

Abstract
The viability of living systems depends inextricably on enzymes that catalyze phosphoryl transfer reactions. For many enzymes in this class, including several ribozymes, divalent metal ions serve as obligate cofactors. Understanding how metal ions mediate catalysis requires elucidation of metal ion interactions with both the enzyme and the substrate(s). In the Tetrahymena group I intron, previous work using atomic mutagenesis and quantitative analysis of metal ion rescue behavior identified three metal ions (MA, MB, and MC) that make five interactions with the ribozyme substrates in the reaction's transition state. Here, we combine substrate atomic mutagenesis with site-specific phosphorothioate substitutions in the ribozyme backbone to develop a powerful, general strategy for defining the ligands of catalytic metal ions within RNA. In applying this strategy to the Tetrahymena group I intron, we have identified the pro-S P phosphoryl oxygen at nucleotide C262 as a ribozyme ligand for MC. Our findings establish a direct connection between the ribozyme core and the functionally defined model of the chemical transition state, thereby extending the known set of transition-state interactions and providing information critical for the application of the recent group I intron crystallographic structures to the understanding of catalysis.
A combination of substrate atomic mutagenesis with site-specific substitutions in the ribozyme backbone allow the ligands of catalytic metal ions to be identified.
Introduction
Phosphoryl transfer reactions occur ubiquitously in biology, playing roles in gene replication, recombination, and expression. To orchestrate these central biological processes, biomacromolecules have harnessed the catalytic power of divalent metal ions [1–10]. The chemistry underlying these cellular events hinges critically on the metal ion interactions that occur during function. However, there exists little functional data defining the coordination environment of individual metal ions during catalysis and correspondingly few methods for obtaining such information.
Structural analyses can serve as a powerful starting point for investigation of catalytic metal ions, but these approaches provide no direct information about transition-state interactions. Reflecting this uncertainty, for a given enzyme or enzyme family the number of active-site metal ions and their proposed interactions during catalysis can vary with both enzyme and observer ([2–10] and references therein). For example, restriction enzymes have been crystallized with zero, one, two, or three metal ions in their active sites [7,8,10], and different RNase H crystal structures support either a one or two metal ion mechanism [3]. This variability in metal ion binding also occurs for RNA. In structures of the hammerhead ribozyme and tRNA, Mg2+ and Mn2+ occupy different sites [11,12], and recent group I intron structures exhibit differences in the number, location, and identity of active-site metal ions [13–15]. Thus, only the combination of structural and functional studies can establish metalloenzyme mechanisms unambiguously.
To understand how metalloenzymes utilize the catalytic power of metal ions, we must identify individual catalytic metal ions functionally, determine their relationships to each other, define their coordination environment, and establish the network of interactions that position the coordinating groups. Metal ion rescue experiments using substrate mutations offer a strategy to identify specific catalytic metal ion interactions within enzyme active sites, a particularly formidable challenge in ribozymes due to the sea of metal ions that interact electrostatically with the anionic phosphodiester backbone. Functionally deleterious sulfur or nitrogen perturbations that exhibit rescue upon increasing cation softness (e.g., replacing Mg2+ with Cd2+ or Mn2+) suggest direct metal ion coordination during catalysis [16,17]. This approach has revealed catalytic metal ion interactions with enzyme substrates in the Tetrahymena group I ribozyme [18–21], the ai5γ group II intron [22,23], RNase P [24], the hammerhead ribozyme [25,26], the human spliceosome [27,28], and many protein enzymes (e.g., [16,29–32] and references therein).
The Tetrahymena group I ribozyme catalyzes nucleotidyl transfer from an oligonucleotide substrate that mimics the natural 5′-splice site to an exogenous guanosine (G) that serves as the nucleophile in a reaction analogous to the first step of group I intron self-splicing (Equation 1) [33,34].
Metal ion rescue experiments have identified four atoms within the oligonucleotide substrate and G nucleophile that interact with metal ions in the chemical transition state [18–21]. To determine whether one or several distinct metal ions mediate these interactions, Shan et al. developed thermodynamic fingerprint analysis, quantitatively analyzing the reactivity of modified substrates relative to unmodified substrates over a range of rescuing metal ion concentrations [35]. In this approach, the reactions for both modified and native substrates start from the same ground state and monitor the same elementary reaction steps. The resulting rescue profiles serve as distinctive “fingerprints” for the rescuing metal ion(s), revealing by comparison whether the same or distinct metal ions interact with the identified substrate ligands. Thermodynamic fingerprint analysis and related analyses [36] using a series of substrates bearing single or multiple atomic perturbations have provided functional evidence for a network of three distinct metal ions within the Tetrahymena ribozyme active site (Figure 1), making a total of five interactions with the reaction's transition state. Metal ions coordinate to the 3′-oxygen leaving group (MA), the 3′-oxygen on the G nucleophile (MB), and the 2′-hydroxyl of the G nucleophile (MC). Two of these metal ions (MA and MC) also contact the pro-S P oxygen of the scissile phosphate [35,36]. However, the ligands within the ribozyme core architecture that bind and position these metal ion cofactors remain largely unknown, leaving the catalytic metal ion coordination environments undefined.
Figure 1. Model of the Tetrahymena Ribozyme Transition State during the First Step of Splicing.

The three identified catalytic metal ions (MA, MB, and Mc) and their transition-state interactions with the U(-1) 3′-oxygen, G nucleophile 3′-oxygen, and G 2′-OH, respectively, along with the MA and MC interactions with the scissile phosphate pro-S P oxygen, are shown by red dots [35,36]. The 2′-OH of U(-1) participates in a hydrogen-bonding network with the 2′-OH of A207 and the exocyclic amine of the G·U wobble pair [58,72] and donates a hydrogen bond to the adjacent 3′-oxygen in the transition state [71]; the hydrogen bonds are shown as hashed lines.
The non-bridging phosphate oxygens of the RNA backbone commonly serve as ligands for divalent metal ions. For the Tetrahymena group I ribozyme and other RNA enzymes, phosphorothioate interference studies have generated a plethora of ligand candidates for metal ions [17,26,37–52]. However, there have been few attempts to link these putative ligands to metal ions directly involved in catalysis [42,53,54]. Using the Tetrahymena group I ribozyme as a model system, we have combined thermodynamic fingerprint analysis with an array of atomically perturbed substrates and ribozyme site- and stereo-specific phosphorothioate mutations to develop a general functional approach for identifying ligands for the catalytic metal ions. Our findings establish a direct connection between the ribozyme core and the functionally defined model of the chemical transition state, thereby providing information critical for the application of the recent group I intron crystallographic structures to the understanding of catalysis.
Results
Choosing Sites for Phosphorothioate Substitution within the Ribozyme Core
Backbone mutation sites were chosen prior to the release of the recently reported group I intron structures [13–15]. To guide our choice of substitution sites, we focused on previously reported interferences arising from random R P-phosphorothioate incorporation into the phylogenetically conserved core regions of the Tetrahymena group I intron. As Mg2+ coordinates poorly to sulfur, the R P-phosphorothioate interferences could reflect direct disruption of a metal ion interaction with the pro-R P phosphate oxygen, indirect disruption of a metal interaction with the geminal pro-S P phosphate oxygen, or other effects. A literature survey identified 14 sites of R P-phosphorothioate interference within the ribozyme's conserved core [17,47,52,55]. Herein we analyze ribozymes containing site-specific phosphorothioate incorporation at six sites within the J6/7 and P7 regions of the ribozyme (Figure 2A). We constructed these mutant ribozymes semi-synthetically with both R P- and S P-phosphorothioate mutations at these six sites (Figure 2B), resulting in 12 variant ribozymes.
Figure 2. Construction of Ribozymes Containing Site-Specific Phosphorothioate Substitutions.

(A) Secondary structure of the Tetrahymena group I ribozyme. The ribozyme conserved core is highlighted in blue, and the six positions of phosphorothioate substitution within the J6/7 and P7 regions are labeled in green. The internal guide sequence at the 5′-terminus of the ribozyme is labeled “IGS.” (B) Ligation strategy for constructing mutant ribozymes. Following ion exchange HPLC purification of the phosphorothioate diastereomers (C), the phosphorothioate-substituted mutation oligonucleotides were ligated into full-length ribozymes by two successive-splint mediated ligations using T4 DNA ligase [77], as described in Materials and Methods .
The variant ribozymes were characterized kinetically within the known framework of the Tetrahymena ribozyme reaction (Figure 3; [33,56,57] and references therein). The oligonucleotide substrate (S; Table 1) binds to the ribozyme (E) in two steps. First, S forms Watson–Crick base pairs with the ribozyme's internal guide sequence (see Figure 2A) to give the open complex (E·S)O. The resulting P1 helix then “docks” into the ribozyme core via tertiary interactions, forming the closed complex (E·S)C ([33,57–59] and references therein). G binds to give the ternary (E·S·G)C complex, and the reaction proceeds through the phosphoryl transfer step (k chem), resulting in cleavage of the oligonucleotide substrate.
Figure 3. The Tetrahymena Ribozyme Reaction Pathway.

The ribozyme binds the oligonucleotide substrate (in two steps) and the exogenous G that serves as the nucleophile in the ribozyme reaction as described in the text ([33,56,57] and references therein). K G d(E), K G d(E·S)O, and K G d(E·S)c are G dissociation constants from free E, (E·S)O, and (E·S)C, respectively. K IGS d is the dissociation constant for the oligonucleotide substrate from the internal guide sequence, and K dock and K′dock are the docking equilibria for the E·S and E·S·G complexes, respectively. K S d(E) and K S d(E·G) are the observed dissociation constants for the oligonucleotide substrate from free E and the E·G complex, respectively, and k chem is the observed rate of oligonucleotide substrate cleavage.
Table 1. Oligonucleotide Substrates Used Herein.
