

Abstract
The low-density lipoprotein (LDL) receptor-related protein (LRP) is a multiligand endocytic receptor and a member of the LDL receptor family. Here we show that sorting nexin 17 (Snx17) is part of the cellular sorting machinery that regulates cell surface levels of LRP by promoting its recycling. While the phox (PX) domain of Snx17 interacts with phosphatidylinositol-3-phosphate for membrane association, the FERM domain and the carboxyl-terminal region participate in LRP binding. Immunoelectron microscopy shows that the membrane-bound fraction of Snx17 is localized to the limiting membrane and recycling tubules of early endosomes. The NPxY motif, proximal to the plasma membrane in the LRP cytoplasmic tail, is identified as the Snx17-binding motif. Functional mutation of this motif did not interfere with LRP endocytosis, but decreased LRP recycling from endosomes, resulting in increased lysosomal degradation. Similar effects are found after knockdown of endogenous Snx17 expression by short interfering RNA. We conclude that Snx17 binds to a motif in the LRP tail distinct from the endocytosis signals and promotes LRP sorting to the recycling pathway in the early endosomes.
Keywords: endosome, LRP, recycling, sorting, sorting nexin 17
Introduction
The LDL receptor-related protein (LRP) is a multiligand endocytic receptor that belongs to the LDL receptor family (Herz and Bock, 2002). Ligands of LRP include proteins that are involved in lipid metabolism, proteinase regulation, blood coagulation, growth regulation, and the pathogenesis of Alzheimer's disease. A unique feature of LRP is its rapid endocytosis when compared to other members of the LDL receptor family. The 100-amino-acid cytoplasmic tail of LRP contains two NPxY motifs, two di-leucine motifs, and an YxxL motif. Mutagenesis studies have shown that the YxxL motif and the di-leucine motif, distal to the plasma membrane, are the primary endocytosis signals (Li et al, 2000). LRP endocytosis is further regulated by a cyclic AMP-dependent protein kinase A-mediated serine phosphorylation (Li et al, 2001). For many years, LRP was considered a prototypic endocytic receptor for the delivery of macromolecules into cells; however, recent studies suggest that LRP is also a signaling receptor (Schneider and Nimpf, 2003). LRP signaling has been linked to cellular events such as resistance to apoptosis, synaptic plasticity, focal adhesion disassembly, and cell migration (Herz and Bock, 2002). The dual function of LRP in endocytosis and signal transduction requires strict regulation of its availability at the cell surface. Indeed, several studies have shown that cell surface LRP levels can be regulated by growth factor receptor signaling (Weaver et al, 1996; Bu et al, 1998) and the proteasome (Melman et al, 2002). In 3T3-L1 adipocytes, insulin treatment stimulates recycling of LRP from an endosomal pool to the plasma membrane in a phosphatidylinositol 3-kinase (PI3K)-dependent manner (Ko et al, 2001). However, the molecular component(s) that participate in this sorting and are subjected to PI3K regulation are unknown.
Recently, sorting nexin (Snx) 17 was identified as a binding partner of several members of the LDL receptor family (Stockinger et al, 2002). Sorting nexins are a family of proteins involved in intracellular protein trafficking (Worby and Dixon, 2002). The hallmark of this family is the presence of a phox (PX) homology domain, which has been shown to bind to phosphatidylinositol phosphates (PtdInsPs) (Cheever et al, 2001; Kanai et al, 2001; Xu et al, 2001). This domain is thought to play a role in targeting these proteins to specialized membrane domains enriched in specific phospholipids. Snx17 was initially identified as an intracellular binding protein for P-selectin (Florian et al, 2001). This binding was recently shown to accelerate P-selectin internalization and inhibit its lysosomal degradation (Williams et al, 2004). The protein consists of an N-terminal PX domain, followed by a B41 (band 4.1 or FERM) domain. No additional known protein domains are present in the C-terminus. Stockinger et al (2002) showed that Snx17 colocalizes with markers of early endosomes and that overexpression of the protein in mouse embryonic fibroblasts enhances LDL endocytosis. More recently, it was suggested that Snx17 plays a role in the cellular trafficking of the LDL receptor through interaction with the NPVY motif in the LDL receptor tail (Burden et al, 2004). Because this motif also serves as the endocytosis signal for the LDL receptor (Chen et al, 1990), it was not clear whether Snx17 participates in LDL receptor endocytosis or intracellular sorting.
In this study, we found that Snx17 binds to the proximal NPxY motif of the LRP tail distinct from its endocytosis signals. We also found that Snx17 interacts preferentially with PtdIns(3)P through its PX domain and is localized to the limiting membrane and recycling tubules of the early endosomal structures. Functional studies indicate that Snx17 promotes LRP recycling, likely by preventing its sorting to the lysosomal degradation pathway. Together, our studies define a novel signal-mediated endosomal sorting mechanism that favors the recycling pathway.
Results
Snx17 interacts with LRP cytoplasmic tail independently of its endocytosis signals
Figure 1A depicts the 100-amino-acid cytoplasmic tail of LRP. To examine whether LRP's dominant endocytosis motif Y63ATL is involved in LRP–Snx17 interaction, we produced fusion proteins between GST and the 100-amino-acid tail of LRP without (GSTT100) or with mutation at the Y63 position (GSTT100Y63A). Extracts of 293 cells expressing myc-tagged Snx17 (Stockinger et al, 2002) were incubated with GST-LRP tail fusion proteins bound to glutathione beads. Precipitated proteins were analyzed by Western blotting using anti-myc antibody. As seen in Figure 1B, myc-tagged Snx17 interacted with GST-LRP tail fusions both without or with mutation at the Y63 position, but not with GST alone. To determine if the interaction between Snx17 and LRP tail was direct or bridged by other proteins, we produced [35S]methionine-labeled myc-Snx17 in an in vitro transcription/translation reaction and incubated it with GST or GST-LRP tail fusions (Figure 1C). The product of the in vitro transcription/translation reaction (Lysate) shows two bands, of which the upper band (open arrowhead) comigrates with the myc-tagged Snx17 expressed in 293 cells. The lower band is not detectable with anti-myc antibody and likely represents Snx17 translated from the first methionine of Snx17. In vitro-produced Snx17 interacted with GST-LRP tail (GSTT100) but not with GST alone, suggesting a direct interaction between Snx17 and LRP. LRP endocytosis motifs do not play a role in this interaction because mutations at individual residues critical to each endocytosis motif (Y63A, S76A, or L86L87A) did not abolish binding (Figure 1C). To examine in vivo interaction between LRP and Snx17 by co-immunoprecipitation, human glioblastoma U87 cells, which express abundant LRP (Bu et al, 1994), were stably transfected with myc-tagged Snx17 (U87/Snx) and lysates were immunoprecipitated with an antibody against LRP or normal rabbit IgG. The immunoprecipitates were separated by SDS–PAGE and detected on a Western blot with either anti-LRP antibody (upper panel) or anti-myc antibody (lower panel). Figure 1D shows that anti-LRP (lane 1), but not control IgG (lane 2), co-immunoprecipitated myc-Snx17. Lanes 3 and 4 show negative controls in which pcDNA3 vector-transfected U87 cells were used for immunoprecipitation. From these cells, LRP can be immunoprecipitated (lane 3, upper panel) but no signal for myc-Snx17 is detected (lane 3, lower panel). Lanes 5 and 6 show direct lysates of the U87/Snx and U87/pcDNA3 cells.
Figure 1.
