

Abstract
The most common neuropathy associated with diabetes mellitus is a distal sensory polyneuropathy. The relative importance of the direct effects of prolonged glycaemia on nervous tissue compared with indirect damage resulting from changes in blood vessels is not known. Although the importance of glycaemia is confirmed by a study showing that the incidence of neuropathy is greatly reduced by strict glycaemic control, many of the details of the deleterious effects of glycaemia on the peripheral nervous system (PNS) are not understood. These may be the result of direct damage to any of the cells in the PNS or the disruption of neuronal metabolism, axonal transport mechanisms, or repair capabilities; in addition, they may result from the effects of glycation on PNS connective tissue or a combination of some or all of the above mentioned mechanisms. The relative importance of these various mechanisms by which diabetes damages the PNS is a matter of conjecture. Therapeutic approaches targeting a specific mechanism such as those utilising aldose reductase inhibitors, or advanced glycation endproduct inhibitors have met with limited success. Clearly, it is difficult to design a treatment for diabetic neuropathy while its pathogenesis is still poorly understood.
Keywords: diabetes mellitus, glycation, peripheral nerve
Abnormalities of glucose metabolism in diabetes mellitus lead to an increased risk of atherosclerosis and neurological and microvascular complications. Distal sensory symmetric polyneuropathy (DPN) is the most common of the various peripheral nerve disorders associated with diabetes. This syndrome affects the extremities in a “glove and stocking” distribution and is usually more severe in the legs. The symptoms are rather variable and can be either motor, sensory, or autonomic, or a combination of any of these. Similarly, the nerve fibres involved may be of a specific calibre or all fibres may be affected equally. The pathogenesis is currently uncertain, but the hypothesis that chronic hyperglycaemia plays an important role is supported by the findings of the diabetes control and complications trial (DCCT) research group, 1993, which found that strict glycaemic control reduces the incidence of neuropathy.1 A more recent trial emphasises a multifactorial basis for the neuropathy in type II diabetes.2 Changes in the vasculature in particular may play an important part in producing nerve damage.
There are various possible mechanisms by which glycaemia could have an adverse effect on the peripheral nervous system (PNS) and it is difficult to disentangle the importance of the different insults. Not only do the nerve fibres degenerate, but attempts at regeneration by the damaged fibres, although vigorous, are short lived, and the numerous regenerative sprouts produced (fig 1 ▶) fail to survive.3 Therefore, the neuropathy becomes progressively worse. This worsening occurs in a dying back pattern (distal–proximal direction) that is characteristic of failure in fast axonal transport. It is unlikely to result from a failure of Schwann cells to support their axons because this would be expected to affect nerve fibres equally along their length. Among the various alternative causes of the failure of the sprouting axons to persist and mature are metabolic failure of the neurone, ischaemic effects caused by vascular abnormalities, or deleterious effects of glycation on the Schwann cells or extracellular matrix. There is also the additional possibility that abnormally glycated collagen in the endoneurium of the nerve trunks might act as a physical barrier to elongation of the axonal sprouts. Glycated collagen is less susceptible to the protease digestion that is a necessary adjunct to axonal elongation and hence may not be removed to allow new axons to penetrate the extracellular matrix. Glycation of the Schwann cell basal laminal components may also have the effect of reducing the ability of the new axons to recognise and/or adhere to the original Schwann cell basal lamina, which remains as a tube when the myelinated fibre that it had initially surrounded degenerates. If undamaged, this basal laminal tube connects the damaged region and the end organ and can therefore act as a guide for regenerating axon sprouts. However, in diabetes, even if the new axons reach an appropriate end organ, reconnection may be hindered by abnormalities in the axonal growth cones as a result of glycation of the cytoskeleton, plasma membrane, or extracellular matrix.
Figure 1.

Transverse section of a radial nerve from a 42 year old woman with distal sensory symmetric polyneuropathy. There are circular clusters of regenerative sprouts (arrows) and the endoneurial microvessels are encircled by thickened basal lamina. Resin section, stained with thionin and acridine orange. Original magnification, ×200.
Non-enzymatic glycosylation of any tissue involves the covalent linkage of glucose, primarily to lysine residues, producing a Schiff-base intermediate. This then undergoes an Amadori rearrangement to a stable ketoamine derivative that is then further rearranged to a hemiketal structure.4 The net result is the formation of insoluble and irreversible advanced glycation endproducts (AGEs). The AGE pentosidine is formed by glucose auto-oxidation,5 and Nɛ-(carboxymethyl)lysine (CML) is an AGE formed by both glucose auto-oxidation and lipid peroxidation.6 Pentosidine is relatively easy to quantify and is frequently used as a measure of total AGE products in experimental studies of the effects of glycation on the PNS.
It has been shown that increased intracellular AGE formation occurs in cytoskeletal and myelin proteins in nerve specimens from patients with diabetes.7 Some earlier experiments on retinal blood vessels seem to have underestimated the degree to which AGE formation occurs by measuring only fluorescent AGEs.8 This may be misleading because later work on the lens and renal cortex found that non-fluorescent AGEs predominate and that AGE formation increases disproportionately with the degree of the increase in blood glucose concentrations.9 AGE formation is considerably faster with intracellular sugars such as fructose than with glucose. Despite the slow rate of glycation by glucose, after only one week in high glucose medium the AGE content of endothelial cell cultures increased by 13.8%. In this experiment there was also a 70% reduction in mitogenic activity, which was thought to be caused by the 6.1 fold increase in AGE formation on basic fibroblast growth factor.10 A possible explanation for the considerable rise in intracellular AGEs is that hyperglycation induces the formation of glycolytic intermediates, which are much more reactive than glucose.
There are several possible pathways by which AGE formation could be involved in the development of diabetic complications.11 First, both intracellular and extracellular AGE may be directly pathogenetic. Second, AGE induced alterations of DNA and nuclear proteins may also occur.12 Third, extracellular AGEs may interfere with cellular adhesion and interaction, and intracellular AGEs may alter protein transport and function. These indirect effects provide a mechanism by which diabetes may damage cells such as microvascular endothelial cells and neurones that do not require insulin for glucose transport. In addition, the increased transport of glycated serum albumin across the blood–nerve barrier may induce deleterious osmotic changes in the endoneurium.13 Preferential transport of glycated immunoglobulins may be the explanation for the considerable increase in trapped IgG and IgM in diabetic peripheral nerves both in the perineurium14 and on the myelin sheaths.15 It seems reasonable to postulate that trapping of IgM on myelin may contribute to peripheral nerve damage. Although demyelination is not the major pathological change seen in most diabetic nerve biopsies this could be because of supervening axonal damage.
Experiments to demonstrate IgM trapping have not been performed on streptozotocin (STZ) induced diabetic rat nerves, but it has been shown that there is non-enzymatic glycosylation of both peripheral and central nervous system (CNS) myelin.16 The pathogenic importance of this is unclear because the CNS is less affected by diabetes.
