

Abstract
Arachidonic acid and its metabolites are implicated in regulating endothelial cell proliferation. Cytosolic phospholipase A2-α (cPLA2α) is responsible for receptor-mediated arachidonic acid evolution. We tested the hypothesis that cPLA2α activity is linked to endothelial cell proliferation. The specific cPLA2α inhibitor, pyrrolidine-1, inhibited umbilical vein endothelial cell (HUVEC) proliferation in a dose-dependent manner. Exogenous arachidonic acid addition reversed this inhibitory effect. Inhibition of sPLA2 did not affect HUVEC proliferation. The levels of cPLA2α did not differ between subconfluent and confluent cultures of cells. However, using fluorescence microscopy we observed a novel, confluence-dependent redistribution of cPLA2α to the distal Golgi apparatus in HUVECs. Association of cPLA2α with the Golgi was linked to the proliferative status of HUVECs. When associated with the Golgi apparatus, cPLA2α activity was seen to be 87% inhibited. Relocation of cPLA2α to the cytoplasm and nucleus, and cPLA2α enzyme activity were required for cell cycle entry upon mechanical wounding of confluent monolayers. Thus, cPLA2α activity and function in controlling endothelial cell proliferation is regulated by reversible association with the Golgi apparatus.
INTRODUCTION
Endothelial cells line the luminal surface of all blood vessels and are essential for vascular processes including blood clotting, blood pressure, and blood flow (Vane et al., 1990). New blood vessel formation mediated by endothelial cells (angiogenesis) is also a crucial step in wound healing and the establishment of tumors (Folkman and Shing, 1992; Carmeliet, 2000). Antiangiogenic therapy is seen as one of the most promising strategies to restrict tumor growth and prevent metastases (Brem et al., 1993). During the process of angiogenesis endothelial cells migrate, proliferate, and differentiate to form new capillaries (Carmeliet, 2000). Numerous reports have implicated arachidonic acid (AA) in the control of proliferation in a variety of cell types. Actively proliferating endothelial cells in culture liberate elevated levels of AA and its metabolites (Evans et al., 1984; Whatley et al., 1994) and stimulation of endothelial cells with the potent mitogen, basic fibroblast growth factor, induces AA release (Fafeur et al., 1991; Sa et al., 1995). Eicosanoids, the bioactive metabolites of AA, also play a major role in the proliferation and migration of endothelial cells (Dethlefsen et al., 1994; Nie et al., 2000; Dormond et al., 2001; Antoniotti et al., 2003).
Cytosolic phospholipase A2α (cPLA2α) is an 85-kDa, Ca2+-sensitive member of the phospholipase A2 family of enzymes (Clark et al., 1995) that includes the calcium-independent (iPLA2) and secretory (sPLA2) phospholipases A2. These are responsible for hydrolysis of the sn-2 fatty-acyl bond of phospholipids to simultaneously generate free fatty acid and lysophospholipid (Dennis, 1997). There are at least four cPLA2 paralogs (cPLA2α, cPLA2β, cPLA2γ, and cPLA2δ), of which cPLA2α is most characterized. On cell stimulation and intracellular Ca2+ elevation, cPLA2α translocates from the cytosol to intracellular membrane substrates utilizing an N-terminal Ca2+-dependent lipid binding (CalB) domain (Channon and Leslie, 1990; Nalefski et al., 1994; Schievella et al., 1995; Bittova et al., 1999; Evans et al., 2001). cPLA2α preferentially cleaves phospholipids containing AA at the sn-2 position (Dennis, 1997) and is thus considered the key enzyme responsible for receptor-mediated AA liberation and subsequent eicosanoid synthesis (Kramer and Sharp, 1997). As such, it is a pivotal enzyme in AA-mediated cell proliferation and migration. At confluence endothelial cells undergo contact inhibition of growth, cease proliferating, and enter the G0 phase of quiescence. Correspondingly there is a significant decline in AA output and eicosanoid synthesis (Evans et al., 1984; Whatley et al., 1994), which has been attributed to the reduced cPLA2α activity (Whatley et al., 1994). Because of the importance of endothelial cell proliferation in human retinopathies and cancers, there is great emphasis on developing therapies that modulate endothelial cell growth. Despite this, the exact mechanisms by which cPLA2α activity is differentially regulated in nonconfluent and confluent endothelial cells and the role of cPLA2α in endothelial cell proliferation remain unclear.
In this study we demonstrate that cPLA2α activity is required for endothelial cell proliferation and cell cycle control. We also describe a novel redistribution of cPLA2α to the Golgi apparatus of human umbilical vein endothelial cells (HUVECs) in a confluence-dependent manner. The subcellular localization of cPLA2α is closely linked to the proliferative status of HUVECs. When associated with the Golgi apparatus, cPLA2α activity is 87% inhibited. After mechanical wounding, cPLA2α redistribution is linked to the induction of Ki67 expression and entry into the cell cycle. These observations shed light on the mechanisms by which cPLA2α localization, activity, and function are modulated in quiescent and proliferating endothelial cells.
MATERIALS AND METHODS
Materials
Alexafluor 594–conjugated concanavalin A (ConA) and secondary anti-goat, anti-mouse, and anti-rabbit Alexafluor 488– or 594–conjugated antibodies were purchased from Molecular Probes (Eugene, OR). Rhodamine-conjugated wheat germ agglutinin (WGA) was purchased from Sigma (Poole, United Kingdom). Affinity-purified goat polyclonal anti-cPLA2α antibodies were purchased from Santa Cruz Biotechnology (sc-1724, Santa Cruz, CA). Rabbit anti-GM130 antibodies were from M. Lowe (University of Manchester, United Kingdom). Mouse monoclonal anti-ERGIC-53 and β-1,4-galactosyltransferase (GalT) antibodies were provided by H. P. Hauri (Basel, Switzerland) and T. Suganuma (Miyazaki, Japan), respectively. Rabbit polyclonal anti-TGN46 was supplied by S. Ponnambalam (University of Leeds, United Kingdom). Rabbit polyclonal anti-mannosidase II (ManII) antibodies were purchased from Serotec Ltd (Oxford, United Kingdom). HRP-conjugated anti-goat secondary antibodies were purchased from Pierce (Tattenhall, United Kingdom). 2-(p-amylcinnamoyl)amino-4-chlorobenzoic acid (ONO-RS-082) was purchased from BioMol (Plymouth Meeting, PA). Pyrrolidine-1 was supplied by M. H. Gelb (University of Washington, Seattle,WA). All other reagents were obtained from Sigma or Life Technologies (Paisley, United Kingdom) unless otherwise stated.
Cell Culture
HUVECs were isolated from human umbilical cords as previously described (Jaffe, 1984; Howell et al., 2004). Cells were cultured in endothelial cell basal medium supplemented with human recombinant epidermal growth factor (EGF; 5 ng/ml), hydrocortisone (0.2 μg/ml), vascular endothelial growth factor (VEGF, 0.5 ng/ml), human recombinant basic fibroblast growth factor (bFGF, 10 ng/ml), recombinant long R3 insulin-like growth factor-1 (IGF-1, 20 ng/ml), ascorbic acid (1 μg/ml), heparin (22.5 μg/ml), amphotericin B (50 ng/ml), gentamicin (50 μg/ml), and 2% (vol/vol) fetal calf serum (PromoCell, Heidelberg, Germany). All HUVEC cultureware was coated with 0.1% (wt/vol) pigskin gelatin unless stated otherwise. HUVECs were never used in excess of three passages and expressed the characteristic endothelial markers von Willebrand factor and platelet-endothelial cell adhesion molecule-1. All cells were grown at 37°C in a humid incubator containing 5% (vol/vol) CO2.
Cell Proliferation ELISA
HUVEC proliferation rates in the presence or absence of varying concentrations of ONO-RS-082 or pyrrolidine-1 were compared using a 5-bromo-2′-deoxyuridine (BrdU) incorporation-based ELISA (Roche Diagnostics, Lewes, East Sussex, United Kingdom). Cells were seeded at 1 × 103 cells per well (0.55 × 103 cells/cm2) in 96-well plates and cells, grown for 24 h, and then processed according to manufacturer's instructions. The BrdU incorporation period was fixed at 16 h.
SDS-PAGE and Immunoblotting
Samples (20 or 10 μg protein) were resolved for 60 min at 30 mA/gel on 10% SDS-PAGE mini-gels using a discontinuous buffer system (Laemmli, 1970). For immunoblotting, protein was transferred onto nitrocellulose membranes for 3 h at 300 mA (Towbin et al., 1979). Membranes were blocked in 5% (wt/vol) nonfat milk in phosphate-buffered saline (PBS) for 30 min and then incubated overnight with primary antibody (1:500) at room temperature. After incubation with HRP-conjugated anti-goat IgG (1:3000) for 1 h, immunoreactive bands were visualized using a West Pico–enhanced chemiluminescence (ECL) detection kit (Pierce, Rockford, IL). A Fuji Film Intelligent dark box II image reader using Fuji Las-1000 Prosoftware (Tokyo, Japan) was used to capture images. Band intensities were determined densitometrically using Aida (Advanced Image Data Analyzer, Raytest, Straubenhardt, Germany) 2.11 software. For comparison of nonconfluent and confluent HUVEC protein expression, cells were grown on noncoated plastic to control for misleading protein estimation due to gelatin-coated surfaces.