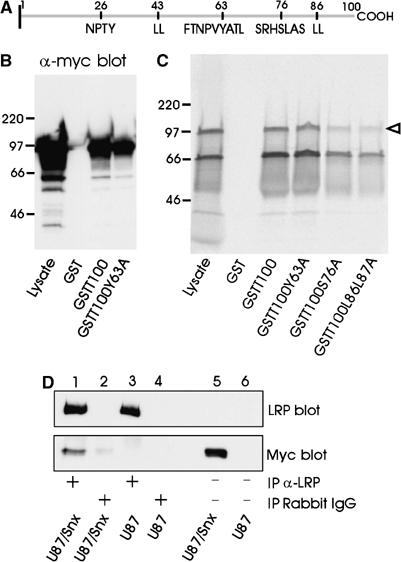
Snx17 interacts with LRP cytoplasmic tail independent of the endocytosis signal. (A) Schematic drawing of the 100-amino-acid LRP cytoplasmic tail. Residue numbered 1 is the first amino acid following the transmembrane domain. Depicted in one-letter amino-acid code are the tyrosine-based and di-leucine motifs present in the tail as well as a serine phosphorylation site. (B) Myc-Snx17 pull-down by LRP cytoplasmic tail. GST or GST fusion proteins of the LRP cytoplasmic tail without (GSTT100) or with a mutation in the endocytosis motif (GSTT100Y63A) were incubated with cell lysate of myc-Snx17-transfected HEK293 cells. Bound proteins were detected with anti-myc antibody on a Western blot. The lane marked with ‘Lysate' contains 10% of cell lysate used for pull-down assays. Relative molecular size standards (Mr × 10−3) in this and subsequent figures are shown on the left. (C) GST fusion proteins of the LRP cytoplasmic tail (GSTT100) or several LRP cytoplasmic tail constructs mutated at individual endocytosis motifs (GSTT100Y63A, GSTT100S76A, or GSTT100L86L87A) were incubated with [35S]methionine-labeled myc-Snx17 from in vitro transcription/translation reaction. Bound proteins were separated by SDS–PAGE and radioactivity was detected using a STORM 820 imaging system. The lane marked with ‘Lysate' contains 20% of cell lysate used for pull-down assays. (D) Co-immunoprecipitation of myc-Snx17 with LRP. Cell lysates of U87 cells stably transfected with myc-Snx17 (U87/Snx) or vector alone (U87) were immunoprecipitated with anti-LRP antibody (lanes 1 and 3) or normal rabbit IgG (lanes 2 and 4). The immunoprecipitated proteins were separated on SDS–PAGE and the upper part of the gel was detected with anti-LRP antibody (LRP blot) while the lower part of the same gel was detected with anti-myc antibody (Myc blot). Lanes 5 and 6 contain cell lysates representing 5% of those used for immunoprecipitation. Note that the signal for endogenous LRP is only visible after immunoprecipitation due to the limited sensitivity of the Western blotting technique.
LRP membrane-proximal NPxY motif interacts with Snx17
To identify the motif in LRP that interacts with Snx17, we performed deletion and mutation analysis of the LRP cytoplasmic tail. The right panel of Figure 2A shows that the first 53 amino acids of the LRP tail (GSTT4–53) are required and sufficient for binding in vitro-produced [35S]methionine-labeled myc-Snx17. No interaction was detected with amino acids 54–100 of the LRP tail (GSTT54–100). Deletion of the N-terminal 23 amino acids from the cytoplasmic tail (GSTT24–100) did not affect binding of myc-Snx17, while removal of residues 1–33 (GSTT34–100) resulted in a complete loss of interaction (Figure 2B). These results demonstrate the importance of residues 24–34 of the LRP tail in binding to Snx17. Next, we replaced either one or two residues in the full-length LRP tail with alanine (Figure 2C) and the resulting GST fusion proteins were incubated with extracts of 293 cells expressing myc-tagged Snx17. In Figure 2D and E, the results of the pull-down assays are summarized. Single point mutations of I24, N26, P27, or Y29 to alanine resulted in a complete loss of interaction between LRP cytoplasmic tail and Snx17, indicating that the I24xN26P27xY29 motif in LRP tail is essential for interaction with Snx17. Some residues downstream of this motif may also contribute to the interaction because double mutations of Y32 and E33, or D38 and D39, to alanine resulted in a reduced binding of Snx17.
Figure 2.
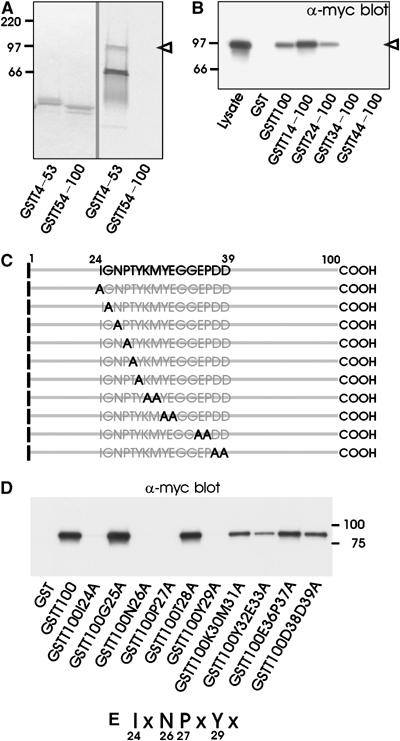
LRP membrane-proximal NPxY motif is required for interaction with Snx17. (A) GST fusion proteins of the N-terminal half (GSTT4–53) or the C-terminal half (GSTT54–100) of the LRP tail were incubated with in vitro-translated [35S]methionine-labeled myc-Snx17 as in Figure 1. The left panel shows a Coomassie blue staining of the gel, while the right panel shows a phosphorimager visualization of the radioactivity in the same gel. The open arrowhead points to the ∼90 kDa protein that comigrates with myc-tagged Snx from cell lysates. (B) GST fusion proteins of the full-length (GSTT100) or N-terminally truncated LRP cytoplasmic tail (GSTT14–100, GSTT24–100, GSTT34–100, and GSTT44–100) were incubated as in Figure 1. Bound proteins were detected with anti-myc antibody on a Western blot. The lane marked with ‘Lysate' contains 10% of cell lysate used for pull-down assays. (C) Schematic overview of site-directed mutations within the LRP cytoplasmic tail designed to localize the Snx17-binding site. (D) GST fusion proteins of the LRP cytoplasmic tail (GSTT100) or single or double point mutations (see panel C) were incubated as in panel B. (E) Summary of the individual residues in one-letter amino-acid code that are essential for Snx17 binding.
Snx17 binding to LRP is important for its cell surface distribution and stability
To establish the role of this newly identified Snx17 interacting motif in the LRP tail, we made use of LRP minireceptors, which mimic the function and trafficking of full-length endogenous LRP (Li et al, 2000, 2001). These LRP minireceptors contain an N-terminal HA tag, followed by the fourth ligand-binding domain, the transmembrane domain and the full-length cytoplasmic tail of LRP (mLRP4T100). The mutant of the proximal NPxY motif, mLRP4T100Y29A, contains a Y29A mutation. Our previous studies have shown that this mutation does not affect LRP endocytosis (Li et al, 2000). We generated stable U87 cell lines expressing mLRP4T100 (WT) and mLRP4T100Y29A (Y29A) and performed cell surface staining with anti-HA antibody. The flow cytometry results presented in Figure 3A (right panel) demonstrate the purity of these stable clones. When cell lysates were analyzed by Western blotting (Figure 3A, left panel), three specific bands representing different forms of LRP minireceptor are detected, which include the ER precursor form (∼200 kDa), the Golgi form (∼210 kDa with complex glycosylation), and the furin-processed mature form (∼120 kDa, representing the fourth ligand-binding domain; see Li et al, 2000, 2001). The Western blot analysis shows that, although both cell lines display comparable amounts of the ER precursor form, the Y29A mutant minireceptor has considerably less mature form. To examine whether this is caused by a faster turnover of the Y29A mutant, we performed metabolic pulse–chase labeling experiments. Results shown in Figure 3B demonstrate a faster turnover rate of both 120 kDa N-terminal and 85 kDa noncovalently associated C-terminal furin-cleaved subunits of the Y29A mutant when compared to the wild-type minireceptor. Next, we analyzed the relative cell surface distribution of the Y29A mutant and the wild-type receptor by antibody binding assays. 125I-anti-HA IgG was allowed to bind to U87 cells stably expressing wild-type or Y29A mutant either on ice (to determine cell surface receptors) or at 37°C (to measure all receptors recycling through the endocytic pathway). As seen in Figure 3C, the wild-type LRP minireceptor shows much higher surface expression than the Y29A mutant.