Effects of diabetes on dorsal root ganglion neurones
Diabetic polyneuropathy is often predominantly sensory but there is little evidence to suggest the loss of dorsal root ganglion cells. Few studies have attempted to measure this, but an early study by Dolman (1963) found no significant losses,17 and a more recent study on a single patient reported only mild dorsal root ganglion neurone loss.18 Some experimental studies have shown a reduced size of the dorsal root ganglion neurones,19 and it is possible that some aspects of their function may be reduced.18 Further support for the dying back nature of the axonopathy suggested by Said and colleagues20 has been provided by studies of myelinated fibre density at different levels in the PNS. These showed an increased loss in the most distal parts of the nerves.21 High glucose concentrations could damage sensory neurones preferentially because of their location in the dorsal root ganglia, where the blood–nerve barrier is less complete. This is the result of fenestration of a proportion of the blood vessels within the capsule, making it easier for proteins to leak out of the blood vessels into the endoneurium22; fenestrations are very rare in the endoneurial microvessels of the peripheral nerve trunks.23 Work on congenitally diabetic Bio-Breeding (BB) Wistar rats with a selective sensory neuropathy found a reduction in blood flow in the dorsal root ganglion but not in the sciatic nerve.24 In the spinal cord and brain where motor neurones are situated, the blood–brain barrier could be expected to protect them against high circulating glucose concentrations.
Recent experimental work suggests that the PNS cytoskeleton is more vulnerable to non-enzymatic glycosylation than the CNS cytoskeleton.25 This difference could also contribute to preferential damage of sensory dorsal root ganglion neurones compared with motor neurones. Additional support for the importance of AGEs in the development of PNS damage is provided by experiments on STZ diabetic rats using AGE inhibitors, such as aminoguanidine. Sensory and motor nerve conduction velocities are reduced in this animal model of diabetes and are improved by treatment with aminoguanidine.26
Some tissue culture studies also support the hypothesis that glycation has a direct effect on the neurone. Recent work by N Yagihashi et al (personal communication, 2000) found that the injection of AGEs into rat nerves produced similar neuropathic changes to those found in STZ diabetic rats. Other experiments on growing dorsal root ganglion neurones from STZ induced diabetic rats in vitro show a reduction in survival and growth compared with normal neurones,26a but this could be the result of some effect of diabetes other than glycation.
Axonal dysfunction in diabetes
Disruption of neural function by AGE formation may affect the cytoskeleton directly and may also involve intracellular messengers and protein phosphorylation. Ryle and Donaghy7 detected increased concentrations of pentosidine in both myelin and cytoskeletal fractions from human diabetic nerves, but there were no changes in the concentration of the early soluble glycation adduct furosine. AGEs cause protein crosslinking, resulting in the formation of insoluble aggregates.27 In vivo it seems that the most important pathway leading to the formation of AGE products is via the Amadori product. Amadori glycation products have been demonstrated in the spinal cord of patients with amyotrophic lateral sclerosis and spinobulbar muscular atrophy, and may be related to glycation of cytoskeletal proteins.28 Non-enzymatic glycosylation of intracellular proteins, particularly tubulin29 and actin,30 occurs readily. This inhibits GTP dependent polymerisation of tubulin and produces aggregates resistant to disruption by detergents or reducing agents. The mechanism for fast axonal transport (200–400 mm/day) of vesicles and mitochondria along the axon uses microtubule associated proteins and a kinesin motor to drive them along microtubules aligned parallel to the long axis of the axon. A similar process using a dynein motor provides retrograde axonal transport of effete proteins for recycling in the perikaryon. The process at the distal end of the axon, where proteins are packaged for return to the cell body, is known as turnaround. A very small change in fast axonal transport could disrupt turnaround, despite having little effect on transport times.31 Glycation seems to affect a subset of proteins differentially; in STZ induced diabetic rats, leucine transport was affected by diabetes but glucosamine was unaltered.32 Similar changes in axonal transport were found in galactosaemic rats, suggesting that glucose or its derivatives are important in the development of diabetic neuropathy.33
In support of the importance of changes in the axonal cytoskeleton in human diabetic neuropathy, experimental work on diabetic rats has shown a relatively small reduction in the rate of fast axonal transport34,35 and a greater reduction in retrograde transport.36 Changes found in the dorsal root ganglion in the expression of nerve growth factor (NGF)37 and insulin-like growth factor (IGF)38 could be explained by impaired axonal transport, particularly the retrograde flow of neurotrophins.39 Growth factor abnormalities could be implicated both in the development of diabetic neuropathy40 and also in the impairment of axonal regeneration. The relative importance of the glycation of cytoskeletal proteins and metabolic changes in the neurone is unknown.
Although the animal models of diabetic neuropathy show very few morphological changes and do not replicate the extensive degeneration often seen in human diabetic polyneuropathy, it has been confirmed that amino acids, mainly lysine, in diabetic rat nerves show almost a threefold increase in non-enzymatic glycosylation.41 Axonal regeneration is reduced in both STZ induced diabetic and galactosaemic rats.42,43
A protein that may be particularly important in the development of diabetic neuropathy is the small protein known as growth associated protein 43 (GAP-43). GAP-43 is normally only important in development but is upregulated in regeneration.
In vitro GAP-43 binds calmodulin only at low calcium ion concentrations and dissociates when concentrations are high. This calcium dependant property is eliminated by phosphorylation by a protein kinase. Biologically, the function of GAP-43 may be to localise calmodulin to specific sites on the cell membrane under resting conditions. When the neurone is stimulated, a rise in calcium ions releases calmodulin, which is then available as an activator for calmodulin dependent processes in the presynaptic region. Simultaneously, GAP-43 is available as a substrate for calcium/phospholipid dependent protein kinase and hence cannot reassociate with calmodulin.44,45 GAP-43 can then be dephosphorylated by the action of calcineurin, which abolishes the calcium signal.46 One could speculate that if this process were disrupted in diabetes, this could result in a dying back neuropathy and also produce a deleterious effect on axonal growth cones. Growth cones are the growing tips of regenerating axons so that abnormalities in these structures could inhibit regenerative success. In vitro experiments have strengthened this theory by confirming that the depletion of GAP-43 leads to growth cone abnormalities.47
GAP-43 is manufactured in the cell body and transported by fast axonal transport in vesicles.48 Calmodulin is transported separately in slow component b (∼ 2 mm/day),49 the same mechanism by which components of the cytoskeleton are moved. Concentrations of GAP-43 in the dorsal root ganglion in normal animals are increased after peripheral nerve injury, but not after dorsal root injury,50 suggesting that the signal for upregulation is derived from the periphery. In addition, ligature plus crush experiments in STZ diabetic rats have shown a reduction of immunostaining for GAP-43 proximal to the obstruction; the amounts of mRNA in the cell bodies were similar to those found in normal animals.51 If this is the result of the effect of diabetes on transport or turnaround, it must be specific to GAP-43, because concentrations of vasoactive intestinal polypeptide, which is carried by the same system, were not reduced. Slow axonal transport is also altered in diabetes, so this could affect the supply of calmodulin to the presynaptic region and possibly also disrupt the GAP-43 related mechanisms.