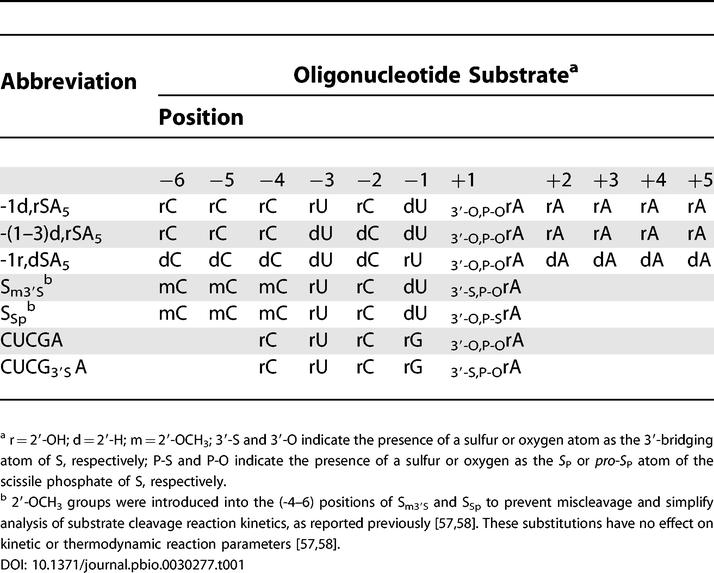
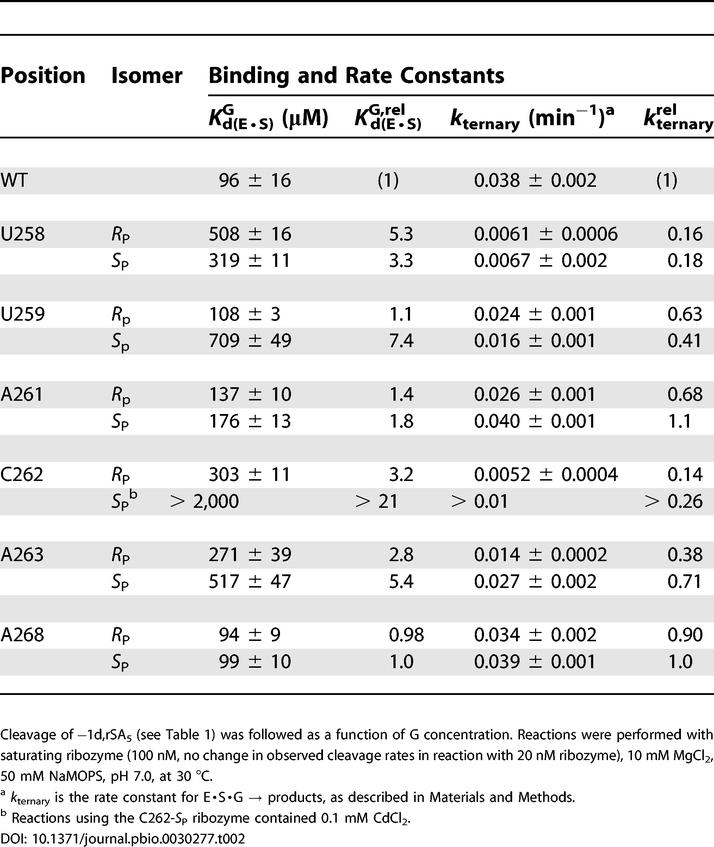
We first tested whether Cd2+, a thiophilic metal ion that can adopt octahedral coordination geometry like Mg2+ [60–62], stimulates the ability of the phosphorothioate containing ribozymes to catalyze oligonucleotide substrate cleavage (Figure 4). Under conditions of saturating ribozyme and G (10 mM MgCl2), several of the phosphorothioates affected catalysis significantly (data not shown, and see Table 2 below), but upon addition of 0.1–1.0 mM Cd2+, only one of the variant ribozymes, the C262-S P variant, experienced significant stimulation (Figure 4 and data not shown). This stimulation suggested that a functionally important Cd2+ phosphorothioate interaction may occur at the C262-S P position, leading us to focus predominantly on this variant ribozyme. The other positions tested, in principle, remain viable ligand candidates as the absence of rescue cannot be taken as evidence that the modification site does not serve as a metal ion ligand. However, metal ion rescue experiments analogous to those described below for the C262-S P ribozyme suggest that none of the other phosphorothioate substitutions have large effects on binding of the known catalytic metal ions (see Figure S1).
Figure 4. The Effect of Cd2+ on Activity of Phosphorothioate-Containing Ribozymes.

Reactions contained saturating G and ribozyme, and monitored cleavage of −1d,rSA5 (see Table 1) in the presence or absence of 0.1 mM Cd2+ (10 mM Mg2+ background). The largest Cd2+ stimulation occurred with the C262-S P variant ribozyme. Significant Cd2+ stimulation remained unique to this ribozyme at all Cd2+ concentrations tested (0.1–10 mM; data not shown). Concentrations of Cd2+ above 1 mM inhibit the ribozyme reaction with a steep concentration dependence (see, e.g., Figures S4 and S5A). The small Cd2+ stimulation observed for the U258 variant ribozymes is consistent with coordination of a metal ion important for structural stability, as proposed by Lindqvist et al. [78].
Table 2. G Binding and Reactivity for Ribozymes with Site-Specific Phosphorothioate Substitutions within J6/7 and P7.
Cd2+ Accelerates a Non-Chemical Step in the C262-S P Ribozyme Reaction
Metal ion rescue experiments provide definitive information about transition-state metal ion-ligand interactions only when conducted under conditions in which the same reaction steps are monitored and the chemical step limits the reaction rate [35]. To learn more about the apparent Cd2+ stimulation and to establish appropriate reaction conditions under which the chemical step could be monitored, we undertook basic characterization of the C262-S P ribozyme. We first present data concerning the rate-limiting step of the C262-S P ribozyme reaction, and then assess the effects of this mutation on individual reaction steps.
Previous work established that, in the wild-type (WT) ribozyme, the chemical step follows a pre-equilibrium loss of a proton, presumably from the 3′-hydroxyl of the attacking G (see Figure 1). This leads to a log-linear pH dependence with a slope of one under conditions of rate-limiting chemistry [33,63–65]. However, in the absence of Cd2+, the pH dependence for cleavage of the −1d,rSA5 substrate (Table 1) by the C262-S P variant ribozyme is essentially flat above pH 5 (Figure 5A, closed circles), in contrast to the slope of one for the WT ribozyme [63,65]. This difference suggests that, under the reaction conditions, a conformational change limits the rate for the variant ribozyme. In the presence of low concentrations of Cd2+, the variant behaves more like the WT ribozyme, exhibiting a log-linear pH dependence with slope one (Figure 5A, open circles). This Cd2+-induced change in pH dependence suggests that Cd2+ accelerates the non-chemical step sufficiently to render the chemical step rate-limiting throughout the entire pH range.
Figure 5. The pH Dependence of C262-S P Ribozyme Activity Reveals a Non-Chemical Rate-Limiting Step.

(A) Cleavage of −1d,rSA5 under saturating conditions, as given in Figure 3. In the absence of Cd2+ (·), the pH dependence flattens above pH ∼4.7; the data are fit to k max = 1/(1+10n(pKa-pH)). Addition of 1 mM Cd2+ (s○) leads to log-linear pH dependence (slope = 1.1 ± 0.1) over the pH range of 4–7, consistent with the chemical step being rate-limiting [33,63–65]. The Cd2+ rescue observed in Figure 3 results from acceleration of the non-chemical step, as indicated by the vertical arrow, such that this non-chemical step is no longer rate-limiting. The increased slope observed at low pH for reactions both with and without Cd2+ is consistent with ribozyme inhibition due to multiple independent protonations [65].
(B) Cleavage of −1r,dSA5 under saturating ribozyme and G conditions. Cleavage of −1r,dSA5 is log-linearly dependent on pH with and without added Cd2+ (0 mM Cd2+, • ; 1 mM Cd2+, s○) (best fit slopes of 0.8 in both cases), consistent with the chemical step being rate-limiting in both cases.
Although the molecular basis for the rate-limiting, non-chemical step and its stimulation by Cd2+ in the C262-S P ribozyme reaction remains undefined, comparison to the reaction of −1r,dSA5 offers some insight. The −1r,dSA5 substrate reacts from the ternary complex (E·S·G) with a log-linear pH dependence whose slope approximates one, even in the absence of Cd2+ (Figure 5B, closed circles). Addition of Cd2+ has no effect on the reaction rate or the pH-dependence (Figure 5B, open circles). These results suggest that the chemical step limits the reaction of −1r,dSA5 both in the presence and absence of Cd2+. As −1r,dSA5 binds primarily in the open complex with the WT ribozyme (see Figure 3) [36,66] and −1d,rSA5 binds in the closed complex [57,58], the rate-limiting non-chemical step observed with −1d,rSA5 may reflect phosphorothioate-induced formation of an altered closed complex that must rearrange prior to the chemical step. Acceleration of this non-chemical step depends on Cd2+; increasing the Mg2+ concentration does not accelerate this step, nor does Mn2+ or Zn2+ (data not shown). The Cd2+ dependence suggests that this non-chemical step may require metal ion coordination to the backbone phosphorothioate. Future study of this metal-ion-dependent non-chemical step revealed by phosphorothioate incorporation may provide an opportunity for deeper understanding of the role of metal ion coordination in conformational rearrangements within RNA.
This initial characterization of the C262-S P ribozyme allowed us to choose appropriate reaction conditions for further characterization. For analyzing the binding of G and oligonucleotide substrates to the variant ribozyme, we included Cd2+ in sufficient but subsaturating concentrations to render the chemical step rate-limiting without influencing the measured binding constants (i.e., subsaturating Cd2+ in order to have no significant effect on substrate affinities; [Cd2+] ≤ 1 mM) [35]. For metal ion rescue reactions in which a constant Cd2+ concentration was not feasible, we chose oligonucleotide substrates (Table 1) or reaction pHs that render the chemical step rate-limiting.
Phosphorothioate Substitution Effects on Reactivity and Binding
We analyzed the 12 phosphorothioate-substituted variants within the J6/7 and P7 regions of the ribozyme (see Figure 2A) under a variety of reaction conditions. As noted above, reactions with the C262-S
P variant contained Cd2+ to maintain rate-limiting chemistry. Cd2+ had no significant effect on the behavior of the other ribozymes (data not shown). Representative data from this survey—phosphorothioate substitution effects on the reaction E·S·G → products (k
ternary) and G binding to the E·S complex  —are given in Table 2. This survey revealed one position at which phosphorothioate substitution strongly perturbs (greater than 10-fold) G binding: the S
P phosphorothioate at nucleotide C262, the same variant that exhibited Cd2+ stimulation.
—are given in Table 2. This survey revealed one position at which phosphorothioate substitution strongly perturbs (greater than 10-fold) G binding: the S
P phosphorothioate at nucleotide C262, the same variant that exhibited Cd2+ stimulation.
