1998; Ziętkiewicz et al. 1998; Kaessmann et al. 1999; Kidd et al. 2000; Yu et al. 2001).
The excess of African genetic variation has been taken as evidence of an older age of African populations (Cann et al. 1987; Chen et al. 1995), a greater long-term effective population size in Africa (Relethford 1995), or a severe out-of-Africa demographic bottleneck (Tishkoff et al. 1996). Each of these models, which perceive the partitioning of contemporary genetic diversity as resulting from a prehistoric rift between sub-Saharan Africa and the rest of the world, can provide a nonexclusive explanation of the historical conditions that led to contemporary population characteristics. On the basis of our earlier analysis of dys44 haplotypes (also see Baird et al. 2000; Labuda et al. 2000), we proposed a more subtle and complex scenario that can unite the above aspects. In our model, the genetic pool of sub-Saharan Africans reflects the contribution of at least two lineages, which had evolved separately for some period of time and eventually hybridized. One of these lineages existed in the population(s) that underwent an out-of-Africa expansion, whereas the other lineage(s) were present in groups that remained in Africa and/or contributed locally to the diversity of the emerging worldwide population. The former lineage is represented by the frequent, continentally shared haplotypes and their structurally related variants, and the latter is represented by diverse haplotypes, most of them carrying several of the 14 new alleles not found on chromosomes from outside Africa (fig. 1).
The NJ trees of populations (as in fig. 3) illustrate a “classical” out-of-Africa topology, as previously observed in population trees from other genetic systems (e.g., Bowcock et al. 1994; Cavalli-Sforza et al. 1994; Fan et al. 2002). Most published trees of sequences (or haplotypes) have also presented an out-of-Africa topology, with Sub-Saharan, African-only haplotypes emerging closest to, or even containing, the ancestral sequence. This has been observed for mitochondrial (Cann et al. 1987; Chen et al. 1995) and Y-chromosome (Hammer 1995; Underhill et al. 2000) haplotype trees, as well as for the trees of many non-Y-chromosome nuclear DNA haplotypes (e.g., Harding et al. 1997; Harris and Hey 1999; Jaruzelska et al. 1999; Kaessmann et al. 1999; Verrelli et al. 2002). However, the topology of the haplotype (sequence) tree based on NJ clustering of dys44 haplotypes (fig. 2) did not match the pattern typical for other DNA segments. The B006 haplotype, which is virtually absent in sub-Saharan Africa, was closest to the root, carrying only four new alleles on the ancestral background (as defined by the ancestral haplotype). The haplogroups endemic to sub-Saharan Africa, such as B010, B007, and B012, appeared in the middle of the tree. Although structurally diverse, these haplotypes carrying African-specific alleles are sufficiently distinct from the non-African-specific haplotypes to form a relatively well-defined cluster. The fact that the new, African-specific alleles are distributed on a variety of poorly related and structurally diverse haplotypes (fig. 1) indicates the contribution of historical recombination. This suggests the relatively old age of the African-specific haplotypes (by the same token, the contributing mutations must also be old, in spite of their low new-allele frequencies).
African-specific haplotypes are almost exclusively associated with Tn alleles representing the long T20/22 mode. This further distinguishes them from the non-African-specific haplotypes, which, except for the haplotype/haplogroup B005, are associated with Tn alleles representing the short T15 mode. It is among the African-specific haplotypes, in spite of their small number in the analyzed sample, that the S2 variance of the Tn allele length is the highest, indicating again the relatively old age of these haplotypes (table 4). On the other hand, the frequent, continentally shared haplotypes, such as B001, B002, B003, and B005, are also well represented in sub-Saharan Africa, thus supporting the split ancestry of modern African populations, as previously suggested by Labuda et al. (2000).
Detailed inspection of the individual haplotype lineages can reveal information concerning their origins. There is little doubt about the African origin of B002 and B003. On the basis of the variance in their short-mode Tn repeat, these haplotypes appear very old when analyzed in Africa but not when studied outside of Africa (in contrast to the African B002 and B003 haplotypes, their frequent non-African copies are almost monomorphic for the allele T15). Conspicuously, even in Africa, there are no B002 or B003 chromosomes found associated with the long-mode Tn alleles, although such alleles could have been acquired by recombination with the numerous T20/22-length-mode haplotypes found in the same sub-Saharan populations. The study of recombination within the haplotype lineages can also aid in the search for their origins. For example, recombination is a plausible mechanism by which the T15-associated B005 (from the otherwise T22-linked haplotype) was created in Asia (tables 1 and 4); in addition, a recombination event exchanging the 3′ end of B002 was also responsible for creating B004, which is very rare in Africa, frequent outside of Africa, and completely monomorphic for T15. The virtual absence of 3′ terminal crossovers between B002 and B003, on the one hand, and the exclusively African haplotypes associated with the T20/22 length mode, on the other, suggest that their cohabitation started only recently and that they evolved separately for a significant period of time. B005, with S2 variance at similarly moderate levels within and outside Africa, seems to represent an old lineage, which did not necessarily go through the bottleneck. Although its position within the haplotype tree in figure 2, as well as its association with the T20/22 length mode, suggests a structural connection of B005 with the African-specific lineage, its geographic origin remains elusive.