Figure 3.
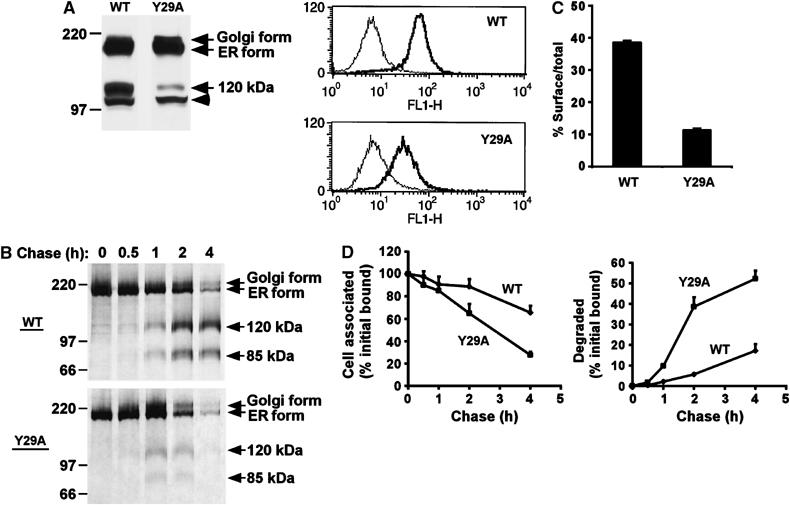
Snx17-binding motif in LRP tail is important for receptor cell surface expression and stability. (A) (Left panel) Western blot of equal amounts of cell lysate of U87 cells expressing wild-type LRP minireceptor (WT) or the Y29A mutant detected with anti-HA antibody. The ER precursor form, the glycosylated Golgi form, and the furin-processed mature 120 kDa form are indicated. The arrowhead marks a nonspecific band that is also present in nontransfected cells. (Right panel) Flow cytometric analysis of cell surface LRP minireceptor expression. The same cell lines as used in the left panel were labeled with anti-HA antibody and detected with goat anti-mouse Ig-FITC. Background fluorescence was assessed in the absence of anti-HA (thin line). The x-axis represents relative fluorescence intensity and the y-axis represents cell number. (B) U87 cells expressing wild-type LRP minireceptor (WT) or the Y29A mutant were metabolically labeled with [35S]cysteine for 30 min and chased for the indicated times. After each chase, cell lysates were immunoprecipitated with anti-HA antibody, separated by SDS–PAGE and visualized using a phosphorimager. (C) LRP minireceptor cell surface expression measured with 125I-anti-HA IgG. U87 cells expressing wild-type LRP minireceptor (WT) or the Y29A mutant were incubated with 125I-anti-HA for 1 h on ice to measure cell surface binding, or for 1 h at 37°C to measure total binding. The percentage of surface binding as of total binding is plotted. (D) Degradation of 125I-anti-HA-labeled LRP minireceptors. U87 cells expressing wild-type LRP minireceptor (WT) or the Y29A mutant were labeled with 125I-anti-HA for 20 min at 37°C and chased for the indicated times at 37°C. After each chase, cell-associated and degraded radioactivity in the media was determined as described in Materials and methods. The percentage of cell-associated (left) or degraded (right) 125I-anti-HA is plotted at each time point normalized to the total radioactivity at time 0.
LRP is an endocytic receptor that recycles to undergo multiple rounds of endocytosis (Herz and Bock, 2002). Since the endocytosis rate of the Y29A mutant is identical to that of the wild-type minireceptor (Li et al, 2000), a less efficient recycling of the Y29A mutant could be a possible explanation for its shorter half-life and decreased cell surface expression. To monitor the degradation rate of receptors within the endocytic pathway, we pulse-labeled U87 cells expressing LRP minireceptors with 125I-anti-HA IgG at 37°C and measured the amount of cell-associated and degraded antibody over time. Since the antibody does not dissociate from the receptor at endosomal pH (Touret et al, 2003), the trafficking and degradation of the 125I-anti-HA reflect those of bound receptors. Figure 3D shows that the 125I-anti-HA is more stably associated with cells when bound to the wild-type LRP minireceptor (left panel), while it is more susceptible to degradation when bound to the Y29A mutant (right panel). Taken together, these results show that a disruption of Snx17 binding to LRP results in a decreased receptor distribution at the plasma membrane and an increased receptor degradation, consistent with an impaired recycling.
The PX domain of Snx17 binds to PtdIns(3)P and is not required for LRP binding
The presence of the PX domain is the hallmark of the Snx family and the ability of this domain to bind various PtdInsPs is thought to target these proteins to specific cellular membranes enriched in specific phospholipids (Worby and Dixon, 2002). Because PX domains were found to participate in both protein–lipid and protein–protein interactions, we sought to determine whether the PX domain of Snx17 was involved in LRP binding. Figure 4A depicts the different myc-tagged Snx17 domain constructs that were expressed in 293 cells. Cell extracts from transfected 293 cells were incubated with GST alone or GST-LRP tail fusions without (GSTT100) or with a mutation in the Snx17-binding motif (GSTT100N26A). The Western blots in Figure 4B show that full-length Snx17 (Snx 1–470) interacted, as expected, with the full-length LRP tail but not with the N26A mutant or GST alone. Both the N-terminal half (Snx 2–250) and the C-terminal half of Snx17 (Snx 250–470) failed to interact with the LRP tail fusion protein. Truncation of the N-terminal 105 (Snx 105–470) or 150 (Snx 150–470) amino-acid residues of Snx17 did not affect LRP tail binding. However, further truncation within the B41 domain of Snx17 up to the N-terminal 200 amino acids (Snx 200–470) resulted in a nearly complete loss of LRP tail binding. These results suggest that the C-terminal region, but not the PX domain, of Snx17 contributes to LRP binding.
Figure 4.

Snx17 PX domain mediates phospholipid binding but is not required for LRP interaction. (A) Schematic representation of myc-Snx17 and the truncated constructs. (B) Myc-Snx17 pull-down by LRP cytoplasmic tail. GST or GST-LRP tail fusions without (GSTT100) or with a mutation in the Snx17-binding motif (GSTT100N26A) were immobilized on glutathione beads and incubated with cell lysates of myc-Snx17-transfected HEK293 cells. Bound proteins were immunoblotted using anti-myc antibody. The lane marked with ‘Lysate' contains 5% of cell lysate used for pull-down assays. (C) Protein–lipid overlay assay using PIP strips. Commercial nitrocellulose strips loaded with equal amounts of known lipids were incubated with GST fusion protein of full-length Snx17 (GST-Snx17) and detected with anti-GST antibody. ECL detection was used to show binding of GST-Snx17 to individual lipids. LPA: lysophosphatidic acid; LPC: lysophosphocholine; PtdIns: phosphatidylinositol; PtdIns(3)P: phosphatidylinositol-3-phosphate; PE: phosphatidylethanolamine; PC: phosphatidylcholine; PA: phosphatidic acid; PS: phosphatidylserine.