Based on the G-binding results, the Cd2+ rescue of a non-chemical step, and screens for the effects of J6/7 and P7 phosphorothioates on rescue by the catalytic metal ions (see Figure S1), we decided to characterize the C262-S P ribozyme further, focusing on how the phosphorothioate affects the individual reaction steps (see Figure 3). We found that the C262-S P substitution had no significant effect on oligonucleotide substrate association or dissociation (Table S1). We then further investigated the observed effect of the C262-S P phosphorothioate substitution on G binding. In reactions with the −(1–3)d,rSA5 substrate, which binds to the WT ribozyme in the open complex (E·S)O (based on observed G-binding affinity [56,67] and Mn2+ stimulation of cleavage activity [66] [data not shown]), G binds to the C262-S P variant and the WT ribozyme with the same affinity. Thus, in the absence of docked oligonucleotide substrate, the C262-S P phosphorothioate has no effect on G binding (Table 3). The C262-S P phosphorothioate affects subsequent reaction steps, however. The ternary open complex, (E·S·G)O, for the variant reacts about 40-fold slower than that for the WT ribozyme, suggesting possible effects on oligonucleotide substrate docking into the G-bound active site or on the chemical step.
Table 3. G Binding and Reactivity with WT and C262-S P Ribozymes.

In the presence of WT ribozyme, G and S bind cooperatively to give the ternary closed complex, (E·S·G)C. G binds to (E·S)C five- to ten-fold tighter than to (E·S)O or free enzyme [56,67]. Using −1d,rSA5 to form (E·S)C and −(1–3)d,rSA5 to form (E·S)O, we reproduced this coupling for the WT ribozyme (Table 3). In contrast, the C262-S P ribozyme exhibited no coupling. Indeed, the presence of docked oligonucleotide substrate with the C262-S P ribozyme weakens G binding. Overall, the C262-S P ribozyme closed complex has an approximately 40-fold weaker affinity for G than does the WT ribozyme closed complex (Table 3). Reaction chemistry from the ternary closed complex remains unaffected, as (E·S·G)C formed with −1d,rSA5 has the same rate for the WT and C262-S P ribozymes (Table 3).
Previous work has linked the coupled binding of G and the oligonucleotide substrate to MC, which mediates contacts to the G 2′-hydroxyl and the scissile phosphate pro-S P oxygen (see Figure 1) [68,69]. The loss of coupled binding between the oligonucleotide substrate and G induced upon C262-S P phosphorothioate substitution therefore raised the possibility that the C262 pro-S P phosphoryl oxygen resides near the MC binding site in the ribozyme tertiary structure.
A Linkage between C262-S P and One of the Catalytic Metal Ions
The apparent functional connection between the C262 pro-S P phosphoryl oxygen atom and the metal ion bound at site C (MC) could occur through direct interaction of this oxygen with MC, or indirectly through a chain of interactions. We used the C262-S P ribozyme to ascertain whether direct coordination occurs, analyzing the metal ion rescue behavior for each of the previously identified metal ion sites. If the C262-S P phosphorothioate directly coordinates to a Cd2+ ion bound at one of the known catalytic metal ion binding sites, we expect the stronger Cd2+–sulfur interaction to shift the metal ion rescue profile toward lower Cd2+ concentration relative to the WT ribozyme.
We determined the MA, MB, and MC profiles for the WT and C262-S P ribozymes according to approaches described previously [35,36,68]. Table 4 lists the substrates and kinetic regimes for the reactions used to obtain each rescue profile. For each rescue profile, WT and mutant ribozyme reactions with both modified and unmodified substrates must start from the same ground state and monitor the chemical step. Unless indicated otherwise, we chose substrates and conditions so that the substrate bearing the modification is not bound within the active site in the starting ground state. This ensures that substrate modifications have no effect on ground-state binding of the rescuing metal ion to the WT and mutant ribozymes. The supporting information provides full details of the methods, models, and equations used to fit the Cd2+ rescue dependencies.
Table 4. Metal Rescue Assays Used to Probe Catalytic Metal Ions in the Tetrahymena Ribozyme.

To monitor Cd2+ binding at the metal ion site A, we followed the reactivity of an oligonucleotide substrate containing a 3′-thiophosphoryl linkage at the cleavage site, Sm3′S (Figure 6A) [21,35]; i.e., Cd2+ specifically rescues the cleavage rate of Sm3′S relative to the unmodified 3′-oxygen oligonucleotide substrate (k rel, k 3′S/k 3′O). The C262-S P and WT ribozymes exhibit nearly identical profiles for rescue at metal ion site A (Figure 6A). The small deviation at high Cd2+ is beyond experimental error (as indicated by error bars in Figure 6A) and may reflect an indirect effect from Cd2+ occupancy at a different site (see Protocol S1 ). The lack of significant change in Cd2+ rescue at metal site A provides no evidence for direct contact between the S P-phosphorothioate at residue 262 and a Cd2+ ion binding at metal site A. Therefore, we conclude that the C262 pro-S P phosphoryl oxygen in the WT ribozyme is not a ligand for MA.
Figure 6. The C262-S P Phosphorothioate Perturbs the Rescue Profile of MC .

Cd2+ rescue profiles for MA, MB, and MC with WT (s○) and C262-S P (▪) ribozymes are displayed. In each case, the specific metal ion-substrate contact being probed is indicated by closed red dots.
(A) MA rescue of Sm3′S cleavage. In the Tetrahymena ribozyme transition-state model, the oligonucleotide substrate 3′-thiophosphoryl modification at U(-1) is shown in green. MA rescue reactions monitoring cleavage of −(1–3d)rSA5 and Sm3′S were carried out under E·S·G → products (k ternary) conditions as described in Materials and Methods . The Cd2+ profiles for MA rescue are fit to a Hill equation, facilitating comparison between the WT and C262-S P ribozyme profiles (see Protocol S1). The fits give Hill constants of 1 and 1.2 for the WT and C262-S P ribozymes, respectively. Error bars not seen are obscured by the data symbols.
(B) MB rescue of CUCG3′SA cleavage. In the transition-state model, the G 3′-thio modification is shown in green. MB rescue reactions monitoring cleavage of CUCGA and CUCG3′SA were performed under E·P + CUCG3′XA → products [(k c/K m)CUCG(3′X)A] conditions, as described in Materials and Methods. MB rescue is fit to a model in which one Cd2+ ion binds to E·P and stimulates reaction of CUCG3′SA.
(C) MC rescue of −1r,dSA5 cleavage by subsaturating GN ((k c/K m)G(or GN ) conditions). In the transition-state model, the 2′-amino group of the G nucleophile is shown in blue. MC rescue of −1r,dSA5 cleavage under E·S + G(or GN) → products ((k c/K m)G(or GN ) conditions) was performed as described in Materials and Methods. k rel data were fit to a model in which one Cd2+ ion binds and rescues reaction with GN.
(D) MC rescue of −1r,dSA5 cleavage by saturating GN. In the transition-state model, the 2′-amino group of the G nucleophile is again shown in blue. MC rescue of −1r,dSA5 cleavage under E·S·G/GN→ products (k
ternary) conditions was measured as described in Materials and Methods. k
rel data were fit to a model in which one Cd2+ ion binds and rescues reaction with GN; with the C262-S
P ribozyme, the fit to this model gives  = 0.22 ± 0.08 mM. Cd2+ dependencies of observed cleavage rates in the MA, MB, and MC rescue reactions are displayed in Figures S2, S3, and S4, respectively.
= 0.22 ± 0.08 mM. Cd2+ dependencies of observed cleavage rates in the MA, MB, and MC rescue reactions are displayed in Figures S2, S3, and S4, respectively.
To follow Cd2+ binding to the MB site, we monitored Cd2+ rescue of the reactivity of CUCG3′SA relative to CUCGA in the reverse reaction with E·P (Equation 2) [20,35]. CUCG3′SA contains a 3′-thiophosphoryl linkage at the cleavage site.
In reactions catalyzed by both the WT and C262-S P ribozymes, Cd2+ specifically stimulates the reactivity of CUCG3′SA more than 500-fold. This rescue fits well to a model in which a single Cd2+ ion binds to the MB site and rescues the reaction (Figure 6B). As C262-S P phosphorothioate incorporation exhibits no effect on the MB Cd2+ rescue profile, we conclude that the C262 pro-S P phosphoryl oxygen in the WT ribozyme is not a ligand for MB.
To follow Cd2+ binding to the MC site, we monitored Cd2+ rescue of oligonucleotide substrate cleavage by 2′-aminoguanosine (GN) relative to G [19,35,68]. We first probed Cd2+ binding to the MC site in the E·S complex, conducting reactions at subsaturating G or GN concentrations ((k
c/K
m)G or (k
c/K
m
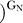 conditions). Specific Cd2+ stimulation of cleavage with GN occurs with both the WT and C262-S
P ribozymes, giving linear rescue curves throughout the Cd2+ concentration range tested with slopes that reflect a single Cd2+ stimulating cleavage by GN (Figure 6C). In contrast to the MA and MB rescue profiles discussed above, the MC rescue curve for the C262-S
P variant shifts to the left, with 16-fold lower Cd2+ concentrations required to achieve the same level of rescue as the WT. This shift suggests that the S
P-phosphorothioate at nucleotide C262 interacts directly with the Cd2+ ion binding at the MC site, a model we test further below.
conditions). Specific Cd2+ stimulation of cleavage with GN occurs with both the WT and C262-S
P ribozymes, giving linear rescue curves throughout the Cd2+ concentration range tested with slopes that reflect a single Cd2+ stimulating cleavage by GN (Figure 6C). In contrast to the MA and MB rescue profiles discussed above, the MC rescue curve for the C262-S
P variant shifts to the left, with 16-fold lower Cd2+ concentrations required to achieve the same level of rescue as the WT. This shift suggests that the S
P-phosphorothioate at nucleotide C262 interacts directly with the Cd2+ ion binding at the MC site, a model we test further below.
The MC rescue profiles for both the WT and C262-S P ribozymes increase linearly up to the highest experimentally accessible Cd2+ concentrations (Figure 6C), indicating that the rescuing Cd2+ ion binds to the E·S complexes of these ribozymes under these conditions with a dissociation constant that exceeds 10 mM. Without saturation behavior, we cannot definitively ascertain whether the C262 phosphorothioate-induced shift in the MC rescue profile emanates from tighter Cd2+ binding. Consequently, we probed Cd2+ binding to the MC site in the presence of saturating GN. Bound GN provides MC with an additional ligand, the nitrogen of the 2′-amino group, which should interact strongly with Cd2+ [70].
