

Abstract
By using time-resolved x-ray crystallography at room temperature, structural relaxations and ligand migration were examined in myoglobin (Mb) mutant L29W from nanoseconds to seconds after photodissociation of carbon monoxide (CO) from the heme iron by nanosecond laser pulses. The data were analyzed in terms of transient kinetics by fitting trial functions to integrated difference electron density values obtained from select structural moieties, thus allowing a quantitative description of the processes involved. The observed relaxations are linked to other investigations on protein dynamics. At the earliest times, the heme has already completely relaxed into its domed deoxy structure, and there is no photodissociated CO visible at the primary docking site. Initial relaxations of larger globin moieties are completed within several hundred nanoseconds. They influence the concomitant migration of photodissociated CO to the Xe1 site, where it appears at ≈300 ns and leaves again at ≈1.5 ms. The extremely long residence time in Xe1 as compared with wild-type MbCO implies that, in the latter protein, the CO exits the protein from Xe1 predominantly via the distal pocket. A well-defined deligated state is populated between ≈2 μs and ≈1 ms; its structure is very similar to the equilibrium deoxy structure. Between 1.5 and 20 ms, no CO is visible in the protein interior; it is either distributed among many sites within the protein or has escaped to the solvent. Finally, recombination at the heme iron occurs after >20 ms.
Keywords: kinetics, Laue crystallography, protein relaxation
Proteins are not rigid molecules but fluctuating entities. They can adopt a large number of different conformations that can be depicted as local minima on a rugged energy surface (1). The dynamics of proteins shows strong analogies to the dynamics of viscous liquids. Motions occur on time scales from ≈10–14 s to several thousands of seconds. Time and temperature are important determinants of protein motions on the energy landscape. At ambient temperature, a protein might be able to explore the entire landscape within a certain time interval, whereas it will become increasingly confined in a small region of its conformational space as the temperature is decreased (2, 3). Above a characteristic temperature Tc (≈180 K), quasidiffusive motions become important (4, 5). A significant fraction of conformational transitions occurs between 100 ps and a few nanoseconds in the case of myoglobin (Mb). Below Tc, these fluctuations become more and more arrested. Proteins where larger motions are required for their function will cease to work.
One method of studying protein dynamics is to generate a nonequilibrium state, for example, by changing the sample pressure, temperature, the concentrations of reaction components in fast mixing experiments, or by photodissociating ligands from proteins using short laser pulses. In a crystalline sample, the subsequent relaxation can be followed on the atomic scale by time-resolved x-ray crystallography (6–10) and interpreted kinetically after collection of a sufficiently large number of time points (7, 11–13). Numerous time-resolved experiments have been performed on Mb, a small heme protein that binds diatomic ligands such as carbon monoxide (CO) and dioxygen (O2) to a ferrous iron in the center of a heme prosthetic group.
The bond between the ligand and the heme iron can be broken by light (14). The subsequent ligand migration and recombination process has been investigated in great detail, both with spectroscopy and x-ray diffraction (6–10, 15–31). Three intermediate ligand-docking sites have been described. Site B is found on the distal side (labeled BI in Fig. 1B), with the CO oriented parallel to the heme plane (6, 21–23, 25, 32). Additionally, the ligand can be trapped in the so-called xenon cavities Xe4 (33) on the distal side and Xe1 on the proximal side of the heme (7, 10, 29–31, 34, 35) (Fig. 1B).
Fig. 1.

Architecture of myoglobin. (A) Ribbon diagram of L29W MbCO superimposed on a difference map obtained 4 s after photolysis. Structural moieties (heme as well as helices) are marked. (B) Close-up of the active site showing the following: BI, initial docking site suggested from low temperature spectroscopic investigations (29); Xe1 and Xe4, positions of CO in x-ray structures determined by photolysis experiments at low temperatures (26, 37); site W29, dotted circle, weakly populated site close to the tip of Trp-29 (see text). Figure was prepared with ribbons (57).
Mb mutant L29W has a very small CO association rate coefficient at ambient temperature, ≈130 times smaller than that of wild-type (WT) MbCO (29, 36). Therefore, this mutant is particularly well-suited for time-resolved crystallography studies because the slow rebinding enables us to follow structural relaxations to completion. To describe the relaxation processes in L29W MbCO after photolysis in atomic detail, we collected 20 x-ray data sets over 9 orders of magnitude in time by using time-resolved crystallography. This work extends previous studies on WT MbCO and mutants L29F and YQR (8–10) because the large number of time points permits us to fit the observed time courses with analytical functions and enables a quantitative analysis and detailed description of both the CO migration process and the accompanying structural rearrangements in various globin moieties (11, 12). A similar approach was used to fit the time courses of CO occupancy at select docking sites in WT MbCO (7).
Materials and Methods
Sample Preparation. L29W MbCO crystals (typical volume of ≈0.03 mm3; space group P6 with unit cell size a = b = 91.54 Å, c = 45.87 Å, α = β = 90°, γ = 120°) were grown from genetically engineered protein (37). Crystals were mounted in glass capillaries in a CO atmosphere.
