

Abstract
The three-dimensional structure of CD4M33, a mimic of the host-cell receptor-antigen CD4 and a powerful inhibitor of CD4–gp120 (viral envelope glycoprotein 120) interaction and HIV-1 entry into cells [Martin, Stricher, Misse, Sironi, Pugniere, Barthe, Prado-Gotor, Freulon, Magne, Roumestand et al. (2003) Nat. Biotechnol. 21, 71–76], was solved by 1H-NMR and its structure was modelled in its complex with gp120. In this complex, CD4M33 binds in a CD4-like mode and inserts its unnatural and prominent Bip23 (biphenylalanine-23) side-chain into the gp120 interior ‘Phe43 cavity’, thus filling its volume. CD4M33 was specifically labelled with fluorescein and shown by fluorescence anisotropy to bind to different gp120 glycoproteins with dissociation constants in the nanomolar range. Fluorescent CD4M33 was also used in a miniaturized 384-well-plate assay to study direct binding to a large panel of gp120 glycoproteins and in a competition assay to study binding of CD4 or other ligands targeting the CD4 binding site of gp120. Furthermore, by using the fluorescently labelled CD4M33 and the [Phe23]M33 mutant, which possesses a natural Phe23 residue and thus cannot penetrate the gp120 Phe43 cavity, we show that a recently discovered small-molecule-entry inhibitor, BMS-378806, does not target the CD4 binding site nor the Phe43 cavity of gp120. The fluorescently labelled CD4M33 mimic, its mutants and their derivatives represent useful tools with which to discover new molecules which target the CD4 binding site and/or the Phe43 cavity of gp120 glycoproteins in a high-throughput fluorescence-polarization assay and to characterize their mechanism of action.
Keywords: CD4 binding, drug discovery, fluorescence polarization, HIV inhibitors, protein–protein interaction, three-dimensional structure
Abbreviations: 2D, two-dimensional; Bip, biphenylalanine; CD4M33-Fl, fluorescein–CD4M33 derivative; DQF, double quantum filtered scalar; EGT, ecdysteroid glycosyltransferase; Fmoc, fluoren-9-ylmethoxycarbonyl; gp120, viral envelope glycoprotein 120; IC50, concentration causing 50% inhibition; ivDde, 4,4-dimethyl-2,6-dioxocyclohex-1-ylidene; NOE, nuclear Overhauser enhancement; r.m.s., root mean square; Tpa, thiopropionic acid; wt, wild-type
INTRODUCTION
According to the Joint United Nations Programme on HIV/AIDS (http://www.unaids.org/Unaids/EN/Resources/epidemiology.asp), about 40 million people worldwide are infected with HIV-1 as of the end 2004, and 20 million have already died. Drugs used in therapy are molecules that target HIV protease or reverse transcriptase enzymes, and only one new fusion inhibitor, enfuvirtide, has been recently approved [1,2]. Combinations of these inhibitors in the highly active antiretroviral therapy (‘HAART’) can control viral load and slow disease progression in many cases [1]. However, the emergence of new HIV isolates resistant to existing drugs, in addition to difficulties in compliance with drug regimens because of pill burden and adverse side effects, suggests that new therapies with new drugs targeting different steps of the HIV-1 life cycle are urgently needed.
HIV-1 entry is a multiple-step process that firstly involves binding of the gp120 (viral envelope glycoprotein 120) to the host-cell receptor CD4, then binding to CCR5 or CXCR4 chemokine receptors, insertion of the envelope fusion domain into the host-cell membrane and finally fusion of virus-cell membranes. Each of these steps can represent a potential target for new drugs [3,4].
The three-dimensional structure of the gp120 ‘core’ protein has been determined in the CD4-bound conformation [5] and very recently also in the unliganded form [6]. This structural information may help in the design of entry inhibitors. In the CD4-bound conformation, gp120 consists in an inner and outer domain connected by a four-stranded β-sheet (bridging sheet), whereas, in the unliganded conformation, although it maintains this two-domain organization, the inner domain is significantly different and the β-sheet is not formed [6]. CD4 binding creates a cavity of roughly 0.15 nm3 (150 Å3), which extends deeply in the interior of gp120 at the intersection between the inner and outer domain, whereas this cavity is absent in the unliganded form. Residue Phe43 of CD4 engages the entrance of this cavity, hence the name of ‘Phe43 cavity’, but leaves its interior free.
Only a handful of small molecules targeting HIV-1 envelope glycoproteins and inhibiting the entry step have been reported [3,7,8]. Stilbenedisulphonic acid [9], bathophenanthroline acid [10] and the polyanionic dye FP-21399 [11] have been reported to inhibit entry, but do not target the CD4 binding site. BMS-378806 [12–14] is a recently discovered low-molecular-mass organic compound that inhibits entry and infection of a large panel of HIV-1 viruses using the CCR5 and CXCR4 co-receptors and clinical isolates with median EC50 values of 50 nM. Elucidation of the mechanism of action shows that BMS-378806 binds to gp120 and inhibits the interaction of the HIV-1 envelope with cellular CD4 receptors [12,13]. However, data from a different laboratory suggested that its target might be distinct from the CD4 binding site [15,16] and, indeed, the recently determined structure of unliganded gp120 could identify a binding site in this structure [6]. This molecule was discovered during an indirect cell-based assay. The development of more direct and specific binding assay is required, not only to elucidate the mechanism of action of drug candidates such as this, but also to screen compound libraries for the discovery of new drug candidates.
We have engineered a small fully functional CD4 mimic, designated CD4M33, which binds the CD4-binding site of recombinant gp120, gp140 glycoproteins and HIV-1 particles with CD4-like affinity [17]. This mimic was designed to reproduce the structure of the gp120 glycoprotein binding ‘hot spot’ of the CD4 surface on to the scaffold consisting of the scorpion (Leiurus quinquestriatus hebraeus) toxin scyllatoxin. This small (31-residue) toxin was selected as the host structure for the CD4 epitope, since its structure, formed by an antiparallel β-sheet linked to a short helix by three disulphide bridges, contains an exposed positions-18–29 β-hairpin, which could superimpose its backbone atoms on those of the positions-36–47 CDR2-like loop of CD4 with an r.m.s. (root mean square) deviation of only 0.11 nm (1.10 Å). On the basis of this structural similarity, the scaffold permissiveness in sequence mutations and stability, even after sequence replacements [18,19], critical functional residues of the CDR2-like loop of CD4 could be grafted on to the β-hairpin of scyllatoxin. Then, by subsequent optimization steps, the initial low-affinity mimic (CD4M3) was first modified to increase CD4-likeness, yielding a 100-fold more potent CD4M9 mimic, which was then further improved to increase complementary interactions at the mimic/gp120 interface into CD4M33 [19], which presented CD4-like binding affinity. Notably, in order to increase the interaction surface between residue 23 (equivalent to Phe43 of CD4) and the interior Phe43 cavity of gp120, a longer Bip (biphenylalamine) side chain, instead of a regular phenylalanine one, was engineered in CD4M33. Hence, in constrast with the CD4 phenylalanine side chain, which could only engage the entrance of the Phe43 cavity, the additional phenyl ring of the CD4M33 biphenyl moiety could penetrate more deeply within the interior hydrophobic gp120 cavity and fill its empty space.
In the present study we determined the three-dimensional structure of CD4M33 by 1H-NMR. Our structure reveals a close structural similarity between the mimic β-hairpin and the CD4 CDR2 loop, which is central in gp120 binding. By using the CD4-bound structure of gp120 [5] as a template, we modelled CD4M33 in complex with gp120 and show that this miniprotein interacts with gp120 in a CD4-like mode and its protruding Bip23 residue penetrates into the Phe43 cavity. We then synthesized a fluorescently labelled CD4M33 derivative and, using fluorescence anisotropy, we determined its binding affinity for a number of gp120 glycoproteins from HIV-1 isolates that use CCR5 or CXCR4 co-receptors. Furthermore, by using this fluorescent CD4M33 as a tracer, a competition assay was developed and used to characterize different gp120 ligands and determine their dissociation constants. Then our fluorescence-anisotropy method using a mutant [Phe23]M33, which does not possess a protruding Bip23 but rather a ‘natural’ Phe23, permitted us to ascertain whether the newly discovered BMS-378806 inhibitor could bind the CD4 binding site or the Phe43 cavity of gp120 glycoproteins. Finally, the assay was then miniaturized and adapted to test multiple inhibitors in a short time and to screen small-molecule libraries. The fluorescent CD4M33, its mutants or derivatives may find applications in high-throughput screening of chemical libraries, thus may contribute to the discovery of inhibitors of HIV-1 entry, potential new antiviral drugs and the elucidation of their mechanism of action.