We conducted the rescue experiment as described above but included GN at saturating concentration to form the E·S·GN ternary complex. As with subsaturating GN, both the WT and C262-S
P ribozymes experienced significant specific Cd2+ rescue (Figure 6D). The rescue profile for the WT E·S·GN complex remains linear throughout the Cd2+ concentration range tested, indicating that even in the presence of bound GN, Cd2+ binds to the MC site with an apparent affinity of greater than 10 mM (50 mM Mg2+ background). In striking contrast, the Cd2+ rescue profile for reaction of the C262-S
P E·S·GN complex exhibits saturation behavior, with an apparent dissociation constant of 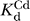 ∼ 0.2 mM. The C262-S
P E·S·GN ternary complex therefore binds the rescuing Cd2+ ion more than 50-fold tighter than does C262-S
P ribozyme E·S binary complex, suggesting that the 2′-amino group of GN interacts with MC in the ground state. Moreover, the C262-S
P E·S·GN complex binds the rescuing Cd2+ ion more than 50-fold tighter than does the corresponding WT ternary complex, showing that the phosphorothioate substitution at C262 enhances Cd2+ binding to metal ion site C. Taken together, these data further support a model in which the C262-S
P phosphorothioate interacts directly with the Cd2+ ion in the MC site and strongly implicate the C262 pro-S
P oxygen as a ligand for MC in the WT ribozyme.
∼ 0.2 mM. The C262-S
P E·S·GN ternary complex therefore binds the rescuing Cd2+ ion more than 50-fold tighter than does C262-S
P ribozyme E·S binary complex, suggesting that the 2′-amino group of GN interacts with MC in the ground state. Moreover, the C262-S
P E·S·GN complex binds the rescuing Cd2+ ion more than 50-fold tighter than does the corresponding WT ternary complex, showing that the phosphorothioate substitution at C262 enhances Cd2+ binding to metal ion site C. Taken together, these data further support a model in which the C262-S
P phosphorothioate interacts directly with the Cd2+ ion in the MC site and strongly implicate the C262 pro-S
P oxygen as a ligand for MC in the WT ribozyme.
An Independent Test of the C262 pro-S P Phosphoryl Oxygen as a MC Ligand
The reaction of an oligonucleotide substrate bearing a S P-phosphorothioate at the cleavage site (SSp, Table 1) also experiences Cd2+ stimulation with the WT ribozyme. The Cd2+ rescue profile for this modified oligonucleotide substrate exhibits a slope of two, suggesting that two Cd2+ ions contact the non-bridging sulfur atom in the transition state [36]. Thermodynamic fingerprint analysis established that these rescuing metal ions bind at sites A and C (Figure 7A) [36]. The SSp oligonucleotide substrate therefore offers another strategy by which to test whether the C262-S P phosphorothioate enhances Cd2+ binding to the MC site. If the MC site in the C262-S P ribozyme becomes saturable in the presence of bound GN, as described in the previous section, then the rescue profile for SSp cleavage should transition from an apparent dependence on two Cd2+ ions to a dependence on one Cd2+ ion. The results described below meet this prediction, thereby providing additional quantitative support for the assignment of the pro-S P phosphoryl atom of residue C262 as a ligand for MC.
Figure 7. Cd2+ Rescue of SSp Cleavage by the C262-S P Ribozyme.

Reactions were performed under E·S·G/GN → products (k ternary) conditions, as described in Materials and Methods, with −(1–3)d,rSA5 serving as the unmodified control oligonucleotide substrate. Cd2+ dependencies for oligonucleotide substrate cleavage by G and GN are displayed in Figure S5.
(A) Model of the Tetrahymena ribozyme transition state for rescue of SSp reaction by Cd2+ ions bound at MA and Mc. Closed red circles indicate metal ion coordinations considered important for rescue, and the S P phosphorothioate modification at the scissile phosphate is denoted in green.
(B) k rel versus Cd2+ for cleavage of SSp with G. The data are fit to a model in which two Cd2+ ions rescue reaction, with neither Cd2+ binding site saturating (see Protocol S1).
(C) Model of the Tetrahymena ribozyme transition state for rescue of SSp cleavage by two Cd2+ ions bound at MA and MC in the presence of saturating GN. Metal ion coordinations important for rescue are denoted by closed red circles, and the GN amino group is shown in blue.
(D) k rel versus Cd2+ for cleavage of SSp with GN. The dotted line is a fit to the model used in (B), for two Cd2+ ions rescuing reaction and neither Cd2+ binding site saturating. The solid line is a fit to a model in which two Cd2+ ions rescue reaction, with one site saturating with a K Mc d,app = 0.2 mM (see Protocol S1).
The Cd2+ rescue profile for SSp cleavage by C262-S
P ribozyme with saturating G fits well to a model with two Cd2+ ions rescuing SSp cleavage with neither metal ion saturating, analogous to that for the WT ribozyme (Figure 7B) [36]. This profile matches expected rescue behavior, given that neither the MA nor MC (subsaturating GN) individual rescue profiles exhibit saturation with the C262-S
P ribozyme (see Figures 6A and 6C, respectively). In contrast, with saturating GN, Cd2+ rescue of SSp cleavage by the C262-S
P ribozyme no longer fits well to a dependence on two nonsaturating Cd2+ ions (see Figure 7C and the dashed line in Figure 7D). Rather, the best fit gives a dependence on only one Cd2+ ion, suggesting that one of the two metal ion sites involved in rescuing the SSp reaction saturates. Assuming that one Cd2+ ion binds to the C262-S
P ribozyme with 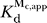 = 0.2 mM, the value determined for Cd2+ binding to metal site C in the presence of saturating GN (see Figure 6D), we obtain the solid line in Figure 7D. In contrast, for the WT ribozyme, Cd2+ rescue of the SSp reaction in the presence of saturating GN exhibits a dependence on two Cd2+ ions up to the highest accessible Cd2+ concentration [36]. Therefore, as predicted, the C262-S
P phosphorothioate substitution enhances binding of one of the rescuing Cd2+ ions in the reaction of SSp, strongly supporting the conclusion that MC interacts directly with the pro-S
P atom of the C262 phosphate.
= 0.2 mM, the value determined for Cd2+ binding to metal site C in the presence of saturating GN (see Figure 6D), we obtain the solid line in Figure 7D. In contrast, for the WT ribozyme, Cd2+ rescue of the SSp reaction in the presence of saturating GN exhibits a dependence on two Cd2+ ions up to the highest accessible Cd2+ concentration [36]. Therefore, as predicted, the C262-S
P phosphorothioate substitution enhances binding of one of the rescuing Cd2+ ions in the reaction of SSp, strongly supporting the conclusion that MC interacts directly with the pro-S
P atom of the C262 phosphate.
Discussion
Metalloenzymes that catalyze phosphoryl transfer play multiple roles throughout biology, serving as kinases, phosphatases, polymerases, and nucleases, among other functions [1–10]. We have established a powerful new experimental paradigm with which to identify ligands that coordinate to catalytic metal ions. Our analysis of simultaneous atomic perturbations within the Tetrahymena ribozyme core and its substrates, under conditions that allow valid thermodynamic comparisons, provides strong evidence that the pro-S P oxygen at residue C262 serves as a ligand for metal ion C (MC, Figure 8A). A single phosphorothioate substitution at this site, alone and in combination with GN, changes the metal ion affinity and specificity at the MC site while having little or no effect on the MA and MB sites. Although we cannot determine unambiguously whether the interaction between the pro-S P oxygen at C262 and MC occurs via outer sphere or inner sphere coordination in the natural ribozyme, the presence of a direct, inner sphere metal ion-ligand interaction provides the simplest model to account for our observations. More extensive rearrangement by the ribozyme active site to accommodate the backbone mutation appears less likely, as the metal ion that contacts C262-S P satisfies all the transition-state contacts proposed for MC. Further demonstrating the efficacy of this approach, detailed analysis of phosphorothioate mutations in the P4 and J5/4 regions of the ribozyme core identified the pro-S P oxygen at position C208 as a ligand for MA (AVK, JLH, JAP, and DH, unpublished data), consistent with the previous proposal of Szewczak et al. [53].
Figure 8. Functional and Structural Models of Group I Intron Active Sites.

(A) Model of the Tetrahymena ribozyme transition state from functional data with the C262 pro-S P phosphoryl oxygen and C208 pro-S P oxygen coordinating to MC and MA, respectively ([35,36,53,58,71,72, 74], data herein, and AVK, JLH, JAP, and DH, unpublished results).
(B) Model of Mg2+ binding in the crystal structure of a thermostable variant of the Tetrahymena group I ribozyme (derived from PDB file 1X8W) [14]. The model shown is derived from molecule C, although the position of the metal ion appears to vary among the four molecules observed in the asymmetric unit. The putative Mg2+ ion (dashed green circle) is 2.4 Å from the pro-S P oxygen of C262 and 2.1 Å from the terminal G (ωG) 2′-OH. U(-1) is not shown, as the crystallized form of the thermostable Tetrahymena variant ribozyme lacks this nucleotide.
(C) Model of K+ binding from the crystal structure of the Azoarcus group I ribozyme (derived from PDB file 1T42) [15]. The putative K+ ion (dashed green circle) is 2.4 Å from the pro-S P oxygen of G128 (C262 homologue) and 2.8 Å from the modeled position for the terminal G (ωG) 2′-OH; the Azoarcus intron construct that was crystallized contained 2′-deoxyguanosine at ωG, and electron density for the K+ ion is observed only in the presence of this 2′-deoxyguanosine modification at ωG.
(D) Model of proposed Mg2+ binding site derived from the crystal structure of the Twort group I ribozyme [13]. The crystallographic data lack clear density for a metal ion in the region expected for the MC binding site but show the pro-S P phosphoryl oxygen of A120 (C262 homologue) and the 2′-OH of ωG appropriately juxtaposed to coordinate a single Mg2+ ion (purple circle).
Under some conditions, the C262-S P ribozyme E·S·G ternary complex must undergo a rate-limiting conformational change en route to the chemical transition state. Cd2+ accelerates this non-chemical step in this C262-S P ribozyme, suggesting that MC, which interacts with the phosphorothioate during the reaction, mediates this conformational change. Currently, we lack sufficient information to speculate further on the molecular basis of this conformational change. The ability to induce a new or existing conformational change may provide a future opportunity to investigate the relationship between RNA function and dynamics.