Immunofluorescence Microscopy
This technique was adapted from previous protocols (Heggeness et al., 1977; Barwise and Walker, 1996). Cells were seeded on 0.1% (wt/vol) gelatin-coated coverslips and grown to the required level of confluence. Medium was aspirated and cells were fixed in 10% (vol/vol) Formalin (in neutral buffered saline; Sigma) for 5 min at 37°C. Before fixation some cells were stimulated for 1 min with 5 μM A23187 as previously described (Grewal et al., 2003, 2004). All ensuing steps were performed at 25°C. After permeabilization with 0.1% (vol/vol) Triton X-100 in PBS for 5 min, cells were refixed for 5 min, washed three times with PBS, and then incubated in 50 mM ammonium chloride in PBS for 10 min. After three further PBS washes, nonspecific binding sites were blocked with 5% (vol/vol) donkey serum in PBS for 3 h. Cells were incubated overnight with primary antibody followed by the appropriate Alexafluor-conjugated secondary antibodies, Alexafluor 594–conjugated ConA or rhodamine-conjugated WGA for 3 h. For antigenic adsorption, antibodies to cPLA2α were incubated with blocking peptide (1:5 ratio of μg antibody to μg peptide) for 30 min before incubation, as previously described (Grewal et al., 2003). Finally cells were washed eight times in PBS and mounted on microscope slides in prolong mounting medium (Molecular Probes).
Deconvolution Imaging and Quantitation
Deconvolution fluorescence microscopy was performed using an Olympus IX-70 inverted fluorescence microscope and DeltaVision deconvolution system (Applied Precision, Issaquah, WA). Individual optical sections of 0.2 μm were generated from 15 iterative cycles of deconvolution. Quantification of colocalization was determined using the IMARIS software suite (Bitplane AG, Zurich, Switzerland) on selected Golgi regions. Background was eliminated by excluding gray scale values lower than 10% of the maximum pixel intensity. Colocalized pixels were recorded and expressed as percentages of the total pixels selected. Mean pixel intensities were determined using Image J software (http://rsb.info.nih.gov/ij/). Regions of interest were selected, mean pixel intensity calculated then mean background pixel intensity subtracted from this. No images analyzed were pixel saturated.
Brefeldin A Treatment
HUVECs were grown to confluence on 0.1% (wt/vol) gelatin-coated coverslips. Cells were washed twice with prewarmed (37°C) PBS and then incubated with 5 μg/ml brefeldin A (BFA; Roche Diagnostics, Sussex, United Kingdom) in serum-free media for 30 min and processed for immunofluorescence microscopy as described above.
AA Release
This technique was adapted from previous protocols (Whatley et al., 1994; Bailleux et al., 2004). HUVECs were cultured to the required cell density in six-well culture dishes and labeled for 24 h with 1 μCi/ml [3H]AA. Cells were washed three times with PBS and then incubated with 10 μM bromoenol lactone for 30 min to inhibit iPLA2 activity. Medium was then aspirated and cells were incubated with serum-free medium supplemented with 5 μM A23187 and 0.3% (wt/vol) fatty acid–free bovine serum albumin. Aliquots of media were removed at times indicated, centrifuged at 16,000 × g for 5 min, and supernatant was counted for radioactivity by liquid scintillation. Cells were lysed in 0.1% Triton X-100 for 5 min and also counted by liquid scintillation for radioactivity.
RESULTS
cPLA2α Mediates Endothelial Cell Proliferation
cPLA2α activity but not sPLA2 activity was essential for maximal HUVEC proliferation (Figure 1). The contribution of cPLA2α to HUVEC proliferation was assessed by determining BrdU incorporation in the presence of varying concentrations of pyrrolidine-1 (Figure 1A). Pyrrolidine-1 specifically inhibits cPLA2α and has no effect on iPLA2 or sPLA2 (type IIA, X and V) activity (Ghomashchi et al., 2001). Pyrrolidine-1 acted in a dose-dependent manner, inhibiting proliferation by 28.7 ± 4.6, 49.9 ± 4.2, and 70.7 ± 2.9% at concentrations of 5, 10, and 20 μM (Figure 1A). At concentrations above 20 μM, pyrrolidine-1 was toxic, whereas at concentrations of 10 μM and below, cell viability was unaffected, as determined by trypan blue exclusion. Varying concentrations of the sPLA2 inhibitor, ONO-RS-082 (Hashimoto et al., 2003), had no significant effect on BrdU incorporation (Figure 1B). The inhibitory effects of pyrrolidine-1 could be significantly reversed by addition of exogenous free AA (Figure 1C). AA, 10 μM, reversed the effect of 10 μM pyrrolidine-1, reaching 75.1 ± 2% of control. We decided to investigate further the properties of cPLA2α under different growth conditions.
Figure 1.
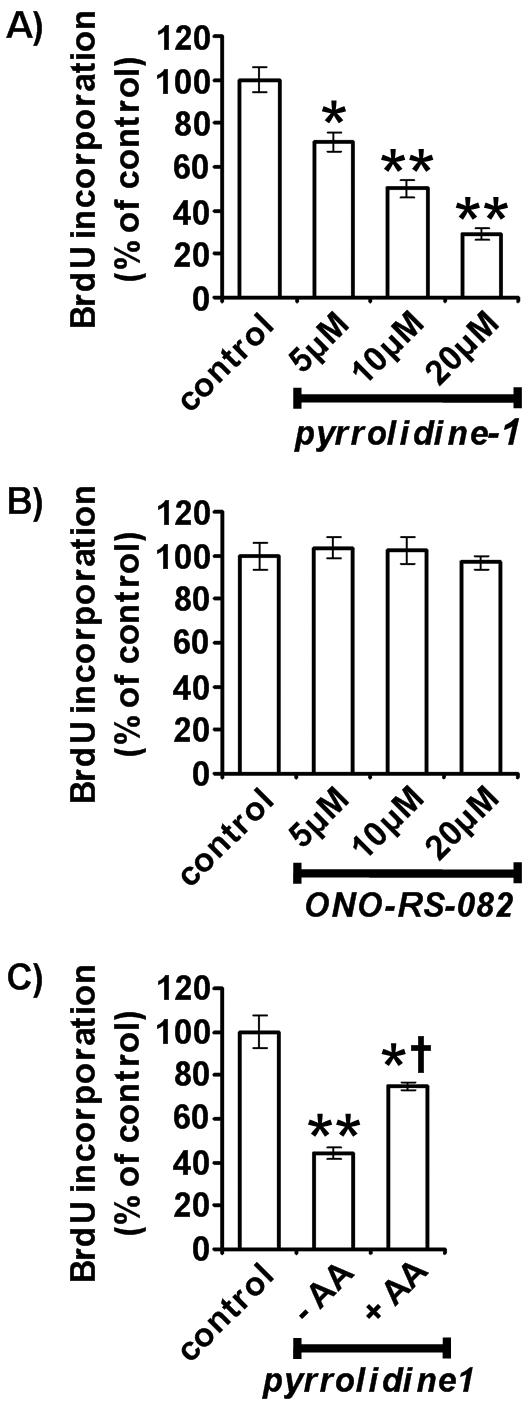
Inhibition of HUVEC proliferation by cPLA2α inhibition. Quantitation of HUVEC growth in the presence of varying concentrations of pyrrolidine-1 (A) and ONO-RS-082 (B) for 16 h (n = 6, ±SEM). Quantitation of HUVEC growth in the presence of 10 μM pyrrolidine-1 with and without 10 μM arachidonic acid supplementation (C; n = 5, ±SEM). Proliferation was determined using a colorimetric ELISA based on BrdU incorporation, as described in Materials and Methods. * p < 0.02 versus control. ** p < 0.001 versus control. † p < 0.001 versus 10 μM pyrrolidine-1 without AA.
cPLA2α Expression and Localization in Confluent and Nonconfluent HUVECs
The lower levels of AA release seen with confluent endothelial cells are not due to reduced cPLA2α expression (Figure 2, A and B). It is well established that nonconfluent endothelial cells release much greater levels of AA and eicosanoids than confluent endothelial cells and that the mechanical wounding of confluent monolayers results in elevated AA production (Evans et al., 1984; Whatley et al., 1994). Lower levels of AA release could be due to reduced cPLA2α expression because it is the rate-limiting enzyme in AA liberation. The protein levels of cPLA2α were determined in subconfluent proliferating and in confluent nonproliferating HUVECs (Figure 2, A and B). cPLA2α levels were assessed by Western blotting of HUVEC total protein using a well-characterized affinity-purified antibody specific to the C-terminal region of cPLA2α (Grewal et al., 2002, 2003, 2004). Densitometric analysis demonstrated no significant difference in total cPLA2α levels between subconfluent and confluent cells when using 10 μg (Figure 2B) and 20 μg (unpublished data) of total protein. The amounts of total cell protein used for Western blot detection of cPLA2α gave responses that reside in the linear region of a saturation curve. Antigenic absorption with a blocking peptide, corresponding to the C-terminal 20 amino acids unique to cPLA2α, eliminated detection by immunoblotting (unpublished data).