EXPERIMENTAL
Materials
All Fmoc (fluoren-9-ylmethoxycarbonyl)-protected amino acids, N,N′-dicyclohexylcarbodi-imide and 1-N-hydroxy-7-benzotriazole were from Nova Biochem (via VWR International, Fontenay-sous-Bois, France). Fmoc PAL-PEG-PS resin was from Applied Biosystems (via Applera Corporation, Norwalk, CT, U.S.A.). Fmoc-Bip was from Advanced Chemtech (Louisville, KT, U.S.A.). All other reagents and solvents used in the synthesis and purification were from Fluka (St Quentin Fallavier, France) or SDS (Solvants Documentation Syntheses) (Peypin, France). 5- and 6-carboxyfluorescein succinimidyl ester was from Molecular Probes (Leiden, The Netherlands). PBS and Tween 20 were from Sigma.
Chemical synthesis
Compound BMS-378806 was synthesized as described in [14]. The CD4 mimic CD4M33, the [Phe23]M33 mutant and fluorescently labelled derivatives were synthesized on an ABI-433 automated peptide synthesizer (Applied Biosystems, Applera Corporation) by using the stepwise solid-phase method and ABI small-scale Fmoc chemistry protocol, with protected amino acids suggested by the manufacturer. Synthesis was performed on a 100 μmol scale with 10 mol-equiv. of Fmoc-amino acids, single coupling and systematic capping with acetic anhydride. N-terminal thiopropionic acid (Tpa) was introduced as its disulphide. Fluorescein was specifically incorporated at Lys11 by using N-α-Fmoc-Nϵ-1-(ivDde-3-methylbutyl)-L-lysine (where ivDde is 4,4-dimethyl-2,6-dioxocyclohex-1-ylidene) during peptide synthesis and subsequent coupling of 5- and 6-carboxyfluorescein succinimidyl ester, after ivDde was removed by four treatments of 3 min with 2% hydrazine. Peptide was cleaved from the resin with simultaneous removal of side-chain protecting groups by treatment with reagent K′ [81.5% trifluoroacetic acid/5% water/5% phenol/5% thioanisole/2.5% ethanedithiol/1% tri-isopropylsilane (all v/v)] for 2.5 h at room temperature. The resin was then filtered off and the fully deprotected peptide was precipitated in methyl t-butyl ether at 4 °C. After centrifugation and washing with methyl t-butyl ether, the peptide was dissolved in 20% (v/v) acetic acid and freeze-dried. To form disulphide bonds, the crude reduced peptide was dissolved (0.1 mg/ml) in 0.1 M Tris/HCl buffer, pH 7.8, containing 5.0 mM GSH; after 30 min, 1.0 mM GSSG was added and, after 1 h, oxidation was stopped by acidification to pH 3.0 with HCl. The solution was loaded directly on a Vydac C18 column (2.5 cm×25 cm), and, at 10 ml/min flow rate, the oxidized peptide was then eluted using a linear gradient 0–50% acetonitrile in aq. 0.1% trifluoroacetic acid over 90 min. The identity of the purified products was verified by amino acid analysis and electrospray MS.
Structure determination of CD4M33
Standard pulse sequences and parameters were employed to record 2D (two-dimensional) homonuclear DQF-COSY (double quantum filtered scalar COSY), TOCSY (isotropic mixing period 80 ms) and NOESY (mixing times 80 and 200 ms) spectra. NMR experiments were collected on a 600 MHz spectrometer (Bruker AMX600) with 4 mM CD4M33 as the sample (pH 3.5, 25 °C). A standard strategy was used for assignment [20]. Residual overlaps were resolved from analogous spectra recorded at 15 and 35 °C. The data sets were processed with the Gifa software [21]. Nuclear-Overhauser-enhancement (NOE) intensities used for the structure calculations were obtained from the NOESY spectra recorded with an 80 ms mixing time on the fully protonated sample. NH-CαH and CαH-CβH coupling constants used to define angular restraints were measured by gradient enhanced double band filtered scalar COSY spectroscopy [22] and DQF-COSY respectively.
The 307 collected NOEs were partitioned into three categories of intensities that were converted into distance upper limits of 2.8 Å, 3.6 Å and 4.8 Å (1 Å=0.1 nm). Additionally, some cross-peaks present only in 200 ms 2D NOESY maps were converted into distance constraints of 5.5 Å. When no stereospecific assignment was possible, pseudo-atoms were defined and corrections added as described by Wüthrich et al. [23]. A total of 31 angular restraints (φ and χ1) obtained from J-coupling-constants analysis were added as the usual distance restraints used to enforce the three disulphide bridges.
Final structure calculations using the torsion-angle-dynamics protocol of DYANA (Dynamics algorithm for NMR applications) [24] were started from 999 randomized conformers. The 30 conformers with the lowest final target function value (<0.02 Å2) presented no distance violation larger than 0.2 Å and no dihedral violation. These conformers were energy-minimized with the AMBER 5 program [25,26], using the AMBER all-atom force field with parameters from Cornell et al. [27], including improved torsional-angle parameters [28]. All calculations were performed on a Silicon Graphics Origin 200 (biprocessor) workstation.
Molecular modelling of the complex CD4M33–gp120
The structure of the complex gp120–CD4M33 was determined by molecular modelling. The crystallographic structure of the complex gp120–CD4 (PDB code 1g9m) was used as a template to dock CD4M33 mimic on gp120. All molecular-mechanics and molecular-dynamic calculations were performed with the AMBER 5 package [25,26]. The CD4M33 NMR structure with the lowest energy was used to replace the CD4 on the gp120–CD4 complex structure by superimposing the backbone atoms of CD4M33 (residues 17–26) on those of CD4 (residues 37–46). A total of 5000 cycles of energy minimization were carried out on all CD4M33 side chains and on the gp120 side chains at a maximum of 30 Å around CD4M33 mass centre, to reduce any bad contacts. A 25-ps-long simulated annealing procedure followed, in which the temperature was raised to 700 K for 15 ps and gradually lowered to 300 K. During this stage, the relative weights for the non-bonded energy terms and for the torsion energy terms were gradually increased from 0.1 to 1.0, to permit the side-chain rearrangement to take place. Finally, the full mini-protein and gp120 side chains were minimized for 5000 cycles. This procedure was repeated ten times and the complex with the minimal energy was saved.
Production of wt (wild-type) and mutant HIV-1 YU2 gp120 glycoproteins in the baculovirus expression system
As previously described for the expression of HxB2 gp120 [29], the natural signal peptide sequence of YU2 gp120 was replaced by a new signal sequence isolated from the ecdysteroid glycosyltransferase (EGT) gene of the Autographa californica (alfalfa looper) baculovirus. Briefly, the HIV-1 YU2 gp120 wt DNA sequence was amplified by PCR from codon 30 to codon 498 of native gp120 using the following primers: sense primer (5′-GCTGTTAACGCCGCAGAACAATTGTGGGTCACAGTC-3′) and antisense primer (5′-CCGCTGCATTGGTTCTGCAGTTATCTTTTTTCTCTCTGCACCA-3′) and the pTZ-YU2, a plasmid containing the entire sequence of HIV-1 YU2 as a template. The PCR product obtained presents at its 5′ end a unique HpaI site (bold and underlined) followed by one codon of the EGT signal-peptide sequence and, at the 3′ end, a unique PstI site (bold and underlined) downstream of a stop codon introduced in the codon position 499. This fragment of 1453 bp was digested with HpaI and PstI, gel purified and inserted into the HpaI–PstI sites of pUC-EGT, allowing the fusion of the native coding sequence of gp120 in-frame with the EGT signal peptide sequence present in the pUC-EGT. The resulting construct, pUC-EGT-gp120yu2, was digested with BamHI and HindIII, and the 1492-bp fragment encoding the full-length gp120 was cloned into the baculovirus transfer vector p119L [30] between the BglII and HindIII sites to produce the p119L-g120y2 plasmid. Sf9 [Spodoptera frugiperda (fall armyworm)] cells were co-transfected with viral DNA purified from the modified baculovirus AcSLP10 expressing the polyhedrin gene under the control of the P10 promoter and with DNA from p119L-g120y2. After incubation for 5 days at 28 °C, recombinant viruses were plaque-purified by standard methods [29].