To determine the lipid-binding specificity of Snx17, we performed a protein/lipid overlay assay using nitrocellulose strips loaded with equal amounts of different lipids. The lipid strip was incubated with bacterially produced GST-Snx17 or GST and were detected with anti-GST antibody. While GST showed no affinity for any of the lipids (not shown), GST-Snx17 clearly bound most strongly to PtdIns(3)P (Figure 4C). Weaker interactions were detected in the following order: PtdIns(5)P>PtdIns(4)P>PtdIns(3,5)P2>PtdIns(4,5)P2. The protein/lipid overlay was also performed with a lysate of 293 cells expressing myc-snx17 as the source for Snx17. Incubation of the lysate with lipid strips, followed by detection of the retained protein with anti-myc antibody, also revealed a binding of Snx17 to PtdIns(3)P (not shown).
Snx17 localizes to early endosomal limiting membrane and recycling tubules
To assess the localization of Snx17 at the ultrastructural level, ultrathin cryosections of U87 cells stably expressing myc-Snx17 were immunogold labeled with anti-myc (Figure 5). Snx17 was clearly found on the limiting membrane of endosomal vacuoles as well as on the emerging recycling tubules (Figure 5A). The endosomes were of early endosomal origin because they contained only few internal vesicles (Figure 5A) and could be labeled with the early endosomal marker Alexa-Tf that was internalized for 30 min (Figure 5C and D). Large parts of these early endosomes are coated with a bilayered clathrin coat involved in the sorting of growth factor receptors into the internal vesicles prior to degradation (Raiborg et al, 2002; Sachse et al, 2002). Double labeling of Snx17 with Hrs, a major component of this coat, showed only marginal overlap (Figure 5B). Double labeling of myc-Snx17 with LRP (Bu et al, 1995) showed colocalization of the two proteins in both the early endosomal vacuoles and within the recycling tubules (Figure 5E and F). This observation shows a direct connection between Snx17 and the LRP recycling pathway.
Figure 5.
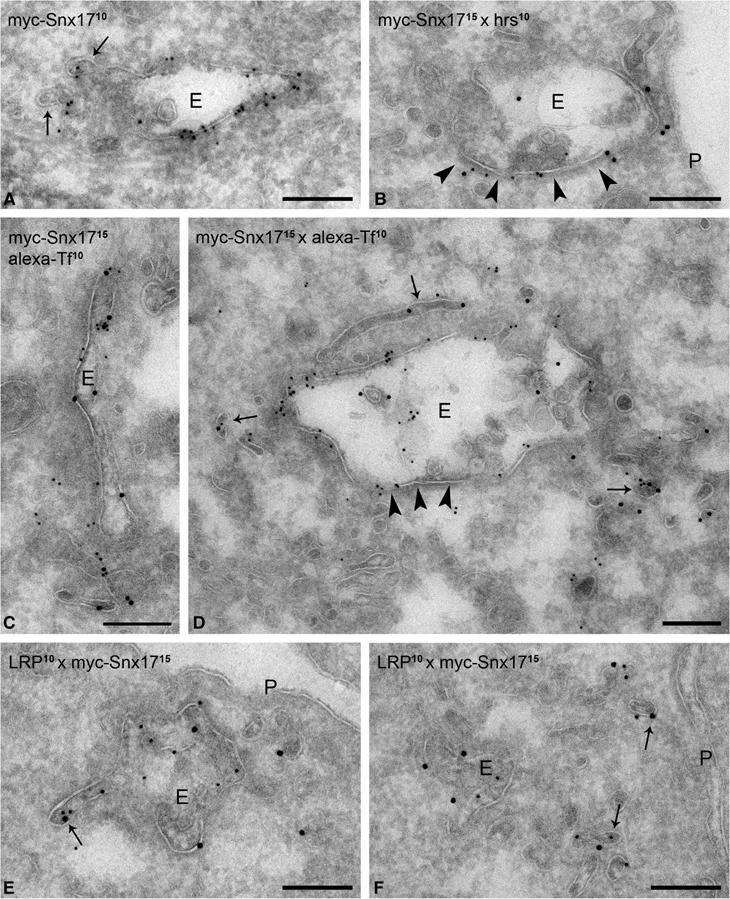
Snx17 localizes to early endosomal vacuoles and recycling tubules. U87 cells expressing myc-Snx17 were labeled with anti-myc for ultrastructural immuno-EM localization studies. (A) Myc-Snx17 (10 nm gold particles) is associated with an early endosomal compartment (E). The arrows point to forming recycling tubules that are positive for myc-Snx17. (B) Double labeling of myc-Snx17 (15 nm gold particles) and Hrs (10 nm gold particles). Hrs marks the bilayered coat (arrowheads) involved in protein sorting into internal vesicles. Myc-Snx17 prevails mostly in the noncoated domains of the endosome. (C, D) Cells were incubated with Alexa-Tf for 30 min and labeled with anti-Alexa to mark early endocytic compartments. Myc-Snx17 (15 nm gold particles) and Alexa-Tf (10 nm gold particles) colocalize on early endosomal vacuoles as well as on recycling tubules (arrows in panel D). The arrowheads indicate the bilayered coat. (E, F) LRP (10 nm gold particles) and myc-Snx17 (15 nm gold particles) colocalize on the endosomal vacuole and emerging recycling tubules (arrow in panel E) as well as with endosome-associated tubulo-vesicular recycling profiles (arrows in panel F). P: plasma membrane. Bars, 200 nm.
In order to detect endogenous Snx17, we generated an Snx17-specific antibody directed against the last 14 amino acids of Snx17, which are identical between human and mouse. This antibody, which specifically recognizes Snx17 when analyzed by Western blotting or immunofluorescence staining (not shown), was used for immunoelectron microscopy (immuno-EM) localization of the endogenous Snx17 in U87 cells (Supplementary Figure 1). Notably, these nontransfected U87 cells labeled with anti-Snx17 antibody showed a similar endosomal morphology and displayed identical labeling patterns as the myc-Snx17-transfected cells.
Snx17 is not involved in LRP endocytosis
Figure 6A shows a Western blot of Snx17 from U87 cells (left lane) or U87 cells overexpressing myc-tagged mouse Snx17 (U87/Snx) detected with Snx17 antibody. A clear band of ∼50 kDa is visible in both lanes, representing the endogenous human Snx17. In cells overexpressing myc-tagged Snx17, an additional band at ∼90 kDa was detected, which represents the myc-tagged mouse Snx17. The nature of this band was confirmed with anti-myc antibody (not shown). The molecular size of this 9xmyc-tagged Snx17 detected by SDS–PAGE is ∼20 kDa larger than the calculated size. Interestingly, when a 9xmyc tag was placed at the N-terminus of another protein, Cue1P, it also migrated ∼20 kDa larger on SDS–PAGE than the calculated size (Gauss et al, 2005). Therefore, the size discrepancy is likely caused by the electrophoretic property of the 9xmyc tag.
Figure 6.
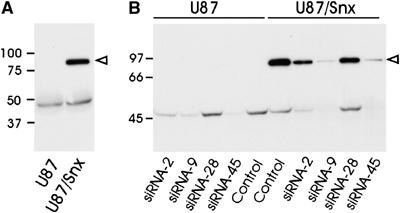
Snx17 knockdown by siRNA in U87 cells. (A) Cell lysates of U87 cells (U87) or myc-Snx17 stably transfected U87 cells (U87/Snx) were subjected to Western blotting with an anti-Snx17 antibody. The open arrowhead indicates the position of the overexpressed myc-tagged Snx17 and the lower band represents the endogenous Snx17. (B) Four different siRNAs (siRNA-2, siRNA-9, siRNA-28, or siRNA-45) designed to target both human and mouse Snx17 were transfected into U87 cells (U87) or U87 cells stably overexpressing mouse myc-Snx17 (U87/Snx). Control cells were transfected with Lipofectamine2000 alone without siRNA. Cell lysates were blotted as in panel A.