Data Collection. Time-resolved crystallography was performed at the BioCARS Laue beamline 14-ID at the Advanced Photon Source at 15°C by using the pump-probe technique (38). For reaction initiation, we used 615-nm light from a Nd:YAG pumped dye laser (Continuum, Santa Clara, CA), with a pulse width (full width at half maximum) of 7 ns. The structure was probed by an intense, polychromatic x-ray beam from the Advanced Photon Source Undulator A. The duration of the x-ray pulses was chosen according to the time delay between pump and probe pulses (for additional experimental details see Table 2, which is published as supporting information on the PNAS web site). The time delay refers to the center-to-center delay between pump laser and probe x-ray pulse. For each time point, dark and light exposures were collected in an alternating mode until a sufficiently large volume of reciprocal space was sampled. For a single diffraction image, multiple x-ray exposures were typically superimposed on the detector, with a 6-s wait time between exposures. Images from 24 crystal orientations separated by 3° comprised a data set.
Data Reduction. The Laue images were indexed, integrated and scaled with precognition and epinorm (RenzResearch, Westmont, IL) or laueview (39). At ambient temperature, the reference structure L29W MbCOL was determined from a dark Laue data set (see below). Weighted time-dependent difference maps (40, 41) were calculated from weighted difference structure factor amplitudes 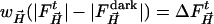 and phases derived from the reference L29W MbCOL structure.
and phases derived from the reference L29W MbCOL structure. 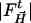 are the time-dependent (light) structure factor amplitudes obtained after photolysis and
are the time-dependent (light) structure factor amplitudes obtained after photolysis and 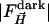 are the corresponding reference structure factor amplitudes at
are the corresponding reference structure factor amplitudes at 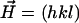 . The weighting factor
. The weighting factor 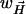 was calculated, as
was calculated, as 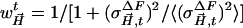 , where
, where 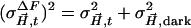 is the squared error of a particular difference structure factor amplitude and
is the squared error of a particular difference structure factor amplitude and 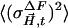 is the mean square error of all
is the mean square error of all 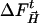 in a data set.
in a data set.
Data Analysis. Difference maps were integrated in selected regions by the program promsk (available from the authors): within a mask, which is automatically generated by supplying atomic coordinates of a structural moiety (Fig. 1), the program adds up negative and positive difference electron density values above or below a chosen threshold that was set to ±2.0σ throughout. Absolute values were summed to capture the total integrated density of both positive and negative features associated with structural changes. To estimate the error (error bars in Fig. 2), a similar integration was performed on a difference map free of signal (4-s time delay). To partially correct for variations in the extent of reaction initiation, each difference map was multiplied by a factor E(t) =〈EL〉/EL(t), with the actual laser pulse energy used for the Laue data set at time delay t, EL(t), and the average pulse energy 〈EL〉 = 2.8 mJ. In the time range from 1 ns to 1.3 ms, the maps were further multiplied by a factor S(t), derived by assuming that no rebinding had occurred. This finding is justified by the observation that the integrated CO difference density did not show any decay within this time interval; only noise is present that can be attributed in part to differences in the extent of photolysis from crystal to crystal. The factor S(t) corrects for these differences and normalizes the negative, integrated electron counts at the CO binding site to about the same value. The corrected maps were used for the subsequent analysis.
Fig. 2.

Integrated difference electron density values from different structural moieties of L29W Mb plotted as a function of (log) time. To be comparable, the values are divided by the number of residues (molecules) in the moieties. (A) •, heme features; solid line, bimolecular rebinding; ▴, negative CO density at the heme iron, COd, uncorrected maps; ▵, corrected maps; solid line, bimolecular rebinding. (B) Comparison of negative COd density corresponding to photolyzed CO and positive density corresponding to CO found at docking sites. •, COp, density in the proximal (Xe1) site; solid line, fit by exponential build-up and decay functions; □ COW29 density feature close to Trp-29; dashed line, fit by exponential build-up and bimolecular decay functions; ▵, COd; dotted line, fit by bimolecular function. Note the different ordinate scales for negative and positive values. (C) Relaxation of helices E and F. •, E-helix; solid line, initial phase stretched exponential, final relaxation follows bimolecular rebinding; dashed line, initial, fast phase as well as final, slow phase exponential; ▪, F-helix; dotted line to guide the eye. (D) Relaxation of helices B and H. •, B-helix; solid line, initial phase stretched exponential, final relaxation follows bimolecular rebinding; ▪, H-helix; dotted line to guide the eye. (E) Relaxation of side chains. •, His-64 and Trp-29; solid line, initial phase stretched exponential, final relaxation follows bimolecular rebinding; dashed line, subnanosecond phase assumed, nanosecond time points fit by exponential; ▪, Phe-43 and Phe-46, scale expanded twofold; dotted line, initial phase stretched exponential; final relaxation follows bimolecular rebinding. (F) Normalized fit results. Solid line, heme and COd; dotted line, averaged relaxations of the structural moieties (M); dashed line, COp.
The time-dependent, integrated difference electron densities were plotted as a function of time and fitted with a sum of two exponential functions
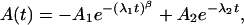 |
[1] |
where the first term accounts for the fast initial phase and the second accounts for the slow final relaxation phase. The first exponential can be stretched by varying the parameter β between 0 and 1. Alternatively, we have used a sum of a (stretched) exponential function and a term corresponding to bimolecular rebinding
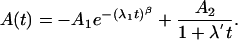 |
[2] |
The second term in Eq. 2 was calculated by assuming that the concentrations of deligated Mb and free CO are equal at all times (42). The amplitudes A1, A2, the stretching parameter β, and the apparent rate coefficients λ1, λ2, and λ′ as well as the times t1/2 when half of the molecules have relaxed into or from a particular state are summarized in Table 1. A global analysis of the difference density for the entire molecule was performed by using singular value decomposition (11) whose time courses also were fitted by Eq. 1 or 2.