Crystal structures of three different group I introns have emerged in the past several months, providing a structural context for these functionally identified catalytic metal ligands [13–15]. The three structures converge beautifully with respect to the global architecture and reveal the highly electrostatic character of the active site, consistent with functional studies. These structures also underscore the difficulty of defining catalytic metal ion interactions, exhibiting limited agreement regarding the active-site conformation and the number, charge, and location of bound metal ions. This variability in metal ion number and location notwithstanding, the C262 pro-S P oxygen (or its equivalent) lies in proximity to the ωG 2′-hydroxyl in all three structures (Figure 8). The Azoarcus and Tetrahymena crystals contain electron density for a metal ion within coordination distance of this phosphoryl oxygen [14,15], whereas the Twort ribozyme structure lacks such electron density [13]. The data presented herein establish the importance of this proximity for function and suggest that the active-site configuration in the crystals bears at least some relevance to the active-site configuration in the transition state. This agreement between structural and functional data for metal ion C supports the assignment of a conserved catalytic metal ion binding site at the top of the P7 helix in group I introns (see Figure 2A).
Extensive group I intron biochemical investigations, conducted for two decades in the absence of atomic resolution structures, have revealed an intricate network of interactions surrounding the reaction center in the transition state interconnected by hydrogen bonds, metal ion coordinations, and the atomic configuration of the reactants (see Figure 1). The results suggest a constellation of three metal ions at the active site [35,36], making five atomic interactions to the transition state. In addition to these catalytic metal ion coordinations, the 2′-hydroxyl group of U(-1) donates a hydrogen bond to the adjacent 3′-oxygen leaving group in the transition state [71]. The 2′-hydroxyl group of A207 donates a hydrogen bond to the 2′-OH of U(-1) and accepts a hydrogen bond from the exocyclic amine of G22, thereby bridging the cleavage site 2′-OH and the G·U wobble pair [58,72]. The recent group I intron crystal structures, though unable to define the number, location, or catalytic interactions of metal ions unambiguously, provide an opportunity to visualize how these functionally defined networks extend deeper into the ribozyme core.
Based on the recent crystal structure of the Azoarcus group I intron, an alternative model for the transition state was proposed in which only two metal ions interact with the reaction center [15]. In this model, one metal ion coordinates to the 3′-oxygen leaving group of the cleavage site uridine and the pro-S P oxygen of the scissile phosphate, the same interactions proposed for MA in the functional model (Figure 8A). The other metal ion coordinates directly with the 2′-hydroxyl group of G as does MC in the functional model, but coordinates indirectly to the pro-Sp oxygen of the scissile phosphate, in contrast to the direct interaction implicated by the functional data. A metal ion interaction with the 3′-oxygen of G, mediated by MB in the functional model, is absent from the crystallographic model. No direct evidence for a MB-binding site was obtained from the X-ray structure. The resting structure of the RNA in the crystal could adopt a conformation that differs from the active structure and thereby exclude MB from the crystals. Alternatively, the MB rescue profile could reflect the recruitment of a thiophilic metal ion to the active site during the reaction of the sulfur-containing G analogue. As the 3′-oxygen of the G undergoes a large charge rearrangement in the transition state, we suggest that a direct metal ion interaction with the 3′-oxygen of G is likely, such that MB is present in the normal reaction or metal ion C makes both 2′- and 3′-interactions with G [73]. These different proposals for specific metal ion coordination configurations during the ribozyme reaction highlight the need for further structural and functional tests and refinements of the catalytic models.
To achieve a unified description of catalytic function that integrates the biochemically defined interaction networks and the crystallographically defined three-dimensional structure, we must establish “anchor points”—functionally verifiable linkages between transition-state interactions and the enzyme's core. Our analysis establishes the C262 pro-S P oxygen as such an anchor point. Anchor points provide critical information about the spatial arrangement of catalytic groups within the global architecture of the enzyme. Together, the transition state model, anchor points, and the recent X-ray structures establish a powerful foundation to build toward an in-depth understanding of how cooperative structure adopted by this RNA and other enzymes engenders enormous catalytic power and exquisite specificity.
Materials and Methods
Materials
WT Tetrahymena ribozyme was prepared as described previously [34]. All oligonucleotide substrates (see Table 1) were prepared and 5′-end-labeled using standard methods [34,35,54,74]. Oligonucleotides with thio substitutions were prepared by published procedures [75]. Oligonucleotides containing phosphorothioate diastereomers were separated by anion exchange HPLC [42,54]. Reverse phase HPLC of purified diastereomers, under conditions in which the R P diastereomer elutes before the S P, allowed assignment of each diastereomer's configuration [76].
Ribozyme preparation
Variant ribozymes were constructed semisynthetically using successive splint-mediated ligations [77]. Synthetic oligonucleotides corresponding to nucleotides 255–274 of the ribozyme, each containing a single phosphorothioate modification at the desired mutation site, were purchased from Dharmacon (Lafayette, Colorado, United States). Following phosphorothioate separation, synthetic oligonucleotides were 5′-phosphorylated with T4 polynucleotide kinase and cold ATP. Constructs corresponding to nucleotides 22–254 and 274–409 of the Tetrahymena ribozyme were transcribed using DNA templates produced by PCR truncation of the plasmid-encoded ribozyme sequence, with excess GMP present in the transcription of the 3′-construct to yield a 5′-monophosphate. The transcripts were ligated to the synthetic oligonucleotide via two successive splint-mediated ligations with T4 DNA ligase to yield full-length ribozyme containing a single R P- or S P-phosphorothioate mutation at the desired site.
Ligated ribozymes appear to contain approximately 40% inactive enzyme fraction, as indicated by biphasic kinetics under conditions in which oligonucleotide substrate cleavage occurs faster than oligonucleotide substrate dissociation (data not shown). Evidence suggests that the inactive ribozyme fraction in these semisynthetic ribozymes may be due to errors at the ligation junctions resulting from ligation of a subset of transcribed RNA constructs with incorrect termini (K. Travers, V. Diankov, and DH, unpublished data). Under conditions used in this work to characterize variant ribozyme reactivity, the inactive fraction did not contribute to the cleavage activity monitored in our assays of ribozyme activity. The inactive fraction did not affect association and dissociation rate constants measured by pulse chase experiments, as evidenced by monophasic binding behavior (data not shown). The activity of WT ribozyme constructed by ligation varied less than 2-fold from that of transcribed WT ribozyme (Table S2) after accounting for the presence of the inactive ribozyme fraction. The inactive ribozyme fraction does not affect the conclusions in this work.
General kinetic methods
All cleavage reactions were single turnover, with ribozyme in excess of radiolabeled S (S*), and were carried out at 30 °C in 50 mM buffer and 50 mM MgCl2 unless noted otherwise. All reactions without Cd2+ contained 0.1 mM EDTA. The buffers used were NaOAC (pH 4.0–5.2), NaMES (pH 5.6–6.7), and NaMOPS (pH 7.0–7.5). Reaction mixtures containing all components except Cd2+, EDTA, and radiolabeled oligonucleotide substrate were pre-incubated at 50 °C for 30 min to renature the ribozyme. Reactions were followed and analyzed as described previously [33,36,74].
Determination of rate and equilibrium constants
For oligonucleotide substrates that bind to the WT ribozyme in the closed complex, e.g. −1d,rSA5 (see Table 1), we define k
c as the first-order rate constant for the reaction of the ternary complex (E·S·G)C → products. Values of k
c were determined at pH 7.0, with ribozyme saturating with respect to oligonucleotide substrate (20–100 nM E, 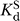 < 1 nM) and with saturating G (2 mM,
< 1 nM) and with saturating G (2 mM, 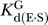 as reported in Table 2).
as reported in Table 2).
For cleavage of oligonucleotide substrates that are known to bind in the open complex to the WT ribozyme, e.g., −1r,dSA5 and SSp [36,66], we define k
ternary as the first-order rate constant for the reaction E·S·G/GN → products. We also used k
ternary to describe cleavage reactions catalyzed by variant ribozymes for which the oligonucleotide substrate binding mode has not been assigned definitively. Values of k
ternary were determined at pH 7.0, with ribozyme saturating with respect to oligonucleotide substrate (20–50 nM E, ribozyme concentration at least 4-fold above , 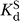 data not shown) and with saturating G or GN (2 mM,
data not shown) and with saturating G or GN (2 mM, 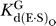 ∼ 500 μM;
∼ 500 μM; 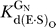 ∼ 100–150 μM) ([68], Tables 2 and 3, and data not shown).
∼ 100–150 μM) ([68], Tables 2 and 3, and data not shown).
(k
c/K
m)G and (k
c/K
m
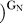 are the second-order rate constants for the reaction E·S + G (or GN) → products. In these experiments, we used the −1r,dSA5 oligonucleotide substrate that binds to the WT and variant ribozymes in the open complex [36,66]. Values of (k
c/K
m)G and (k
c/K
m
are the second-order rate constants for the reaction E·S + G (or GN) → products. In these experiments, we used the −1r,dSA5 oligonucleotide substrate that binds to the WT and variant ribozymes in the open complex [36,66]. Values of (k
c/K
m)G and (k
c/K
m
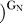 were determined at pH 7.0, with ribozyme saturating with respect to oligonucleotide substrate (20–50 nM E, ribozyme concentration at least 4-fold above ,
were determined at pH 7.0, with ribozyme saturating with respect to oligonucleotide substrate (20–50 nM E, ribozyme concentration at least 4-fold above , 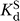 data not shown) and with subsaturating G or GN (30 μM G or GN,
data not shown) and with subsaturating G or GN (30 μM G or GN, 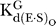 ∼ 500 μM;
∼ 500 μM; 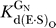 ∼ 100–150 μM) ([68], Table 3, and data not shown).
∼ 100–150 μM) ([68], Table 3, and data not shown).
(k
c/K
m)CUCG(3′X)A (where X = O or S) is the second-order rate constant for the reaction E·P + CUCGA → products. Values of (k
c/K
m)CUCG(3′X)A were determined at pH 6.5 with trace amounts of radiolabeled CUCG3′XA and E·P subsaturating with respect to CUCG3′XA (X = O, 10–20 nM E·P, 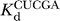 > 100 nM; X = S, 100–200 nM E·P,
> 100 nM; X = S, 100–200 nM E·P, 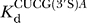 > 400 nM) (data not shown). To maintain the chemical step as rate-limiting in reactions catalyzed by the WT ribozyme, an oligonucleotide product with a 2′-deoxyribothymidine at the 3′-terminus was used (CCCUCdT); 2′-deoxyribose incorporation at this site slows the chemical step approximately 103-fold [74]. Reactions in the presence of the C262-S
P variant ribozyme used an all-ribose oligonucleotide product (CCCUCU).