In contrast to B002/B004 and B003, the ubiquitous B001 haplotype seems to be young both within and outside of Africa (table 4). B001 is almost exclusively associated with the predominant allele T15, as is observed in all continental populations, including Africa. B001 could have been recently derived from B003 by elimination of the new allele A at site 65 (fig. 1); if such an event happened recently, it would explain the low associated Tn diversity. Its time of origin, however, had to precede the out-of-Africa expansion (since it was the B001-carrying population that spread most widely, as is reflected in the observation that a quarter of all chromosomes observed worldwide are of type B001), and the out-of-Africa spread of B002 and B003 haplotypes appears concomitant with B001 expansion, thus providing good evidence for the African origin of the latter lineage. The fact that B001 constitutes 10% of present-day sub-Saharan African chromosomes suggests that expansion not only was directed out of Africa but also occurred within Africa itself; understandably, the “expanding” chromosomes on this continent were diluted within the existing gene pool and, hence, did not reach the high frequencies observed outside Africa.
Evidence for an ancient within-Africa expansion has also been observed in mtDNA (Watson et al. 1997). Watson et al. (1997) proposed that most of the African mitochondrial sequences originated from demographic expansions that started ∼60–80 kya, the earliest of which led to the colonization of Eurasia. The same expansion presumably involved haplotypes B002 and B003. An expansion signature, reflected by the lack of the B002 and B003 Tn variability, would only be visible outside of Africa because of a bottleneck and the subsequent population growth, whereas any similar effect within Africa would likely have been masked by the presence of endemic B002 and B003 copies polymorphic at the Tn site. By the same token, the fact that the old, African-specific alleles (characteristic of African-specific haplotypes) occur at a relatively low frequency could be explained by their “dilution” as a result of hybridization with the expanding lineage. This, again, is in agreement with earlier observations by Watson et al. (1997), who noted that a minority of mtDNA sequences (13%) fell outside the presumed out-of-Africa expansion clusters. It was postulated that these sequences echoed the time before the out-of-Africa expansions, when the human mitochondrial gene pool was more diverse. In our data, this “minority” is reflected by the local African-specific lineages.
Although B001 appears to be the best marker of the recently expanded lineage, it does not aid our understanding of the expansion route(s) out of Africa. The clues here may come from the analysis of other frequent dys44 haplotypes, which together suggest that the impact of modern human expansion on human diversity in Africa and other continents does not reflect a single demographic event. The frequency distributions of B002 and B003 outside Africa are skewed: B002 is poorly represented in Europe and is relatively abundant in Southeast Asia, in contrast to B003, which is frequent in Europe and relatively rare among Asiatic populations. Do these distributions mark two separate expansion events, or do they result from genetic drift (due to the initial population bottleneck) and subsequent reinforcement as a result of their geographic separation? Although there are insufficient data to examine possible temporal differences in the time of expansion of the two lineages, the geographic separation of the haplotype frequency gradients suggests independent expansions, towards Europe and towards Southeast Asia, that could correspond to the northern and southern routes referred to in the literature (Lahr and Foley 1994; Kivisild et al. 1999; Quintana-Murci et al. 1999).
In our data, the northern route seems to be locally enriched by the addition of another, very ancient and distinct lineage, represented by a single haplotype, B006. This haplotype is not only structurally the simplest but is also the one that lies closest to the root, as defined by the hypothetical haplotype with ancestral alleles at all positions (fig. 2). Two of B006’s four derived alleles, at sites 10 and 85, are private (restricted to B006 and its closest recombinants), whereas the remaining two, with worldwide frequencies of 0.7–0.9 at sites 30 and 95, are among the oldest polymorphisms of the dys44 segment (Ziętkiewicz et al. 1998; Ziętkiewicz and Labuda 2001). The Tn allele variance associated with B006 independently suggests the haplotype’s relatively ancient origin. In spite of the otherwise substantial role of recombination in the dys44 segment, there is no record of extensive B006 interaction with haplotypes from other lineages, and only a few recent recombinant haplotypes are observed (fig. 1). These characteristics are reminiscent of the African-specific haplotypes that are structurally distinct, old, continentally restricted, and that carry private polymorphisms. B006 is practically absent in Africa and is poorly represented in Southeast Asia, in contrast to its relative abundance in Europe, central Asia and the Americas. Where, then, could haplotype B006 and the lineage it represents have originated?