Glycation of the extracellular matrix
The extracellular matrix (ECM) within the nerve trunk comprises mainly fibrous collagens I and III, arranged predominantly longitudinally, parallel to the nerve fibres, plus smaller quantities of other connective tissue proteins, and the basal laminal sheaths around Schwann cells, perineurial cells, and blood vessel endothelial cells.
It is difficult to separate the effects of glycation on the cytoskeleton from glycation of the extracellular environment because they are interrelated. If connections between the axon and its end organs were damaged by glycation, this could produce alterations in transport, and thus the observed growth factor changes. Direct observations on nerve biopsies from patients with diabetic neuropathy suggest that the changes in the endoneurial microenvironment produce morphological alterations that may be very important. A chain of Schwann cells in a cylindrical basal laminal tube surrounds each myelinated axon. When an axon degenerates, the Schwann cells multiply and form columns of cells within this tube (bands of Büngner).52–54 Regenerating sprouts are produced by the intact part of the axon and, if the basal lamina is not disrupted, they will track along this column of Schwann cells inside the basal laminal tube to reach the original end organ of the axon. In non-diabetic nerves, the original basal laminal tube later breaks down55 and is rarely seen by the time the regenerating sprouts have become myelinated.54 In a high proportion of diabetic nerves there are numerous mature regenerative sprouts with well developed myelin sheaths within a prominent and often circular persisting basal laminal tube (fig 2 ▶). It has been suggested that the abnormal persistence and circularity of this tube is the result of glycation of its components.56
Figure 2.

Electron microscopy of a regenerative cluster shows the persistent basal laminal tube (arrow) around myelinated and unmyelinated axonal sprouts (asterisks). Radial nerve, 47 year old man with distal sensory symmetric polyneuropathy. Contrasted with lead and uranyl acetate. Bar, 1 μm.
Several reports describe the deleterious effects of glycation of the ECM on the growth of a variety of cells, including endothelial,57 mesangial,58 and human glomerular epithelial cells.59 The most relevant to diabetic neuropathy are experiments showing reduced growth of neuroblastoma cells on glycated laminin.60
Non-enzymatic glycosylation of collagens produces crosslinkages and hence may produce physical alterations in the properties of the ECM. Chemical alterations may render the environment unattractive to the growing axons by reducing the ability of the growth cones to bind to the ECM, hence leading to the observed regenerative failure. In addition to changes in the basal lamina itself, the basal laminal tubes surrounding axonal sprouts in diabetic nerves are often densely packed with fibrillar collagen (fig 2 ▶). Glycation renders collagen less digestible by proteases so it may act as a physical barrier to axonal growth.61 Measurement of collagen fibrils inside the Schwann tubes as compared with that in the endoneurium and epineurium showed an increase in their diameter, but this was also found in other chronic neuropathies.62 Recently, scanning force microscopy has been used to show that rat tail collagen from spontaneously diabetic BB/WOR/MOL\BB rats has a larger diameter than that from non-diabetic rats. This work also showed that these physical changes correlated with increased concentrations of fructosamine and pentosidine, both in diabetic rats and glucose incubated collagen.63
There has been considerable discussion over the years about the time course of axon extension versus Schwann cell multiplication, and whether axons precede or follow Schwann cells in the outgrowth from a transected stump. Recent experiments on regenerating axons in a film model in vivo showed that there was a lag of three days between axon sprouts appearing and Schwann cells following them. After this period, however, the Schwann cells were needed for further axonal extension.64 During the initial growth phase axonal growth cones are directly attached to the extracellular matrix. This is mediated by several different mechanisms at the basal laminal interface. The rapid movements of actin filaments in the growth cone may be regulated via integrin mediated interactions involving F-actin, talin, vinculin, and α-actinin with the substrate.65 It has been shown that actin and tubulin undergo non-enzymatic glycosylation readily.29,30 These proteins are essential components of the growth cone and the rearrangement of actin allows changes in shape and movement of the growth cone. Therefore, it seems likely that glycation would reduce the mobility of the growth cone.
Details of the linkages between the basal lamina and axon or Schwann cell differ for the various components. Laminin is the largest molecule and has several sites either via galactosyltransferase, proteoglycan, or a variety of non-integrin receptors. It has recently been shown that vinculin is specifically associated with integrin at the points of the filopodia and in the central domains of growth cones.66 AGE formation on laminin causes decreased polymer self assembly and decreased binding to other ECM components. It has been shown to modify the neurite promoting sequence and inhibit neurite outgrowth considerably.60
Another component of the basal lamina, fibronectin, may be bound via heparan sulphate proteoglycan (HPSG) or tissue plasminogen activator.67 Binding to HPSG is also reduced by AGE formation. Anionic HPSG is absent from the glomerular basement membrane in STZ rats with prolonged duration of diabetes.68 Fibronectin production in mesangial cell basal lamina is increased by glycation, possibly because of an altered responsiveness to cytokines. This is associated with a 50% reduction in cell proliferation.69 Schwann cells can deposit fibronectin as part of their basal lamina, using a mechanism involving collagen IV.70 AGE formation inhibits the development of the normal network structure of collagen IV by interfering with the binding of the non-collagenous NCl domain to the helix rich domain.71 Heparin binding to collagen IV regulates polymerisation.72 It has been shown that glycation of the heparin binding domain results in decreased endothelial cell adhesion,73 but it is not yet known whether Schwann cells or neurones are similarly effected.
The integrins that are involved in Schwann cell linkage to collagen IV differ from those involved in fibronectin polymerisation. Where there are several different attachment types on one component of the ECM, it is possible that glycation could affect one site but leave others undamaged. A further complication is that both the filopodia in axonal growth cones and Schwann cells express the same integrin initially,74 but the integrins on Schwann cells change in the presence of neurones and with myelination. It seems probable that β4 integrin is involved in axon–Schwann cell interactions and is downregulated in Wallerian degeneration.75
Cell surface proteoglycans may also be implicated in mediating Schwann cell–laminin reactions.76 There is evidence that Schwann cells can synthesise laminin-1 and use it to support their migration on a laminin free substrate.77 This could mean that Schwann cells may prove to be less affected by glycation of the ECM. They only mature into myelin forming cells when β1-integrins are functional,78 and the regenerating axon sprouts seen in diabetic nerves are often well myelinated (figs 1, 2 ▶ ▶). In addition, laminin-2 is needed to promote neurite outgrowth after Wallerian degeneration.79 Although neurones do not produce a basal lamina, they normally express mRNA for laminin genes,80 and these are upregulated during regeneration.81 Neuronal growth on laminin is severely affected by glycation.60 It is not yet known whether glycation affects the separate components of laminin components differentially, but this is quite possible because AGE formation on laminin not only decreases polymer self assembly but also binding to collagen IV and HSPG.82 This last protein is a component of basal laminae that is postulated to play an important part in the filtration properties of glomerular basement membrane. Reduction in binding to laminin has been suggested to lead to the overproduction of other basement membrane components in the vessel walls of diabetic kidneys.83 It could be postulated that a similar mechanism leads to the widening of the basal laminae of endoneurial microvessels (figs 3, 4 ▶ ▶).84 Alternatively, or in addition, there may be an accumulation of basal laminal components as a result of the increased resistance of AGE proteins to protease digestion.61
Figure 3.