Mutations were introduced into the gp120 coding sequence using the PCR procedure. Briefly two PCRs were performed with p119L-g120y2 as a matrix and two sets of primers: (i) the forward primer p1 (5′-CTCTTTCAATATCACCACAA-3′) and the reverse primer p1* 5′-GAATTGTAACAGCCAGTTTTAATTGT-3′, which bears the mutation or (ii) the forward primer p2* 3′-CTTTAACATTGTCGGTCAAAATTAACA-5′ (complementary of primer p1* and bearing the same mutation) and the reverse primer p2 (5′-TTGTCCCTCATATCTCCTCC-3′). The high-fidelity polymerase Pfu from the hyperthermophilic archaeon Pyrococcus furiosus (Stratagene) was used. The two overlapping fragments generated were mixed (20 ng of each) and used as a template for a third PCR with forward primer p1 and reverse primer p2 to generate the whole fragment NheI–BsaBI. The PCR product was digested with NheI–BsaBI and inserted into NheI–BsaBI sites of p119L-g120y2. The mutant was then sequenced to confirm the presence of the desired mutation and the absence of any other mutation. Sf9 cells were co-transfected with this new construct as described above. Production of the recombinant gp120 was analysed by Western blotting using 5 μl of crude cellculture supernatant.
Purification of wt and mutant HIV-1 YU2 gp120 glycoprotein
YU2 gp120 and mutant were purified in a two-step chromatographic run. Briefly, 800 ml of cell culture was centrifuged at 3000 g for 10 min. The supernatant was adjusted to pH 7.4, and the saline composition was modified to obtain the following final concentrations: 500 mM NaCl, 0.1 mM MgCl2, 0.1 mM CaCl2 and 0.1 mM MnCl2. The solution was filtered, then loaded on to a concanavalin A–Sepharose gel column (Amersham Biosciences) equilibrated with 3 vol. of 50 mM Tris/HCl buffer, pH 7.4, containing 500 mM NaCl, 0.1 mM MgCl2, 0.1 mM CaCl2, 0.1 mM MnCl2. The gel was then washed with 3 vol. of the Tris buffer, and YU2 gp120 was eluted with the same buffer containing 750 mM α-methyl D-mannoside. The eluent was diluted 6-fold with PBS and loaded on to a dextran sulphate gel column equilibrated with 3 vol. of PBS. The gel was washed with 5 vol. of PBS, and YU2 gp120 was eluted with phosphate buffer, pH 7.4, containing 200 mM NaCl. The eluent was then dialysed against PBS and concentrated with a Centricon membrane (30 kDa cut-off) to obtain a final volume of 2 ml. Concentration of purified wt and mutant YU2 gp120 was determined by measuring the A280, using 80310 and 86000 litre·mol−1·cm−1 as molar absorption coefficients.
ELISA analysis
Competition binding ELISAs were performed as described in [17]. Briefly, the wells of 96-well plates (Maxisorb; Nunc) were coated overnight at 4 °C with antibody D7324 (Aalto Bio Reagents, Dublin, Republic of Ireland; 50 ng/50 μl per well); wells were saturated with PBS/3% BSA buffer, washed three times, then gp120 (4–15 ng/well) was added, followed by the addition of 62 pg of sCD4 (Progenics, Tarrytown, NY, U.S.A.) and different concentrations of soluble competitors. For revelation, anti-CD4 mAb L120 [Centralized Facility for AIDS Reagents, National Institute for Biological Standards and Control (NIBSC), South Mimms, Potters Bar, Herts., U.K.] was then added, followed by the addition of goat anti-mouse peroxidase-conjugated antibody (Jackson ImmunoResearch, West Grove, PA, U.S.A.) and substrate (3,3′,5,5′-tetramethylbenzidine; Sigma). After measuring the A450, the IC50 (concentration causing 50% inhibition) values were calculated from the results of triplicate experiments.
Anisotropy measurements
Fluorescence measurements were made using a Beacon 2000 fluorescence polarization spectrometer (PanVera, Madison, WI, U.S.A.) with fixed excitation (490 nm) and emission (530 nm) wavelength filters. All measurements were taken at room temperature (25 °C) in 10 mM sodium phosphate/0.1 M NaCl, pH 7.0, after 20 min of equilibration. In gp120 titration experiments, typically, to a 200 μl solution, containing 6.0 nM fluorescein-labelled CD4M33 and 410 nM gp120, 40 μl of solution was removed and the same volume of 6.0 nM CD4M33 solution successively added. In this way, 0.1 log serial dilutions of gp120 were obtained. In competition experiments, 1μl of various dilutions of unlabelled inhibitors were added to 200 μl of 6.0 nM fluorescein–CD4M33 and 20 nM gp120 solution, and fluorescence anisotropy was measured after each inhibitor addition. It was verified that fluorescence intensity remained constant throughout the anisotropy measurements.
Miniaturized 384-well assays
Measurements were made in a LJL Analyst (LJL Biosystems, Sunnyvale, CA, U.S.A.) microplate reader, using the fluoresecence polarization detection mode and 485 nm excitation and 530 nm emission filters, and an additional 505 nm dichroic filter to limit blank response. A 1.2 G parameter was used. Samples were in 10 mM sodium phosphate buffer, pH 7.0, containing 135 mM NaCl and 0.05% Tween 20. The binding assay was performed in a final volume of 20 μl (titration experiments) or 21 μl (competitive experiments) in a 384-well small-volume black microtitration plate by using 1 nM fluorescein–CD4M33. Titration assays were performed in quadruplicate by the addition of 10 μl of fluorescein ligand (1 nM final concn.) to 10 μl of gp120 (two-fold dilutions in 16 wells, starting at 200 nM). Competition assays were performed in triplicate by mixing 7.0 μl of competing unlabelled ligand, 7.0 μl of fluorescein-labelled ligand (1 nM final concn.) and 7.0 μl of gp120 (12.5 nM final concn.). Fluorescence anisotropy was determined after 40 min equilibration at 25 °C.
Anisotropy data analysis
Fluorescence-polarization measurements were expressed in anisotropy, because its values can be combined additively. Anisotropy (A) values were obtained from polarization (P) as follows:
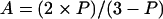 |
1 |
Binding data were analysed using the non-linear least-squares numerical solver-based binding data global analysis program BIOEQS, in which the calculated binding surface is obtained using a numerical constrained optimization chemical equilibrium solver [31–33]. On the basis of the ΔG (change in Gibbs free energy) value of complex dissociation, the program relates the concentration vectors of the species present in the complexes to the determined anisotropy value at each point of the titration and fits for the free energy by adjusting these floating parameters using the Marquardt–Levenberg non-linear least-squares algorithm. In the competition experiments the value of the free energy of interaction of the labelled CD4M33 with gp120 was fixed, and the value of the free energy of interaction of the competitor ligand was allowed to float in the fit.
Some of the fluorescence experiments were analysed as well with closed-form analytical expression describing equilibrium binding interactions considering ligand depletion. The binding interaction between a ligand and its receptor is described at equilibrium by:
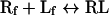 |
2 |
where Rf is free receptor, Lf is the free ligand and RL is the receptor–ligand complex. Also:
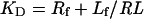 |
3 |
Where KD is the thermodynamic dissociation constant, Rf is the concentration of free receptor, Lf is the concentration of free ligand and RL is the concentration of the ligand–receptor complex. If Rt and Lt denote the total receptor and ligand concentrations respectively, we can re-write eqn (3) as:
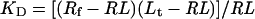 |
4 |
The concentration of ligand bound to receptor, RL, is then simply the solution of the quadratic equation:
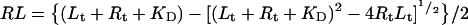 |
5 |
By using the relationship:
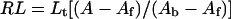 |
6 |
where Lt is the total ligand concentration, A is the measured anisotropy, Af is the anisotropy of the free ligand, Ab is the anisotropy of the fully bound ligand, we can re-write eqn (6) in the form:
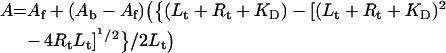 |
7 |
which can then be used by a non-linear regression program to evaluate KD, Af (anisotropy of the free ligand) and Ab (anisotropy of the fully bound ligand). The total ligand concentration is assumed to be equal to the free ligand concentration in these closed-form approximations.