To define the function of endogenous Snx17 in LRP trafficking, we used small interfering RNA (siRNA) to silence Snx17 expression. The siRNAs were designed to target sequences that were 100% homologous between human and mouse Snx17. U87 and U87 cells stably expressing myc-Snx17 were transfected with four different siRNAs or with vehicle Lipofectamine2000 only and cell lysates were blotted with Snx17 antibody. The Western blot showed that both siRNA-45 and siRNA-9 suppressed endogenous Snx17 expression almost completely while siRNA-2 was less effective and siRNA-28 was completely ineffective (Figure 6B, left panel). The same silencing pattern by these siRNAs for the overexpressed myc-Snx17 was observed (Figure 6B, right panel).
Our previous studies have established that the LRP proximal NPTY motif, which binds Snx17, is not required for LRP endocytosis, as mutations of this motif did not change its endocytosis rate (Li et al, 2000). To further examine potential involvement of Snx17 in LRP endocytosis, we performed an internalization assay with Cy3-labeled LRP-specific ligand, alpha-2-macroglobulin (α2M), in U87 cells (Supplementary Figure 2). As seen in the figure, the kinetics of Cy3-labeled α2M were identical between U87 cells transfected with functional (panel e–h) or nonfunctional (panels a–d) Snx17 siRNA. These results confirm that Snx17 is not involved in LRP rapid endocytosis.
Snx17 modulates cell surface LRP levels by promoting its recycling
To gain further insights into the function of Snx17 in LRP intracellular trafficking, we silenced the Snx17 expression with siRNA in wild-type LRP minireceptor expressing cells. Silencing of Snx17 expression resulted in a decrease in the ratio of furin-processed mature form to ER form (Figure 7A), indicating a more rapid degradation of LRP minireceptor when Snx17 expression is knocked down. To analyze the effect of altered Snx17 expression on endogenous LRP trafficking, we compared the relative cell surface LRP levels in U87 cells stably transfected with either vector alone or myc-Snx17. Cell surface LRP levels were detected by flow cytometry (Melman et al, 2002) using a monoclonal antibody directed against the heavy chain of LRP. As seen in Figure 7B (left panel), overexpression of myc-Snx17 resulted in more than doubling of cell surface LRP. To analyze the effects of Snx17 loss of function on LRP trafficking, we compared the cell surface LRP levels in U87 cells transfected with either control or Snx17 siRNA. As seen in Figure 7B (right panel), we observed an ∼40% decrease in cell surface LRP in Snx17 silenced cells. To verify this result, we performed the same experiment but detected cell surface LRP with its ligand, 125I-α2M (Figure 7C, left panel). Consistent with the LRP antibody data, we found a similar decrease in LRP ligand-binding capacity in cells transfected with Snx17 siRNA. In order to assess whether this decrease in cell surface LRP levels also affects LRP ligand degradation efficiency, we compared 125I-α2M degradation in cells transfected with either control siRNA or Snx17 siRNA. Consistent with a lower number of receptors at the cell surface, degradation of 125I-α2M was clearly diminished in Snx17 siRNA-transfected cells (Figure 7C, right panel).
Figure 7.
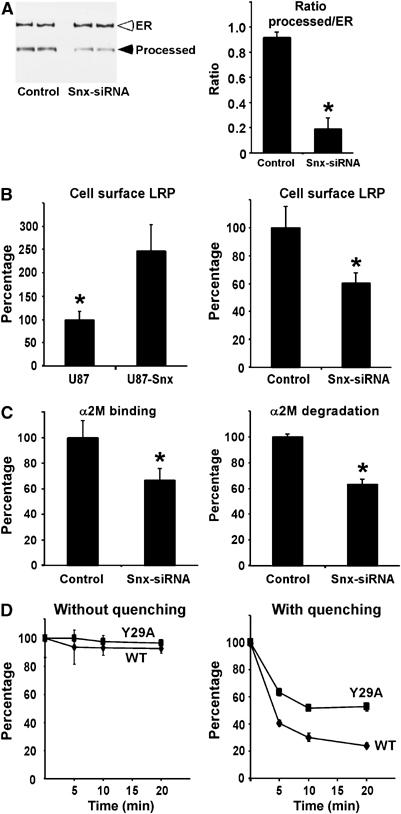
Snx17 modulates LRP cell surface levels by promoting its recycling. (A) U87-mLRP4 cells were transfected with control or Snx17 siRNA and analyzed via Western blotting with anti-HA antibody (left panel). The intensity of the bands was quantified and plotted as a ratio of furin-processed form over ER precursor form. In all experiments, s.e.m. values are given as error bars, and Western blots were performed to confirm knockdown of endogenous Snx17. *P<0.05. (B) (Left panel) U87 cells stably transfected with vector alone (U87) or myc-Snx17 (U87-Snx) were analyzed for cell surface LRP by flow cytometry. Results in U87-Snx cells are expressed as a percentage of those in vector-transfected U87 cells. (Right panel) U87 cells transfected with control or Snx17 siRNA were analyzed for cell surface LRP levels by flow cytometry. Results with Snx17 siRNA are expressed as a percentage of those with control siRNA. (C) U87 cells were transfected with control or Snx17 siRNA and analyzed for cell surface LRP by measuring 125I-α2M ligand binding at 4°C for 1 h (left panel), or LRP endocytic capacity by measuring 125I-α2M ligand degradation at 37°C for 4 h (right panel). Results with Snx17 siRNA are expressed as a percentage of those with control siRNA. (D) U87 cells expressing wild-type LRP minireceptor (diamonds) or the Y29A mutant (squares) were pulse labeled with Alexa488 anti-HA antibody for 20 min at 37°C and chased for the indicated time in the absence (left panel) or presence of quenching anti-Alexa488 IgG (right panel). Cells were processed for flow cytometry and the percentage of pulsed fluorescence remaining was calculated as described in Materials and methods. Each data point is a mean of two experiments measured in triplicate with s.e.m. values given as error bars.
The above results with LRP Y29A mutant and with Snx17-specific siRNA indicate that an absence of Snx17 binding to LRP results in a faster receptor turnover. Because silencing of Snx17 or mutation of the Snx17-binding motif does not influence LRP endocytosis, we hypothesize that Snx17 is involved in an endosomal sorting event favoring the recycling pathway. To test this hypothesis directly, we made use of a cell surface fluorescence quenching recycling assay as described in recently published work (Austin et al, 2004; Schapiro et al, 2004). U87 cells stably expressing HA-tagged wild-type LRP minireceptor (mLRP4T100) or the Snx17-binding mutant (Y29A) were incubated with Alexa488-conjugated anti-HA antibodies for 20 min and either processed directly or chased for 5–20 min in the presence of anti-Alexa488 antibody. If the internalized receptor is recycled, it will be delivered to the plasma membrane where the Alexa488 fluorescence will be quenched by the anti-Alexa488 antibody. The left panel in Figure 7D shows that there is a negligible loss of fluorescence if the chase is performed in the absence of quenching antibody, indicating that receptor degradation is minimal within the chase period. When the same experiment was repeated in the presence of quenching anti-Alexa488 antibody during the chase (Figure 7D, right panel), we observed a faster loss of fluorescence in wild-type LRP minireceptor cells when compared to those of Y29A mutant, indicating a less efficient recycling for the Y29A mutant receptor. To ensure that expression of the Y29A mutant did not affect the recycling machinery, we performed similar experiments in both cell lines with Alexa488-labeled transferrin. The loss of fluorescence, due to recycling of internalized transferrin receptor, was indistinguishable in both cell lines (not shown). Together, these results support an important role for Snx17 in LRP recycling.