Light micrograph of resin section of sural nerve from 48 year old woman with distal sensory symmetric polyneuropathy. The endoneurial microvessels have extensively enlarged basal laminal ensheathment. Few myelinated fibres have survived in this case. Stained with thionin and acridine orange. Original magnification, ×400.
Figure 4.
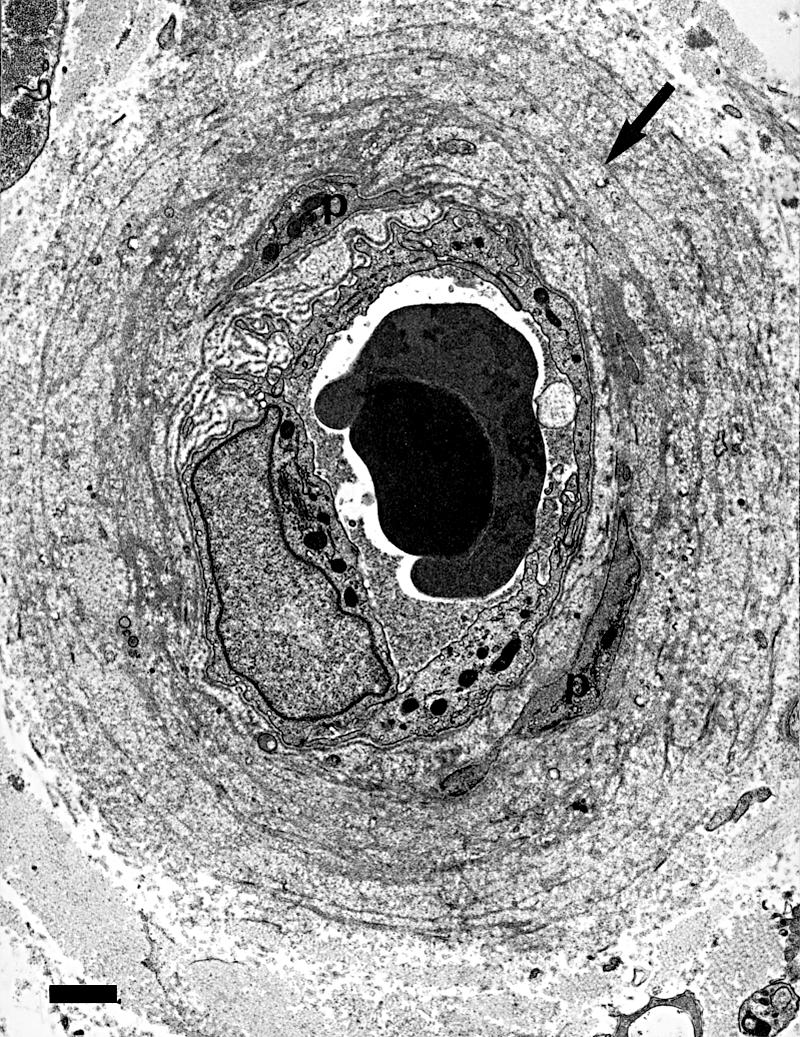
Electron micrograph showing that the widened basal laminal ensheathment around endoneurial blood vessels consists of basal lamina (arrow) and collagen fibrils, and contains pericyte processes (p). Same case as fig 2 ▶. Bar, 1 μm.
Unfortunately, no studies on alterations of the different basal laminal components in diabetic peripheral nerves have been detailed enough to confirm whether there is an alteration in the proportions of the various components. Morphometric and morphological studies on human nerves suggest that the factors causing thickening of vascular basal lamina differ from those producing widened basal lamina in the perineurium.85 These studies suggested that the alterations in perineurial basal lamina were more characteristic of diabetic neuropathy than thickening of the basal laminal zone around endoneurial capillaries. Perineurial basal lamina also widens with age. Differences in the basal laminal components and their differing susceptibility to glycation may be one factor in producing an increase in basal laminal thickness, but another may be the function of the perineurium as a filter between the epineurium and endoneurium and its ability to trap glycated proteins.14,86 Morphologically, the appearances of the widened basal laminal zone differ. In the perineurium it takes the form of a smooth, amorphous layer wider than normal with maximum width on the central layers of the perineurium, whereas around blood vessels there are multiple thin layers of basal lamina often separated by fibrous collagen (fig 5A,B ▶). Basal laminal changes of both perineurial cells and blood vessels differ from the Schwann cell basal laminal changes, where the abnormality is its abnormal shape and persistence during regeneration; on undamaged fibres it appears to be normal.
Figure 5.

(A) Electron microscopy of a section through the whole thickness of the perineurium of a radial nerve fascicle from a 39 year old man with distal sensory symmetric polyneuropathy; the epineurial face is at the top. The basal lamina of the perineurial cells is extensively widened (asterisks). The small electron dense bodies are deposits of calcium apatite. Bar, 1 μm. (B) Electron microscopy of the perineurium from a normal control subject with approximately the same numbers of perineurial laminae. The basal laminae are much thinner. Bar, 1 μm.
Extracellular calcium deposits may be found associated with the basal lamina in the perineurium of abnormal nerves and these are particularly common in diabetic neuropathy. The details of the process leading to the formation of these deposits are unknown.87
Diabetes and the Schwann cell
The direct effects of diabetes on Schwann cells themselves rather than their basal laminal envelopment has received little attention. Diabetes could lead to a primary deleterious effect and hence to segmental demyelination and/or to alterations to injury related changes that could hinder regeneration. However, the demyelinating changes and onion bulb formations reported in some nerve biopsy studies of diabetic polyneuropathy (for example, Ballin and Thomas88) might not result from primary Schwann cell abnormalities, but could be secondary to axonal abnormalities or represent a coexisting chronic progressive inflammatory demyelinating polyneuropathy.
Investigations of the changes in protein expression of denervated Schwann cells show that they upregulate the expression of NGF and the NGF receptor, brain derived neurotrophic factor (BDNF), GAP-43,89 and the adhesion molecules L1 and neural cell adhesion molecule (N-CAM). It has been proposed that the role of GAP-43 in these circumstances is to enable the cell membrane to change its shape90; this would suggest that it has a similar function in both growth cones and Schwann cells. In support of this notion, it has been noted that Schwann cells at the neuromuscular junction extend long process after nerve injury.91 However, the re-establishment of axonal contact does not immediately reduce GAP-43 concentrations. In addition, many Schwann cell specific proteins are reduced in migrating Schwann cells compared with those resident in the denervated distal stump.92 It remains to be shown how diabetes alters these injury related changes and how AGE formation is involved.