In competition binding assays, the binding interactions can be described by:
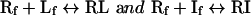 |
8 |
where If is free inhibitor and RI is the receptor–inhibitor complex, and the dissociation constants can be described by the relationships:
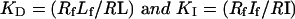 |
9 |
The bound/free ratio, X, which is measured by fluorescence polarization, could be expressed as:
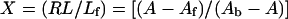 |
10 |
where Af and Ab are obtained by previous titration experiments. Combining these equations with the Conservation of Mass equation, the following equation can be obtained for It, the total concentration of inhibitor as a function of X:
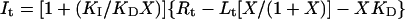 |
11 |
In order to obtain the IC50 in competitive assays, the signal of bound fluorophore (A) is plotted against the logarithm of the competitor concentrations (Z=log It), resulting in a sigmoid curve, which is fitted by a three-parameter equation:
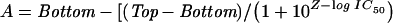 |
12 |
The experimental data were fitted to these equations using a commercially available non-linear regression program (Prism; GraphPad software, Inc., San Diego, CA, U.S.A.).
RESULTS AND DISCUSSION
Structure of CD4M33 and its complex with gp120
The CD4M33 solution structure was solved by 1H NMR spectroscopy and molecular modelling. Complete 1H resonance assignments were obtained by standard two-dimensional NMR techniques. A total of 338 structural (distance and dihedral) constraints were collected and taken into account for structure calculations using the torsion-angle-dynamics protocol of DYANA. The 30 three-dimensional structures with the lowest DYANA target value (<0.02 Å2) were further refined by simulated annealing and restrained energy minimization using the AMBER force field. They satisfied all experimental restraints (no distance violation larger than 0.2 Å and no dihedral violation) and had good energies (Etotal=−232.1±4.6 kcal, Etotal,non-bond=−416.5±6.3 kcal, Ebond=9.5±0.3 kcal, Eangle=47.4±1.7 kcal, EvdW=−162.3±4.4 kcal, Eelect=−674.3±9.9 kcal and Econst=2.3±0.5 kcal; 1 kcal=4.184kJ; vdW=van der Waals; elect=electronic). The average global r.m.s. deviation calculated for backbone and heavy atoms decreases to 0.42±0.15 Å and 1.36±0.24 Å respectively after refinement. The high number of NOEs, i.e. 20.5 per residue, in conjunction with the low r.m.s. deviation values, indicates a good sampling of the conformational space during the simulated annealing procedure. The final 30 structures (Figure 1A) possessed the expected scyllatoxin α/β-fold, with a helix in the region 2–13, an antiparallel β-sheet in the region 16–26 and a rootmean-square deviation of 0.21 Å between the average structure and the corresponding region of scyllatoxin. In particular, the Bip23 side chain is well defined in the 30 structures because of many long-range contacts and because it bends over the miniprotein structure out of the solvent. The structure of the site transferred from CD4 (Figure 1B) was well defined and superimposed on the native CD4 site with striking precision (Figure 1C): the r.m.s. deviation between the backbone atoms of the 17–26 sequence of CD4M33 average structure and the 37–46 sequence of CD4 [34] was only 0.61 Å (0.66 Å with CD4 structure [5] bound to gp120).
Figure 1. Three-dimensional structure of CD4M33 and its computed complex with gp120.

(A) Ensemble of 30 low energy NMR structures, superposed by using backbone atoms (in black); disulphide bridges are in yellow and side chains in blue. (B) Sequence alignment of the CDR2-like loop of CD4 and the engineered CD4M33 in the one-letter amino acid code. Critical residues are in red. (C) Structure comparison of the CD4 CDR2-loop (sequence 36–47) and the CD4M33 β-hairpin (sequence 16–27). (D) Three-dimensional model of CD4M33 (cyan) in complex with gp120 (green), computed according to the CD4–gp120 complex crystallographic structure (PDB code: 1g9m). Protruding Bip side chain engaging gp120 Phe43 cavity is represented in space-filling model and the interface in red.
Since CD4M33 closely mimicked the CDR2 loop of CD4, central to gp120 binding, the CD4M33 structure was used to model its complex with gp120, which was computed by using the crystallographic structure of the gp120–CD4 complex [5] as a template. Briefly, CD4 in the crystallographic structure was replaced by CD4M33, and molecular dynamics was used to optimize atomic interactions. Analysis of this complex shows that the CD4M33 miniprotein binds gp120 by using 463.3 Å2 of its surface, mostly including the β-sheet region. The backbone atoms of CD4M33 residues 18–28 superimpose on those of CD4 residues 38–48 with 0.67 Å r.m.s. deviation. Interestingly, the Bip side chain, which was packed on to the miniprotein, moved into the gp120 Phe43 cavity during the dynamics. In this modelled complex, the CD4M33 Bip23 side chain superimposes its proximal phenyl ring on to the phenyl side chain of CD4 Phe43 in the CD4–gp120 complex with a 0.52 Å r.m.s. deviation, while its distal phenyl ring occupies the interior of the cavity (Figure 1D). In addition, the Bip side chain deeply engages the Phe43 cavity, and 181.0 Å2 of its surface is buried upon binding, which represents 39.1% of the CD4M33 total binding surface. In comparison, Phe43 of CD4 buries 106.5 Å2 of its surface upon gp120 binding.
In order to confirm the mode of CD4M33 binding to the gp120 envelope protein and the mode of Bip23 interaction with the Phe43 cavity, we synthesized a [Phe23]M33 miniprotein and a [Trp375]gp120YU2 envelope glycoprotein. The [Phe23]M33 mutant presents a phenylalanine instead of a Bip residue in position 23, which is structurally equivalent to position 43 of CD4 [5]. Thus the [Phe23]M33 mutant, like CD4, possesses a natural Phe23 side chain which cannot engage the Phe43 cavity interior but only its entrance. The [Trp375]gp120YU2 protein, containing the Ser375→Trp mutation, has been previously shown to fill the Phe43 cavity, which therefore cannot any longer accommodate propan-2-ol or other solvent molecules [35]. When tested in ELISA competition experiments, CD4M33 bound wt gp120 with an IC50 of 3 nM and bound [Trp375]gp120YU2 with a 1000-fold-decreased affinity (Figures 2A and 2B). Interestingly, the [Phe23]M33 mutant could bind both wt and [Trp375]gp120YU2 protein with comparable affinity (Figures 2A and 2B). This suggests that the additional phenyl ring of the CD4M33 Bip residue was occluded from the Phe43 cavity of [Trp375]gp120YU2, because this cavity is occupied by the bulkier Trp375 residue. Similar results were obtained by Biacore, by analysing binding of wt and [Trp375]gp120YU2 protein solutions with CD4M33 or [Phe23]M33 immobilized on biochips (results not shown).
Figure 2. Binding of CD4M33 and [Phe23]M33 to gp120YU2 wt and S375W mutant.

ELISA of inhibition of CD4 binding to gp120 YU2 wt (A) and S375W mutant (B) by CD4M33 (□), [Phe23]M33 (‘[F23]M33’) (△), and sCD4(D1D2 domain) (○).
This demonstrates that CD4M33 can bind with high, CD4-like, affinity to gp120 proteins, which present an interior cavity, whereas it can only weakly bind envelope proteins that do not contain such an interior cavity or present a bound molecule within this cavity. Accordingly, the [Phe23]M33 mutant, which, with its Phe23, can only engage the entrance of the Phe43 cavity, cannot differentiate between an envelope gp120 protein possessing or not possessing an interior cavity or a cavity already engaged by exogenous bound molecule. Thus both the binding assays and the modelled CD4M33–gp120 complex suggest that CD4M33 and the [Phe23]M33 mutant represent highly useful molecules that can be used to detect and distinguish ligands which can bind to the CD4 binding site, the Phe43 cavity or both.