Given its structural uniqueness and its large Tn allele variance, B006 is unlike any of the haplotypes associated with the main, out-of-Africa expanding lineages (i.e., B002/B004 and B003/B001). We can therefore postulate two scenarios regarding the origin of B006. First, it may represent an ancient African lineage that made its way out of Africa, spreading within the newly colonized regions and becoming extinct in its place of origin. The arid climatic conditions (due to the glacial maxima) between 50 and 100 kya were likely to have induced population fragmentation (Lahr and Foley 1994), thereby allowing sister populations of modern H. sapiens to evolve in Africa prior to the Upper Paleolithic expansion. Therefore, in a way similar to the structurally unique African-specific lineage(s), B006 could represent a vestige of an independent northern lineage. Alternatively, B006 may represent an as-yet-unknown non-African contribution to the human gene pool. Such a “northern” contribution to the contemporary human gene pool could have escaped detection in studies of mitochondrial or Y lineages, because of the shorter “evolutionary memory” of these systems as compared with nuclear loci. It is notable, however, that B006 has been found at very low frequency in one sub-Saharan population, the West African Rimaibe. The question of whether this occurrence is (i) suggestive of the haplotype’s ancient African roots, (ii) a record of back migration from Eurasia, as postulated by a number of authors (Hammer et al. 1997; Harding et al. 1997; Cruciani et al. 2002), or (iii) a result of more recent admixture with northern populations across the Sahara remains unanswered. However, the presence of another, essentially non-African haplotype (B004) in the Rimaibe would lend support to the back migration and/or recent admixture scenario(s). On the other hand, we have to emphasize that our results were obtained with a limited sub-Saharan African sample, mostly representative for West Africans and Pygmies. Among other important groups, such as the Khoisan from South Africa and/or the East Africans, which were not included in our sample, the latter could be of particular interest in this context. For example, mtDNA haplogroup M was long considered typical of Asians, until it was observed that a specific and very limited subset of it (now termed “Ml”) was present in East Africans (but not in other African groups); this lead to the final conclusion that M was indeed among the mtDNA haplogroups that were involved in the out-of-Africa exit (Quintana-Murci et al. 1999). It is therefore necessary to extend the analysis of the dystrophin locus to include other African populations. It is possible that such studies will not only provide evidence for the origin of B006 but also shed more light on a number of ancient lineages that contributed to the human gene pool (also see Watson et al. 1997).
A similar scenario, of the hybridization of distinct lineages ultimately giving rise to modern H. sapiens, was previously suggested by Baird et al. (2000), to explain the coexistence of deeply diverged haplotypes adjacent to the Xp/Yp and 2q telomeres in humans. The study suggested that the lineages could have remained separate for as long as 1.9 million years, although the time of their fusion might have been as recent as the out-of-Africa expansion of modern humans. The results of Baird et al. (2000) parallel those from dys44. Although estimating the time to the most recent common ancestor of the dys44 haplotypes is complicated by a number of historical recombination and gene conversion events, the divergent structure of the African-specific and continentally shared haplotypes argues for their independent evolution for a significant period of time (Labuda et al. 2000). A similar conclusion can be reached for the lineage represented by B006. Taken together, these data suggest a mosaic origin of modern human populations; this amalgamation of lineages did not precede but rather accompanied the great expansion of one of these lineages represented by haplotypes B003/B001 and B002/B004.
We estimated the time since the bottleneck that accompanied the out-of-Africa expansion by two independent approaches: one based on population frequencies of dys44 recombinants, and the other based on variance in the associated Tn microsatellite, both of which provided relatively consistent results. The recombination-based estimates presented in table 2 were obtained under the assumption that continentally shared haplotypes represented the ancestral population before expansion, whereas region-specific recombinants were acquired after the initial expansion. There are a few factors that may render these estimates uncertain. The first of these is the recombination rate, which was determined at a “macro” scale from mapping data (Kong et al. 2002) but may differ locally because of micro-scale fluctuations (such as the presence of recombination hotspots, as reported by Harding et al. [1997]). The second is that some of the region-specific haplotypes, rather than being the result of postexpansion recombination, might in fact represent old but rare sequences; such a phenomenon would lead to an overestimation of the time since expansion. However, this can be at least partly controlled by inspecting the structure of the new haplotypes and ensuring that they are genealogically simply related to their assumed ancestral haplotypes (i.e., those associated with the expansion), as is the case in dys44. Third, the overall proportion of informative recombinants, estimated from our total data at 30%, actually fluctuates regionally, depending on the local haplotype diversity and structural complexity. Because of this, the level of informative recombinations within dys44 is notably higher in Africa than outside of Africa. However, African samples were not analyzed using this method, because of difficulties in assigning cohorts of ancestral haplotypes. Finally, the variance associated with the estimated time since expansion is affected by population history; strong expansion reduces variance and will be discussed further, in the case of PNG populations.
The second approach we took to estimating the time since expansion utilized the variance at the Tn microsatellite. Its mutation rate, 0.23×10-4, was assessed from the observed variance of two Tn length modes and from the effective population size (estimated previously by Ziętkiewicz et al. 1998). We justified the separate analysis for each Tn length mode because we observed (i) the persistent association of a single allele length mode with a particular B haplotype and (ii) a simple mutation mechanism that changed the allele length by one repeat unit at a time (table 1; figs. 4 and 5). The two length modes could have been created either by a rare mutational jump, over many repeat units, or by a duplication of the short ancestral allele (11–13 units); once formed, the two modes could have coexisted and evolved independently. Alternatively, the much greater effective population size of ancient hominids (Chen and Li 2001) might have resulted in a broader spread of Tn allele lengths in the ancestral human population; only the lineages representing the short and long alleles could have persisted after population fragmentation and size reduction (e.g., Takahata 1994; Stiner et al. 1999; Satta and Takahata 2002). However, regardless of the underlying model, the estimation of variance (S2) for each length mode proved reasonable and provided consistent results.