Although glycation of the Schwann cell cytoskeleton could also be expected to be deleterious, a study using in vitro experiments with Schwann cells extracted from human nerves found no alterations in those from patients with diabetes: they had normal phenotypic characteristics, mitotic abilities, and antigenic properties.93 These authors did not take into account the type of diabetes or the extent of the pathological changes in the nerves. Recent work has failed to find staining for the AGE adduct, CML, in Schwann cells from diabetic human nerves (A Bierhaus et al, personal communication, 2000). On the other hand, an experimental study on STZ diabetic rats showed that they were more susceptible to tellurium induced demyelination than normal animals. This could be the result of diabetic Schwann cells having an increased sensitivity to stress, which could be a possible mechanism for myelin breakdown.94
A less direct pathway for the production of segmental demyelination has been suggested by findings of macrophage recognition of abnormally glycosylated myelin proteins in diabetes.95 However, the rarity of reports of active myelin breakdown in diabetic neuropathy suggests that this is not an important factor.
Although several studies have investigated the existence of axonal atrophy, which could produce secondary demyelination in diabetic neuropathy, there is little direct evidence that this is an important factor in the aetiology of the neuropathy,96–98 apart from a teased fibre study showing atrophy above a distally degenerating axon.20
Vascular abnormalities in diabetes
Damage as a result of vascular changes in the nerve trunks may also be a contributory factor in the evolution of diabetic neuropathy, particularly in older patients.99 Because ischaemia would be expected to affect motor and sensory nerves equally, this may be the cause of the motor deficit that can also be found. On morphological examination, the loss of fibres caused by ischaemia is typically patchy and often greater in the centre of nerve fascicles. Examination of biopsy material from cases of diabetic polyneuropathy has produced conflicting results. Dyck et al were convinced that they had found evidence for focal fibre loss,21,100 whereas Llewelyn and colleagues101 found no difference in patchyness between diabetic nerves and those from inherited neuropathies.
Direct examination of the endoneurial blood vessels has failed to show convincingly that the endothelial cells are abnormal in patients with diabetes. One light microscopic study showed that the blood vessel lumens were closed,102 but this was not confirmed by a later electron microscopic investigation.103 However, widening and reduplication of the basal laminal sheath that surrounds these endoneurial blood vessels has been described frequently (fig 3 ▶).104,105 The effects of non-enzymatic glycosylation on basal laminal components have already been discussed in the preceding section. Changes in blood vessel basal laminae may in part be the result of non-enzymatic glycosylation occurring over many years. This may occur even when blood sugar values are normal,106 so that the effect of diabetes is to produce an accelerated version of the age changes seen in normal nerves. Similar widened basal laminal ensheathment is often seen in nerves from cases of neuropathy associated with paraproteinaemia. This may be related to the age of these patients and it may also be seen in other nerve biopsies from older patients.
Although it has been suggested that the effect of non-enzymatic glycosylation on the growth and adhesion properties of various cell types, including endothelial cells,57 could be related to the loss of pericytes reported in diabetic retinopathy,107 the numbers of pericytes are slightly increased in diabetic polyneuropathy.103
Summary
The aetiology of diabetic neuropathy is still poorly understood but it is clear that it is very complex. Glycation is probably a major factor but dissecting its influence on the various components of peripheral nerves is a very complicated problem. To date, attempts to modify the progression of the disease by drug treatment targeted at specific pathways (such as the use of the AGE inhibitor aminoguandine) have had little success.
References
- 1.Diabetes Control and Complications Trial Research Group. The effect of intensive treatment for diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N Engl J Med 1993;329:977–86. [DOI] [PubMed] [Google Scholar]
- 2.Gaede P, Vedel P, Parving HH, et al. Intensified multifactorial intervention in patients with type 2 diabetes mellitus and microalbuminuria: the Steno type 2 randomised study. Lancet 1999;353:617–22. [DOI] [PubMed] [Google Scholar]
- 3.Bradley JL, Thomas PK, King RHM, et al. Myelinated fibre regeneration in diabetic sensory polyneuropathy: correlation with type of diabetes. Acta Neuropathol (Berl) 1995;90:403–10. [DOI] [PubMed] [Google Scholar]
- 4.Shaklai N, Garlick RL, Bunn HF. Nonenzymatic glycosylation of human serum albumin alters its conformation and function. J Biol Chem 1984;259:3812–17. [PubMed] [Google Scholar]
- 5.Sell DR, Nagaraj RH, Grandhee SK, et al. Pentosidine: a molecular marker for the cumulative damage to proteins in diabetes, aging, and uremia. Diabetes Metab Rev 1991;7:239–51. [DOI] [PubMed] [Google Scholar]
- 6.Fu M, Requena J, Jenkins A, et al. The advanced glycation end-product, Nɛ-(carboxymethyl)lysine, is a product of both lipid peroxidation and glycoxidation reactions. J Biol Chem 1996;271:9982–6. [DOI] [PubMed] [Google Scholar]
- 7.Ryle C, Donaghy M. Non-enzymatic glycation of peripheral nerve proteins in human diabetics. J Neurol Sci 1995;129:62–8. [DOI] [PubMed] [Google Scholar]
- 8.Hammes HP, Martin S, Federlin K, et al. Aminoguanidine treatment inhibits the development of experimental diabetic retinopathy. Proc Natl Acad Sci U S A 1991;88:11555–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brownlee M. Lilly lecture 1993. Glycation and diabetic complications. Diabetes 1994;43:836–41. [DOI] [PubMed] [Google Scholar]
- 10.Giardino I, Edelstein D, Brownlee M. Nonenzymatic glycosylation in vitro and in bovine endothelial cells alters basic fibroblast growth factor activity. A model for intracellular glycosylation in diabetes. J Clin Invest 1994;94:110–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brownlee M. Glycation products and the pathogenesis of diabetic complications. Diabetes Care 1992;15:1835–43. [DOI] [PubMed] [Google Scholar]