Table 1. Fitting parameters corresponding to time courses of different structural moieties in L29W MbCO.
|
t1/2
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Fast,‡ | Slow,‡ | ||||||||
| Moiety* | A1,e- | λ1, 1/s | β | A2,e- | λ2, 1/s | λ′, 1/s | Type† | ns | ms |
| COd corrected | 0 | 0 | 0 | 5.2 ± 0.3 | 50 ± 20* | B | <1 | 19 | |
| Heme | 0 | 0 | 0 | 8.3 ± 0.3 | 50 ± 20 | B | <1 | 19 | |
| COp | 1.67 ± 0.01 | (2.60 ± 0.03) × 106 | 0.8 | 1.7 ± 0.1 | 455 ± 5 | S + E | 243 | 1.5 | |
| COp | 1.62 ± 0.01 | (2.30 ± 0.09) × 106 | 1.0 | 1.7 ± 0.1 | 454 ± 2 | E + E | 301 | 1.5 | |
| COW29 | 0.5 ± 0.1 | 159.2 ± 0.6 | 1.0 | 0.5 ± 0.02 | 15 ± 0.1 | E + E | 4000 | 47 | |
| E-helix | 1.75 ± 0.01 | (1.00 ± 0.01) × 108 | 0.36 ± 0.12 | 1.75 ± 0.06 | 36.2 ± 0.4 | S + B | 4 | 28 | |
| E-helix | 1.68 ± 0.02 | (1.7 ± 0.1) × 108 | 1.0 | 1.68 ± 0.02 | 25 ± 8 | E + E | 6 | 27 | |
| B-helix | 1.23 ± 0.06 | (1.00 ± 0.01) × 108 | 0.36 ± 0.12 | 1.23 ± 0.06 | 36.2 ± 0.4 | S + B | 3 | 28 | |
| CD-turn | 1.61 ± 0.50 | (1.4 ± 0.1) × 108 | 0.36 | 1.61 ± 0.50 | 68 ± 9 | S + B | 4 | 15 | |
| H64 and W29 | 21.7 ± 0.4 | (4.0 ± 1.0) × 108 | 0.36 | 21.7 ± 0.4 | 45 ± 10 | S + B | 2 | 22 | |
| H64 and W29 | 11.1 ± 1.9 | (1.1 ± 0.5) × 108 | 1.0 | 21.5 ± 0.4 | 44 ± 10 | E + B | 6 | 23 | |
| F46 and F43 | 5.0 ± 0.4 | (2.2 ± 1.1) × 108 | 0.36 | 5.0 ± 0.4 | 50 ± 10 | S + B | 2 | 22 | |
| 1st rSV§ | -392 ± 9 | (8.3 ± 4.1) × 108 | 0.5 ± 0.1 | -392 ± 9 | 20 ± 6 | S + E | 0.6 | 35 | |
| 1st rSV§ | -393 ± 11 | (8.2 ± 3.6) × 108 | 0.5 ± 0.1 | -393 ± 11 | 27 ± 8 | S + B | 0.6 | 37 | |
In most cases, one relaxation phase is assumed (-A1 = A2).
E, exponential; S, stretched exponential; B, bimolecular rebinding (according to Eq. 1 or 2).
Exponential, t1/2 = 1/λi In 2; stretched exponential: t1/2 = 1/λ1(In 2)1/β; bimolecular, t1/2 = 1/λ′, t1/2 smaller than our time-resolution (3 ns) estimated by extrapolating the fit to faster times.
rSV, right-singular vector.
Structure Determination. Because the orientation of the Trp-29 side chain is slightly temperature dependent (37), the reference structure L29W MbCOL was determined from dark Laue data. For the refinement, the room temperature L29W MbCOM structure previously obtained from monochromatic data (37) was used as a start. To determine the Trp-29 conformation, a simulated annealing omit electron density map was calculated, omitting Trp-29 from the refinement. Two slightly different Trp-29 conformations were inserted in the elongated electron density and refined in cns (43) by using standard techniques. The final values of Rcryst and Rfree were 21.5% and 24.9%, respectively.
To determine the structure of the photoproduct MbTR (TR for time-resolved), six difference maps were averaged in the time interval between 2 and 110 μs. Difference structure factors 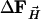 were calculated by Fourier inversion. A multiple n = 3.5 of the
were calculated by Fourier inversion. A multiple n = 3.5 of the 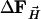 was added to the structure factors
was added to the structure factors 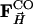 derived from the MbCOL model, yielding extrapolated structure factors
derived from the MbCOL model, yielding extrapolated structure factors 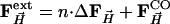 . From the extrapolated structure factors, a map was calculated into which a model was built which was refined against the extrapolated amplitudes by using cns (43).
. From the extrapolated structure factors, a map was calculated into which a model was built which was refined against the extrapolated amplitudes by using cns (43).
Results and Discussion
The difference electron density maps for the entire protein and close-ups of the heme pocket are available for all 20 time points in Fig. 5 and Movies 1 and 2, which are published as supporting information on the PNAS web site. Substantial signal appears already at the shortest time delays, as shown by the red (negative) and blue (positive) difference electron densities. To describe the time evolution of the signal in different structural moieties (compare Fig. 1 A), the difference density features were integrated and fitted with Eqs. 1 and 2 (Table 1 and Fig. 2).