Figure 2.
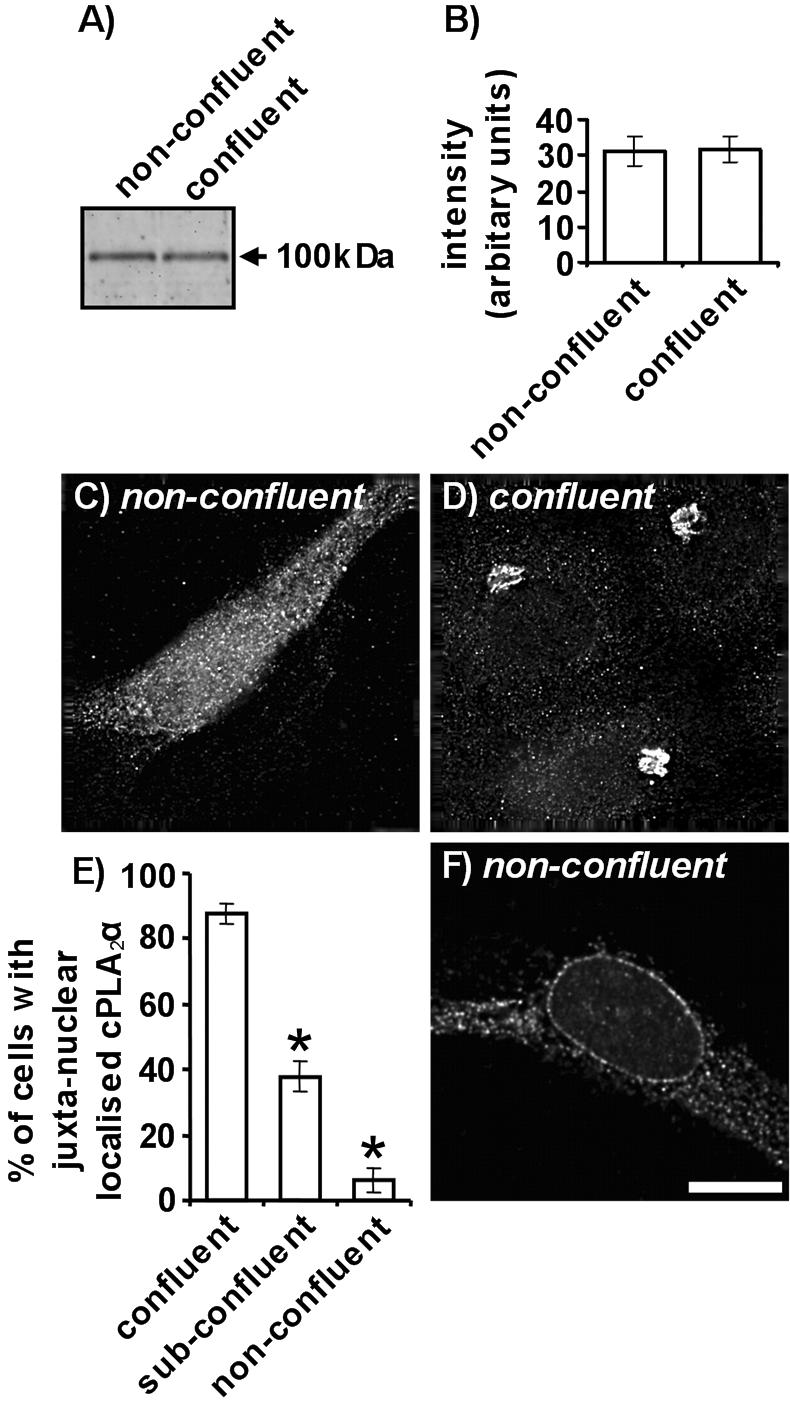
cPLA2α expression and localization in nonconfluent and confluent HUVECs. cPLA2α was detected in nonconfluent and confluent HUVEC total protein (10 μg) by immunoblotting using a goat polyclonal antibody (A). Relative amounts of cPLA2α immunoreactivity were determined using Aida densitometry software and plotted (±SEM) (B). The localization of cPLA2α in nonconfluent (C) and confluent cells (D) was determined by immunofluorescence microscopy. HUVECs were grown to nonconfluence or confluence on 0.1% gelatin coated coverslips, fixed, permeabilized, and cPLA2α detected with a goat antibody. In E, the percentage of total cells with juxta-nuclear cPLA2α localization was quantified for confluent, subconfluent, and nonconfluent cells (n = 3, ±SEM). The localization of cPLA2α in nonconfluent cells upon A23187 stimulation was determined by immunofluorescence microscopy (F). HUVECs were grown to nonconfluence or confluence on 0.1% gelatin-coated coverslips, fixed, and permeabilized, and cPLA2α was detected with a goat antibody. All images represent 0.2-μm sections through cell nuclei. Scale bar, 25 μm. * p < 0.001 versus confluent cells. All results are representative of at least 3 separate experiments.
cPLA2α redistributes to a compact juxtanuclear reticulum in confluent HUVECs (Figure 2, C–E). Indirect immunofluorescence microscopy was used to determine the subcellular localization of cPLA2α in HUVECs exhibiting differing levels of confluence (Figure 2, C and D). As previously described for endothelial cells (Sierra-Honigmann et al., 1996; Grewal et al., 2002), nonconfluent HUVECs displayed homogeneous cPLA2α staining throughout the cytoplasm and nucleus (Figure 2C). High-resolution deconvolution microscopy shows that cPLA2α was present both as diffuse and structured pools of staining in nonconfluent cells, similar to observations with fibroblasts (Bunt et al., 1997). At confluence, cPLA2α was redistributed to a compact juxtanuclear reticulum (Figure 2D). HUVECs of differing levels of confluence were scored for the presence of juxtanuclear cPLA2α staining (Figure 2E). Our criteria defined confluent HUVECs as those completely surrounded by other cells. Cells not contacting any other were categorized as nonconfluent. Cells contacting one or more cells but not entirely surrounded were classed as subconfluent. A distinct correlation between confluence and the presence of juxtanuclear cPLA2α staining was evident. A high percentage of confluent cells (87.5 ± 3.2%) displayed juxtanuclear cPLA2α staining, whereas only 37.8 ± 4.7% of subconfluent and 6.2 ± 3.5% of nonconfluent cells displayed this localization pattern.
On stimulation of nonconfluent cells with the Ca2+ ionophore, A23187, cPLA2α translocates to the peri-nuclear membrane and endoplasmic reticulum (Figure 2F). These regions are classical sites of cPLA2α-mediated AA release in response to Ca2+ elevation (Hirabayashi et al., 1999). Stimulation of confluent endothelial cells with A23187 did not induce cPLA2α translocation and cPLA2α staining remained restricted to the juxtanuclear region (unpublished data). Antigenic absorption with a blocking peptide, corresponding to the C-terminal 20 amino acids unique to cPLA2α, eliminated detection by immunofluorescence microscopy (unpublished data).
cPLA2α Redistributes to Membrane Components of the Distal Golgi Apparatus at HUVEC Confluence
cPLA2α redistributes to the Golgi apparatus in confluent endothelial cells under standard cell culture conditions (Figure 3, A and B). Because the juxtanuclear staining pattern for cPLA2α resembled the location of the Golgi apparatus, possible colocalization was investigated by counterstaining confluent HUVECs with rhodamine-conjugated WGA (Figure 3A). This lectin specifically binds N-acetyl-β-d-glucosaminyl residues found predominantly in the Golgi apparatus, with lower levels at the plasma and nuclear membranes (Virtanen et al., 1980). cPLA2α was seen to colocalize extensively with WGA-positive juxtanuclear structures corresponding to the Golgi apparatus (yellow indicates overlap), but no significant overlap was seen with the endoplasmic reticulum (ER) marker lectin, ConA (Virtanen et al., 1980; Figure 3B). A smaller pool of cPLA2α staining that did not colocalize with WGA-positive or ConA-positive structures was also evident (Figure 3, A and B).