- 12.Lornezi M, Montisano DF, Toledo S, et al. High glucose and DNA damage in endothelial cells. J Clin Invest 1986;77:322–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Poduslo J, Curran GL. Increased permeability across the blood–nerve barrier of albumin glycated in vitro and in vivo from patients with diabetic polyneuropathy. Proc Natl Acad Sci USA 1992;89:2218–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Graham AR, Johnson PC. Direct immunofluorescence findings in peripheral nerve from patients with diabetic neuropathy. Ann Neurol 1985;17:450–4. [DOI] [PubMed] [Google Scholar]
- 15.Brownlee M, Vlassara H, Cerami A. Trapped immunoglobulins on peripheral nerve myelin from patients with diabetes mellitus. Diabetes 1986;35:999–1003. [DOI] [PubMed] [Google Scholar]
- 16.Brownlee M, Vlassara H, Cerami A. Excessive nonenzymatic glycosylation of peripheral and central nervous system myelin components in diabetic rats. Diabetes 1983;37:670–4. [DOI] [PubMed] [Google Scholar]
- 17.Dolman CL. The morbid anatomy of diabetic neuropathy. Neurology 1963;13:135–42. [DOI] [PubMed] [Google Scholar]
- 18.Watkins PJ, Gayle C, Alsanjari N, et al. Severe sensory-autonomic neuropathy and endocrinopathy in insulin-dependent diabetes. Q J Med 1995;88:795–804. [PubMed] [Google Scholar]
- 19.Sidenius P, Jakobsen J. Reduced perikaryal volume of lower motor and primary sensory neurons in early experimental diabetes. Diabetes 1980;29:182–6. [DOI] [PubMed] [Google Scholar]
- 20.Said G, Slama G, Selva J. Progressive centripetal degeneration of axons in small fibre diabetic neuropathy. Brain 1983;106:791–807. [DOI] [PubMed] [Google Scholar]
- 21.Dyck PJ, Karnes JL, O'Brien CP, et al. The spatial distribution of fiber loss in diabetic polyneuropathy suggests ischaemia. Ann Neurol 1986;19:440–9. [DOI] [PubMed] [Google Scholar]
- 22.Allen DT, Kiernan JA. Permeation of proteins from the blood into peripheral nerves and ganglia. Neuroscience 1994;59:755–64. [DOI] [PubMed] [Google Scholar]
- 23.Olsson Y. Studies on vascular permeability in peripheral nerves. 4. Distribution of intravenously injected protein tracers in the peripheral nervous system of various species. Acta Neuropathol 1971;17:114–26. [DOI] [PubMed] [Google Scholar]
- 24.Zochodne DW, Ho LT, Allison JA. Dorsal root ganglia microenvironment of female BB Wistar diabetic rats with mild neuropathy. J Neurol Sci 1994;127:36–42. [DOI] [PubMed] [Google Scholar]
- 25.Ryle C, Leow CK, Donaghy M. Nonenzymatic glycation of peripheral and central nervous system proteins in experimental diabetes mellitus. Muscle Nerve 1997;20:577–84. [DOI] [PubMed] [Google Scholar]
- 26.Cameron NE, Cotter MA, Dines K, et al. Effects of aminoguanidine on peripheral nerve function and polyol pathway metabolites in streptozotocin-diabetic rats. Diabetol 1992;35:946–50. [DOI] [PubMed] [Google Scholar]
- 26a.Luo Z-J, King RHM, Lewin J, et al. Effects of nonenzymatic glycosylation of extracellular matrix components on cell survival and sensory neurite extension in cell culture. J Neurol [In press.] [DOI] [PubMed]
- 27.Smith MA, Sayre LM, Monnier VM, et al. Radical AGEing in Alzheimer's disease. Trends Neurosci 1995;18:172–6. [DOI] [PubMed] [Google Scholar]
- 28.Kikuchi S, Ogata A, Shinpo K, et al. Detection of an Amadori product, 1-hexitol-lysine, in the anterior horn of the amyotrophic lateral sclerosis and spinobulbar muscular atrophy spinal cord: evidence for early involvement of glycation in motoneuron diseases. Acta Neuropathol 2000;99:63–6. [DOI] [PubMed] [Google Scholar]
- 29.Williams SK, Howarth NL, Devenny JJ, et al. Structural and functional consequences of increased tubulin glycosylation in diabetes mellitus. Proc Natl Acad Sci U S A 1982;79:6546–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pekiner C, Cullum NA, Hughes JN, et al. Glycation of brain actin in experimental diabetes. J Neurochem 1993;61:436–42. [DOI] [PubMed] [Google Scholar]
- 31.Brimijoin S. Axonal transport in diabetic neuropathy. In: Dyck PJ, Thomas PK, eds. Diabetic neuropathy. London: WB Saunders, 1999:341–51.
- 32.Sidenius P, Jakobsen J. Axonal transport in early experimental diabetes. Brain Res 1979;173:315–30. [DOI] [PubMed] [Google Scholar]
- 33.Sidenius P, Jakobsen J. Axonal transport in rats after galactose feeding. Diabetologia 1980;19:229–33. [DOI] [PubMed] [Google Scholar]
- 34.Meiri KF, Mclean WG. Axonal transport of protein in motor fibers of experimentally diabetic rats—fast anterograde transport. Brain Res 1982;238:77–88. [DOI] [PubMed] [Google Scholar]
- 35.Mendell JR, Sahenk Z, Warmolts JR, et al. The spontaneously diabetic BB Wistar rat: morphologic and physiologic studies of peripheral nerve. J Neurol Sci 1981;52:103–15. [DOI] [PubMed] [Google Scholar]
- 36.Kilgour RD, Gardiner K, Gardiner PF. Diabetes affects retrograde but not anterograde transport of sciatic nerve phosphofructokinase in Sprague-Dawley rats. Can J Physiol Pharmacol 1990;68:1317–21. [DOI] [PubMed] [Google Scholar]
- 37.Tomlinson DR, Fernyhough P, Diemel LT. Role of neurotrophins in diabetic neuropathy and treatment with nerve growth factors. Diabetes 1997;46:S43–9. [DOI] [PubMed] [Google Scholar]
- 38.Zhuang HX, Wuarin L, Fei ZJ, et al. Insulin-like growth factor (IGF) gene expression is reduced in neural tissues and liver from rats with non-insulin-dependent diabetes mellitus, and IGF treatment ameliorates diabetic neuropathy. J Pharmacol Exp Ther 1997;283:366–74. [PubMed] [Google Scholar]
- 39.Tomlinson DR, Mayer JH. Defects of axonal transport in diabetes mellitus—a possible contribution to the etiology of diabetic neuropathy. J Auton Pharmacol 1984;4:59–72. [DOI] [PubMed] [Google Scholar]
- 40.Sidenius P, Jakobsen J. Retrograde axonal transport: a possible role in the development of neuropathy. Diabetologia 1981;20:110–12. [DOI] [PubMed] [Google Scholar]
- 41.Vlassara H, Brownlee M, Cerami A. Nonenzymatic glycosylation of peripheral nerve protein in diabetes mellitus. Proc Natl Acad Sci U S A 1981;78:5190–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Longo FM, Powell HC, Lebeau J, et al. Delayed nerve regeneration in streptozotocin diabetic rats. Muscle Nerve 1986;9:365–93. [DOI] [PubMed] [Google Scholar]