Fig. 2 A shows the negative difference density of CO at the heme iron due to photolysis (COd). As is apparent from the full triangles, the electron count is nearly constant up to ≈1.3 ms and decreases with CO rebinding, with t1/2 = 19 ms. The open triangles are the values from maps corrected further by the factor S(t). Because 5.3 electrons are photolyzed and a CO molecule has 14 electrons, the photolysis yield is ≈38%. The integrated absolute (total) difference density values at and close to the heme closely track the values obtained for CO (Fig. 2 A).
After ≈100 ns, a positive electron density feature appears at the secondary docking site Xe1 on the proximal side of the heme (Fig. 1B). The time course of this feature (COp) is shown in Fig. 2B. The occupancy of the Xe1 site reaches a constant value of 1.7 electrons after 1 μs, which corresponds to only 12% of the total and 32% of the photodissociated CO. CO exits Xe1 after ≈1 ms with a time course that can be fitted by Eq. 1, with β = 1.0. Depopulation of Xe1 is significantly faster than rebinding of CO at the heme iron (Fig. 2B and Table 1). We also have identified an accumulation of density near Trp-29 (COW29; Fig. 2B, open squares and dashed line; Fig. 1B, dashed circle) on a time scale comparable to the depletion of Xe1. Therefore, this site may be populated by CO shortly before rebinding.
Fig. 2 C and D show results from the integration of difference density in various parts of the globin: helices B, E, F, and H. The difference densities for helices B and E were fitted with the same rate parameters (Table 1). The dashed line in Fig. 2C results from a fit of the initial relaxation phase of helix E with an exponential (β = 1), whereas the solid line was obtained with β = 0.36. This value was kept in the fits of all other time courses. Helices B and E show strong relaxations (Fig. 2 C and D). By contrast, the structural changes of helices F and H are only minor (Fig. 2 C and D, with dotted lines to guide the eye). In all cases, the initial relaxation is followed by a plateau region during which no relaxations occur. It is reached after ≈100 ns and prevails up to a few milliseconds. The final decay happens in concert with CO rebinding, with t1/2 ≈ 25 ms (Table 1). The integrated difference densities for the His-64 (helix E) and Trp-29 (helix B) side chains (Fig. 2E) show the same temporal behavior and thus were analyzed together. Similar arguments hold for the CD-turn residues Phe-43 and Phe-46, which also were treated in conjunction. Their time courses closely follow the global relaxations of the corresponding helices (Table 1).
Normalization of the maximum amplitudes of the various relaxation processes to 1 allows a direct comparison of the time courses (Fig. 2F). It is obvious that the time scales of the initial relaxation vary substantially, with the heme relaxation being much faster than those of other globin moieties. After the initial relaxation, the various parts of the protein remain fully relaxed up to several milliseconds. This result is corroborated by the global singular value decomposition analysis, from which only a single significant right-singular vector is obtained that assumes a constant value from 800 ns to 1 ms (see Fig. 6, which is published as supporting information on the PNAS web site). The positive feature at the Xe1 site, corresponding to photolyzed CO molecules, remains constant from ≈1 to 110 μs (Fig. 2F). Therefore, only the maps between 2 and 110 μs represent a fully relaxed state. Return to the MbCO state is initiated by the escape of COp from the Xe1 site after 1.5 ms. However, CO rebinding at the heme iron is an order of magnitude slower, with t1/2 = 19 ms (Table 1).
In their pioneering flash photolysis experiments at room temperature, Gibson and Ainsworth (14) observed surprisingly simple ligand rebinding kinetics in heme proteins. Only much later, the actual complexity of this process was demonstrated by flash photolysis experiments on Mb at low temperatures (15). In low-temperature kinetic studies, different active-site conformations were apparent on time scales shorter than the time it takes for a protein to fluctuate between these substates (44). Time-resolved crystallography on WT MbCO was used to establish a structural basis for the observed rebinding kinetics (6, 7, 10). The CO ligand was shown to access different transient docking sites on its migration pathway through the protein matrix. However, in WT MbCO crystals, CO rebinds on the microsecond time scale (7), which may be faster than the time it takes the protein to completely relax to a deoxy-like structure. In Mb mutant YQR, recombination is considerably slower, but in a recent study, only five time-points were collected from 3 ns to 3 ms, which did not permit a quantitative kinetic analysis (8). In Mb mutant L29W, rebinding is even slower, which enables us to observe complete protein relaxation after CO dissociation. Below, we discuss the structural transitions of L29W MbCO after photolysis on various time scales, from the fastest times through completion of the reaction.
Events Faster than Our Experimental Time Resolution: Subnanosecond Time Range. Photolysis of CO and heme relaxation occur faster than the time resolution of our experiment (Fig. 2 A). This observation is consistent with spectroscopic investigations on WT MbCO (25) and time-resolved crystallographic studies on WT and mutant Mbs (7–9). In WT MbCO, the heme group relaxes within a time range spanning from hundreds of femtoseconds to a few picoseconds (45, 46). L29W Mb is most likely similar in this respect. The photodissociation of the CO from the heme iron and the subsequent very fast relaxation of the heme trigger substantial relaxations of the entire protein. As a consequence, kinetic traces for the different protein moieties start at roughly half their maximum amplitude (Fig. 2).