Discussion
The signaling and endocytic capacity of LRP is dictated by its cell surface availability. In this study, we show that Snx17 specifically recognizes the membrane-proximal NPxY motif of LRP, which does not participate in receptor endocytosis. More importantly, we demonstrate that Snx17 modulates LRP cell surface distribution by promoting its endocytic recycling.
Snx17 binds directly to the proximal NPxY motif of LRP and thus appears to be involved in direct recognition of the cargo receptor. Previous studies have demonstrated a role for NPxY motifs in receptor endocytosis by serving as a recognition site for endocytic adaptor proteins (Bonifacino and Traub, 2003). Here we show that interaction of Snx17 with the LRP proximal NPxY motif does not mediate the internalization step but an intracellular sorting event leading to recycling. LRP contains two NPxY motifs, of which the distal motif is not required and cannot replace the proximal motif for binding to Snx17. The most likely differences between these two NPxY motifs are the surrounding residues. For example, the isoleucine residue essential for Snx17 binding, two positions N-terminal to the critical asparagine, is replaced by a phenylalanine residue in the distal motif. Furthermore, we found that several residues downstream of the proximal NPxY motif also contribute to Snx17 binding. In addition to linear sequence elements, residues from different regions of the LRP tail may also convened together within the three-dimensional structure and contribute to SNX17 binding. Future mutagenesis studies should address these possibilities.
The PX domains of Snx family members play important roles in the membrane localization of these proteins through their binding to various PtdInsPs (Kanai et al, 2001). Specifically, the PX domain is required for the proper localization of Vam7, a yeast protein involved in vacuolar morphogenesis (Cheever et al, 2001), and the regulation of the endosomal sorting function of Snx3 (Xu et al, 2001). The PX domain of Snx17 is required for its endosomal localization (Burden et al, 2004). Here we show that Snx17 binds preferentially to PtdIns(3)P, with lower affinity to PtdIns(5)P, PtdIns(4)P, PtdIns(3,5)P2, and PtdIns(4,5)P2. This preferential binding to PtdIns(3)P was also demonstrated in a recently published work (Knauth et al, 2005). PtdIns(3)P is a well-characterized lipid component of endosomal membranes (Simonsen et al, 2001). In MEF cells, overexpressed Snx17 resides on distinct vesicular structures, partially overlapping with endosomal compartments characterized by the presence of EEA1 and rab4 (Stockinger et al, 2002). Localization of sorting nexins can be determined by both the PX domain and additional domains in the protein as was shown for Snx16, which distributes to both early and late endosomes through a combined action of the PX domain and the coiled-coil domain (Hanson and Hong, 2003). By immuno-EM, we clearly showed that Snx17 associates with the limiting membrane of the early endosomes as well as with emerging recycling tubules. Within the early endosomes, Snx17 prevailed on the membrane domains that were not coated by the Hrs-containing bilayered coat involved in protein sorting into intraendosomal vesicles, leading to lysosomal degradation. This localization is fully consistent with a role of Snx17 in recycling from early endosomes.
Our results provide the first evidence that Snx17 is involved in the intracellular sorting of LRP to the recycling pathway. Although receptor recycling is generally believed to be a default pathway, recent studies have provided evidence that it can also be a signal-mediated event (Tanowitz and von Zastrow, 2003; Tran et al, 2003; Vargas and Von Zastrow, 2004). In this study, we have provided several lines of evidence that implicate a role for Snx17 in LRP recycling. First, mutation of the Snx17-binding motif (Y29A) within the LRP tail significantly decreased LRP cell surface distribution and increased its degradation rate. Second, a reduced recycling efficiency of the LRP Y29A mutant when compared to the wild-type receptor was observed by a fluorescent quenching-based recycling assay. Third, knockdown of endogenous Snx17 expression by siRNA approach resulted in a decreased LRP cell surface distribution and a reduced stability of mature LRP. Fourth, a distribution of the Snx17 in the limiting membrane and recycling tubules of the early endosomes suggests that Snx17 likely functions by retaining and preventing its cargo receptors from entering the lumen of MVBs via double-layered clathrin coats. In this paper, we did not study a possible role for Snx17 in other endosome-derived pathways (e.g. recycling of the cation-independent mannose-6-phosphate receptor, CI-MPR, from endosomes to TGN). Therefore, it remains possible that Snx17 functions in multiple pathways from the endosomes. Although we found a direct interaction between Snx17 and LRP, it is likely that other components exist within a sorting complex that includes Snx17. In yeast, the PX domain-containing proteins Vps5 and Vps17 are part of the retromer complex that functions in the endosome to Golgi retrieval of the vacuolar protein sorting receptor Vps10p (Seaman and Williams, 2002). A similar role for Snx1 in tubular endosome-to-TGN sorting of the CI-MPR has recently been demonstrated in mammalian cells (Carlton et al, 2004). Our initial analysis of Snx17 by gel filtration has detected myc-tagged Snx17 from cell lysates in both monomeric form and within large molecular size complexes eluted in the void volume (P van Kerkhof et al, unpublished results). Future identification of Snx17 binding partners should provide further insights into how Snx17 achieves its role in receptor recycling.
In a recent study, we reported that the differential distribution of LRP and another large member of the LDL receptor family, megalin, in polarized epithelial cells is determined by their cytoplasmic domains (Marzolo et al, 2003). Megalin, which does not bind Snx17 (Stockinger et al, 2002), was found to be apically distributed, while LRP was basolaterally sorted. Interestingly, it was found that the LRP proximal NPTY motif, identified as the Snx17-binding motif in this study, was important for basolateral sorting; mutation of the tyrosine residue in this motif to alanine resulted in a mis-sorting of LRP to the apical surface. Together, these results suggest a possible role for Snx17 as part of the molecular machinery that drives LRP basolateral sorting in polarized cells. Defining the binding partners of Snx17 and establishing a possible role for Snx17 in basolateral sorting should provide important mechanistic insight into the complex process of polarized sorting.
Materials and methods
Plasmids and fusion proteins
Mouse Snx17 cDNA tagged at the 5′ end with a 9xmyc epitope and cloned in pCIneo was kindly provided by Dr J Nimpf (University of Vienna, Austria; see Stockinger et al, 2002). Constructs to produce subdomains of 9myc-Snx17 were prepared by PCR-based subcloning of Snx17 fragments into the same vector. Snx17 and a fragment corresponding to amino acids 105–470 were amplified using Advantage cDNA polymerase (Clontech) and subcloned into pGEX-2T (Pharmacia) to yield GST-Snx17. GST-LRP tail constructs were also produced by PCR and subcloned into pGEX-2T. Mutations in the LRP tail were introduced by site-directed mutagenesis using the Quick Change Mutagenesis kit (Stratagene). All constructs were verified by sequencing. GST fusion proteins were produced in Escherichia coli BL21 strain (Novagen) and purified as described previously (Bu et al, 1995). Constructs of LRP minireceptor in pcDNA3 (mLRP4T100) and the mutant Y29A were described previously (Li et al, 2000).
Plasmid transfection
Human glioblastoma U87 cells (Bu et al, 1994) or HEK293 cells were transfected with various plasmid DNA using Lipofectamine2000 (Invitrogen). Stably transfected U87 cells were selected with 600 μg/ml G418 (Sigma) and maintained with 450 μg/ml G418 in the medium.