Figure 3.

Association of cPLA2α with membrane components of the Golgi apparatus. HUVECs were grown to confluence on 0.1% gelatin-coated coverslips, fixed, and then permeabilized, and cPLA2α was detected with a goat antibody (A and B). Cells were then incubated with Alexafluor 488–conjugated anti-goat antibodies and either rhodamine-conjugated WGA (A) or Alexafluor 594–conjugated ConA (B). Images represent 0.2-μm sections through cell nuclei. Disruption of the Golgi apparatus with brefeldin A (BFA; C and D). Confluent HUVECs were grown on 0.1% gelatin-coated coverslips and directly fixed (C) or treated for 30 min with 5 μg/ml the fungal metabolite BFA (D) before being fixed and permeabilized. Cells were then incubated with goat anti-cPLA2α and rabbit anti-ManII antibodies. Then cells were incubated with Alexfluor 488–conjugated anti-goat and Alexfluor 594–conjugated anti-rabbit antibodies. Images represent projected stacks of whole cells. Scale bar, 10 μm. All results are representative of three separate experiments.
cPLA2α is associated with membrane components of the Golgi apparatus in confluent HUVECs and does not simply reside in close proximity (Figure 3, C and D). Cells were treated with BFA, a fungal metabolite that inhibits trafficking through the secretory pathway and redistributes the Golgi stack to the ER. Protein transport through the Golgi apparatus is blocked and Golgi- and trans-Golgi network (TGN)-resident proteins accumulate in the ER and endosomal systems, respectively (Lippincott-Schwartz et al., 1990, 1991; Wood et al., 1991; Klausner et al., 1992). Mannosidase II (ManII), a Golgi-resident protein (Novikoff et al., 1983), and cPLA2α-positive Golgi structures were fully dispersed (Figure 3D) by 5 μg/ml BFA compared with control cells (Figure 3C). ManII- and cPLA2α-positive Golgi structures reform upon BFA washout and recovery (unpublished data).
In confluent HUVECs, cPLA2α colocalized with components of the distal Golgi apparatus, i.e., the medial-Golgi, trans-Golgi, and trans-Golgi network (Figure 4). The Golgi complex consists of five spatially distinct subcompartments: the ER-Golgi intermediate compartment (ERGIC), cis-Golgi, medial-Golgi, trans-Golgi, and TGN. Antibodies specific to subcompartment resident proteins were used in costaining microscopy experiments to determine more specifically the localization of cPLA2α in confluent HUVECs (Figure 4). ERGIC-53 (Figure 4A), GM130 (Figure 4B), ManII (Figure 4C), β-1,4-galactosyltransferase (GalT; Figure 4D) and TGN46 (Figure 4E) are specific markers for the ERGIC (Schweizer et al., 1988, 1990), cis-Golgi (Nakamura et al., 1995), medial-Golgi (Novikoff et al., 1983), trans-Golgi (Roth et al., 1985; Nilsson et al., 1993), and TGN (Prescott et al., 1997), respectively. cPLA2α appeared to colocalize most extensively with ManII- and GalT-positive structures (yellow indicates overlap; Figure 4, C and D). Extensive overlap was also seen with TGN46 (Figure 4E) but not to the extent of ManII and GalT. cPLA2α only appeared closely associated and not superimposed with ERGIC-53 (Figure 4A) and GM130 (Figure 4B). Quantification of the colocalization of cPLA2α with Golgi subcompartment markers was determined (Figure 4F). Percentage colocalization of cPLA2α with ManII, GalT, and TGN-46 was seen to be 68.4 ± 3.5, 59.7 ± 3.8, and 42.2 ± 4.5%, respectively, whereas ERGIC-53 and GM130 displayed only 19.8 ± 5.6 and 23.8 ± 3.1% colocalization. All secondary antibody controls gave no staining (unpublished data).
Figure 4.

Colocalization of cPLA2α with Golgi compartment markers. Confluent HUVECs were grown on 0.1% gelatin-coated coverslips, fixed, and permeabilized. Cells were incubated with goat anti-cPLA2α antibody and either mouse anti-ERGIC-53 (A), rabbit anti-GM130 (B), rabbit anti-ManII (C), mouse anti-GalT (D), or rabbit anti-TGN46 (E) antibodies. Cells were then incubated with anti-goat Alexafluor 488–conjugated and either anti-rabbit or anti-mouse Alexafluor 594–conjugated antibodies. Images represent 0.2-μm sections through cell nuclei. Scale bar, 10 μm. Results are representative of three separate experiments. Quantification of cPLA2α overlap with ERGIC-53, GM130, ManII, GalT, and TGN46 (F). Percentage colocalization was calculated using the IMARIS computer package as described in Materials and Methods (n = 9, ±SEM). * p < 0.02 versus GM130. ** p <0.001 versus GM130.
The Subcellular Localization of cPLA2α Is Associated with HUVEC Proliferation
HUVEC confluence and proliferation are directly linked (Figure 5A). Subconfluent cultures of endothelial cells proliferate until they reach confluence then cell-cell contacts inhibit cellular proliferation (Chen et al., 2000). HUVECs were seeded at equivalent densities (0.55 × 103/cm2) and cultured for 7 d. From days 2–7, cell numbers and proliferation per cell were determined and compared (Figure 5A). Cell density rose exponentially to reach a plateau after 5 d. BrdU incorporation per cell declined in correlation to also plateau at 5 d. At this point a confluent HUVEC monolayer is formed, cell-cell contacts inhibit proliferation, cells enter the G0 phase of quiescence, and cell numbers cease to increase.
Figure 5.

The subcellular localization of cPLA2α is associated with HUVEC proliferation. HUVEC cell density and growth were quantified over a culture period of 7 d (A). HUVECs (0.55 × 103/cm2) were seeded in 0.1% gelatin-coated 24-well culture dishes. They were counted after trypsinization each day from day 2 to day 7 after seeding (logarithmic scale, solid line with diamonds). Results represent the number of cells per cm2 (n = 3, ±SEM). HUVEC proliferation was determined using a colorimetric ELISA based on BrdU incorporation, as described in Materials and Methods (dotted line with squares). HUVECs (0.55 × 103/cm2) were seeded in 0.1% gelatin-coated 96-well culture dishes and grown for varying periods of time. Cells were labeled for 16 h with BrdU before processing. Results represent the BrdU incorporation per cell (n = 6, ±SEM). The subcellular localization of cPLA2α was quantified over a culture period of 7 d and compared with the BrdU incorporation per cell (B). HUVECs were seeded in 0.1% gelatin-coated coverslips at an equivalent density to those seeded in 96-well culture dishes. From day 2 to day 7 they were fixed, permeabilized, and processed for immunofluorescence microscopy and the localization of cPLA2α determined (solid line with diamonds). Results represent the percentage of total cells with juxta-nuclear localized cPLA2α at each time point (n = 3, ±SEM). Results were plotted alongside the quantification of BrdU incorporation per cell from Figure 5A. Arachidonic acid release in response to A23187 stimulation was determined for confluent (dotted line) and nonconfluent (solid line) HUVECs (C). Cells were loaded with 1 μCi/ml [3H]AA, stimulated with 5 μM A23187, and released AA was determined at the times indicated (n = 3, ±SEM). Results are expressed as a percentage of the total [3H]AA incorporated. * p < 0.01 versus confluent.
cPLA2α redistributed to the Golgi apparatus upon inhibition of HUVEC proliferation (Figure 5B). The subcellular localization of cPLA2α is confluence-dependent (Figure 2E), and endothelial cell confluence is directly linked to proliferation (Figure 5A). As cPLA2α mediates HUVEC proliferation, we hypothesized that the subcellular localization of cPLA2α was also associated with HUVEC proliferation. From days 2–7, indirect immunofluorescence microscopy was used to determine the subcellular localization of cPLA2α. HUVECs were scored for Golgi-localized cPLA2α staining, and results again plotted against proliferation per cell (Figure 5B). A direct correlation between BrdU incorporation per cell and the percentage of total cells with Golgi-localized cPLA2α was observed. The percentage of cells displaying Golgi-localized staining again reached a plateau after 5 d of growth. As cPLA2α mediates HUVEC proliferation, it is significant that its subcellular localization is closely linked to the proliferative status of these cells.
It was important to determine whether association of cPLA2α with the Golgi apparatus affected its enzyme activity. Consequently, nonconfluent, proliferating, and confluent nonproliferating cultures of cells were assayed for AA release in response to elevation of cytosolic Ca2+ (Figure 5C). cPLA2α activity was reduced by 87% in confluent endothelial cells (Figure 5C); therefore, Golgi associated cPLA2α was inactivated.