The separation time between Africans and non-Africans, estimated from Tn variance in non-Africans, was 101–107 kya. This likely represents an overall figure summarizing the contributions from both the older (B005 and B006) and younger (B001, B003, and B002/B004) haplotypes. Using the Tn variance estimated from non-African populations for these haplotype lineages, we arrived at the out-of-Africa bottleneck time estimates of 5–12 kya. After applying a correction for the effect of a growing population, the estimated time since the out-of-Africa expansion increases to 27–56 kya, placing it within a historically plausible time range that corresponds to the documented Upper Paleolithic expansions of 35–50 kya. However, the above estimates have to be treated with caution, because of the large variance inherently associated with microsatellite markers. The use of the go correction, which depends on the rate of population growth rate d, brings an additional source of uncertainty to the above estimates, since the variance within different haplogroups could be also explained by different values of d. For example, had B001 and B003 been equally represented in the original expanding population, then their present-day occurrence would have indicated different growth dynamics (B001 having expanded to its present-day frequency of 0.25, and B003 having expanded to a frequency just under 0.1). Accordingly, the population growth rate d and, hence, the derived go correction, would have been different for each of the two haplotypes, possibly explaining the lower variability associated with the less frequent haplotype (see eq. [6]). These considerations also suggest that, under the assumption of a founder effect followed by rapid population growth, the time-since-expansion values in table 2 are also underestimates. Although easily applicable in the cases of B001, B002/B004, and B003, this model is less suitable when used with B005, B006, and other haplotypes.
The estimates of time since expansion from the two independent methods discussed above provide a reasonable time frame of past events that compares well with other estimates from the literature (Rogers and Harpending 1992; Torroni et al. 1998; Richards et al. 2000). They both indicate that major migrations and population expansions coincided with the demographic events of the Upper Paleolithic. Although the absolute time estimates need necessarily be treated with caution, the relative values are illuminating. They indicate that a mosaic of lineages, of different time depths and likely of different geographic provenience, within Africa and perhaps including western Eurasia, contributed to contemporary human diversity. Although it is likely that the origin of haplotype B006 is in Africa, the question remains as to whether its frequent, non-African presence dates from the time of the great expansion, or whether it left Africa earlier and was then admixed into the major expanding lineage on its northern route into Eurasia. To further address this question, we will need to expand our sample pool and study further polymorphisms, adjacent to dys44, to increase temporal resolution and accuracy.
Acknowledgments
We are grateful to Alan Lovell for his comments on the final version of this article, as well as to Maidar Jamba for providing Mongolian samples and for his contribution to their genotyping. We are also indebted to J. Jaruzelska, K. Kidd, M. Labuda, L. Osipova, M. Stoneking, and S. Tishkoff, who have shared samples from their earlier collections, as well as to all individuals who generously donated their DNA, making this study possible. The present study was supported by Louisiana Board of Regents Millennium Trust Health Excellence Fund grants HEF (2000–05)-05 and (2000–05)-01 (to M.A.B.) and primarily by a grant from the Canadian Institutes of Health Research (to D.L.).
Appendix A: Fraction of Informative Recombinations
The extent of genome diversity is due to the cumulative effect of mutations and recombinations. Mutations such as nucleotide substitutions almost always lead to new variants, since, given a mutation rate on the order of 10−8 per generation, recurrence is extremely rare. In contrast, a significant fraction of recombination events have no effect on haplotype diversity; they occur but do not result in an outcome that can be perceived as a new recombinant. This is the case of recombinations occurring between homologous genomic segments that are identical or that differ at a single site only (i.e., in homozygous or single-site heterozygous individuals). The fraction of such “silent”—or noninformative—recombinations depends on the density of sequence polymorphisms (Hudson and Kaplan 1985; Stephens 1986). Indeed, an informative (i.e., potentially detectable) recombination has to occur within a segment delimited by heterozygous sites, such that the resulting recombinant haplotype will differ from the parental ones.
The fraction of informative recombinations, FIR, for a segment with K haplotypes in a population, can be written as
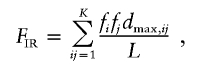 |
where fifj is the population frequency of a diploid genotype combining haplotype variants i and j, such that 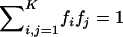 represents the sum of all possible genotypes; dmax,ij represents the distance (in number of nucleotides) between maximally separated pairs of heterozygous sites for a combination of the ij haplotypes (note that if the combination ij has less than two heterozygous sites, dmax,ij=0, such that noninformative recombinations do not contribute to FIR); and L denotes haplotype length, which corresponds to the distance between maximally separated, potentially informative positions (for the dys44 haplotype, L corresponds to 7,917 bp between sites “2” and “96”).
represents the sum of all possible genotypes; dmax,ij represents the distance (in number of nucleotides) between maximally separated pairs of heterozygous sites for a combination of the ij haplotypes (note that if the combination ij has less than two heterozygous sites, dmax,ij=0, such that noninformative recombinations do not contribute to FIR); and L denotes haplotype length, which corresponds to the distance between maximally separated, potentially informative positions (for the dys44 haplotype, L corresponds to 7,917 bp between sites “2” and “96”).