GST pull-down assay
Lyophilized GST fusion proteins were dissolved in PBS and centrifuged to remove insoluble material. Glutathione beads were added to yield a final concentration of 1 mg of fusion protein per ml of beads. Following incubation for 2 h at 4°C, beads were washed twice with PBS and resuspended in the same buffer. HEK293 cells transfected with myc-Snx17 plasmids were lysed in HUNT buffer (20 mM Tris–HCl pH 8.0, 100 mM NaCl, 1 mM EDTA, 0.5% NP-40, 50 mM NaF, 1 mM Na3VO4, 1 mM PMSF, and 1 × Complete protease inhibitor). Cell lysates were cleared by centrifugation and aliquots of the supernatant were incubated with 25 μg of fusion protein bound to glutathione beads in HUNT buffer for 2 h at 4°C. Beads were washed twice and boiled in SDS sample buffer.
Co-immunoprecipitation
Cells were lysed on ice in HUNT buffer. Cell lysates were cleared by centrifugation and immunoprecipitated with anti-LRP antibody for 2 h at 4°C (Bu et al, 1995). Immune complexes were isolated using protein A-agarose beads (Repligen) and subjected to SDS–PAGE and Western blotting using anti-LRP or anti-myc antibodies.
In vitro transcription/translation
The TNT Quick coupled transcription/translation system (Promega) was used to produce [35S]methionine-labeled protein with T7 RNA polymerase. Radioactive proteins were visualized after SDS–PAGE using a STORM 820 phosphoimager system from Amersham Biosciences.
Metabolic pulse–chase labeling and immunoprecipitation
Metabolic pulse–chase labeling of mLRP4 with [35S]cysteine was performed as described before (Melman et al, 2002). Cell lysates were incubated with excess anti-HA antibody (Babco) followed by recovery of the immune complexes with protein A-agarose beads. Immunoprecipitated protein was released from the beads by boiling in sample buffer under reducing conditions and analyzed by SDS–PAGE. Radioactive proteins were visualized using a STORM 820 phosphoimager system.
Ligand and antibody iodination, internalization and degradation
α2M or anti-HA IgG was iodinated (50 μg) by the IODOGEN method as described previously (Li et al, 2000). Cells were incubated in serum-free DMEM containing 6 mg/ml BSA and 0.2 nM 125I-α2M or 1 nM 125I-anti-HA and either incubated for 1 h on ice to measure cell surface binding, or for 0.5–4 h at 37°C to measure total endocytic binding and/or degradation. Internalized and degraded ligand was determined as described (Li et al, 2000) with nonspecific/background signal measured in the presence of 500 nM receptor-associated protein (RAP) or excess unlabeled anti-HA antibody and subtracted from the total signal.
Flow cytometric analysis of cell surface LRP
U87 cells were detached by incubation with nonenzymatic cell dissociation solution (Sigma). Detection of cell surface LRP, using monoclonal LRP antibody (clone 8G1, RDI) or monoclonal anti-HA antibody, followed by incubation with goat anti-mouse Ig-FITC (Biosciences-Pharmingen) was described previously (Li et al, 2000). Background fluorescence intensity was assessed in the absence of primary antibody and subtracted. Mean fluorescence values were obtained in triplicate with a FACScalibur (BD Biosciences-Pharmingen), and data were analyzed with Cell Quest software.
Cell surface fluorescence quenching recycling assay
U87 cells stably expressing LRP minireceptors were incubated with Alexa488-labeled anti-HA antibodies for 20 min at 37°C. Following removal of fluorescent antibody in the medium, cells were incubated for indicated times in the absence or presence of 24 μg/ml anti-Alexa488 IgG (Molecular Probes). At the end of each incubation, cells were rapidly chilled, detached, and analyzed by flow cytometry for fluorescence intensity. Percentage of the initial fluorescence (pulse) remaining at each time point was calculated as the difference between nonchased (time 0) and chased cell fluorescence, normalized to the nonchased value.
Protein–lipid overlay assay
The protein–lipid overlay assay has been described previously (Dowler et al, 2002). Briefly, nitrocellulose-immobilized phospholipids at 100 pmol per spot were obtained from Echelon Biosciences Inc. and blocked with 3% fatty acid-free BSA (Sigma) in TBS containing 0.1% Tween 20 for 1 h at room temperature. Membranes were incubated overnight at 4°C with 0.5 μg/ml of the GST fusion protein, followed by washing and detection with anti-GST antibody.
SiRNA and Snx17 knockdown
Four independent siRNAs were designed to investigate their effectiveness on knocking down Snx17 expression. The target sequences were CAUUCACGUGAAUGGAGUC (siRNA2), GUACAUGCAAGCUGUUCGG (siRNA9), GAGUUAUUGGGACUCUGCC (siRNA28), and CUGGCUUUUGAAUACCUCA (siRNA45). All the siRNAs were designed according to the manufacturer's instructions and sense and antisense oligonucleotides were produced by Ambion Inc. Double-stranded siRNA was transfected into U87 cells for 48 h using Lipofectamine2000 (Invitrogen).
Immuno-EM
Cells were fixed by adding 4% freshly prepared formaldehyde in 0.1 M phosphate buffer (pH 7.4) to an equal volume of culture medium for 10 min, followed by postfixation in 4% formaldehyde without medium, and were stored at 4°C. Processing of cells for ultrathin cryosectioning and immuno-labeling by the protein A-gold method was performed as described (Slot et al, 1991).
Supplementary Material
Supplementary Figure 1
Supplementary Figure 2
Acknowledgments
We thank Dr Johannes Nimpf for providing the Snx17 cDNA. We also thank Drs Alan Schwartz, Maria Paz Marzolo, and Jane Knisely for critical reading of the manuscript and members of the Bu laboratory for stimulating discussions. This work was supported by grants from the National Institutes of Heath to GB. GB is an Established Investigator of the American Heart Association. PvK was supported by a travel grant from the Netherlands Organization for Scientific Research (NWO).