- 43.Powell HC, Longo FM, LeBeau JM, et al. Abnormal nerve regeneration in galactose neuropathy. J Neuropathol Exp Neurol 1986;45:151–60. [DOI] [PubMed] [Google Scholar]
- 44.Alexander KA, Cimler BM, Meier K, et al. Regulation of calmodulin binding to P-5777. J Biol Chem 1987;262:6108–13. [PubMed] [Google Scholar]
- 45.Alexander KA, Wakim BT, Doyle GS, et al. Identification and characterization of the calmodulin-binding domain of neuromodulin, a nonspecific calmodulin-binding protein. J Biol Chem 1988;263:7544–9. [PubMed] [Google Scholar]
- 46.Schrama LH, Heemskerk FJ, De GP. Dephosphorylation of protein kinase C phosphorylated B-50/GAP-43 by the calmodulin-dependent phosphatase calcineurin. Neurosci Res Commun 1989;5:141–7. [Google Scholar]
- 47.Aigner L, Caroni P. Depletion of 43-kD growth-associated protein in primary sensory neurons leads to diminished formation and spreading of growth cones. J Cell Biol 1993;123:417–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Skene JHP, Virag I. Posttranslational membrane attachment and dynamic fatty acetylation of a neuronal growth cone protein, GAP-43. J Cell Biol 1989;108:613–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Verkade P, Verkleij AJ, Gispen WH, et al. Ultrastructural evidence for the lack of co-transport of B-50/GAP-43 and calmodulin in myelinated axons of the regenerating rat sciatic nerve. J Neurocytol 1996;25:583–95. [DOI] [PubMed] [Google Scholar]
- 50.Woolf CJ, Reynolds ML, Molander C, et al. The growth-associated protein GAP-43 appears in dorsal root ganglion cells and in the dorsal horn of the rat spinal cord following peripheral nerve injury. Neuroscience 1990;34:465–78. [DOI] [PubMed] [Google Scholar]
- 51.Pekiner C, Dent EW, Roberts RE, et al. Altered GAP-43 immunoreactivity in regenerating sciatic nerve of diabetic rats. Diabetes 1996;45:199–204. [DOI] [PubMed] [Google Scholar]
- 52.Büngner Ov. Versuche zum Studium der histologischen Degeneration und Regeneration verletzer Nerven, ihre Anordnung und ihre Ergebnisse. Zieglers Beitrage 1891;10:321–87. [Google Scholar]
- 53.Nathaniel EJH, Pease DC. Degenerative changes in rat dorsal roots during Wallerian degeneration. J Ultrastruct Res 1963;9:511–15. [DOI] [PubMed] [Google Scholar]
- 54.Thomas PK. Changes in the endoneurial sheaths of peripheral nerve fibres during Wallerian degeneration. J Anat 1964;98:175–82. [PMC free article] [PubMed] [Google Scholar]
- 55.Giannini C, Dyck PJ. Basement membrane reduplication and pericyte degeneration precede development of diabetic polyneuropathy and are associated with its severity. Ann Neurol 1995;37:498–504. [DOI] [PubMed] [Google Scholar]
- 56.King RHM, Llewelyn JG, Thomas PK, et al. Diabetic neuropathy: abnormalities of Schwann cell and perineurial basal laminae. Implications for diabetic vasculopathy. Neuropathol Appl Neurobiol 1989;15:339–55. [DOI] [PubMed] [Google Scholar]
- 57.Haitoglou CS, Tsilibary EC, Brownlee M, et al. Altered cellular interactions between endothelial cells and nonenzymatically glucosylated laminin/type IV collagen. J Biol Chem 1992;267:12404–7. [PubMed] [Google Scholar]
- 58.Anderson SS, Kim Y, Tsilibary EC. Effects of matrix glycation on mesangial cell adhesion, spreading and proliferation. Kidney Int 1994;46:1359–67. [DOI] [PubMed] [Google Scholar]
- 59.Krishnamurti U, Rondeau E, Sraer JD, et al. Alterations in human glomerular epithelial cells interacting with nonenzymatically glycosylated matrix. J Biol Chem 1997;272:27966–70. [DOI] [PubMed] [Google Scholar]
- 60.Federoff HJ, Lawrence D, Brownlee M. Nonenzymatic glycosylation of laminin and the laminin peptide CIKVAVS inhibits neurite outgrowth. Diabetes 1993;42:509–13. [DOI] [PubMed] [Google Scholar]
- 61.Lubec G, Pollak A. Reduced susceptibility of nonenzymatically glucosylated glomerular basement membrane to proteases: is thickening of diabetic glomerular basement membranes due to reduced proteolytic degradation? Renal Physiology 1980;3:4–8. [DOI] [PubMed] [Google Scholar]
- 62.Bradley JL, King RHM, Muddle JR, et al. The extracellular matrix of peripheral nerve in diabetic polyneuropathy. Acta Neuropathol (Berl) 2000;99:539–46. [DOI] [PubMed] [Google Scholar]
- 63.Odetti P, Aragno I, Rolandi R, et al. Scanning force microscopy reveals structural alterations in diabetic rat collagen fibrils: role of protein glycation. Diabetes Metab Res Rev 2000;16:74–81. [DOI] [PubMed] [Google Scholar]
- 64.Torigoe K, Tanaka HF, Takahashi A, et al. Basic behavior of migratory Schwann cells in peripheral nerve regeneration. Exp Neurol 1996;137:301–8. [DOI] [PubMed] [Google Scholar]
- 65.Reichardt LF, Tomaselli KJ. Extracellular matrix molecules and their receptors: functions in neural development. Annu Rev Neurosci 1991;14:531–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Renaudin A, Lehmann M, Girault JA, et al. Organization of point contacts in neuronal growth cones. J Neurosci Res 1999;55:458–71. [DOI] [PubMed] [Google Scholar]
- 67.Letourneau PC, Condic ML, Snow DM. Extracellular matrix and neurite outgrowth. Curr Opin Genet Dev 1992;2:625–34. [DOI] [PubMed] [Google Scholar]
- 68.Klein DJ, Oegema TR, Brown DM. Release of glomerular heparan-32 SO4 proteoglycan by heparin from glomeruli of streptozotocin-induced diabetic rats. Diabetes 1989;38:130–9. [DOI] [PubMed] [Google Scholar]
- 69.Crowley ST, Brownlee M, Edelstein D, et al. Effects of nonenzymatic glycosylation of mesangial matrix on proliferation of mesangial cells. Diabetes 1991;40:540–7. [DOI] [PubMed] [Google Scholar]
- 70.Chernousov MA, Stahl RC, Carey DJ. Schwann cells use a novel mechanism for fibronectin fibril assembly. J Cell Sci 1998;111:2763–77. [DOI] [PubMed] [Google Scholar]
- 71.Tsilibary EC, Charonis AS, Reger LA, et al. The effect of nonenzymatic glycosylation on the binding of the main noncollagenous NC1 domain to type IV collagen. J Biol Chem 1988;263:4302–8. [PubMed] [Google Scholar]
- 72.Tsilibary EC, Koliakos GG, Charonis AS, et al. Heparin type IV collagen interactions: equilibrium binding and inhibition of type IV collagen self-assembly. J Biol Chem 1988;263:19112–18. [PubMed] [Google Scholar]
- 73.Tsilibary EC, Charonis AS. The effect of nonenzymatic glucosylation on cell and heparin-binding microdomains from type IV collagen and laminin. Diabetes 1990;39(suppl 1):194A. [Google Scholar]
- 74.Yanagida H, Tanaka J, Maruo S. Immunocytochemical localization of a cell adhesion molecule, integrin alpha5beta1, in nerve growth cones. J Orthop Sci 1999;4:353–60. [DOI] [PubMed] [Google Scholar]