FIR represents the fraction of all recombinations leading to haplotypes that differ from the parental haplotypes. Such recombinants will always be detected in family studies, when both parents and their child are analyzed. In population studies, however, only the recombinants that represent structurally new, distinct haplotypes will be discerned. Recombinants IBS with the haplotypes already observed in the analyzed population will be perceived as IBD with the latter rather than as being due to a separate crossover event. Therefore, among potentially informative recombinations, FIR, we need to distinguish the fraction of events leading to new recombinants, FNR, from that of back recombinations “recreating” already observed haplotypes, FBR (FIR=FBR+FNR). FNR and FBR can be calculated by simulating recombination between the set of observed haplotypes and cataloguing all outcomes of informative recombinations as either new or already observed haplotypes, respectively. The apparent recombination rate of the haplotype, rapp, to be used in a population study for time and/or recombinational divergence estimations is therefore rapp=rFNR, where r represents the recombination rate of the haplotype estimated from the genetic map.
Using our sample of 1,343 chromosomes from around the world and assuming panmixia, we obtained an FIR of 0.62 (FBR=0.21 and FNR=0.41). The histogram in figure A1 shows the observed haplotype frequencies (fig. A1A) compared with the contribution of each of the haplotypes to the overall FBR (fig. A1B). Figure A1C depicts the distribution of the probabilities of occurrence of 6,857 distinct, novel recombinants that could arise through single crossovers from all pairwise combinations of the 86 observed haplotypes (i.e., individual contributions of these new haplotypes to the overall FNR). The cumulative probability of the recombinations that would result in 86 (i.e., equivalent to the number of the observed haplotypes) most probable new recombinants corresponds to two-thirds of the overall FNR (0.26 out of 0.41). The overall FNR values vary among populations, from 0.54 in Africa to 0.26 in Indonesia and PNG; nevertheless, using FNR=0.3 to calculate the apparent recombination rate for non-African populations is a good approximation. With <30 novel haplotypes observed outside of Africa, a similar count of back recombinants might be expected (FBR and FNR are similar), and, therefore, even haplotypes with individual contributions to FBR as high as 1% have little chance to reoccur through back recombination. For most of the existing haplotypes, the individual probability of their reappearance resulting from back recombination is lower than the collective probability of the appearance of the potential newcomers (fig. A1B and A1C; note different scaling of the Y-axes). Furthermore, there is no correlation between population frequency and the recurrence rate recreating a particular haplotype through independent crossovers, suggesting that new recombinants originate from high and low probability values of the recombinations’ probability spectrum, illustrated in figure A1C.
Figure A1.

Frequencies of 86 observed haplotypes (A) compared with their individual probabilities of back recombination (B). C, Probability distribution of new recombinants resulting from pairwise recombination of the observed haplotypes from the whole data set. Note that the scale of the ordinate in C is reduced by a factor of 10 compared with A and B, that the total count of new haplotypes is 6,857 (C), and that the left and right parts of chart C depict the 86 most frequent novel haplotypes versus the remaining 6,771 relatively infrequent novel haplotypes.
The population frequency of haplotype B004 exceeds the probability of its reappearance through a recurrent recombination. However, a reciprocal solution of this recombination leading to B004 and engaging the most frequent combination of ubiquitous haplotypes B001 and B002 is the haplotype b054, which, conspicuously, was observed only once. Haplotypes B030 and B034 appear to have the highest rate of recurrence through back recombinations (fig. A1B), yet only few copies of these haplotypes were seen in our sample. Therefore, although the rate of back recombinations is not negligible, their confounding effect on interpreting the geographic distribution and frequencies of haplotype variants can be largely neglected for the dys44 data set. A few haplotypes especially likely to be recreated through back recombinations are easily identifiable through the analysis of possible recombination outcomes, as shown in fig. A1B, which may assist data interpretation. For example, the individual probability of reappearance of B004 through back recombination was found to be 2.6% worldwide, 2.4% in Africa, 1.5% in Europe, 3.5% in Asia, and 4.6% in Indonesia and PNG, whereas, in the Americas, where 80% of B004 chromosomes reside, the probability was only 0.05%. This strongly suggests that all Amerindian copies of B004 are IBD. Their high frequency on this continent is most probably due to a local founder effect, which, at the same time, presumably eliminated haplotype B002, an essential ingredient needed to recreate B004 by de novo recombination.
The program used to evaluate FNR and FBR is available upon request from D.G. (Dominik.Gehl@UMontreal.ca).