Mechanical Wounding Induces cPLA2α Relocation in HUVECs
On mechanical wounding of confluent HUVEC monolayers, cPLA2α reversibly redistributes from the Golgi apparatus to the diffuse cytoplasmic and nuclear location of nonconfluent HUVECs (Figure 6A–D). Mechanical wounding of confluent endothelial monolayers induces exit from G0, entry into the cell cycle, proliferation, and migration of cells to fill denuded areas (Chen et al., 2000). HUVECs were mechanically wounded to investigate the effects on Golgi-localized cPLA2α upon conversion from a confluent, quiescent to a subconfluent, proliferating state. Cells were fixed at various time points after initial wounding and cPLA2α was detected by immunofluorescence microscopy (Figure 6A–C). Areas to the right side of the panels depict wound borders at the edge of denuded zones. At 0 h after wounding, HUVECs retained Golgi localized cPLA2α (Figure 6A). However, 8 h (Figure 6B) and 24 h (Figure 6C) after wounding, HUVECs at the wound border displayed progressively less Golgi-associated cPLA2α. Cells invading the wound revert to the diffuse nuclear and cytoplasmic localization characteristic of nonconfluent cells. From 2 h after wounding, the percentage of HUVECs at wound borders displaying Golgi-localized cPLA2α was significantly reduced (Figure 6D). After 24 h, only 11.4 ± 1.7% of cells at the wound border retained Golgi-localized cPLA2α. The Golgi apparatus remained intact (ManII immunofluorescence staining) throughout the experiment. Association of cPLA2α with the Golgi apparatus is thus reversible upon conversion from confluent to subconfluent cells.
Figure 6.

The Golgi association of cPLA2α is reversible upon wounding of confluent HUVEC monolayers. HUVECs were grown to confluence on 0.1% gelatin-coated coverslips and subjected to mechanical wounding. HUVECs were allowed to recover for varying periods of time before fixation. The localization of cPLA2α in cells at the wound border (right of panels) after 0 h (A), 8 h (B), and 24 h (C) of recovery was determined by immunofluorescence microscopy using a goat polyclonal antibody. Images represent projections of whole cells; scale bar, 100 μm. Results are representative of three separate experiments. In D, the percentage of cells at the wound border with Golgi-localized cPLA2α were quantified after 0, 1, 2, 4, 8, and 24 h of recovery (n = 3, ±SEM). * p < 0.05 versus 0 h, ** p < 0.001 versus 0 h.
cPLA2α Activity Is Required for Induction of Ki67 Expression and Entry Into the Cell Cycle
After mechanical wounding, entry into the cell cycle occurs upon cPLA2α relocation (Figure 7). Ki67 is a nuclear protein vital for proliferation (Schluter et al., 1993) and is expressed in the G1, S, G2, and mitotic phases of the cell cycle but not in the G0 phase of quiescent cells (Gerdes et al., 1984). Confluent monolayers of endothelial cells undergo contact inhibition of cellular proliferation and enter G0. On mechanical wounding, cells migrate and proliferate to fill the denuded zone and Ki67 expression is induced upon entry into the cell cycle. By immunofluorescence microscopy, only 11 ± 0.1% of HUVECs in a confluent monolayer expressed Ki67 because most cells are in the G0 phase of quiescence (Figure 7, A and C). On mechanical wounding and recovery for 24 h, 72 ± 1.8% of cells at the wound border expressed Ki67 as they migrate and proliferate into the wound (Figure 7, B and C). A large proportion of HUVECs expressing Ki67 at wound borders (88.8 ± 0.7%) also displayed relocation of cPLA2α from the Golgi apparatus to the nucleus and cytosol (Figure 7, B and D). In correlation, 87.8 ± 0.72% of cells that have lost their Golgi-localized cPLA2α are Ki67 positive (unpublished data). Thus upon mechanical wounding, Ki67 expression, entry into the cell cycle and proliferation are induced in cells where cPLA2α has relocated to the cytosol and nucleus.
Figure 7.

Ki67 upregulation induced by mechanical wounding is associated with cPLA2α relocation. Confluent HUVECs were grown on 0.1% gelatin-coated coverslips and either directly fixed (A) or mechanically wounded and allowed to recover for 24 h before fixing (B). Cells were permeabilized and incubated with goat anti-cPLA2α antibody and rabbit anti-Ki67 antibodies. Cells were then incubated with anti-goat Alexafluor 488–conjugated and either anti-rabbit Alexafluor 594–conjugated antibodies. Images represent a confluent monolayer (A) or wound border (B) and are projections of whole cells. Scale bar, 100 μm. In C, the percentage of total cells with nucleoli localized Ki67 was quantified in confluent monolayers of cells and at wound borders after mechanical wounding and 24 h of recovery (n = 3; ±SEM). In D, the subcellular localization of cPLA2α in cells at the wound border with nucleoli localized Ki67 was determined. * p < 0.001 versus confluent monolayer. † p < 0.001 versus Golgi. All results are representative of at least three separate experiments
cPLA2α activity is vital for exit from G0 and entry into the S phase of the cell cycle as pyrrolidine-1 inhibits Ki67 induction upon wounding (Figure 8). Confluent monolayers of HUVECs were mechanically wounded and recovered for 24 h in the absence or presence of 10 μM pyrrolidine-1. Cells that underwent cPLA2α relocation were analyzed by immunofluorescence microscopy to assess the level of cPLA2α and Ki67 expression (Figure 8, A and B). Levels of cPLA2α expression do not significantly change (Figure 8C) but a 48.2% (±4.7%) reduction in Ki67 expression was observed after recovery in pyrrolidine-1 (Figure 8D). Ki67 expression is essential for cell cycle progression and proliferation. Thus, induction of HUVEC proliferation requires cPLA2α activity and occurs upon relocation of cPLA2α to the cytoplasm and nucleus.
Figure 8.
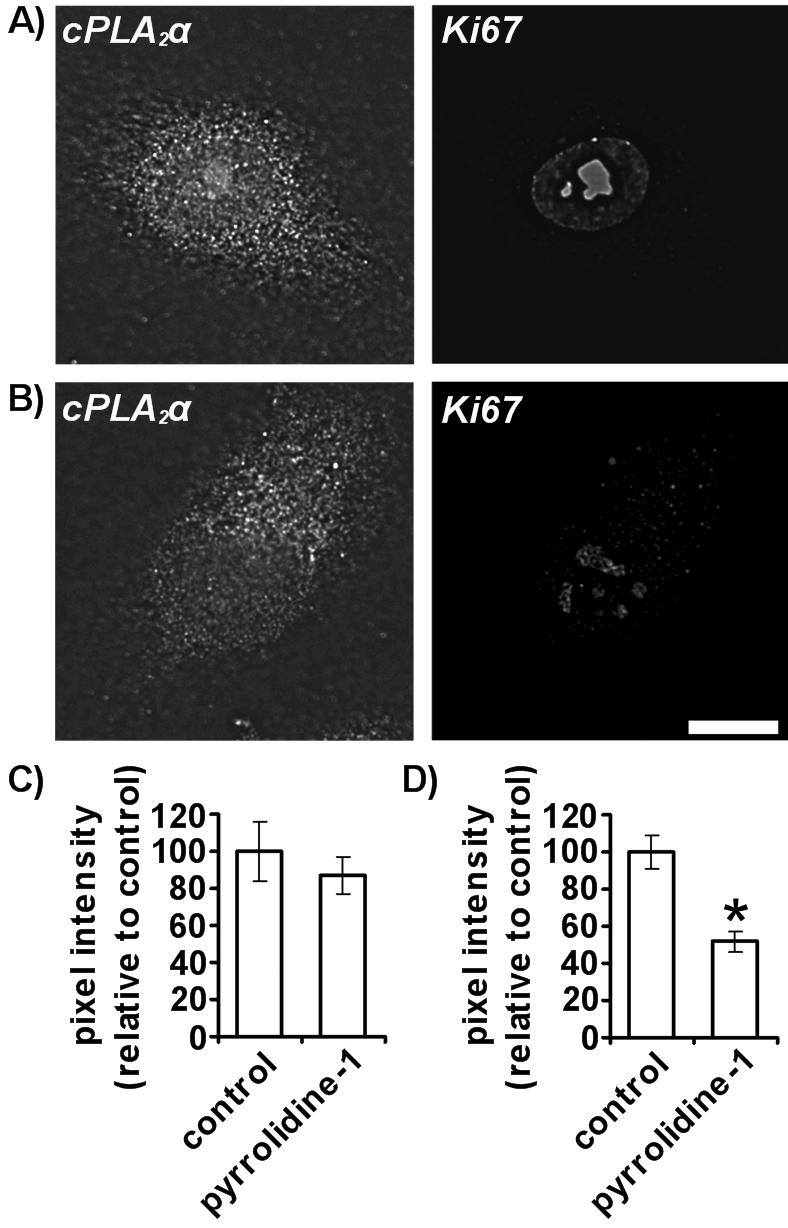
cPLA2α inhibition prevents Ki67 induction in response to mechanical wounding. Confluent HUVECs were grown on 0.1% gelatin-coated coverslips, mechanically wounded, and recovered for 24 h in the absence (A) and presence (B) of 10 μM pyrrolidine-1. Cells were fixed, permeabilized, and incubated with goat anti-cPLA2α antibody and rabbit anti-Ki67 antibodies. Cells were then incubated with anti-goat Alexafluor 488–conjugated and either anti-rabbit or anti-mouse Alexafluor 594–conjugated antibodies. Only cells displaying non-Golgi localized cPLA2α at wound borders were imaged. Images represent 0.2-μm sections through cell nuclei. Scale bar, 25 μm. Mean cellular cPLA2α pixel intensity (C) and nuclear Ki67 pixel intensity (D) were determined using Image J software as described in Materials and Methods. Results represent cells displaying non-Golgi localized cPLA2α at wound borders for those recovered with and without 10 μM pyrrolidine-1 (n = 20). * p < 0.001 versus control. All results are representative of at least three separate experiments.