Interestingly, ELISA binding experiments provided evidence for a limited difference in gp120 binding affinity between CD4M33 and its [Phe23]M33 mutant. This suggests that, in spite of the fact that the Bip23 side chain more deeply penetrates within the Phe43 cavity than does Phe23, this larger surface of interaction is not translated into a large increase in binding affinity. Indeed, increasing binding surface between an apolar ligand and a hydrophobic cavity is expected to provide a significant increase of binding energy [36,37]. However, it is possible that the biphenyl moiety is too rigid and too long, and thus it may not fit appropriately inside the envelope cavity. Some adjustments must have occurred both on the biphenyl side chain and the internal envelope cavity to fit this long side chain into the viral glycoprotein cavity. These adjustments may be of limited entity, but energetically costly, thus may affect binding energy. There may be a loss in rotational entropy as well, since the ring is constrained within the cavity. However, because of the modelling procedure utilized, no differences between gp120 structure in complex with CD4 and CD4M33 could be observed. Thus our modelled complex cannot reveal any gp120 conformational adaptations that are produced by CD4M33 binding. Only the experimentally determined structure of this miniprotein in complex with gp120 may eventually reveal structural differences within the structure of CD4–gp120 complex.
Synthesis of a CD4M33–fluorescein derivative
The Lys11 residue of CD4M33 is located in the helical region, opposite the CD4 binding site and is solvent-exposed in the miniprotein–gp120 complex (Figures 1A and 1D). Thus a fluorescein probe attached to this position is predicted not to interfere with gp120 binding. Specific and quantitative incorporation of the fluorescent label at the side chain of Lys11 was performed by peptide synthesis, which uses the orthogonal ivDde protecting group on Lys11 during polypeptide chain assembly, selective cleavage of this protection after polypeptide chain completion and final fluorescein coupling. Fluorescein labelling did not perturb miniprotein folding significantly, since the fluorescein–CD4M33 derivative (hereafter called ‘CD4M33-Fl’) could fold with yields comparable with the unlabelled CD4M33 (results not shown). CD4M33-Fl was finally obtained in a pure form with 5.5% total yield. Excitation and emission spectra exhibited maxima at 498 and 522 nm respectively (results not shown), as expected. Tested in gp120 competition ELISA, CD4M33-Fl exhibited gp120-binding affinity similar to that shown by unlabelled CD4M33 (Table 1), demonstrating that the labelling reaction did not modify its gp120 affinity, as expected by the appropriate and specific labelling position. Similarly, we also synthesized [Phe23]M33-Fl. This mutant is designed to mimic CD4 more closely, since does not contain the longer biphenyl moiety which has been shown to fill the Phe43 cavity, but rather a short and native phenyl ring, which is intended to engage the entrance of the cavity like residue Phe43 of CD4.
Table 1. Binding parameters for different CD4 miniproteins obtained using competition experiments.
S.D. values were 15% of the mean in ELISA and less than 5% in fluorescence polarization assays.
| IC50 (M) | KI (M) | ||||
|---|---|---|---|---|---|
| Competitor | HxB2* | LAI† | LAI‡ | SF162† | LAI† |
| CD4M9 | 1.6×10−6 | 1.1×10−6 | 3.6×10−7 | 4.6×10−7 | 6.9×10−7 |
| [Tpa1]CD4M9 | 2.2×10−7 | 8.4×10−8 | 2.6×10−8 | 2.7×10−8 | 5.7×10−8 |
| [Val27]CD4M9 | 9.6×10−7 | 6.1×10−7 | 3.2×10−7 | 1.4×10−7 | 5.0×10−7 |
| [Bip23]CD4M9 | 9.0×10−7 | 3.4×10−7 | 9.7×10−8 | 1.7×10−7 | 3.1×10−7 |
| CD4M32 | 3.8×10−8 | 3.7×10−9 | 2.3×10−8 | 3.8×10−9 | 1.1×10−8 |
| CD4M33 | 7.5×10−9 | 2.6×10−9 | 1.4×10−8 | 3.2×10−9 | 5.3×10−9 |
| sCD4 | 2.0×10−9 | 2.0×10−9 | 9.0×10−9 | 2.8×10−9 | 4.5×10−9 |
| CD4M33-Fl | 1.2×10−9 | ||||
* Values obtained using ELISA.
† Values obtained using fluorescence polarization experiments in 384-well plates.
‡ Values obtained using fluorescence polarization assays on Beacon 2000 spectrophotometer.
CD4M33 and [Phe23]M33 contain Tpa1 and five cysteine residues (Figure 1B), which must be linked by disulphide groups with a connectivity 1–4, 2–5, 3–6, in order to stabilize the characteristic α/β motif of scorpion toxins. In CD4M33, CD4M33-Fl, [Phe23]M33 and [Phe23]M33-Fl, folding and disulphide formation proceed with great efficiency, similar to that observed with native scyllatoxin. Furthermore, direct structure resolution of CD4M33 by NMR confirms the presence of native disulphide groups in this miniprotein. Thus the presence of Tpa does not affect folding and formation of an α/β structural motif, which is essential to stabilize the miniprotein functional β-hairpin and allow the proper positioning of Bip23 or Phe23 side chain to engage the Phe43 cavity or its entrance in gp120 glycoprotein.
Fluorescence polarization binding studies
Gp120 proteins from HIV-1HxB2, HIV-1SF2 and HIV-1US4 isolates were titrated using CD4M33-Fl, in a Beacon 2000 fluorescence polarization analyser. The anisotropy of free CD4M33-Fl (6 nM) was 80 mA (anisotropy×1000), whereas in the presence of a large excess of gp120 (410 nM), the anisotropy of fully bound CD4M33-Fl reached 250 mA (Figure 3A).
Figure 3. Binding of CD4M33-Fl to different gp120 glycoproteins, obtained by fluorescence polarization.

(A) Assays were performed in 200 μl tubes (Beacon 2000 polarizer) and (B) in 20 μl wells of 384-well microtitration plates.
It is known that fluorescence-anisotropy values are inversely proportional to the rate of fluorophore rotational diffusion in solution according to:
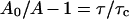 |
where τ is the lifetime of the excited state and τc is the rotational correlation time (the time for the anisotropy to decrease to 1/e of its limiting value, A0, which is a photophysical constant which depends upon the probe used). Since:
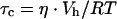 |
where η is the solution viscosity and Vh the hydrated volume of the fluorescent molecule or complex, it follows that mimic CD4M33-Fl (3 kDa) free in solution exhibits a high rotational diffusion rate and hence fluorescence anisotropy is low. However, when CD4M33-Fl binds gp120, which is a large glycoprotein with a molecular mass of over 100 kDa, fluorophore rotational diffusion decreases and the anisotropy increases over 3-fold. Thus the observed variation in fluorescence anisotropy translates the direct binding of CD4M33 to viral envelope glycoproteins in solution and allows calculation of binding dissociation constants without need to separate free and bound ligand or radioactive labelling.
Data from these titrations have been analysed with the non-linear least-squares program BIOEQS and the dissociation constants, KD, obtained were 8.0 nM (±2), ≤1.0 nM and 8.0 nM (±2) for gp120HxB2, gp120SF2 and gp120US4 respectively. These values are similar to those previously determined using a completely different technique, such as Biacore, which is based on plasmon resonance, and is also similar to those reported in the literature for CD4 binding [38].
This large change in anisotropy affords a large dynamic range and thus a very high signal-to-noise ratio in the anisotropy data. Assays are highly reproducible and can be carried out with very small quantities of labelled target. These characteristics of the assay provide a broad concentration range over which competition experiments can be carried out. For example, the high sensitivity allows for screening for very-high- affinity compounds, whereas the large dynamic range allows for screening for relatively lowaffinity molecules. This is true because competition, even beginning at a level of 20% saturation, corresponds to a significant change in anisotropy.