Electronic-Database Information
The accession number and URL for data presented herein are as follows:
- GenBank, http://www.ncbi.nlm.nih.gov/Genbank/ (for the human dystrophin gene [accession number U94396])
References
- Baird DM, Coleman J, Rosser ZH, Royle NJ (2000) High levels of sequence polymorphism and linkage disequilibrium at the telomere of 12q: implications for telomere biology and human evolution. Am J Hum Genet 66:235–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batzer MA, Stoneking M, Alegria-Hartman M, Bazan H, Kass DH, Shaikh TH, Novick GE, Ioannou PA, Sheer DW, Herrera RJ, Deininger PL (1994) African origin of human-specific polymorphic Alu insertions. Proc Natl Acad Sci USA 91:12288–12292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi NO, Catanesi CI, Bailliet G, Martinez-Marignac VL, Bravi CM, Vidal-Rioja LB, Herrera RJ, Lopez-Camelo JS (1998) Characterization of ancestral and derived Y-chromosome haplotypes of New World native populations. Am J Hum Genet 63:1862–1871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowcock AM, Ruiz-Linares A, Tomfohrde J, Minch E, Kidd JR, Cavalli-Sforza LL (1994) High resolution of human evolutionary trees with polymorphic microsatellites. Nature 368:455–457 [DOI] [PubMed] [Google Scholar]
- Cann RL, Stoneking M, Wilson AC (1987) Mitochondrial DNA and human evolution. Nature 325:31–36 [DOI] [PubMed] [Google Scholar]
- Cavalli-Sforza LL, Menozzi P, Piazza A (1994) The history and geography of human genes. Princeton University Press, Princeton, NJ [Google Scholar]
- Chen FC, Li WH (2001) Genomic divergences between humans and other hominoids and the effective population size of the common ancestor of humans and chimpanzees. Am J Hum Genet 68:444–456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YS, Torroni A, Excoffier L, Santachiara-Benerecetti AS, Wallace DC (1995) Analysis of mtDNA variation in African populations reveals the most ancient of all human continent-specific haplogroups. Am J Hum Genet 57:133–149 [PMC free article] [PubMed] [Google Scholar]
- Clark AG, Weiss KM, Nickerson DA, Taylor SL, Buchanan A, Stengard J, Salomaa V, Vartiainen E, Perola M, Boerwinkle E, Sing CF (1998) Haplotype structure and population genetic inferences from nucleotide- sequence variation in human lipoprotein lipase. Am J Hum Genet 63:595–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombo R (2000) Age estimate of the N370S mutation causing Gaucher disease in Ashkenazi Jews and European populations: a reappraisal of haplotype data. Am J Hum Genet 66:692–697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruciani F, Santolamazza P, Shen P, Macaulay V, Moral P, Olckers A, Modiano D, Holmes S, Destro-Bisol G, Coia V, Wallace DC, Oefner PJ, Torroni A, Cavalli-Sforza LL, Scozzari R, Underhill PA (2002) A back migration from Asia to sub-Saharan Africa is supported by high-resolution analysis of human Y-chromosome haplotypes. Am J Hum Genet 70:1197–1214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Rienzo A, Donnelly P, Toomajian C, Sisk B, Hill A, Petzl-Erler ML, Haines GK, Barch DH (1998) Heterogeneity of microsatellite mutations within and between loci, and implications for human demographic histories. Genetics 148:1269–1284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dieringer D, Schlötterer C (2003) Microsatellite analyser (MSA): a platform independent analysis tool for large microsatellite data sets. Mol Ecol Notes 3:167–169 [Google Scholar]
- Ewens WJ (1972) The sampling theory of selectively neutral alleles. Theor Popul Biol 3:87–112 [DOI] [PubMed] [Google Scholar]
- Fan JB, Gehl D, Hsie L, Shen N, Lindblad-Toh K, Laviolette JP, Robinson E, Lipshutz R, Wang D, Hudson TJ, Labuda D (2002) Assessing DNA sequence variations in human ESTs in a phylogenetic context using high-density oligonucleotide arrays. Genomics 80:351–360 [DOI] [PubMed] [Google Scholar]
- Felsenstein J (1993) PHYLIP (phylogeny inference package) release 3.5p. University of Washington, Seattle [Google Scholar]
- Hammer MF (1995) A recent common ancestry for human Y chromosomes. Nature 378:376–378 [DOI] [PubMed] [Google Scholar]
- Hammer MF, Karafet T, Rasanayagam A, Wood ET, Altheide TK, Jenkins T, Griffiths RC, Templeton AR, Zegura SL (1998) Out of Africa and back again: nested cladistic analysis of human Y chromosome variation. Mol Biol Evol 15:427–441 [DOI] [PubMed] [Google Scholar]
- Hammer MF, Spurdle AB, Karafet T, Bonner MR, Wood ET, Novelletto A, Malaspina P, Mitchell RJ, Horai S, Jenkins T, Zegura SL (1997) The geographic distribution of human Y chromosome variation. Genetics 145:787–805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding RM, Fullerton SM, Griffiths RC, Bond J, Cox MJ, Schneider JA, Moulin DS, Clegg JB (1997) Archaic African and Asian lineages in the genetic ancestry of modern humans. Am J Hum Genet 60:772–789 [PMC free article] [PubMed] [Google Scholar]