DISCUSSION
Our results link cPLA2α activity to the regulation of proliferation of endothelial cells and also show that cPLA2α activity is required for entry into the cell cycle from G0. The induction of HUVEC proliferation from G0 occurs upon relocation of cPLA2α from the Golgi apparatus to a diffuse cytoplasmic and nuclear location. Thus, the subcellular localization of cPLA2α mediates its function.
Our report is the first to document the importance of cPLA2α activity for controlling endothelial cell proliferation and entry into the cell cycle. Previously AACOCF3, an inhibitor of both cPLA2 and iPLA2 (Ackermann et al., 1995; Street et al., 1993) was seen to inhibit bovine aortic endothelial cell proliferation (Antoniotti et al., 2003) but that study lacked specific inhibitor studies. Here we use the specific cPLA2α inhibitor, pyrrolidine-1 (Ghomashchi et al., 2001), to directly implicate cPLA2α in endothelial cell proliferation. Reduced AA liberation by cPLA2α is at least partially responsible for the inhibitory effects of pyrrolidine-1. Despite this, exogenous AA does not augment the proliferation of nonconfluent HUVECs (unpublished data) although they may proliferate at an optimal rate in our culture conditions so exogenous AA has no effect. In addition, exogenous AA does not induce entry into the cell cycle and proliferation of confluent endothelial cells (unpublished data). This demonstrates that although cPLA2α activity is essential for cell cycle entry, it is insufficient by itself to override the contact-inhibition of proliferation. Our report suggests that inhibition of cPLA2α could be a viable antiangiogenic route because the induction of cell cycle entry and proliferation in resting endothelial cells is crucial to angiogenesis (Carmeliet, 2000) and cPLA2α activity is essential to this process.
We describe the novel confluence-dependent association of cPLA2α with membrane components of the distal Golgi apparatus. When associated with the Golgi apparatus, cPLA2α activity is inhibited by 87%. This is in contrast to previous studies that demonstrate increased cPLA2α activity upon association with the Golgi apparatus (Pettus et al., 2004; Grimmer et al., 2005). Association with the Golgi apparatus and cPLA2α inactivation may occur to differing extents depending on cell type, cell confluence, or the ability of cells to undergo contact inhibition of proliferation. We propose that Golgi association and inactivation of cPLA2α is a feature of the contact-inhibition of endothelial cells.
Our results suggest that the Golgi association and inactivation of cPLA2α is important to the regulation of its function in endothelial cells. cPLA2α associates with the Golgi apparatus at HUVEC confluence when proliferation is inhibited and cells exit the cell cycle. Entry back into the cell cycle occurs upon relocation of cPLA2α back to the diffuse cytoplasmic and nuclear location seen in subconfluent HUVECs. Thus, cPLA2α is mediating HUVEC proliferation when it is found throughout the cytoplasm and nucleus so the localization of cPLA2α in proliferating endothelial cells is essential for targeting it to areas where it functions to aid proliferation. AA is evolved in response to Ca2+ elevation and cPLA2α relocation to membrane substrate (Hirabayashi et al., 1999) as occurs in response to growth factor stimulation. In nonconfluent endothelial cells in response to Ca2+ elevation, cPLA2α associates with perinuclear membrane substrate, a known site of cPLA2α-derived AA production (Hirabayashi et al., 1999). This does not occur in confluent endothelial cells because cPLA2α remains immobilized on the Golgi apparatus and cPLA2α activity remains inhibited. Thus, we outline a novel mechanism for the inhibition of cPLA2α activity. We propose that association with the Golgi apparatus is a mechanism to partition cPLA2α from sites of action in quiescent endothelial cells, excluding it from substrate. This model is also consistent with the regulation of MEK/ERK by Sef, a molecular scaffold that resides in the Golgi complex. Binding to Sef allows MEK/ERK signaling to cytosolic substrates but excludes nuclear targets (Torii et al., 2004).
We are the first to document the confluence-dependent relocation of cPLA2α in any cell type. Association of cPLA2α with distal components of the Golgi apparatus has been documented previously in epithelial cell lines but only in response to agents that elevate intracellular free Ca2+ (Evans et al., 2001; Grewal et al., 2003). Similar experiments using the Ca2+ ionophore, A23187, or histamine to elevate HUVEC intracellular free Ca2+, does not induce cPLA2α relocation to the Golgi apparatus but to the nuclear envelope (unpublished data). Stimulation-dependent relocation of cPLA2α to the Golgi apparatus of epithelial cells is seen to place cPLA2α in close apposition with the AA-metabolizing enzyme COX-1 (Grewal et al., 2003). In HUVECs, costaining revealed no such functional association with COX-1, COX-2, or prostacyclin I synthase upon HUVEC confluence (unpublished data).
Golgi-associated cPLA2α may play a role in Golgi architecture maintenance or protein trafficking. The PLA2 family of enzymes are strongly implicated in these processes, specifically via membrane tubule formation (Brown et al., 2003). Previous reports document disruption of the Golgi apparatus upon treatment of cells with PLA2 inhibitors (de Figueiredo et al., 1998, 1999; Drecktrah and Brown, 1999; Kuroiwa et al., 2001). We see that in HUVECs, the sPLA2 inhibitor, ONO-RS-082 (Hashimoto et al., 2003), induces Golgi dispersal but pyrrolidine-1 does not (unpublished data). A previous study did report selective blocking of constitutive trafficking to the plasma membrane and retention of proteins in the endoplasmic reticulum of canine kidney cells overexpressing cPLA2α (Choukroun et al., 2000). But the possible role of cPLA2α in specific trafficking events in confluent HUVECs has yet to be investigated.
cPLA2α has previously been linked to cellular proliferation, as seen with studies in macrophages (Tashiro et al., 2004). Although no studies of cPLA2α activity were conducted, cPLA2α was seen to regulate expression of the c-myc protooncogene in a B-Myb–dependent manner (Tashiro et al., 2004). AA metabolites also induce endothelial cell proliferation (Dethlefsen et al., 1994; Nie et al., 2000; Antoniotti et al., 2003), and cPLA2α activity is involved in gene regulation (Pawliczak et al., 2002), but little is known of how these processes are regulated. cPLA2α is the rate-limiting enzyme of AA evolution and is thus a prime candidate for controlling the synthesis of proproliferative AA metabolites in proliferating and nonproliferating cells. We find that in HUVECs, cPLA2α activity is essential for proliferation and cell cycle entry and that these functions are mediated by its subcellular localization.
Acknowledgments
We thank M. H. Gelb for providing pyrrolidine-1 and G. J. Howell for help with bioimaging. This work was funded by a Biotechnology and Biological Sciences Research Council PhD studentship (S.P.H), a British Heart Foundation project grant (S.P., J.H.W.), and a Wellcome Trust equipment award to the Leeds Bioimaging Facility (S.P.).
This article was published online ahead of print in MBC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E05-02-0164) on June 1, 2005.