Even at the earliest times, CO was observed neither at the primary docking site B nor in Xe4, although site B is populated at room temperature in WT MbCO (6) and Xe4 at cryogenic temperatures in L29W MbCO (26). Apparently, the initial CO migration away from the distal side happens extremely fast. In mutant L29F, photodissociated ligands are found at site B after 100 ps and escape from there to the Xe4 site within 1 ns where they reside until ≈10 ns (9). In L29W, however, the bulky Trp-29 residue relaxes partially toward the deoxy conformation on the subnanosecond time scale (Fig. 2E). Hence, it may not only expel ligands from site B but also block access to Xe4. Additionally, Trp-29 apparently forces ligands that have rapidly escaped to Xe4 to leave this site and to migrate further. For the same reason, an initial geminate rebinding phase for CO is missing (26, 28–29), whereas a fast geminate phase has been observed for O2 (29). Because no docking site could be identified up to 100 ns, CO must be distributed between multiple locations so that its electron density at an individual site is too low to be observable. To actually resolve the occupancy of sites B and Xe4, it is necessary to follow photoactivation on a time scale shorter than the initial relaxation time of the Trp-29 side chain. However, typical relaxation times for side chains at room temperature are on the order of tens of picoseconds (47), a time scale currently not accessible to time-resolved x-ray diffraction. Note that after the fast relaxation of Trp-29, an additional, slower process is coupled to the backbone relaxation (see below).
Protein Relaxations on the Nanosecond Time Scale. The initial relaxation of the globin is already about half complete at our shortest observation times and, therefore, its t1/2 can only be estimated. If we interpret the data with a single phase, the first term in Eqs. 1 and 2 must be stretched to obtain acceptable fits, with t1/2 values ≈ 5 ns (Table 1). This result is consistent with typical time scales of quasidiffusive motions (diffusive motions in restricted space) of Mb that are on the order of 100 ps to a few nanoseconds (3, 5). The relaxations extend up to 1 μs (Fig. 2), and a number of these motions are necessary. A stretched initial phase also has been reported from time-resolved spectroscopy (20, 28, 48) and indicates a complicated dynamics on fast time scales. In the case of a simple kinetic mechanism (49, 50), the ensemble of molecules populates well-defined states, and interconversion between these states is described by single exponential relaxations. A simple, multistep mechanism was applicable to time-resolved x-ray data from photoactive yellow protein (12, 13) because exponential transient kinetics was observed throughout. For the initial relaxation in Mb, however, a nonexponential (stretched) phase is observed, excluding a simple mechanism. Initial relaxation may involve (i) interconversions between multiple substates on the relaxation pathway across the energy surface. The characteristic times for molecules to fluctuate between substates are comparable to the times they stay in these states. (ii) In addition, one may envision multiple, asynchronous relaxation pathways on different time scales as suggested for YQR Mb (8). However, with the present data, we are not able to distinguish between these two possibilities. One may even speculate that a separate kinetic phase exists on time scales faster than 1 ns and that the phase we observe here only starts at ≈1 ns. In that case, an exponential fits part of the data reasonably well (Fig. 2E, dashed line). To determine t1/2 more accurately, the measurements need to be extended to the picosecond time range.
Residues including His-64, Trp-29, Phe-43, and Phe-46, which are integral parts of the heme pocket, display the largest difference densities (Table 1 and Fig. 2E). Although the side chain relaxation sets in on the subnanosecond time scale, it proceeds until ≈100 ns, because these side chains are part of larger structural entities such as helix E or the CD-turn, which relax more slowly. Only after the global, large-scale motions are completed do the side chains reach their final position.
CO appears at the Xe1 site with t1/2 = 300 ns. The site is populated in an exponential fashion, substantially more slowly than in WT MbCO and mutant YQR (both with t1/2 ∼ 50 ns) (7, 8). For L29F Mb, theoretical calculations have predicted that cavities Xe4 and Xe2 are involved in CO migration (51). Spectroscopic investigations have shown that Xe1 is the primary sink of CO at temperatures above the characteristic temperature where protein dynamics becomes important (29). Taking into account low-and high-temperature dynamics studies on Mb (3, 5, 52) and relating them to our time-resolved results at room temperature, we propose that two fundamental processes are responsible for CO migration in Mb. (i) Protein dynamics enables structural fluctuations and transiently opens and closes channels for the CO to migrate to positions where it is trapped effectively. (ii) The initial (subnanosecond to 100 ns) structural relaxation changes the overall globin structure, thereby significantly affecting the rate coefficients for CO migration. In L29W Mb, it is the fast relaxation of the Trp-29 side chain that markedly affects ligand migration. CO can only effectively migrate to Xe1 in the presence of protein fluctuations and hence above the characteristic temperature Tc (37). However, the same protein relaxations block the pathway to Xe1 by occluding access to Xe4. As a consequence, ligand migration to Xe1 becomes inefficient in L29W, so that 2/3 of the photolyzed CO molecules cannot be found in Xe1 and most likely have escaped into the solvent.
The Intermediate Time Scale: Photoproduct State MbTR. Relaxations of all globin parts toward the deoxy structure and CO migration are complete after 1 μs. Up to ≈100 μs, no additional processes take place. Therefore, the six maps in that time interval (Fig. 3) were averaged to enhance the signal-to-noise ratio (53). The red model in Fig. 3 represents the reference MbCOL structure; the structure of the photoproduct MbTR determined from an extrapolated map is shown in blue. The extra density in the proximal Xe1 cavity is consistent with the presence of a CO molecule. Because 12% of the total CO are accounted for in this site and the factor n is 3.5 (see Materials and Methods), the occupancy is expected to be 42%. Indeed, it refines to 60%. Negative as well as positive density features are explained notably well by these two models (compare difference densities for the heme, Trp-29, His-64, Phe-43, and Phe-46 in Fig. 3).