We also analysed a number of non-fluorescent miniproteins, derived from CD4M9 [18] and synthesized during the optimization steps, that improved this miniprotein activity to a native-like binding affinity [17]. By using CD4M33-Fl as a tracer, we tested these peptides in competition assays and found that CD4M9 bound gp120HxB2 with a KD of 3.5×10−7 M (BIOEQS fits directly for KD values even in competition experiments), while the single mutants [Tpa1]CD4M9, [Bip23]CD4M9 and [Val27]CD4M9 presented higher affinities with KD in the (32–2.6)×10−8 M range (Table 1), demonstrating that the mutations introduced were effective in increasing gp120 binding affinity. CD4M32, which contained Tpa1, Bip23 and Val27 showed a KD of 2.3×10−8 M (Table 1). CD4M33, which contained, in addition, His4 and Phe5, showed a KD of 1.4×10−8 M, close to the KD value obtained in the direct binding assay and comparable with that for CD4 KD, namely 9.0×10−9 M (Table 1). These values are consistent with previously published IC50 values obtained in ELISA (Table 1) [17].
Fluorescence polarization binding studies on microtitration plates
Anisotropy measurements in the Beacon 2000 analyser are quite reproducible, but require 200 μl total volume and 20 min equilibration time after each determination. Therefore, it is rather consuming in terms of material and time. We thus adapted the assay performed in tubes to 384-well microtitre plates, a method that uses a 10-fold smaller total volume and allows multiple determinations in a short time. The fluorescence anisotropy of CD4M33-Fl was evaluated in quadruplicate experiments, with 16 twofold dilutions of 200 nM gp120. CD4M33-Fl was tested at different concentrations (0.1, 0.5, 1.0 and 10 nM) to limit the use of gp120 at low concentrations. At 0.1 nM CD4M33-Fl, the intensity of fluorescence was such that the signal-to-noise ratio was too low (less than 10; results not shown). Using CD4M33-Fl at 1 nM was the best compromise between an acceptable signal-to-noise ratio and the use of gp120.
Miniaturization of the assay allowed us to utilize a very limited amount of material and to titrate several HIV-1 envelope gp120 glycoproteins from a variety of isolates, such as HxB2 (X4), SF2 (X4), LAI (X4), YU2 (R5) and SF162 (R5) (Figure 3B) and JRFL (R5) (not shown). The minimum and maximum anisotropy values were typically 50 mA for the unbound states and 220 mA for the bound state, findings which represent an increase of anisotropy signal up to 340% upon binding. Thus the dynamic range of the microtitre plate assays is comparable with that of the Beacon-based assay, and the sensitivity is only slightly diminished. The dissociation constants determined for the different gp120 range from 0.19 to 15 nM (Table 2).
Table 2. Dissociation constants of CD4M33-Fl for gp120 glycoproteins.
Values were obtained by fluorescence polarization experiments in 384-well plates using a LJL Analyst microplate reader.
| Glycoprotein | KD (nM) |
|---|---|
| SF162 | 0.83±0.04 |
| HxB2 | 6.20±0.20 |
| LAI | 2.71±0.09 |
| W61D | 0.19±0.08 |
| BAL | 6.36±0.26 |
| III B | 3.81±0.15 |
| SF2 | 0.76±0.03 |
| YU2 | 15.2±0.76 |
Using the 384-well plates, we analysed the same CD4 mimics that were previously tested in tubes, this time using three different gp120 glycoproteins from isolates HxB2, LAI and SF162. These unlabelled miniproteins were used to reverse the fluorescence-anisotropy increase observed when CD4M33-Fl is bound to gp120. Thus, based on the titration curves obtained above, we set the gp120 concentration at about 70% labelled CD4M33 bound, e.g. 15 nM gp120HxB2 and 1 nM CD4M33-Fl. gp120 glycoprotein was then added to a mixture containing 1 nM CD4M33-Fl with various serial dilutions of non-fluorescent inhibitors in a 21 μl total volume (Figure 4A). By non-linear regression we obtained IC50 values which correlate with those obtained from the competitive ELISA (Table 1), confirming the relative ranking among the inhibition potency of the different miniproteins.
Figure 4. Binding of different CD4 miniproteins to gp120 LAI.

(A) IC50 determination by non-linear regression. (B) KI determination. Curves were obtained by fitting fluorescence-polarization competition data obtained using 6 nM gp120 LAI, 1 nM CD4M33-Fl and various dilutions of each competitor.
Those data were also used to determine affinities by using eqn (11), corresponding to the analytical approach for equilibrium competition binding described by Dandliker et al. [39] (Figure 4B). The curves fitting yielded a high (more than 0.995) R2 (correlation coefficient), and unlabelled CD4M33 gave a KI close to the KD of its labelled form (Table 1). Furthermore, the KI determined for sCD4 is close to the affinity values already published, providing an estimate of assay accuracy. In addition, affinity constants obtained in competitive assays conducted in 200 μl tubes using the Beacon Polarization instrument or in 21 μl using microtitre plates and the LJL Analyst, yielded comparable affinity constants (Table 1), confirming the reliability of fluorescence anisotropy determinations in different formats.
In the miniaturized 384-well microplate format, the assay was used to screen several thousands of small molecules from the French Chemo-library (results not shown). The assay volume of 20 μl used can be also further decreased, and thus this assay can be easily adapted to high-throughput screening for large library screening.
The high sensitivity of fluorescence (≈1 nM or less depending upon the instrument used) and the large dynamic range of the present assay (over 150 milli-anisotropy units) allows for adaptation of the assay in competition mode to screen for both very-high- and very-low-affinity competitors. This advantage is quite useful for application to all phases of drug discovery from identifying possible hits to improving the properties of lead compounds.
Studies on small-molecule inhibitors
Reported entry inhibitors, stilbenedisulphonic acid [9], bathophenanthroline acid [10] and the related molecules 1,10-phenanthroline and 2-naphthalenesulphonic acid were tested for their ability to inhibit CD4M33-Fl binding to gp120 in a fluorescence polarization assay. These compounds, tested up to 10−4 M concentration, did not exhibit any inhibitory activity in this assay and thus did not bind to the CD4 binding site of gp120, in agreement with their reported specificity. By contrast, the bis-azo polyanionic dye FP-21399, previously reported to block HIV-1 entry and fusion of CD4+ cells [23], was effective in inhibiting CD4M33-Fl binding to gp120 from LAI, HxB2 and SF162 isolates, with IC50 values of 0.1–1 μM (Figure 5A). This compound also inhibits CD4 binding to gp120LAI, gp120HxB2 and gp120YU2 in the ELISA assay (results not shown). In addition, still using ELISA, we find that it may also target gp120 regions proximal to the CD4i epitopes, since it inhibits gp120–antibody 17b binding significantly (results not shown). Polyanions and heparin have been reported to target CD4i epitopes [40], and thus, since FP21399 is highly sulphated, it is not surprising that it may also target CD4i epitopes, affecting gp120 conformation such that CD4 and 17b binding are also affected indirectly.
Figure 5. Inhibitory activity of different small compounds obtained by fluorescence polarization.

Assays were performed using gp120 LAI (6 nM) and (A) CD4M33-Fl (1 nM) or (B) [Phe23]M33-Fl (1 nM).
A recently reported small-molecule entry inhibitor BMS-378806 was also tested for binding to the CD4 binding site of gp120LAI (Figure 5A), gp120HxB2 and gp120SF162 (results not shown). This molecule was found to inhibit CD4M33-gp120 binding only weakly, with an IC50 of 2.0×10−5 M. Weak (IC50>1μM) or absence of inhibition of the CD4–gp120 interaction has also been verified in ELISA, by using gp120 from isolates HxB2, YU2, and SF162 (results not shown). This inhibitory concentration is several orders of magnitude higher than that needed for cell infection inhibition, since BMS-378806 has been reported to inhibit a panel of 11 HIV-1 laboratory strains with a median IC50 of 12 nM [12]. The same rather weak potency was also measured when the [Phe23]M33-Fl derivative was used as a tracer (Figure 5B). Interestingly, when BMS-378806 was preincubated with gp120 for 1 h and CD4M33-Fl binding measured after 5 min, the potency of this inhibitor was increased by more than 10-fold, but decreased to the measured IC50 of 2.0×10−5 M after 40 min. These data demonstrate that the actual target of this small-molecule inhibitor is neither the CD4 binding site nor the Phe43 cavity, but a different site to which it binds with high affinity before CD4M33 binding. To define its target, HIV-1 resistant variants have been isolated and mutations mapped on the HIV-1 envelope [13,15]. When mutated residues are represented on the gp120 structure, they do not map the Phe43 cavity in the CD4-bound structure but rather a deep, hydrophobic channel in the recently determined structure of unliganded gp120 [6]. Our CD4M33-Fl binding data are consistent with the proposed binding site and suggest that BMS-378806 binds the unliganded gp120 protein, stabilizes its structure, thus inhibiting formation of the CD4-bound conformation which is an indispensable step in the entry mechanism, and therefore blocks HIV entry.