Abbreviations used: AA, arachidonic acid; BFA, brefeldin A; cPLA2α, cytosolic phospholipase A2-α; ERGIC-53, endoplasmic reticulum-Golgi intermediate compartment-53; GalT, β-1,4-galactosyltransferase; HUVEC, human umbilical vein endothelial cell; iPLA2, calcium-independent phospholipase A2; ManII, mannosidase II; sPLA2, secretory phospholipase A2
References
- Ackermann, E. J., Conde-Frieboes, K., and Dennis, E. A. (1995). Inhibition of macrophage Ca(2+)-independent phospholipase A2 by bromoenol lactone and trifluoromethyl ketones. J. Biol. Chem. 270, 445-450. [DOI] [PubMed] [Google Scholar]
- Antoniotti, S., Fiorio Pla, A., Pregnolato, S., Mottola, A., Lovisolo, D., and Munaron, L. (2003). Control of endothelial cell proliferation by calcium influx and arachidonic acid metabolism: a pharmacological approach. J. Cell Physiol. 197, 370-378. [DOI] [PubMed] [Google Scholar]
- Bailleux, A., Wendum, D., Audubert, F., Jouniaux, A. M., Koumanov, K., Trugnan, G., and Masliah, J. (2004). Cytosolic phospholipase A2–p11 interaction controls arachidonic acid release as a function of epithelial cell confluence. Biochem. J. 378, 307-315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barwise, J. L., and Walker, J. H. (1996). Annexins II, IV, V and VI relocate in response to rises in intracellular calcium in human foreskin fibroblasts. J. Cell Sci. 109(Pt 1), 247-255. [DOI] [PubMed] [Google Scholar]
- Bittova, L., Sumandea, M., and Cho, W. (1999). A structure-function study of the C2 domain of cytosolic phospholipase A2. Identification of essential calcium ligands and hydrophobic membrane binding residues. J. Biol. Chem. 274, 9665-9672. [DOI] [PubMed] [Google Scholar]
- Brem, H., Gresser, I., Grosfeld, J., and Folkman, J. (1993). The combination of antiangiogenic agents to inhibit primary tumor growth and metastasis. J. Pediatr. Surg. 28, 1253-1257. [DOI] [PubMed] [Google Scholar]
- Brown, W. J., Chambers, K., and Doody, A. (2003). Phospholipase A2 (PLA2) enzymes in membrane trafficking: mediators of membrane shape and function. Traffic 4, 214-221. [DOI] [PubMed] [Google Scholar]
- Bunt, G., de Wit, J., van den Bosch, H., Verkleij, A. J., and Boonstra, J. (1997). Ultrastructural localization of cPLA2 in unstimulated and EGF/A23187-stimulated fibroblasts. J. Cell Sci. 110(Pt 19), 2449-2459. [DOI] [PubMed] [Google Scholar]
- Carmeliet, P. (2000). Mechanisms of angiogenesis and arteriogenesis. Nat. Med. 6, 389-395. [DOI] [PubMed] [Google Scholar]
- Channon, J. Y., and Leslie, C. C. (1990). A calcium-dependent mechanism for associating a soluble arachidonoyl-hydrolyzing phospholipase A2 with membrane in the macrophage cell line RAW 264.7. J. Biol. Chem. 265, 5409-5413. [PubMed] [Google Scholar]
- Chen, D., Walsh, K., and Wang, J. (2000). Regulation of cdk2 activity in endothelial cells that are inhibited from growth by cell contact. Arterioscler. Thromb. Vasc. Biol. 20, 629-635. [DOI] [PubMed] [Google Scholar]
- Choukroun, G. J., Marshansky, V., Gustafson, C. E., McKee, M., Hajjar, R. J., Rosenzweig, A., Brown, D., and Bonventre, J. V. (2000). Cytosolic phospholipase A(2) regulates golgi structure and modulates intracellular trafficking of membrane proteins. J. Clin. Invest. 106, 983-993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark, J. D., Schievella, A. R., Nalefski, E. A., and Lin, L. L. (1995). Cytosolic phospholipase A2. J. Lipid Mediat. Cell Signal. 12, 83-117. [DOI] [PubMed] [Google Scholar]
- de Figueiredo, P., Drecktrah, D., Katzenellenbogen, J. A., Strang, M., and Brown, W. J. (1998). Evidence that phospholipase A2 activity is required for Golgi complex and trans Golgi network membrane tubulation. Proc. Natl. Acad. Sci. USA 95, 8642-8647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Figueiredo, P., Polizotto, R. S., Drecktrah, D., and Brown, W. J. (1999). Membrane tubule-mediated reassembly and maintenance of the Golgi complex is disrupted by phospholipase A2 antagonists. Mol. Biol. Cell 10, 1763-1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis, E. A. (1997). The growing phospholipase A2 superfamily of signal transduction enzymes. Trends Biochem. Sci. 22, 1-2. [DOI] [PubMed] [Google Scholar]
- Dethlefsen, S. M., Shepro, D., and D'Amore, P. A. (1994). Arachidonic acid metabolites in bFGF-, platelet-derived growth factor-, and serum-stimulated vascular cell growth. Exp. Cell Res. 212, 262-273. [DOI] [PubMed] [Google Scholar]
- Dormond, O., Foletti, A., Paroz, C., and Ruegg, C. (2001). NSAIDs inhibit alpha V beta 3 integrin-mediated and Cdc42/Rac-dependent endothelial-cell spreading, migration and angiogenesis. Nat. Med. 7, 1041-1047. [DOI] [PubMed] [Google Scholar]
- Drecktrah, D., and Brown, W. J. (1999). Phospholipase A(2) antagonists inhibit nocodazole-induced Golgi ministack formation: evidence of an ER intermediate and constitutive cycling. Mol. Biol. Cell 10, 4021-4032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans, C. E., Billington, D., and McEvoy, F. A. (1984). Prostacyclin production by confluent and non-confluent human endothelial cells in culture. Prostaglandins Leukot. Med. 14, 255-266. [DOI] [PubMed] [Google Scholar]
- Evans, J. H., Spencer, D. M., Zweifach, A., and Leslie, C. C. (2001). Intracellular calcium signals regulating cytosolic phospholipase A2 translocation to internal membranes. J. Biol. Chem. 276, 30150-30160. [DOI] [PubMed] [Google Scholar]
- Fafeur, V., Jiang, Z.P., and Bohlen, P. (1991). Signal transduction by bFGF, but not TGF beta 1, involves arachidonic acid metabolism in endothelial cells. J. Cell Physiol. 149, 277-283. [DOI] [PubMed] [Google Scholar]
- Folkman, J., and Shing, Y. (1992). Angiogenesis. J. Biol. Chem. 267, 10931-10934. [PubMed] [Google Scholar]
- Gerdes, J., Lemke, H., Baisch, H., Wacker, H. H., Schwab, U., and Stein, H. (1984). Cell cycle analysis of a cell proliferation-associated human nuclear antigen defined by the monoclonal antibody (mAb) Ki-67. J. Immunol. 133, 1710-1715. [PubMed] [Google Scholar]
- Ghomashchi, F., Stewart, A., Hefner, Y., Ramanadham, S., Turk, J., Leslie, C. C., and Gelb, M. H. (2001). A pyrrolidine-based specific inhibitor of cytosolic phospholipase A(2)alpha blocks arachidonic acid release in a variety of mammalian cells. Biochim. Biophys. Acta 1513, 160-166. [DOI] [PubMed] [Google Scholar]
- Grewal, S., Morrison, E. E., Ponnambalam, S., and Walker, J. H. (2002). Nuclear localisation of cytosolic phospholipase A(2)-alpha in the EA.hy. 926 human endothelial cell line is proliferation dependent and modulated by phosphorylation. J. Cell Sci. 115, 4533-4543. [DOI] [PubMed] [Google Scholar]
- Grewal, S., Ponnambalam, S., and Walker, J. H. (2003). Association of cPLA2-alpha and COX-1 with the Golgi apparatus of A549 human lung epithelial cells. J. Cell Sci. 116, 2303-2310. [DOI] [PubMed] [Google Scholar]
- Grewal, S., Smith, J., Ponnambalam, S., and Walker, J. (2004). Stimulation-dependent recruitment of cytosolic phospholipase A2-alpha to EA.hy. 926 endothelial cell membranes leads to calcium-independent association. Eur. J. Biochem. 271, 69-77. [DOI] [PubMed] [Google Scholar]
- Grimmer, S., Ying, M., Walchli, S., van Deurs, B., and Sandvig, K. (2005). Golgi vesiculation induced by cholesterol occurs by a dynamin- and cPLA-dependent mechanism. Traffic 6, 144-156. [DOI] [PubMed] [Google Scholar]
- Hashimoto, T., Kihara, M., Yokoyama, K., Fujita, T., Kobayashi, S., Matsushita, K., Tamura, K., Hirawa, N., Toya, Y., and Umemura, S. (2003). Lipoxygenase products regulate nitric oxide and inducible nitric oxide synthase production in interleukin-1beta stimulated vascular smooth muscle cells. Hypertens. Res. 26, 177-184. [DOI] [PubMed] [Google Scholar]
- Heggeness, M. H., Wang, K., and Singer, S. J. (1977). Intracellular distributions of mechanochemical proteins in cultured fibroblasts. Proc. Natl. Acad. Sci. USA 74, 3883-3887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirabayashi, T., Kume, K., Hirose, K., Yokomizo, T., Iino, M., Itoh, H., and Shimizu, T. (1999). Critical duration of intracellular Ca2+ response required for continuous translocation and activation of cytosolic phospholipase A2. J. Biol. Chem. 274, 5163-5169. [DOI] [PubMed] [Google Scholar]
- Howell, G. J. et al. (2004). Endothelial cell confluence regulates Wiebel-Palade body formation. Mol. Membr. Biol. 21, 413-421. [DOI] [PubMed] [Google Scholar]
- Jaffe, E. A. (1984). Biology of Endothelial Cells, Leiden, The Netherlands: Martinus Nijhoff Publishers.