> 400 nM) (data not shown). To maintain the chemical step as rate-limiting in reactions catalyzed by the WT ribozyme, an oligonucleotide product with a 2′-deoxyribothymidine at the 3′-terminus was used (CCCUCdT); 2′-deoxyribose incorporation at this site slows the chemical step approximately 103-fold [74]. Reactions in the presence of the C262-S
P variant ribozyme used an all-ribose oligonucleotide product (CCCUCU).
Association (k on) and dissociation (k off) rate constants were measured by a gel mobility shift assay using pulse-chase methods [33,56]. Specific conditions and experimental details for these experiments are described in Protocol S2.
Experimental errors
All titrations, binding constants, and rate determinations were repeated at least three times. Error bars, when present, indicate standard deviation from at least three determinations. Error bars may be obscured by data symbols. Plots without error bars display representative data. Reported errors in all tables, both in the text and Supporting Information, are the standard deviation of at least three independent measurements.
Data analysis
The Cd2+ concentration dependencies for rescue of reaction of modified substrates were analyzed according to previously described methods ([35,36] and Protocol S1).
Supporting Information
Stimulation of modified ribozyme substrates was assessed by comparing cleavage activity in 50 mM Mg2+ alone to observed activity in the presence of 1 mM Cd2+ and 50 mM Mg2+. The cleavage rate in the presence of Cd2+ was divided by the observed cleavage rate in Mg2+ alone, resulting in a relative rate (k
rel). These k
rel values were then normalized to the WT ribozyme; i.e.,
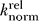 for the WT ribozyme equals one.
for the WT ribozyme equals one.
(A) Normalized stimulation of cleavage of an oligonucleotide substrate containing a 3′-thiophosphoryl linkage by 1 mM Cd2+; reactions performed as described in Figures 6 and S2 and Table 4.
(B) Normalized stimulation of cleavage of a 3′-splice site analogue containing a 3′-thiophosphoryl linkage by 1 mM Cd2+; reactions performed as described in Figures 6 and S3 and Table 4.
(C) Normalized stimulation of oligonucleotide substrate cleavage by subsaturating GN by 1 mM Cd2+; reactions performed as described in Figures 6 and S4 and Table 4.
(2.4 MB PDF).
(A) [Cd2+] dependencies for the reaction E·S·G → products (k ternary) for −(1–3)d,rSA5 (s○) and Sm3′S (•) with the WT ribozyme (see Materials and Methods).
(B) [Cd2+] dependencies of the rate of reaction E·S·G → products (k ternary) for −(1–3)d,rSA5 (□) and Sm3′S (▪) with the C262-S P ribozyme (see Materials and Methods). Error bars not seen are obscured by data symbols.
(2.8 MB PDF).
(A) [Cd2+] dependencies of the rate of the reaction E·P + CUCG3′XA → products ((k c/K m)CUCG(3′X)A) for CUCGA (s○) and CUCG3′SA (•) with the WT ribozyme (see Materials and Methods)
(B) [Cd2+] dependencies of the rate of the reaction E·P + CUCG3′XA → products ((k c/K m)CUCG(3′X)A) for CUCGA (□) and CUCG3′SA (▪) with the C262-S P ribozyme (see Materials and Methods). Error bars not seen are obscured by data symbols.
(3.1 MB PDF).
(A) [Cd2+] dependencies of the rate of the reaction E·S + G (or GN) → products ((k c/K m)G(or GN)) for G(s○) and GN(•) cleavage of −1r,dSA5 with the WT ribozyme (see Materials and Methods).
(B) [Cd2+] dependencies of the rate of the reaction E·S + G (or GN) → products ((k c/K m)G(or GN)) for G (□) and GN(▪) cleavage of −1r,dSA5 with the C262-S P ribozyme (see Materials and Methods).
(C) [Cd2+] dependencies of the rate of the reaction E·S·G/GN → products (k ternary) for G(s○) and GN(•) cleavage of −1r,dSA5 with the WT ribozyme (see Materials and Methods).
D) [Cd2+] dependencies of the rate of the reaction E·S·G/GN → products (k ternary) for G(□) and GN(▪) cleavage of −1r,dSA5 with the C262-S P ribozyme (see Materials and Methods).
(2.9 MB PDF).
(A) [Cd2+] dependencies of the rate of the reaction E·S·G → products [k ternary] for SSp (▪) and −(1–3)d,rSA5 (□) cleavage with G with the C262-S P ribozyme (see Materials and Methods).
(B) [Cd2+] dependencies of the rate of the reaction E·S·G/GN → products [k ternary] for SSp cleavage by GN (▪) and −(1–3)d,rSA5 cleavage by G (□) with the C262-S P ribozyme (see Materials and Methods).
(2.8 MB PDF).
(126 KB DOC).
(26 KB DOC).
(25 KB DOC).
(19 KB DOC).
Accession Numbers
The Protein Data Bank (http://www.rcsb.org/pdb/) accession numbers for the group I intron crystal structures discussed in this paper are Azoarcus (PDB ID 1U6B), Tetrahymena (PDB ID 1X8W), and Twort ribozyme (PDB ID 1Y0Q).
Acknowledgments
We thank members of the Piccirilli and Herschlag labs for helpful discussion and comments on the manuscript, J. Olvera for preparation of T4 DNA ligase, and K. Hougland for assistance with figures. We also thank B. Golden for discussion and sharing unpublished data. JLH was supported in part by the Predoctoral Training Program at the Interface of Chemistry and Biology (2 T32 GM008720–06) at the University of Chicago. This work was supported by NIH Grant GM49243 to DH and a grant from the Howard Hughes Medical Institute to JAP. JAP is an investigator at the Howard Hughes Medical Institute.
Abbreviations
- E
ribozyme
- G
guanosine
- GN
2′-aminoguanosine
- S
oligonucleotide substrate
- WT
wild-type
Conflicts of interest. The authors have declared that no conflicts of interest exist.
Author contributions. JLH, AVK, DH, and JAP conceived and designed the experiments. JLH performed the experiments and analyzed the data. JLH and AVK contributed reagents/materials/analysis tools. JLH, DH, and JAP wrote the paper.
Citation: Hougland JL, Kravchuk AV, Herschlag D, Piccirilli JA (2005) Functional identification of catalytic metal ion binding sites within RNA. PLoS Biol 3(9): e277.
Contributor Information
Daniel Herschlag, Email: herschla@cmgm.stanford.edu.
Joseph A Piccirilli, Email: jpicciri@uchicago.edu.
References
- Dismukes GC. Manganese enzymes with binuclear active sites. Chem Rev. 1996;96:2909–2926. doi: 10.1021/cr950053c. [DOI] [PubMed] [Google Scholar]
- Strater N, Lipscomb WN, Klabunde T, Krebs B. Two-metal ion catalysis in enzymatic acyl- and phosphoryl-transfer reactions. Ang Chem Int Ed. 1996;35:2024–2055. [Google Scholar]
- Cowan JA. Metal activation of enzymes in nucleic acid biochemistry. Chem Rev. 1998;98:1067–1087. doi: 10.1021/cr960436q. [DOI] [PubMed] [Google Scholar]
- Wilcox DE. Binuclear metallohydrolases. Chem Rev. 1996;96:2435–2458. doi: 10.1021/cr950043b. [DOI] [PubMed] [Google Scholar]
- Heikinheimo P, Lehtonen J, Baykov A, Lahti R, Cooperman BS. The structural basis for pyrophosphatase catalysis. Structure. 1996;4:1491–1508. doi: 10.1016/s0969-2126(96)00155-4. [DOI] [PubMed] [Google Scholar]
- Steitz TA, Steitz JA. A general two-metal-ion mechanism for catalytic RNA. Proc Natl Acad Sci U S A. 1993;90:6498–6502. doi: 10.1073/pnas.90.14.6498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galburt EA, Stoddard BL. Catalytic mechanisms of restriction and homing endonucleases. Biochemistry. 2002;41:13851–13860. doi: 10.1021/bi020467h. [DOI] [PubMed] [Google Scholar]
- Pingoud A, Jeltsch A. Structure and function of type II restriction endonucleases. Nucleic Acids Res. 2001;29:3705–3727. doi: 10.1093/nar/29.18.3705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stec B, Holtz KM, Kantrowitz ER. A revised mechanism for the alkaline phosphatase reaction involving three metal ions. J Mol Biol. 2000;299:1303–1311. doi: 10.1006/jmbi.2000.3799. [DOI] [PubMed] [Google Scholar]
- Kovall RA, Matthews BW. Type II restriction endonucleases: Structural, functional and evolutionary relationships. Curr Opin Chem Biol. 1999;3:578–583. doi: 10.1016/s1367-5931(99)00012-5. [DOI] [PubMed] [Google Scholar]
- Shi HJ, Moore PB. The crystal structure of yeast phenylalanine tRNA at 1.93 angstrom resolution: A classic structure revisited. RNA. 2000;6:1091–1105. doi: 10.1017/s1355838200000364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott WG, Murray JB, Arnold JRP, Stoddard BL, Klug A. Capturing the structure of a catalytic RNA intermediate: The hammerhead ribozyme. Science. 1996;274:2065–2069. doi: 10.1126/science.274.5295.2065. [DOI] [PubMed] [Google Scholar]
- Golden BL, Kim H, Chase E. Crystal structure of a phage Twort group I ribozyme-product complex. Nat Struct Mol Biol. 2005;12:82–89. doi: 10.1038/nsmb868. [DOI] [PubMed] [Google Scholar]
- Guo F, Gooding AR, Cech TR. Structure of the Tetrahymena ribozyme: Base triple sandwich and metal ion at the active site. Mol Cell. 2004;16:351–362. doi: 10.1016/j.molcel.2004.10.003. [DOI] [PubMed] [Google Scholar]
- Adams PL, Stahley MR, Kosek AB, Wang JM, Strobel SA. Crystal structure of a self-splicing group I intron with both exons. Nature. 2004;430:45–50. doi: 10.1038/nature02642. [DOI] [PubMed] [Google Scholar]
- Jaffe EK, Cohn M. Divalent cation-dependent stereospecificity of adenosine 5'-O-(2-thiotriphosphate) in hexokinase and pyruvate-kinase reactions-Absolute stereochemistry of diastereoisomers of adenosine 5'-O-(2-thiotriphosphate) J Biol Chem. 1978;253:4823–4825. [PubMed] [Google Scholar]