Conclusion
New drugs targeting different steps of the HIV-1 cycle are needed to compensate for the emergence of new HIV-1 isolates that are resistant to drugs actually used therapeutically, which only target reverse transcriptase and viral protease enzymes. Cell attachment is the first step in HIV-1 entry and a primary target for antiviral therapy. A few small molecules, such as FP21399 and BMS-378806, have been reported to inhibit CD4–gp120 interactions. However, results reported here demonstrate that these small molecules do not target the CD4 binding site in gp120 as a major mechanism of their action.
The CD4M33 fluorescein derivative here described can be used to directly detect molecules that bind the gp120 glycoprotein and target the CD4 binding site in a fluorescence polarization assay. In addition, the [Phe23]M33 fluorescent derivative can be also used to discriminate between molecules occupying the Phe43 cavity or simply engaging its entrance. The assay described here is performed in solution, is rather simple, direct, fast, sensitive and reproducible, and can be miniaturized to microvolumes, and may represent a useful method to characterize new inhibitor candidates and in the discovery of new small-molecule drugs in a high-throughput-screening approach.
Note added in proof (received 13 June 2005)
The structure of CD4M33 in complex with gp120YU2 has been solved recently using crystallography [41]. The experimentally determined structure confirms our modelled complex with a 0.73 Å r.m.s. deviation between the CD4M33 backbone atoms in the two structures.
Acknowledgments
We thank Dr Indresh K. Srivastava (Chiron Co., Emeryville, CA, U.S.A.) for recombinant gp120 from the HIV strains R5-tropic SF162, US4 and X4-tropic SF2, Dr Norbert Schulke (Progenics, Tarrytown, NY, U.S.A.) for gp120 from the strain R5-tropic JRFL, Dr S. Leow (Pharmacia, Kalamazoo, MI, U.S.A.) for 2-domain sCD4. We acknowledge the Centralized Facility for AIDS Reagents, NIBSC, South Mimms, Potters Bar, Herts., U.K. [EU (European Union) Programme EVA/MRC] for recombinant gp120 from strain R5 W61D, anti-human CD4 L120.3, 17b and 48d monoclonal antibodies. Support was provided by the French Agence Nationale de Recherches sur le SIDA (Syndrome d'Immuno-Déficience Acquise) (ANRS) and the 6th EU Framework Program EMPRO (European Microbicides project).
References
- 1.Richman D. D. HIV chemotherapy. Nature (London) 2001;410:995–1001. doi: 10.1038/35073673. [DOI] [PubMed] [Google Scholar]
- 2.Lalezari J. P., Henry K., O'Hearn M., Montaner J. S., Piliero P. J., Trottier B., Walmsley S., Cohen C., Kuritzkes D. R., Eron J. J., Jr, et al. Enfuvirtide, an HIV-1 fusion inhibitor, for drug-resistant HIV infection in North and South America. N. Engl. J. Med. 2003;348:2175–2185. doi: 10.1056/NEJMoa035026. [DOI] [PubMed] [Google Scholar]
- 3.Blair W. S., Lin P. F., Meanwell N. A., Wallace O. B. HIV-1 entry – an expanding portal for drug discovery. Drug Discov. Today. 2000;5:183–194. doi: 10.1016/s1359-6446(00)01484-7. [DOI] [PubMed] [Google Scholar]
- 4.Moore J. P., Doms R. W. The entry of entry inhibitors: a fusion of science and medicine. Proc. Natl. Acad. Sci. U.S.A. 2003;100:10598–10602. doi: 10.1073/pnas.1932511100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kwong P. D., Wyatt R., Robinson J., Sweet R. W., Sodroski J., Hendrickson W. A. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature (London) 1998;393:648–659. doi: 10.1038/31405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen B., Vogan E. M., Gong H., Skehel J. J., Wiley D. C., Harrison S. C. Structure of an unliganded simian immunodeficiency virus gp120 core. Nature (London) 2005;433:834–841. doi: 10.1038/nature03327. [DOI] [PubMed] [Google Scholar]
- 7.De Clercq E. New developments in anti-HIV chemotherapy. Curr. Med. Chem. 2001;8:1543–1572. doi: 10.2174/0929867013371842. [DOI] [PubMed] [Google Scholar]
- 8.Meanwell N. A., Kadow J. F. Inhibitors of the entry of HIV into host cells. Curr. Opin. Drug Discov. Devel. 2003;6:451–461. [PubMed] [Google Scholar]
- 9.Cardin A. D., Smith P. L., Hyde L., Blankenship D. T., Bowlin T. L., Schroeder K., Stauderman K. A., Taylor D. L., Tyms A. S. Stilbene disulfonic acids. CD4 antagonists that block human immunodeficiency virus type-1 growth at multiple stages of the virus life cycle. J. Biol. Chem. 1991;266:13355–13363. [PubMed] [Google Scholar]
- 10.Demaria S., Tilley S. A., Pinter A., Bushkin Y. Bathophenanthroline disulfonate and soluble CD4 as probes for early events of HIV type 1 entry. AIDS Res. Hum. Retroviruses. 1995;11:127–139. doi: 10.1089/aid.1995.11.127. [DOI] [PubMed] [Google Scholar]
- 11.Ono M., Wada Y., Wu Y., Nemori R., Jinbo Y., Wang H., Lo K. M., Yamaguchi N., Brunkhorst B., Otomo H., et al. FP-21399 blocks HIV envelope protein-mediated membrane fusion and concentrates in lymph nodes. Nat. Biotechnol. 1997;15:343–348. doi: 10.1038/nbt0497-343. [DOI] [PubMed] [Google Scholar]
- 12.Lin P. F., Blair W., Wang T., Spicer T., Guo Q., Zhou N., Gong Y. F., Wang H. G., Rose R., Yamanaka G., et al. A small molecule HIV-1 inhibitor that targets the HIV-1 envelope and inhibits CD4 receptor binding. Proc. Natl. Acad. Sci. U.S.A. 2003;100:11013–11018. doi: 10.1073/pnas.1832214100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guo Q., Ho H. T., Dicker I., Fan L., Zhou N., Friborg J., Wang T., McAuliffe B. V., Wang H. G., Rose R. E., et al. Biochemical and genetic characterizations of a novel human immunodeficiency virus type 1 inhibitor that blocks gp120-CD4 interactions. J. Virol. 2003;77:10528–10536. doi: 10.1128/JVI.77.19.10528-10536.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang T., Zhang Z., Wallace O. B., Deshpande M., Fang H., Yang Z., Zadjura L. M., Tweedie D. L., Huang S., Zhao F., et al. Discovery of 4-benzoyl-1-[(4-methoxy-1H-pyrrolo[2,3-b]pyridin-3-yl)oxoacetyl]-2-(R)-methylpiperazine (BMS-378806): a novel HIV-1 attachment inhibitor that interferes with CD4–gp120 interactions. J. Med. Chem. 2003;46:4236–4239. doi: 10.1021/jm034082o. [DOI] [PubMed] [Google Scholar]
- 15.Madani N., Perdigoto A. L., Srinivasan K., Cox J. M., Chruma J. J., LaLonde J., Head M., Smith A. B., 3rd, Sodroski J. G. Localized changes in the gp120 envelope glycoprotein confer resistance to human immunodeficiency virus entry inhibitors BMS-806 and #155. J. Virol. 2004;78:3742–3752. doi: 10.1128/JVI.78.7.3742-3752.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Si Z., Madani N., Cox J. M., Chruma J. J., Klein J. C., Schon A., Phan N., Wang L., Biorn A. C., Cocklin, et al. Small-molecule inhibitors of HIV-1 entry block receptor-induced conformational changes in the viral envelope glycoproteins. Proc. Natl. Acad. Sci. U.S.A. 2004;101:5036–5041. doi: 10.1073/pnas.0307953101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Martin L., Stricher F., Misse D., Sironi F., Pugniere M., Barthe P., Prado-Gotor R., Freulon I., Magne X., Roumestand C., et al. Rational design of a CD4 mimic that inhibits HIV-1 entry and exposes cryptic neutralization epitopes. Nat. Biotechnol. 2003;21:71–76. doi: 10.1038/nbt768. [DOI] [PubMed] [Google Scholar]