- Klausner, R. D., Donaldson, J. G., and Lippincott-Schwartz, J. (1992). Brefeldin A: insights into the control of membrane traffic and organelle structure. J. Cell Biol. 116, 1071-1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer, R. M., and Sharp, J. D. (1997). Structure, function and regulation of Ca2+-sensitive cytosolic phospholipase A2 (cPLA2). FEBS Lett. 410, 49-53. [DOI] [PubMed] [Google Scholar]
- Kuroiwa, N., Nakamura, M., Tagaya, M., and Takatsuki, A. (2001). Arachidonyltrifluoromethy ketone, a phospholipase A(2) antagonist, induces dispersal of both Golgi stack- and trans Golgi network-resident proteins throughout the cytoplasm. Biochem. Biophys. Res. Commun. 281, 582-588. [DOI] [PubMed] [Google Scholar]
- Laemmli, U. K. (1970). Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680-685. [DOI] [PubMed] [Google Scholar]
- Lippincott-Schwartz, J., Donaldson, J. G., Schweizer, A., Berger, E. G., Hauri, H. P., Yuan, L. C., and Klausner, R. D. (1990). Microtubule-dependent retrograde transport of proteins into the ER in the presence of brefeldin A suggests an ER recycling pathway. Cell 60, 821-836. [DOI] [PubMed] [Google Scholar]
- Lippincott-Schwartz, J., Yuan, L., Tipper, C., Amherdt, M., Orci, L., and Klausner, R. D. (1991). Brefeldin A's effects on endosomes, lysosomes, and the TGN suggest a general mechanism for regulating organelle structure and membrane traffic. Cell 67, 601-616. [DOI] [PubMed] [Google Scholar]
- Nakamura, N., Rabouille, C., Watson, R., Nilsson, T., Hui, N., Slusarewicz, P., Kreis, T. E., and Warren, G. (1995). Characterization of a cis-Golgi matrix protein, GM130. J. Cell Biol. 131, 1715-1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalefski, E. A., Sultzman, L. A., Martin, D. M., Kriz, R. W., Towler, P. S., Knopf, J. L., and Clark, J. D. (1994). Delineation of two functionally distinct domains of cytosolic phospholipase A2, a regulatory Ca(2+)-dependent lipid-binding domain and a Ca(2+)-independent catalytic domain. J. Biol. Chem. 269, 18239-18249. [PubMed] [Google Scholar]
- Nie, D., Tang, K., Diglio, C., and Honn, K. V. (2000). Eicosanoid regulation of angiogenesis: role of endothelial arachidonate 12-lipoxygenase. Blood 95, 2304-2311. [PubMed] [Google Scholar]
- Nilsson, T., Pypaert, M., Hoe, M. H., Slusarewicz, P., Berger, E. G., and Warren, G. (1993). Overlapping distribution of two glycosyltransferases in the Golgi apparatus of HeLa cells. J. Cell Biol. 120, 5-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novikoff, P. M., Tulsiani, D. R., Touster, O., Yam, A., and Novikoff, A. B. (1983). Immunocytochemical localization of alpha-d-mannosidase II in the Golgi apparatus of rat liver. Proc. Natl. Acad. Sci. USA 80, 4364-4368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawliczak, R., Han, C., Huang, X. L., Demetris, A. J., Shelhamer, J. H., and Wu, T. (2002). 85-kDa cytosolic phospholipase A2 mediates peroxisome proliferator-activated receptor gamma activation in human lung epithelial cells. J. Biol. Chem. 277, 33153-33163. [DOI] [PubMed] [Google Scholar]
- Pettus, B. J. et al. (2004). Ceramide 1-phosphate is a direct activator of cytosolic phospholipase A2. J. Biol. Chem. 279, 11320-11326. [DOI] [PubMed] [Google Scholar]
- Prescott, A. R., Lucocq, J. M., James, J., Lister, J. M., and Ponnambalam, S. (1997). Distinct compartmentalization of TGN46 and beta 1,4-galactosyltransferase in HeLa cells. Eur J. Cell Biol. 72, 238-246. [PubMed] [Google Scholar]
- Roth, J., Taatjes, D. J., Lucocq, J. M., Weinstein, J., and Paulson, J. C. (1985). Demonstration of an extensive trans-tubular network continuous with the Golgi apparatus stack that may function in glycosylation. Cell 43, 287-295. [DOI] [PubMed] [Google Scholar]
- Sa, G., Murugesan, G., Jaye, M., Ivashchenko, Y., and Fox, P. L. (1995). Activation of cytosolic phospholipase A2 by basic fibroblast growth factor via a p42 mitogen-activated protein kinase-dependent phosphorylation pathway in endothelial cells. J. Biol. Chem. 270, 2360-2366. [DOI] [PubMed] [Google Scholar]
- Schievella, A. R., Regier, M. K., Smith, W. L., and Lin, L. L. (1995). Calcium-mediated translocation of cytosolic phospholipase A2 to the nuclear envelope and endoplasmic reticulum. J. Biol. Chem. 270, 30749-30754. [DOI] [PubMed] [Google Scholar]
- Schluter, C., Duchrow, M., Wohlenberg, C., Becker, M. H., Key, G., Flad, H. D., and Gerdes, J. (1993). The cell proliferation-associated antigen of antibody Ki-67, a very large, ubiquitous nuclear protein with numerous repeated elements, representing a new kind of cell cycle-maintaining proteins. J. Cell Biol. 123, 513-522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweizer, A., Fransen, J. A., Bachi, T., Ginsel, L., and Hauri, H. P. (1988). Identification, by a mAb, of a 53-kD protein associated with a tubulo-vesicular compartment at the cis-side of the Golgi apparatus. J. Cell Biol. 107, 1643-1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweizer, A., Fransen, J. A., Matter, K., Kreis, T. E., Ginsel, L., and Hauri, H. P. (1990). Identification of an intermediate compartment involved in protein transport from endoplasmic reticulum to Golgi apparatus. Eur. J. Cell Biol. 53, 185-196. [PubMed] [Google Scholar]
- Sierra-Honigmann, M. R., Bradley, J. R., and Pober, J. S. (1996). “Cytosolic” phospholipase A2 is in the nucleus of subconfluent endothelial cells but confined to the cytoplasm of confluent endothelial cells and redistributes to the nuclear envelope and cell junctions upon histamine stimulation. Lab. Invest. 74, 684-695. [PubMed] [Google Scholar]
- Street, I. P., Lin, H. K., Laliberte, F., Ghomashchi, F., Wang, Z., Perrier, H., Tremblay, N. M., Huang, Z., Weech, P. K., and Gelb, M. H. (1993). Slow- and tight-binding inhibitors of the 85-kDa human phospholipase A2. Biochemistry 32, 5935-5940. [DOI] [PubMed] [Google Scholar]
- Tashiro, S., Sumi, T., Uozumi, N., Shimizu, T., and Nakamura, T. (2004). B-Myb-dependent regulation of c-Myc expression by cytosolic phospholipase A2. J. Biol. Chem. 279, 17715-17722. [DOI] [PubMed] [Google Scholar]
- Torii, S., Kusakabe, M., Yamamoto, T., Maekawa, M., and Nishida, E. (2004). Sef is a spatial regulator for Ras/MAP kinase signaling. Dev. Cell 7, 33-44. [DOI] [PubMed] [Google Scholar]
- Towbin, H., Staehelin, T., and Gordon, J. (1979). Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc. Natl. Acad. Sci. USA 76, 4350-4354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vane, J. R., Anggard, E. E., and Botting, R. M. (1990). Regulatory functions of the vascular endothelium. N. Engl. J. Med. 323, 27-36. [DOI] [PubMed] [Google Scholar]
- Virtanen, I., Ekblom, P., and Laurila, P. (1980). Subcellular compartmentalization of saccharide moieties in cultured normal and malignant cells. J. Cell Biol. 85, 429-434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whatley, R. E., Satoh, K., Zimmerman, G. A., McIntyre, T. M., and Prescott, S. M. (1994). Proliferation-dependent changes in release of arachidonic acid from endothelial cells. J. Clin. Invest. 94, 1889-1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood, S. A., Park, J. E., and Brown, W. J. (1991). Brefeldin A causes a microtubule-mediated fusion of the trans-Golgi network and early endosomes. Cell 67, 591-600. [DOI] [PubMed] [Google Scholar]