Fig. 3.

The short-lived L29W Mb state. (A) Difference electron density averaged from 2 to 110 μs; contour levels: blue, 4σ; red, –4σ. The refined model of the relaxed, deoxy-like photoproduct is shown in blue, and that of the reference L29W MbCO structure is in magenta. (B) Same as A, but rotated ≈90° about the heme normal. Figure was prepared with o (58).
The displacements between Cα atoms of corresponding residues of the refined L29W MbCOL and L29W MbTR structures (Fig. 4, black line) show the strongest deviations of up to 0.8 Å for helices B and E and the CD-turn; the F-helix and parts of the G-helix have only small displacements of ≈0.2 Å on average. The other parts of the structure remain essentially unchanged. Structural changes are concentrated on the distal side of the heme, as already observed from a comparison of the static structures (Fig. 4, red line). The heme iron does not move toward the proximal side. Rather, the entire heme is pushed to the distal side so that the deligated iron can move out of the heme plane. Heme doming and CO removal trigger the protein relaxations. Within the error (≈0.15 Å), the structure of L29W MbTR is identical to the static L29W MbM structure (Fig. 4, blue line) and the L29W MbCryo photoproduct structure obtained by photolysis at ≈180 K (26) (not shown in Fig. 4).
Fig. 4.

Cα–Cα distances for superimposed L29W structures. Black line, L29W MbTR (short-lived photoproduct state) vs. L29W MbCOL (reference structure, from dark Laue data); red line, static L29W MbM vs. static L29W MbCOM, both determined from monochromatic data (37); blue line, comparison of deoxy Mb like structures, L29W MbRT (short-lived photoproduct state, this study) vs. static L29W MbM.
Millisecond Time Scale: Recovery of L29W MbCO. Because the bulky Trp-29 side chain blocks access not only to the heme iron but also to docking site B, rebinding is severely hindered (29, 37). The slow relaxation phase observed for all protein moieties, with t1/2 ∼ 25 ms, matches the ligand rebinding kinetics (Fig. 2 A) and therefore represents the decay in population of the deoxy-like photoproduct state (Table 1). Ligand rebinding is ≈150 times slower than in WT MbCO crystals, where t1/2 is ≈150 μs (7). One CO molecule is bound to each of the six Mbs in the hexagonal unit cell. Given the size of the unit cell (V = 332,875 Å3), the concentration of heme-bound CO in the crystal is 30 mM. After the flash, 38% or 11.4 mM CO is liberated. The bimolecular rate coefficient λS can be estimated from this concentration and the fitted, apparent rate coefficient λ′ as λS = λ′/[CO]. The result is ≈4.6 mM–1·s–1, in excellent agreement with λS = 3.9 mM–1·s–1 in solution (26, 36).
CO leaves the proximal Xe1 site with t1/2 = 1.5 ms (Fig. 2F), much more slowly than in WT Mb (7). If there was a major proximal path to the solvent, depletion of Xe1 should be on a similar time-scale in both WT and L29W MbCO. Hence, in WT Mb, CO may quickly return via the open cavities Xe2 and Xe4 to the distal side (29). It may exit to the solvent presumably through the histidine gate before it rebinds (54).
In L29W, the Trp-29 side chain acts as a dynamic plug that closes the distal cavities shortly (<1 ns) after the CO is photolyzed. A similar role has been suggested for the distal deoxy water, which enters the distal pocket after a few microseconds (55), blocks site B, and may keep CO in the cavities (56). Ligand escape from Xe1 along the Xe2–Xe4 pathway can occur only upon rare transient fluctuations of the Trp-29 side chain that reopen this pathway toward the binding site. Additional sites inside the protein may be populated on the migration pathway to the distal side. We have identified a putative site close to Trp-29 (Fig. 1B); its occupation with time is shown in Fig. 2B. However, the concentration of CO in this site is too low to account for all CO that has left Xe1. Based on the present data, we cannot exclude that CO escapes via a direct proximal pathway from Xe1 in the L29W mutant and then rebinds from the solvent.
Concluding Remarks
By using time-resolved crystallography, we have kinetically and structurally characterized the relaxation processes on multiple time-scales after ligand photodissociation in L29W Mb. In addition, insights were obtained on the ligand migration pathways in both L29W and WT Mb.
Supplementary Material
Acknowledgments
We thank Keith Moffat for reading and commenting on an earlier version of this manuscript. This work was supported by Deutsche Forschungsgemeinschaft Grants SFB533 (to M.S.) and Ni291/3 and SFB596 (to G.U.N.), the Fonds der Chemischen Industrie (G.U.N. and F.P.), and National Institutes of Health/National Center for Research Resources Grant RR07707 (to R.P. and V.Š.).
Author contributions: M.S. and V.Š. designed research; M.S., R.P., A.K., S.A., and V.Š. performed research; M.S., G.U.N., and K.N. contributed new reagents/analytic tools; M.S., A.K., F.P., and V.Š. analyzed data; and M.S., F.P., G.U.N., and V.Š. wrote the paper.
Abbreviation: Mb, myoglobin.
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.pdb.org [PDB ID codes 2bw9 (MbRT) and 2bwh (MbCOL)].