- Christian EL, Yarus M. Metal coordination sites that contribute to structure and catalysis in the group I intron from Tetrahymena . Biochemistry. 1993;32:4475–4480. doi: 10.1021/bi00068a001. [DOI] [PubMed] [Google Scholar]
- Yoshida A, Sun SG, Piccirilli JA. A new metal ion interaction in the Tetrahymena ribozyme reaction revealed by double sulfur substitution. Nat Struct Biol. 1999;6:318–321. doi: 10.1038/7551. [DOI] [PubMed] [Google Scholar]
- Sjogren AS, Pettersson E, Sjoberg BM, Stromberg R. Metal ion interaction with cosubstrate in self-splicing of group I introns. Nucleic Acids Res. 1997;25:648–653. doi: 10.1093/nar/25.3.648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstein LB, Jones B, Cosstick R, Cech TR. A second catalytic metal ion in a group I ribozyme. Nature. 1997;388:805–808. doi: 10.1038/42076. [DOI] [PubMed] [Google Scholar]
- Piccirilli JA, Vyle JS, Caruthers MH, Cech TR. Metal-ion catalysis in the Tetrahymena ribozyme reaction. Nature. 1993;361:85–88. doi: 10.1038/361085a0. [DOI] [PubMed] [Google Scholar]
- Gordon PM, Sontheimer EJ, Piccirilli JA. Kinetic characterization of the second step of group II intron splicing: Role of metal ions and the cleavage site 2'-OH in catalysis. Biochemistry. 2000;39:12939–12952. doi: 10.1021/bi001089o. [DOI] [PubMed] [Google Scholar]
- Sontheimer EJ, Gordon PM, Piccirilli JA. Metal ion catalysis during group II intron self-splicing: Parallels with the spliceosome. Gen Dev. 1999;13:1729–1741. doi: 10.1101/gad.13.13.1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warnecke JM, Furste JP, Hardt WD, Erdmann VA, Hartmann RK. Ribonuclease P (RNase P) RNA is converted to a Cd(2+)ribozyme by a single Rp-phosphorothioate modification in the precursor tRNA at the RNase P cleavage site. Proc Natl Acad Sci U S A. 1996;93:8924–8928. doi: 10.1073/pnas.93.17.8924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott EC, Uhlenbeck OC. A re-investigation of the thio effect at the hammerhead cleavage site. Nucleic Acids Res. 1999;27:479–484. doi: 10.1093/nar/27.2.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peracchi A, Beigelman L, Scott EC, Uhlenbeck OC, Herschlag D. Involvement of a specific metal ion in the transition of the hammerhead ribozyme to its catalytic conformation. J Biol Chem. 1997;272:26822–26826. doi: 10.1074/jbc.272.43.26822. [DOI] [PubMed] [Google Scholar]
- Sontheimer EJ, Sun SG, Piccirilli JA. Metal ion catalysis during splicing of premessenger RNA. Nature. 1997;388:801–805. doi: 10.1038/42068. [DOI] [PubMed] [Google Scholar]
- Gordon PM, Sontheimer EJ, Piccirilli JA. Metal ion catalysis during the exon-ligation step of nuclear pre-mRNA splicing: Extending the parallels between the spliceosome and group II introns. RNA. 2000;6:199–205. doi: 10.1017/s1355838200992069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohn M. Some properties of the phosphorothioate analogs of adenosinetriphosphate as substrates of enzymic reactions. Acc Chem Res. 1982;15:326–332. [Google Scholar]
- Eckstein F. Nucleoside phosphorothioates. Ann Rev Biochem. 1985;54:367–402. doi: 10.1146/annurev.bi.54.070185.002055. [DOI] [PubMed] [Google Scholar]
- Curley JF, Joyce CM, Piccirilli JA. Functional evidence that the 3'-5′ exonuclease domain of Escherichia coli DNA polymerase I employs a divalent metal ion in leaving group stabilization. J Am Chem Soc. 1997;119:12691–12692. [Google Scholar]
- Aubert SD, Li YC, Raushel FM. Mechanism for the hydrolysis of organophosphates by the bacterial phosphotriesterase. Biochemistry. 2004;43:5707–5715. doi: 10.1021/bi0497805. [DOI] [PubMed] [Google Scholar]
- Herschlag D, Cech TR. Catalysis of RNA cleavage by the Tetrahymena thermophila ribozyme. 1. Kinetic description of the reaction of an RNA substrate complementary to the active site. Biochemistry. 1990;29:10159–10171. doi: 10.1021/bi00496a003. [DOI] [PubMed] [Google Scholar]
- Zaug AJ, Grosshans CA, Cech TR. Sequence-specific endoribonuclease activity of the Tetrahymena ribozyme—Enhanced cleavage of certain oligonucleotide substrates that form mismatched ribozyme substrate complexes. Biochemistry. 1988;27:8924–8931. doi: 10.1021/bi00425a008. [DOI] [PubMed] [Google Scholar]
- Shan S, Yoshida A, Sun SG, Piccirilli JA, Herschlag D. Three metal ions at the active site of the Tetrahymena group I ribozyme. Proc Natl Acad Sci U S A. 1999;96:12299–12304. doi: 10.1073/pnas.96.22.12299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan S, Kravchuk AV, Piccirilli JA, Herschlag D. Defining the catalytic metal ion interactions in the Tetrahymena ribozyme reaction. Biochemistry. 2001;40:5161–5171. doi: 10.1021/bi002887h. [DOI] [PubMed] [Google Scholar]
- Basu S, Strobel SA. Thiophilic metal ion rescue of phosphorothioate interference within the Tetrahymena ribozyme P4-P6 domain. RNA. 1999;5:1399–1407. doi: 10.1017/s135583829999115x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudvillain M, Pyle AM. Defining functional groups, core structural features and inter-domain tertiary contacts essential for group II intron selfsplicing: A NAIM analysis. EMBO J. 1998;17:7091–7104. doi: 10.1093/emboj/17.23.7091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cate JH, Hanna RL, Doudna JA. A magnesium ion core at the heart of a ribozyme domain. Nat Struct Biol. 1997;4:553–558. doi: 10.1038/nsb0797-553. [DOI] [PubMed] [Google Scholar]
- Christian EL, Yarus M. Analysis of the role of phosphate oxygens in the group-I intron from Tetrahymena . J Mol Biol. 1992;228:743–758. doi: 10.1016/0022-2836(92)90861-d. [DOI] [PubMed] [Google Scholar]
- Crary SM, Kurz JC, Fierke CA. Specific phosphorothioate substitutions probe the active site of Bacillus subtilis ribonuclease P. RNA. 2002;8:933–947. doi: 10.1017/s1355838202025025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon PM, Piccirilli JA. Metal ion coordination by the AGC triad in domain 5 contributes to group II intron catalysis. Nature Struct Biol. 2001;8:893–898. doi: 10.1038/nsb1001-893. [DOI] [PubMed] [Google Scholar]
- Harris ME, Pace NR. Identification of phosphates involved in catalysis by the ribozyme RNase-P RNA. RNA. 1995;1:210–218. [PMC free article] [PubMed] [Google Scholar]
- Christian EL, Kaye NM, Harris ME. Helix P4 is a divalent metal ion binding site in the conserved core of the ribonuclease P ribozyme. RNA. 2000;6:511–519. doi: 10.1017/s1355838200000042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christian EL, Kaye NM, Harris ME. Evidence for a polynuclear metal ion binding site in the catalytic domain of ribonuclease P RNA. EMBO. 2002;21:2253–2262. doi: 10.1093/emboj/21.9.2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones FD, Strobel SA. Ionization of a critical adenosine residue in the Neurospora Varkud satellite ribozyme active site. Biochemistry. 2003;42:4265–4276. doi: 10.1021/bi020707t. [DOI] [PubMed] [Google Scholar]
- Ortoleva-Donnelly L, Szewczak AA, Gutell RR, Strobel SA. The chemical basis of adenosine conservation throughout the Tetrahymena ribozyme. RNA. 1998;4:498–519. doi: 10.1017/s1355838298980086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oyelere AK, Kardon JR, Strobel SA. pKa perturbation in genomic hepatitis delta virus ribozyme catalysis evidenced by nucleotide analogue interference mapping. Biochemistry. 2002;41:3667–3675. doi: 10.1021/bi011816v. [DOI] [PubMed] [Google Scholar]
- Ryder SP, Oyelere AK, Padilla JL, Klostermeier D, Millar DP. Investigation of adenosine base ionization in the hairpin ribozyme by nucleotide analog interference mapping. RNA. 2001;7:1454–1463. [PMC free article] [PubMed] [Google Scholar]
- Ryder SP, Strobel SA. Nucleotide analog interference mapping of the hairpin ribozyme: Implications for secondary and tertiary structure formation. J Mol Biol. 1999;291:295–311. doi: 10.1006/jmbi.1999.2959. [DOI] [PubMed] [Google Scholar]
- Yean SL, Wuenschell G, Termini J, Lin RJ. Metal-ion coordination by U6 small nuclear RNA contributes to catalysis in the spliceosome. Nature. 2000;408:881–884. doi: 10.1038/35048617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strauss-Soukup JK, Strobel SA. A chemical phylogeny of group I introns based upon interference mapping of a bacterial ribozyme. J Mol Biol. 2000;302:339–358. doi: 10.1006/jmbi.2000.4056. [DOI] [PubMed] [Google Scholar]
- Szewczak AA, Kosek AB, Piccirilli JA, Strobel SA. Identification of an active site ligand for a group I ribozyme catalytic metal ion. Biochemistry. 2002;41:2516–2525. doi: 10.1021/bi011973u. [DOI] [PubMed] [Google Scholar]
- Wang SL, Karbstein K, Peracchi A, Beigelman L, Herschlag D. Identification of the hammerhead ribozyme metal ion binding site responsible for rescue of the deleterious effect of a cleavage site phosphorothioate. Biochemistry. 1999;38:14363–14378. doi: 10.1021/bi9913202. [DOI] [PubMed] [Google Scholar]
- Michel F, Westhof E. Modeling of the 3-dimensional architecture of group-I catalytic introns based on comparative sequence-analysis. J Mol Biol. 1990;216:585–610. doi: 10.1016/0022-2836(90)90386-Z. [DOI] [PubMed] [Google Scholar]