- Harris EE, Hey J (1999) X chromosome evidence for ancient human histories. Proc Natl Acad Sci USA 96:3320–3324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastbacka J, de la Chapelle A, Kaitila I, Sistonen P, Weaver A, Lander E (1992) Linkage disequilibrium mapping in isolated founder populations: diastrophic dysplasia in Finland [erratum in Nat Genet 2:343]. Nat Genet 2:204–211 [DOI] [PubMed] [Google Scholar]
- Hudson RR, Kaplan NL (1985) Statistical properties of the number of recombination events in the history of a sample of DNA sequences. Genetics 111:147–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaruzelska J, Ziętkiewicz E, Batzer M, Cole DE, Moisan JP, Scozzari R, Tavare S, Labuda D (1999) Spatial and temporal distribution of the neutral polymorphisms in the last ZFX intron: analysis of the haplotype structure and genealogy. Genetics 152:1091–1101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin L, Underhill PA, Doctor V, Davis RW, Shen P, Cavalli-Sforza LL, Oefner PJ (1999) Distribution of haplotypes from a chromosome 21 region distinguishes multiple prehistoric human migrations. Proc Natl Acad Sci USA 96:3796–3800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorde LB, Rogers AR, Bamshad M, Watkins WS, Krakowiak P, Sung S, Kere J, Harpending HC (1997) Microsatellite diversity and the demographic history of modern humans. Proc Natl Acad Sci USA 94:3100–3103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaessmann H, Heissig F, von Haeseler A, Pääbo S (1999) DNA sequence variation in a non-coding region of low recombination on the human X chromosome. Nat Genet 22:78–81 [DOI] [PubMed] [Google Scholar]
- Kidd JR, Pakstis AJ, Zhao H, Lu RB, Okonofua FE, Odunsi A, Grigorenko E, Tamir BB, Friedlaender J, Schulz LO, Parnas J, Kidd KK (2000) Haplotypes and linkage disequilibrium at the phenylalanine hydroxylase locus, PAH, in a global representation of populations. Am J Hum Genet 66:1882–1899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimmel M, Chakraborty R, King JP, Bamshad M, Watkins WS, Jorde LB (1998) Signatures of population expansion in microsatellite repeat data. Genetics 148:1921–1930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kivisild T, Bamshad MJ, Kaldma K, Metspalu M, Metspalu E, Reidla M, Laos S, Parik J, Watkins WS, Dixon ME, Papiha SS, Mastana SS, Mir MR, Ferak V, Villems R (1999) Deep common ancestry of Indian and western-Eurasian mitochondrial DNA lineages. Curr Biol 9:1331–1334 [DOI] [PubMed] [Google Scholar]
- Klein RG (1999) The human career: human biological and cultural origins. University of Chicago Press, Chicago [Google Scholar]
- Kong A, Gudbjartsson DF, Sainz J, Jonsdottir GM, Gudjonsson SA, Richardsson B, Sigurdardottir S, Barnard J, Hallbeck B, Masson G, Shlien A, Palsson ST, Frigge ML, Thorgeirsson TE, Gulcher JR, Stefansson K (2002) A high-resolution recombination map of the human genome. Nat Genet 31:241–247 [DOI] [PubMed] [Google Scholar]
- Labuda D, Ziętkiewicz E, Labuda M (1997) The genetic clock and the age of the founder effect in growing populations: a lesson from French Canadians and Ashkenazim. Am J Hum Genet 61:768–771 [PMC free article] [PubMed] [Google Scholar]
- Labuda D, Ziętkiewicz E, Yotova V (2000) Archaic lineages in the history of modern humans. Genetics 156:799–808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labuda M, Labuda D, Korab-Laskowska M, Cole DE, Ziętkiewicz E, Weissenbach J, Popowska E, Pronicka E, Root AW, Glorieux FH (1996) Linkage disequilibrium analysis in young populations: pseudo-vitamin D- deficiency rickets and the founder effect in French Canadians. Am J Hum Genet 59:633–643 [PMC free article] [PubMed] [Google Scholar]
- Lahr MM, Foley RA (1994) Multiple dispersals and modern human origins. Evol Anthropol 3:48–60 [Google Scholar]
- ——— (1998) Towards a theory of modern human origins: geography, demography, and diversity in recent human evolution. Am J Phys Anthropol Suppl 27:137–176 [DOI] [PubMed] [Google Scholar]
- Luria SE, Delbrück M (1943) Mutations of bacteria from virus sensitivity to virus resistance. Genetics 28:491–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta T, Kimura M (1973) A model of mutation appropriate to estimate the number of electrophoretically detectable alleles in a finite population. Genet Res 22:201–204 [DOI] [PubMed] [Google Scholar]
- Pritchard JK, Feldman MW (1996) Statistics for microsatellite variation based on coalescence. Theor Popul Biol 50:325–344 [DOI] [PubMed] [Google Scholar]
- Quintana-Murci L, Semino O, Bandelt HJ, Passarino G, McElreavey K, Santachiara-Benerecetti AS (1999) Genetic evidence of an early exit of Homo sapiens sapiens from Africa through eastern Africa. Nat Genet 23:437–441 [DOI] [PubMed] [Google Scholar]
- Relethford JH (1995) Genetics and modern human origins. Evol Anthropol 4:53–63 [Google Scholar]
- Richards M, Macaulay V, Hickey E, Vega E, Sykes B, Guida V, Rengo C, et al (2000) Tracing European founder lineages in the near eastern mtDNA pool. Am J Hum Genet 67:1251–1276 [PMC free article] [PubMed] [Google Scholar]
- Rogers AR, Harpending H (1992) Population growth makes waves in the distribution of pairwise genetic differences. Mol Biol Evol 9:552–569 [DOI] [PubMed] [Google Scholar]