References
- 1.Fenimore, P. W., Frauenfelder, H., McMahon, B. H. & Parak, F. G. (2002) Proc. Natl. Acad. Sci. USA 99, 16047–16051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Achterhold, K., Sturhahn, W., Alp, E. E. & Parak, G. (2002) Hyperfine Interact. 141/142, 3–12. [Google Scholar]
- 3.Huenges, A., Achterhold, K. & Parak, F. G. (2003) Hyperfine Interact. 144/145, 209–222. [Google Scholar]
- 4.Parak, F., Knapp, E. W. & Kucheida, D. (1982) J. Mol. Biol. 161, 177–194. [DOI] [PubMed] [Google Scholar]
- 5.Parak, F. (2003) Rep. Prog. Phys. 66, 103–129. [Google Scholar]
- 6.Šrajer, V., Teng, T., Ursby, T., Pradervand, C., Ren, Z., Adachi, S., Schildkamp, W., Bourgeois, D., Wulff, M. & Moffat, K. (1996) Science 274, 1726–1729. [DOI] [PubMed] [Google Scholar]
- 7.Šrajer, V., Ren, Z., Teng, T. Y., Schmidt, M., Ursby, T., Bourgeois, D., Pradervand, C., Schildkamp, W., Wulff, M. & Moffat K. (2001) Biochemistry 40, 13802–13815. [DOI] [PubMed] [Google Scholar]
- 8.Bourgeois, D., Vallone, B., Schotte, F., Arcovito, A., Miele, A. E., Sciara, G., Wulff, M., Anfinrud, P. & Brunori, M. (2003) Proc. Natl. Acad. Sci. USA 100, 8704–8709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schotte, F., Lim, M., Jackson, T. A., Smirnov, A. V., Soman, J., Olson, J. S., Phillips, G. N., Jr., Wulff, M. & Anfinrud, P. A. (2003) Science 300, 1944–1947. [DOI] [PubMed] [Google Scholar]
- 10.Schotte, F., Soman, J., Olson, J. S., Wulff, M. & Anfinrud, P. A. (2004) J. Struct. Biol. 147, 235–246. [DOI] [PubMed] [Google Scholar]
- 11.Schmidt, M., Rajagopal, S., Ren, Z. & Moffat, K. (2003) Biophys. J. 84, 2112–2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schmidt, M., Pahl, R., Anderson, S., Srajer, V., Ren, Z., Ihee, H., Rajagopal, S. & Moffat K. (2004) Proc. Natl. Acad. Sci. USA 101, 4799–4804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rajagopal, S., Anderson, S., Srajer, V., Schmidt, M., Pahl, R. & Moffat, K. (2005) Structure (London) 13, 55–63. [DOI] [PubMed] [Google Scholar]
- 14.Gibson, Q. H. & Ainsworth, S. (1957) Nature 180, 1416–1417. [DOI] [PubMed] [Google Scholar]
- 15.Austin, R. H., Beeson, K. W., Eisenstein, L., Frauenfelder, H. & Gunsalus, I. C. (1975) Biochemistry 14, 5355–5373. [DOI] [PubMed] [Google Scholar]
- 16.Henry, E. R., Sommer, J. H., Hofrichter, J. & Eaton, W. A. (1983) J. Mol. Biol. 166, 443–451. [DOI] [PubMed] [Google Scholar]
- 17.Steinbach, P. J., Ansari, A., Berendzen, J., Braunstein, D., Chu, K., Cowen, B., R., Ehrenstein, D., Frauenfelder, H., Johnson, J. B., Lamb, D. C., et al. (1991). Biochemistry 30, 3988–4001. [DOI] [PubMed] [Google Scholar]
- 18.Tian, W.-D., Sage, J. T., Šrajer, V. & Champion, P. M. (1992) Phys. Rev. Lett. 68, 408–411. [DOI] [PubMed] [Google Scholar]
- 19.Nienhaus, G. U., Mourant, J. R. & Frauenfelder, H. (1992) Proc. Natl. Acad. Sci. USA 89, 2902–2906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ansari, A., Jones, C. M., Henry, E. R., Hofrichter, J. & Eaton, W. A. (1994) Biochemistry 33, 5128–5145. [DOI] [PubMed] [Google Scholar]
- 21.Schlichting, I., Berendsen, J., Phillips, G. N., Jr., & Sweet, R. N. (1994) Nature 371, 808–812. [DOI] [PubMed] [Google Scholar]
- 22.Teng, T. Y., Šrajer, V. & Moffat, K. (1994) Nat. Struct. Biol. 1, 701–705. [DOI] [PubMed] [Google Scholar]
- 23.Hartmann, H., Zinser, S., Komninos, P., Schneider, R. T., Nienhaus, G. U. & Parak, F. (1996) Proc. Natl. Acad. Sci. USA 93, 7013–7016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tian, W.-D., Sage, J. T. & Champion, P. (1996) Biochemistry 35, 3487–3502. [DOI] [PubMed] [Google Scholar]
- 25.Lim, M., Jackson, T. A. & Anfinrud, P. A. (1997) Nat. Struct. Biol. 4, 209–214. [DOI] [PubMed] [Google Scholar]
- 26.Ostermann, A., Waschipky, R., Parak, F. G. & Nienhaus, G. U. (2000) Nature 404, 205–208. [DOI] [PubMed] [Google Scholar]
- 27.Chu, K., Vojtechovsky, J., McMahon, B. H., Sweet, R. M., Berendzen, J. & Schlichting, I. (2000) Nature 403, 921–923. [DOI] [PubMed] [Google Scholar]