References
- Austin CD, De Maziere AM, Pisacane PI, Van Dijk SM, Eigenbrot C, Sliwkowski MX, Klumperman J, Scheller RH (2004) Endocytosis and sorting of ErbB2 and the site of action of cancer therapeutics trastuzumab and geldanamycin. Mol Biol Cell 15: 5268–5282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifacino JS, Traub LM (2003) Signals for sorting of transmembrane proteins to endosomes and lysosomes. Annu Rev Biochem 72: 395–447 [DOI] [PubMed] [Google Scholar]
- Bu G, Geuze HJ, Strous GJ, Schwartz AL (1995) 39 kDa receptor-associated protein is an ER resident protein and molecular chaperone for LDL receptor-related protein. EMBO J 14: 2269–2280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bu G, Maksymovitch EA, Geuze H, Schwartz AL (1994) Subcellular localization and endocytic function of low density lipoprotein receptor-related protein in human glioblastoma cells. J Biol Chem 269: 29874–29882 [PubMed] [Google Scholar]
- Bu G, Sun YL, Schwartz AL, Holtzman DM (1998) Nerve growth factor induces rapid increases in functional cell surface low density lipoprotein receptor-related protein. J Biol Chem 273: 13359–13365 [DOI] [PubMed] [Google Scholar]
- Burden JJ, Sun XM, Garcia Garcia AB, Soutar AK (2004) Sorting motifs in the intracellular domain of the low density lipoprotein (LDL) receptor interact with a novel domain of sorting nexin-17. J Biol Chem 279: 16237–16245 [DOI] [PubMed] [Google Scholar]
- Carlton J, Bujny M, Peter BJ, Oorschot VM, Rutherford A, Mellor H, Klumperman J, McMahon HT, Cullen PJ (2004) Sorting nexin-1 mediates tubular endosome-to-TGN transport through coincidence sensing of high-curvature membranes and 3-phosphoinositides. Curr Biol 14: 1791–1800 [DOI] [PubMed] [Google Scholar]
- Cheever ML, Sato TK, de Beer T, Kutateladze TG, Emr SD, Overduin M (2001) Phox domain interaction with PtdIns(3)P targets the Vam7 t-SNARE to vacuole membranes. Nat Cell Biol 3: 613–618 [DOI] [PubMed] [Google Scholar]
- Chen W-J, Goldstein JL, Brown MS (1990) NPXY, a sequence often found in cytoplasmic tails, is required for coated pit-mediated internalization of the low density lipoprotein receptor. J Biol Chem 265: 3116–3123 [PubMed] [Google Scholar]
- Dowler S, Kular G, Alessi DR (2002) Protein lipid overlay assay. Sci STKE 2002: PL6. [DOI] [PubMed] [Google Scholar]
- Florian V, Schluter T, Bohnensack R (2001) A new member of the sorting nexin family interacts with the C-terminus of P-selectin. Biochem Biophys Res Commun 281: 1045–1050 [DOI] [PubMed] [Google Scholar]
- Gauss R, Trautwein M, Sommer T, Spang A (2005) New modules for the repeated internal and N-terminal epitope tagging of genes in Saccharomyces cerevisiae. Yeast 22: 1–12 [DOI] [PubMed] [Google Scholar]
- Hanson BJ, Hong W (2003) Evidence for a role of SNX16 in regulating traffic between the early and later endosomal compartments. J Biol Chem 278: 34617–34630 [DOI] [PubMed] [Google Scholar]
- Herz J, Bock HH (2002) Lipoprotein receptors in the nervous system. Annu Rev Biochem 71: 405–434 [DOI] [PubMed] [Google Scholar]
- Kanai F, Liu H, Field SJ, Akbary H, Matsuo T, Brown GE, Cantley LC, Yaffe MB (2001) The PX domains of p47phox and p40phox bind to lipid products of PI(3)K. Nat Cell Biol 3: 675–678 [DOI] [PubMed] [Google Scholar]
- Knauth P, Schluter T, Czubayko M, Kirsch C, Florian V, Schreckenberger S, Hahn H, Bohnensack (2005) Functions of sorting nexin 17 domains and recognition motif for P-selectin trafficking. J Mol Biol 347: 813–825 [DOI] [PubMed] [Google Scholar]
- Ko KW, Avramoglu RK, McLeod RS, Vukmirica J, Yao Z (2001) The insulin-stimulated cell surface presentation of low density lipoprotein receptor-related protein in 3T3-L1 adipocytes is sensitive to phosphatidylinositide 3-kinase inhibition. Biochemistry 40: 752–759 [DOI] [PubMed] [Google Scholar]
- Li Y, Marzolo MP, van Kerkhof P, Strous GJ, Bu G (2000) The YXXL motif, but not the two NPXY motifs, serves as the dominant endocytosis signal for low density lipoprotein receptor-related protein. J Biol Chem 275: 17187–17194 [DOI] [PubMed] [Google Scholar]
- Li Y, van Kerkhof PP, Marzolo MP, Strous GJ, Bu G (2001) Identification of a major cyclic AMP-dependent protein kinase A phosphorylation site within the cytoplasmic tail of the low-density lipoprotein receptor-related protein: implication for receptor-mediated endocytosis. Mol Cell Biol 21: 1185–1195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzolo MP, Yuseff MI, Retamal C, Donoso M, Ezquer F, Farfan P, Li Y, Bu G (2003) Differential distribution of low-density lipoprotein-receptor-related protein (LRP) and megalin in polarized epithelial cells is determined by their cytoplasmic domains. Traffic 4: 273–288 [DOI] [PubMed] [Google Scholar]
- Melman L, Geuze HJ, Li Y, McCormick LM, Van Kerkhof P, Strous GJ, Schwartz AL, Bu G (2002) Proteasome regulates the delivery of LDL receptor-related protein into the degradation pathway. Mol Biol Cell 13: 3325–3335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raiborg C, Bache KG, Gillooly DJ, Madshus IH, Stang E, Stenmark H (2002) Hrs sorts ubiquitinated proteins into clathrin-coated microdomains of early endosomes. Nat Cell Biol 4: 394–398 [DOI] [PubMed] [Google Scholar]
- Sachse M, Urbe S, Oorschot V, Strous GJ, Klumperman J (2002) Bilayered clathrin coats on endosomal vacuoles are involved in protein sorting toward lysosomes. Mol Biol Cell 13: 1313–1328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schapiro FB, Soe TT, Mallet WG, Maxfield FR (2004) Role of cytoplasmic domain serines in intracellular trafficking of furin. Mol Biol Cell 15: 2884–2894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider WJ, Nimpf J (2003) LDL receptor relatives at the crossroad of endocytosis and signaling. Cell Mol Life Sci 60: 892–903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seaman MN, Williams HP (2002) Identification of the functional domains of yeast sorting nexins Vps5p and Vps17p. Mol Biol Cell 13: 2826–2840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonsen A, Wurmser AE, Emr SD, Stenmark H (2001) The role of phosphoinositides in membrane transport. Curr Opin Cell Biol 13: 485–492 [DOI] [PubMed] [Google Scholar]
- Slot JW, Geuze HJ, Gigengack S, Lienhard GE, James DE (1991) Immuno-localization of the insulin regulatable glucose transporter in brown adipose tissue of the rat. J Cell Biol 113: 123–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockinger W, Sailler B, Strasser V, Recheis B, Fasching D, Kahr L, Schneider WJ, Nimpf J (2002) The PX-domain protein SNX17 interacts with members of the LDL receptor family and modulates endocytosis of the LDL receptor. EMBO J 21: 4259–4267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanowitz M, von Zastrow M (2003) A novel endocytic recycling signal that distinguishes the membrane trafficking of naturally occurring opioid receptors. J Biol Chem 278: 45978–45986 [DOI] [PubMed] [Google Scholar]
- Touret N, Furuya W, Forbes J, Gros P, Grinstein S (2003) Dynamic traffic through the recycling compartment couples the metal transporter Nramp2 (DMT1) with the transferrin receptor. J Biol Chem 278: 25548–25557 [DOI] [PubMed] [Google Scholar]
- Tran DD, Russell HR, Sutor SL, van Deursen J, Bram RJ (2003) CAML is required for efficient EGF receptor recycling. Dev Cell 5: 245–256 [DOI] [PubMed] [Google Scholar]
- Vargas GA, Von Zastrow M (2004) Identification of a novel endocytic recycling signal in the D1 dopamine receptor. J Biol Chem 279: 37461–37469 [DOI] [PubMed] [Google Scholar]
- Weaver AM, Mccabe M, Kim I, Allietta MM, Gonias SL (1996) Epidermal growth factor and platelet-derived growth factor-bb induce a stable increase in the activity of low density lipoprotein receptor-related protein in vascular smooth muscle cells by altering receptor distribution and recycling. J Biol Chem 271: 24894–24900 [DOI] [PubMed] [Google Scholar]
- Williams R, Schluter T, Roberts MS, Knauth P, Bohnensack R, Cutler DF (2004) Sorting nexin 17 accelerates internalization yet retards degradation of P-selectin. Mol Biol Cell 15: 3095–3105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worby CA, Dixon JE (2002) Sorting out the cellular functions of sorting nexins. Nat Rev Mol Cell Biol 3: 919–931 [DOI] [PubMed] [Google Scholar]
- Xu Y, Hortsman H, Seet L, Wong SH, Hong W (2001) SNX3 regulates endosomal function through its PX-domain-mediated interaction with PtdIns(3)P. Nat Cell Biol 3: 658–666 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2