- Rosenberg NA, Pritchard JK, Weber JL, Cann HM, Kidd KK, Zhivotovsky LA, Feldman MW (2002) Genetic structure of human populations. Science 298:2381–2385 [DOI] [PubMed] [Google Scholar]
- Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425 [DOI] [PubMed] [Google Scholar]
- Satta Y, Takahata N (2002) Out of Africa with regional interbreeding? Modern human origins. Bioessays 24:871–875 [DOI] [PubMed] [Google Scholar]
- Schneider S, Kueffer J-M, Roessli D, Exoffier L (1997) Arlequin version 1.1: a software for population genetic data analysis. Genetics and Biometry Laboratory, University of Geneva, Switzerland [Google Scholar]
- Seielstad MT, Minch E, Cavalli-Sforza LL (1998) Genetic evidence for a higher female migration rate in humans. Nat Genet 20:278–280 [DOI] [PubMed] [Google Scholar]
- Slatkin M, Rannala B (2000) Estimating allele age. Annu Rev Genomics Hum Genet 1:225–249 [DOI] [PubMed] [Google Scholar]
- Stephens JC (1986) On the frequency of undetectable recombinant events. Genetics 112:923–926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens M, Smith NJ, Donnelly P (2001) A new statistical method for haplotype reconstruction from population data. Am J Hum Genet 68:978–989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiner MC, Munro ND, Surovell TA, Tchernov E, Bar-Yosef O (1999) Paleolithic population growth pulses evidenced by small animal exploitation. Science 283:190–194 [DOI] [PubMed] [Google Scholar]
- Stoneking M, Fontius JJ, Clifford SL, Soodyall H, Arcot SS, Saha N, Jenkins T, Tahir MA, Deininger PL, Batzer MA (1997) Alu insertion polymorphisms and human evolution: evidence for a larger population size in Africa. Genome Res 7:1061–1071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stringer CB, Andrews P (1988) Genetic and fossil evidence for the origin of modern humans. Science 239:1263–1268 [DOI] [PubMed] [Google Scholar]
- Takahata N (1994) Repeated failures that led to the eventual success in human evolution. Mol Biol Evol 11:803–805 [DOI] [PubMed] [Google Scholar]
- Tattersall I (1995) The fossil trail: how we know what we think we know about human evolution. Oxford University Press, New York [Google Scholar]
- Tattersall I, Schwartz J (2000) Extinct humans. Westview Press, New York [Google Scholar]
- Tishkoff SA, Dietzsch E, Speed W, Pakstis AJ, Kidd JR, Cheung K, Bonne-Tamir B, Santachiara-Benerecetti AS, Moral P, Krings M (1996) Global patterns of linkage disequilibrium at the CD4 locus and modern human origins. Science 271:1380–1387 [DOI] [PubMed] [Google Scholar]
- Torroni A, Bandelt HJ, D’Urbano L, Lahermo P, Moral P, Sellitto D, Rengo C, Forster P, Savontaus ML, Bonne-Tamir B, Scozzari R (1998) mtDNA analysis reveals a major late Paleolithic population expansion from southwestern to northeastern Europe. Am J Hum Genet 62:1137–1152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torroni A, Schurr TG, Cabell MF, Brown MD, Neel JV, Larsen M, Smith DG, Vullo CM, Wallace DC (1993) Asian affinities and continental radiation of the four founding Native American mtDNAs. Am J Hum Genet 53:563–590 [PMC free article] [PubMed] [Google Scholar]
- Underhill PA, Shen P, Lin AA, Jin L, Passarino G, Yang WH, Kauffman E, Bonne-Tamir B, Bertranpetit J, Francalacci P, Ibrahim M, Jenkins T, Kidd JR, Mehdi SQ, Seielstad MT, Wells RS, Piazza A, Davis RW, Feldman MW, Cavalli-Sforza LL, Oefner PJ (2000) Y chromosome sequence variation and the history of human populations. Nat Genet 26:358–361 [DOI] [PubMed] [Google Scholar]
- Verrelli BC, McDonald JH, Argyropoulos G, Destro-Bisol G, Froment A, Drousiotou A, Lefranc G, Helal AN, Loiselet J, Tishkoff SA (2002) Evidence for balancing selection from nucleotide sequence analyses of human G6PD. Am J Hum Genet 71:1112–1128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson E, Forster P, Richards M, Bandelt HJ (1997) Mitochondrial footprints of human expansions in Africa. Am J Hum Genet 61:691–704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watterson GA (1978) The homozygosity test of neutrality. Genetics 88:405–417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White T, Asfaw B, DeGusta D, Gilbert H, Richards G, Suwa G H, C o (2003) Pleistocene Homo sapiens from Middle Awash, Ethiopia. Nature 423:742–747 [DOI] [PubMed] [Google Scholar]
- Yu N, Zhao Z, Fu YX, Sambuughin N, Ramsay M, Jenkins T, Leskinen E, Patthy L, Jorde LB, Kuromori T, Li WH (2001) Global patterns of human DNA sequence variation in a 10-kb region on chromosome 1. Mol Biol Evol 18:214–222 [DOI] [PubMed] [Google Scholar]
- Ziętkiewicz E, Labuda D (2001) Modern human origins in light of the nuclear DNA diversity in world populations. In: Donnelly P, Foley RA (eds) Genes, fossils and behaviour: an integrated approach to human evolution. IOS Press, Amsterdam, pp 79–97 [Google Scholar]
- Ziętkiewicz E, Yotova V, Jarnik M, Korab-Laskowska M, Kidd KK, Modiano D, Scozzari R, Stoneking M, Tishkoff S, Batzer M, Labuda D (1997) Nuclear DNA diversity in worldwide distributed human populations. Gene 205:161–171 [DOI] [PubMed] [Google Scholar]
- ——— (1998) Genetic structure of the ancestral population of modern humans. J Mol Evol 47:146–155 [DOI] [PubMed] [Google Scholar]