- 75.Einheber S, Milner TA, Giancotti F, et al. Axonal regulation of Schwann cell integrin expression suggests a role for alpha 6 beta 4 in myelination. J Cell Biol 1993;123:1223–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Carey DJ, Crumbling DM, Stahl RC, et al. Association of cell surface proteoglycans of Schwann cells with extracellular matrix proteins. J Biol Chem 1990;265:20627–33. [PubMed] [Google Scholar]
- 77.Dubovy P, Svizenska I, Klusakova I, et al. Migration of Schwann cells and regeneration of peripheral nerves. Electronic Journal of Pathology & Histology 1999;5:21–31. [Google Scholar]
- 78.Fernandez-Valle C, Wood PM, Bunge MB. Localization of focal adhesion kinase in differentiating Schwann cell cultures. Microsc Res Tech 1998;41:416–30. [DOI] [PubMed] [Google Scholar]
- 79.Agius E, Cochard P. Comparison of neurite outgrowth induced by intact and injured sciatic nerves: a confocal and functional analysis. J Neurosci 1998;18:328–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sarthy PV, Fu M. Localization of laminin B1 mRNA in retinal ganglion cells by in situ hybridization. J Cell Biol 1990;110:2099–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.LeBeau JM, Liuzzi FJ, Depto AS, et al. Up-regulation of laminin B2 gene expression in dorsal root ganglion neurons and nonneuronal cells during sciatic nerve regeneration. Exp Neurol 1995;134:150–5. [DOI] [PubMed] [Google Scholar]
- 82.Charonis AS, Reger LA, Dege JE, et al. Laminin alterations after in vitro nonenzymatic glycosylation. Diabetes 1990;39:807–14. [DOI] [PubMed] [Google Scholar]
- 83.Ruoslahti E, Yamaguchi H. Proteoglycans as modulators of growth factor activities. Cell 1991;64:867–9. [DOI] [PubMed] [Google Scholar]
- 84.Knecht R, Leber R, Hasslacher C. Degradation of glomerular basement membrane in diabetes. 1. Susceptibility of diabetic and nondiabetic basement membrane to proteolytic degradation of isolated glomeruli. Res Exp Med (Berl) 1987;187:323–8. [DOI] [PubMed] [Google Scholar]
- 85.Bradley JL, Thomas PK, King RHM, et al. A comparison of perineurial and vascular basal laminal changes in diabetic neuropathy. Acta Neuropathol (Berl) 1994;88:426–32. [DOI] [PubMed] [Google Scholar]
- 86.Johnson PC, Brendel K, Meezan E. Human diabetic perineurial cell basement membrane thickening. Lab Invest 1981;44:265–70. [PubMed] [Google Scholar]
- 87.King RHM, Llewelyn JG, Thomas PK, et al. Perineurial calcification. Neuropathol Appl Neurobiol 1988;14:105–23. [DOI] [PubMed] [Google Scholar]
- 88.Ballin RHM, Thomas PK. Hypertrophic changes in diabetic neuropathy. Acta Neuropathol (Berl) 1968;11:93–102. [DOI] [PubMed] [Google Scholar]
- 89.Plantinga LC, Verhaagen J, Edwards PM, et al. The expression of B-50/GAP-43 in Schwann cells is upregulated in degenerating peripheral nerve stumps following nerve injury. Brain Res 1993;602:69–76. [DOI] [PubMed] [Google Scholar]
- 90.Zuber MX, Goodman DW, Karns LR, et al. The neuronal growth-associated protein GAP 43 induces filopodia in non-neuronal cells. Science 1989;244:1193–5. [DOI] [PubMed] [Google Scholar]
- 91.Reynolds LM, Woolf CJ. Terminal Schwann cells elaborate extensive processes following denervation of the motor endplate. J Neurocytol 1992;21:50–66. [DOI] [PubMed] [Google Scholar]
- 92.Popovic M, Sketelj J, Bresjanac M. Changes of Schwann cell antigenic profile after peripheral nerve injury. Pflugers Arch 1996;431:R287–8. [DOI] [PubMed] [Google Scholar]
- 93.Scarpini E, Doronzo R, Baron P, et al. Phenotypic and proliferative properties of Schwann cells from nerves of diabetic patients. Int J Clin Pharmacol Res 1992;12:211–15. [PubMed] [Google Scholar]
- 94.Jaffey PB, Gelman BB. Increased vulnerability to demyelination in streptozotocin diabetic rats. J Comp Neurol 1996;373:55–61. [DOI] [PubMed] [Google Scholar]
- 95.Vlassara H, Brownlee M, Cerami A. Accumulation of diabetic rat peripheral nerve myelin by macrophages increases with the presence of advanced glycosylation endproducts. J Exp Med 1984;160:197–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Llewelyn JG, Gilbey SG, Thomas PK, et al. Sural nerve morphometry in diabetic autonomic and painful sensory neuropathy. Brain 1991;114:867–92. [DOI] [PubMed] [Google Scholar]
- 97.Sugimura K, Dyck PJ. Does axonal atrophy precede segmental demyelination in human diabetic neuropathy? Neurology 1981;31(suppl):129. [Google Scholar]
- 98.Engelstad J, Davies JL, Giannini C, et al. No evidence for axonal atrophy in human diabetic polyneuropathy. J Neuropathol Exp Neurol 1997;56:255–62. [DOI] [PubMed] [Google Scholar]
- 99.Sugimura K, Dyck PJ. Multifocal fiber loss in proximal sciatic nerve in symmetric distal diabetic neuropathy. J Neurol Sci 1982;53:501–9. [DOI] [PubMed] [Google Scholar]
- 100.Dyck PJ, Lais A, Karnes JL, et al. Fiber loss is primary and multifocal in sural nerves in diabetic polyneuropathy. Ann Neurol 1986;19:425–39. [DOI] [PubMed] [Google Scholar]
- 101.Llewelyn JG, Thomas PK, Gilbey SG, et al. Pattern of myelinated fibre loss in the sural nerve in neuropathy related to type 1 (insulin-dependent) diabetes. Diabetologia 1988;31:162–7. [DOI] [PubMed] [Google Scholar]
- 102.Dyck PJ, Hansen S, Karnes J, et al. Capillary number and percentage closed in human diabetic sural nerve. Proc Natl Acad Sci USA 1985;82:2513–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Bradley J, Thomas PK, King RHM, et al. Morphometry of endoneurial capillaries in diabetic sensory and autonomic neuropathy. Diabetologia 1990;33:611–18. [DOI] [PubMed] [Google Scholar]
- 104.Fagerberg SE. Diabetic neuropathy: a clinical and histological study on the significance of vascular affections. Acta Med Scand 1959;164:1–80. [PubMed] [Google Scholar]
- 105.Vital C, LeBlanc AC, Vallat JM, et al. Étude ultrastructurale du nerf périphérique chez 16 diabétiques sans neuropathie clinique. Comparaisons avec 16 neuropathies diabétiques et 16 neuropathies non diabétiques. Acta Neuropathol (Berl) 1974;30:63–72. [DOI] [PubMed] [Google Scholar]
- 106.Monnier VM, Kohn RR, Cerami A. Accelerated age-related browning of human collagen in diabetes mellitus. Proc Natl Acad Sci USA 1984;81:583–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Engerman RL. Pathogenesis of diabetic retinopathy. Diabetes 1989;38:1203–6. [DOI] [PubMed] [Google Scholar]