- 18.Vita C., Drakopoulou E., Vizzavona J., Rochette S., Martin L., Menez A., Roumestand C., Yang Y. S., Ylisastigui L., Benjouad A., Gluckman J. C. Rational engineering of a miniprotein that reproduces the core of the CD4 site interacting with HIV-1 envelope glycoprotein. Proc. Natl. Acad. Sci. U.S.A. 1999;96:13091–13096. doi: 10.1073/pnas.96.23.13091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Martin L., Barthe P., Combes O., Roumestand C., Vita C. Engineering novel bioactive mini-proteins on natural scaffolds. Tetrahedron. 2000;56:9451–9460. [Google Scholar]
- 20.Wüthrich K. New York: Wiley Interscience; 1986. NMR proteins and nucleic acids. [Google Scholar]
- 21.Pons J. L., Malliavin T. E., Delsuc M. A. Gifa V.4: a complete package for NMR data set processing. J. Biomol. NMR. 1996;8:445–452. doi: 10.1007/BF00228146. [DOI] [PubMed] [Google Scholar]
- 22.Roumestand C., Delay C., Gavin J. A., Canet D. A practical approach to the implementation of selectivity in homonuclear multidimensional NMR with frequency selective-filtering techniques. Application to the chemical structure elucidation of complex oligosaccharides. Magn. Reson. Chem. 1999;37:451–478. [Google Scholar]
- 23.Wüthrich K., Billeter M., Braun W. Pseudo-structures for the 20 common amino acids for use of studies for protein conformations by measurements of intramolecular proton–proton distance constraints with nuclear magnetic resonance. J. Mol. Biol. 1983;169:949–961. doi: 10.1016/s0022-2836(83)80144-2. [DOI] [PubMed] [Google Scholar]
- 24.Güntert P., Mumenthaler C., Wüthrich K. Torsion angle dynamics for NMR structure calculation with the new program DYANA. J. Mol. Biol. 1997;273:283–298. doi: 10.1006/jmbi.1997.1284. [DOI] [PubMed] [Google Scholar]
- 25.Case D. A., Pearlman D. A., Caldwell J. W., Cheatham T. E., III, Ross W. S., Simmerling C. L., Darden T. A., Merz K. M., Stanton R. V., Cheng A. L., et al. San Francisco: University of California; 1997. AMBER 5. [Google Scholar]
- 26.Pearlman D. A., Case D. A., Caldwell J. W., Ross W. S., Cheatham T. E., III, DeBolt S., Ferguson D., Seibel G., Kollman P. AMBER, a package of computer programs for applying molecular mechanics, normal mode analysis, molecular dynamics and free energy calculations to simulate the structural and energetic properties of molecules. Comp. Phys. Commun. 1995;91:1–41. [Google Scholar]
- 27.Cornell W. D., Cieplak P., Bayly C. I., Gould I. R., Merz K. M., Ferguson D. R., Jr, Spellmeyer D. C., Fox T., Caldwell J. W., Kollman P. A. A second generation force field for the simulation of proteins and nucleic acids. J. Am. Chem. Soc. 1995;117:5179–5197. [Google Scholar]
- 28.Kollman P. A., Dixon R., Cornell W. D., Fox T., Chipot C., Pohorille A. The development/application of a “minimalist” organic/biochemical molecular mechanic force field using a combination of ab initio calculations and experimental data. In: van Gunsteren W. F., Weiner P. K., Wilkinson A. W., editors. Computer Simulations of Biomolecules Systems, vol. 3. Leiden, The Netherlands: Escom; 1997. pp. 83–96. [Google Scholar]
- 29.Misse D., Cerutti M., Schmidt I., Jansen A., Devauchelle G., Jansen F., Veas F. Dissociation of the CD4 and CXCR4 binding properties of human immunodeficiency virus type 1 gp120 by deletion of the first putative α-helical conserved structure. J. Virol. 1998;72:7280–7288. doi: 10.1128/jvi.72.9.7280-7288.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chaabihi H., Ogliastro M. H., Martin M., Giraud C., Devauchelle G., Cerutti M. Competition between baculovirus polyhedrin and p10 gene expression during infection of insect cells. J. Virol. 1993;67:2664–2671. doi: 10.1128/jvi.67.5.2664-2671.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Royer C. A. Improvements in the numerical analysis of thermodynamic data from biomolecular complexes. Anal. Biochem. 1993;210:91–97. doi: 10.1006/abio.1993.1155. [DOI] [PubMed] [Google Scholar]
- 32.Royer C. A., Beechem J. M. Numerical analysis of binding data: advantages, practical aspects and implications. Methods Enzymol. 1992;210:481–505. doi: 10.1016/0076-6879(92)10025-9. [DOI] [PubMed] [Google Scholar]
- 33.Royer C. A., Smith W. R., Beechem J. M. Analysis of binding in macromolecular complexes: a generalized numerical approach. Anal. Biochem. 1990;191:287–294. doi: 10.1016/0003-2697(90)90221-t. [DOI] [PubMed] [Google Scholar]
- 34.Ryu S. E., Kwong P. D., Truneh A., Porter T. G., Arthos J., Rosenberg M., Dai X., Xuong N., Axel R., Sweet R. W., Hendrickson W. A. Crystal structure of an HIV-binding recombinant fragment of human CD4. Nature (London) 1990;348:419–426. doi: 10.1038/348419a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xiang S. H., Kwong P. D., Gupta R., Rizzuto C. D., Casper D. J., Wyatt R., Wang L., Hendrickson W. A., Doyle M. L., Sodroski J. Mutagenic stabilization and/or disruption of a CD4-bound state reveals distinct conformations of the human immunodeficiency virus type 1 gp120 envelope glycoprotein. J. Virol. 2002;76:9888–9899. doi: 10.1128/JVI.76.19.9888-9899.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sundberg E. J., Urrutia M., Braden B. C., Isern J., Tsuchiya D., Fields B. A., Malchiodi E. L., Tormo J., Schwarz F. P., Mariuzza R. A. Estimation of the hydrophobic effect in an antigen–antibody protein–protein interface. Biochemistry. 2000;39:15375–15387. doi: 10.1021/bi000704l. [DOI] [PubMed] [Google Scholar]
- 37.Wodak S. J., Janin J. Structural basis of macromolecular recognition. Adv. Protein Chem. 2002;61:9–73. doi: 10.1016/s0065-3233(02)61001-0. [DOI] [PubMed] [Google Scholar]
- 38.Myszka D. G., Sweet R. W., Hensley P., Brigham-Burke M., Kwong P. D., Hendrickson W. A., Wyatt R., Sodroski J., Doyle M. L. Energetics of the HIV gp120-CD4 binding reaction. Proc. Natl. Acad. Sci. U.S.A. 2000;97:9026–9031. doi: 10.1073/pnas.97.16.9026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dandliker W. B., Hsu M. L., Levin J., Rao B. R. Equilibrium and kinetic inhibition assays based upon fluorescence polarization. Methods Enzymol. 1981;74:3–28. doi: 10.1016/0076-6879(81)74003-5. [DOI] [PubMed] [Google Scholar]
- 40.Moulard M., Lortat-Jacob H., Mondor I., Roca G., Wyatt R., Sodroski J., Zhao L., Olson W., Kwong P. D., Sattentau Q. J. Selective interactions of polyanions with basic surfaces on human immunodeficiency virus type 1 gp120. J. Virol. 2000;74:1948–1960. doi: 10.1128/jvi.74.4.1948-1960.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huang C. C., Stricher F., Martin L., Decher J. M., Majeed S., Barthe P., Hendrickson W. A., Robinson J., Roumestand C., Sodroshi J., et al. Scorpian toxin mimics of CD4 in complex with human immunodeficiency virus gp120 crystal structures, molecular mimicry, and neutralization breadth. Structure (Camb.) 2005;13:755–768. doi: 10.1016/j.str.2005.03.006. [DOI] [PubMed] [Google Scholar]