- Karbstein K, Carroll KS, Herschlag D. Probing the Tetrahymena group I ribozyme reaction in both directions. Biochemistry. 2002;41:11171–11183. doi: 10.1021/bi0202631. [DOI] [PubMed] [Google Scholar]
- Narlikar GJ, Khosla M, Usman N, Herschlag D. Quantitating tertiary binding energies of 2'OH groups on the P1 duplex of the Tetrahymena ribozyme: Intrinsic binding energy in an RNA enzyme. Biochemistry. 1997;36:2465–2477. doi: 10.1021/bi9610820. [DOI] [PubMed] [Google Scholar]
- Knitt DS, Narlikar GJ, Herschlag D. Dissection of the role of the conserved G*U pair in group I RNA self-splicing. Biochemistry. 1994;33:13864–13879. doi: 10.1021/bi00250a041. [DOI] [PubMed] [Google Scholar]
- Bevilacqua PC, Kierzek R, Johnson KA, Turner DH. Dynamics of ribozyme binding of substrate revealed by fluorescence-detected stopped-flow methods. Science. 1992;258:1355–1357. doi: 10.1126/science.1455230. [DOI] [PubMed] [Google Scholar]
- Venkataraman D, Du YH, Wilson SR, Hirsch KA, Zhang P. A coordination geometry table of the d-block elements and their ions. J Chem Educ. 1997;74:915–918. [Google Scholar]
- Shriver DF, Atkins P, Langford CH. Inorganic chemistry. New York: W.H. Freeman and Co; 1994. 913 pp. [Google Scholar]
- Feig AL, Uhlenbeck OC. The role of metal ions in RNA biochemistry. In: Gesteland RF, Cech TR, Atkins JF, editors. The RNA world. 2nd ed. Cold Spring Harbor (New York): Cold Spring Harbor Press; 1998. pp. 287–320. [Google Scholar]
- Herschlag D, Khosla M. Comparison of pH dependencies of the Tetrahymena ribozyme reactions with RNA 2'-substituted and phosphorothioate substrates reveals a rate-limiting conformational step. Biochemistry. 1994;33:5291–5297. doi: 10.1021/bi00183a036. [DOI] [PubMed] [Google Scholar]
- Herschlag D, Piccirilli JA, Cech TR. Ribozyme-catalyzed and nonenzymatic reactions of phosphate diesters—Rate effects upon substitution of sulfur for a nonbridging phosphoryl oxygen atom. Biochemistry. 1991;30:4844–4854. doi: 10.1021/bi00234a003. [DOI] [PubMed] [Google Scholar]
- Knitt DS, Herschlag D. pH dependencies of the Tetrahymena ribozyme reveal an unconventional origin of an apparent pK(a) Biochemistry. 1996;35:1560–1570. doi: 10.1021/bi9521147. [DOI] [PubMed] [Google Scholar]
- Shan SO, Herschlag D. An unconventional origin of metal-ion rescue and inhibition in the Tetrahymena group I ribozyme reaction. RNA. 2000;6:795–813. doi: 10.1017/s1355838200000649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McConnell TS, Cech TR, Herschlag D. Guanosine binding to the Tetrahymena ribozyme—Thermodynamic coupling with oligonucleotide binding. Proc Natl Acad Sci U S A. 1993;90:8362–8366. doi: 10.1073/pnas.90.18.8362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan SO, Herschlag D. Probing the role of metal ions in RNA catalysis: Kinetic and thermodynamic characterization of a metal ion interaction with the 2'-moiety of the guanosine nucleophile in the Tetrahymena group I ribozyme. Biochemistry. 1999;38:10958–10975. doi: 10.1021/bi990388e. [DOI] [PubMed] [Google Scholar]
- Profenno LA, Kierzek R, Testa SM, Turner DH. Guanosine binds to the Tetrahymena ribozyme in more than one step, and its 2'-OH and the nonbridging pro-Sp phosphoryl oxygen at the cleavage site are required for productive docking. Biochemistry. 1997;36:12477–12485. doi: 10.1021/bi9708895. [DOI] [PubMed] [Google Scholar]
- Martell AE, Smith RM. Critical stability constants. New York: Plenum Press; 1976. [Google Scholar]
- Yoshida A, Shan S, Herschlag D, Piccirilli JA. The role of the cleavage site 2'-hydroxyl in the Tetrahymena group I ribozyme reaction. Chem Biol. 2000;7:85–96. doi: 10.1016/s1074-5521(00)00074-0. [DOI] [PubMed] [Google Scholar]
- Strobel SA, Ortoleva-Donnelly L. A hydrogen-bonding triad stabilizes the chemical transition state of a group I ribozyme. Chem Biol. 1999;6:153–165. doi: 10.1016/S1074-5521(99)89007-3. [DOI] [PubMed] [Google Scholar]
- Hougland JL, Piccirilli JA, Forconi M, Lee J, Herschlag D. How the group I intron works: A case study of RNA structure and function. In: Gesteland RF, Atkins JF, Cech TR, editors. The RNA world, 3rd edition. Cold Spring Harbor (New York): Cold Spring Harbor Press; 2005. [Google Scholar]
- Herschlag D, Eckstein F, Cech TR. The importance of being ribose at the cleavage site in the Tetrahymena ribozyme reaction. Biochemistry. 1993;32:8312–8321. doi: 10.1021/bi00083a035. [DOI] [PubMed] [Google Scholar]
- Sun SG, Yoshida A, Piccirilli JA. Synthesis of 3'-thioribonucleosides and their incorporation into oligoribonucleotides via phosphoramidite chemistry. RNA. 1997;3:1352–1363. [PMC free article] [PubMed] [Google Scholar]
- Slim G, Gait MJ. Configurationally defined phosphorothioate-containing oligoribonucleotides in the study of the mechanism of cleavage of hammerhead ribozymes. Nucleic Acids Res. 1991;19:1183–1188. doi: 10.1093/nar/19.6.1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore MJ, Sharp PA. Site-specific modification of pre-messenger-RNA—The 2'-hydroxyl groups at the splice sites. Science. 1992;256:992–997. doi: 10.1126/science.1589782. [DOI] [PubMed] [Google Scholar]
- Lindqvist M, Sandstrom K, Liepins V, Stromberg R, Graslund A. Specific metal-ion binding P4-P6 triple-helical domain sites in a model of a group I intron. RNA. 2001;7:1115–1125. doi: 10.1017/s1355838201002576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narlikar GJ, Bartley LE, Khosla M, Herschlag D. Characterization of a local folding event of the Tetrahymena group I ribozyme: Effects of oligonucleotide substrate length pH, and temperature on the two substrate binding steps. Biochemistry. 1999;38:14192–14204. doi: 10.1021/bi9914309. [DOI] [PubMed] [Google Scholar]
- Karbstein K, Herschlag D. Extraordinarily slow binding of guanosine to the Tetrahymena group I ribozyme: Implications for RNA preorganization and function. Proc Natl Acad Sci U S A. 2003;100:2300–2305. doi: 10.1073/pnas.252749799. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Stimulation of modified ribozyme substrates was assessed by comparing cleavage activity in 50 mM Mg2+ alone to observed activity in the presence of 1 mM Cd2+ and 50 mM Mg2+. The cleavage rate in the presence of Cd2+ was divided by the observed cleavage rate in Mg2+ alone, resulting in a relative rate (k
rel). These k
rel values were then normalized to the WT ribozyme; i.e.,
for the WT ribozyme equals one.
(A) Normalized stimulation of cleavage of an oligonucleotide substrate containing a 3′-thiophosphoryl linkage by 1 mM Cd2+; reactions performed as described in Figures 6 and S2 and Table 4.
(B) Normalized stimulation of cleavage of a 3′-splice site analogue containing a 3′-thiophosphoryl linkage by 1 mM Cd2+; reactions performed as described in Figures 6 and S3 and Table 4.
(C) Normalized stimulation of oligonucleotide substrate cleavage by subsaturating GN by 1 mM Cd2+; reactions performed as described in Figures 6 and S4 and Table 4.
(2.4 MB PDF).
(A) [Cd2+] dependencies for the reaction E·S·G → products (k ternary) for −(1–3)d,rSA5 (s○) and Sm3′S (•) with the WT ribozyme (see Materials and Methods).
(B) [Cd2+] dependencies of the rate of reaction E·S·G → products (k ternary) for −(1–3)d,rSA5 (□) and Sm3′S (▪) with the C262-S P ribozyme (see Materials and Methods). Error bars not seen are obscured by data symbols.
(2.8 MB PDF).
(A) [Cd2+] dependencies of the rate of the reaction E·P + CUCG3′XA → products ((k c/K m)CUCG(3′X)A) for CUCGA (s○) and CUCG3′SA (•) with the WT ribozyme (see Materials and Methods)
(B) [Cd2+] dependencies of the rate of the reaction E·P + CUCG3′XA → products ((k c/K m)CUCG(3′X)A) for CUCGA (□) and CUCG3′SA (▪) with the C262-S P ribozyme (see Materials and Methods). Error bars not seen are obscured by data symbols.
(3.1 MB PDF).
(A) [Cd2+] dependencies of the rate of the reaction E·S + G (or GN) → products ((k c/K m)G(or GN)) for G(s○) and GN(•) cleavage of −1r,dSA5 with the WT ribozyme (see Materials and Methods).
(B) [Cd2+] dependencies of the rate of the reaction E·S + G (or GN) → products ((k c/K m)G(or GN)) for G (□) and GN(▪) cleavage of −1r,dSA5 with the C262-S P ribozyme (see Materials and Methods).
(C) [Cd2+] dependencies of the rate of the reaction E·S·G/GN → products (k ternary) for G(s○) and GN(•) cleavage of −1r,dSA5 with the WT ribozyme (see Materials and Methods).
D) [Cd2+] dependencies of the rate of the reaction E·S·G/GN → products (k ternary) for G(□) and GN(▪) cleavage of −1r,dSA5 with the C262-S P ribozyme (see Materials and Methods).
(2.9 MB PDF).
(A) [Cd2+] dependencies of the rate of the reaction E·S·G → products [k ternary] for SSp (▪) and −(1–3)d,rSA5 (□) cleavage with G with the C262-S P ribozyme (see Materials and Methods).
(B) [Cd2+] dependencies of the rate of the reaction E·S·G/GN → products [k ternary] for SSp cleavage by GN (▪) and −(1–3)d,rSA5 cleavage by G (□) with the C262-S P ribozyme (see Materials and Methods).
(2.8 MB PDF).
(126 KB DOC).
(26 KB DOC).
(25 KB DOC).
(19 KB DOC).