- 28.Scott, E. S., Gibson, Q. H. & Olson, J. S. (2001) J. Biol. Chem. 276, 5177–5188. [DOI] [PubMed] [Google Scholar]
- 29.Nienhaus, K., Deng, P., Kriegl, J. M. & Nienhaus, G. U. (2003) Biochemistry 42, 9633–9646. [DOI] [PubMed] [Google Scholar]
- 30.Nienhaus, K., Deng, P., Kriegl, J. M. & Nienhaus, G. U. (2003) Biochemistry 42, 9647–9658. [DOI] [PubMed] [Google Scholar]
- 31.Nienhaus, K., Deng, P., Olson, J. S., Warren, J. J. & Nienhaus, G. U. (2003) J. Biol. Chem. 278, 42532–42544. [DOI] [PubMed] [Google Scholar]
- 32.Nienhaus, K., Olson, J. S., Franzen, S. & Nienhaus, G. U. (2005) J. Am. Chem. Soc. 127, 40–41. [DOI] [PubMed] [Google Scholar]
- 33.Tilton, R. F., Jr., Kuntz, I. D., Jr., & Petsko, G. A. (1984) Biochemistry 23, 2849–2857. [DOI] [PubMed] [Google Scholar]
- 34.Tetreau, C., Blouquit, Y., Novikov, E., Quiniou, E. & Lavalette, D. (2004) Biophys. J. 86, 435–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nienhaus, G. U. & Nienhaus, K. (2002) J. Biol. Phys. 28, 163–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li, T., Quillin, M. L., Phillips, G. N., Jr. & Olson, J. S. (1994) Biochemistry 33, 1433–1446. [DOI] [PubMed] [Google Scholar]
- 37.Nienhaus, K., Ostermann, A., Nienhaus, G. U., Parak, F. & Schmidt, M. (2005) Biochemistry 44, 5095–5105. [DOI] [PubMed] [Google Scholar]
- 38.Schmidt, M., Pahl, R., Ihee, H. & Šrajer, V. (2005) in Methods in Molecular Biology, ed. Nienhaus, U. (Humana, Totowa, NJ), Vol. 305, pp. 115–154. [DOI] [PubMed] [Google Scholar]
- 39.Ren, Z. & Moffat, K. (1995) J. Appl. Crystallogr. 28, 461–481. [Google Scholar]
- 40.Ursby, T. & Bourgeois, D. (1997) Acta Crystallogr. A 53, 564–575. [Google Scholar]
- 41.Henderson, R. & Moffat, J. K. (1971) Acta Crystallogr. B 27, 1414–1420. [Google Scholar]
- 42.Perman, B., Anderson, S., Schmidt, M. & Moffat, K. (2000) Cell. Mol. Biol. 46, 895–913. [PubMed] [Google Scholar]
- 43.Brunger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J. S., Kuszewski, J., Nilges, M., Pannu, N. S., et al. (1998) Acta Crystallogr. D 54, 905–921. [DOI] [PubMed] [Google Scholar]
- 44.Johnson, J. B., Lamb, D. C., Frauenfelder, H., Müller, J. D., McMahon, B., Nienhaus, G. U. & Young, R. D. (1996) Biophys. J. 71, 1563–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Franzen, S., Bohn, B., Poyart, C. & Martin, J. L. (1995) Biochemistry 34, 1224–1237. [DOI] [PubMed] [Google Scholar]
- 46.Kitagawa, T., Haruta, N. & Mizutani, Y. (2001) Biopolymers (Biospectroscopy) 67, 207–213. [DOI] [PubMed] [Google Scholar]
- 47.Engler, N., Ostermann, A., Niimura, N. & Parak, F. (2003) Proc. Natl. Acad. Sci. USA 100, 10243–10248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jackson, T. A., Lim, M. & Anfinrud, P. A. (1994) Chem. Phys. 180, 131–140. [Google Scholar]
- 49.Karplus, M. (1999) in Simplicity and Complexity in Proteins and Nucleic Acids, eds. Frauenfelder, H., Deisenhofer, J. & Wolynes, P. (Dahlem Univ. Press, Berlin), p. 139.
- 50.Moffat, K. (2001) Chem. Rev. 101, 1569–1581. [DOI] [PubMed] [Google Scholar]
- 51.Bossa, C., Anselmi, M., Roccatano, D., Amadei, A., Vallone, B., Brunori, M. & Di Nola, A. (2004) Biophys. J. 86, 3855–3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Prusakov, V. E., Steyer, J. & Parak, F. G. (1995) Biophys. J. 68, 2524–2530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Anderson, S., Šrajer, V., Pahl, R., Rajagopal, S., Schotte, F., Anfinrud, P., Wulff, M. & Moffat K. (2004) Structure (London) 12, 1039–1045. [DOI] [PubMed] [Google Scholar]
- 54.Olson, J. S. & Phillips, G. N., Jr. (1996) J. Biol. Chem. 271, 17593–17596. [DOI] [PubMed] [Google Scholar]
- 55.Chao, W., Christian, J. F., Champion, P. M., Rosca, F. & Sage, J. T. (2001) Biochemistry 40, 5728–5737. [DOI] [PubMed] [Google Scholar]
- 56.McNaughton, L., Hernandez, G. & LeMaster, D. (2003) J. Am. Chem. Soc. 125, 3813–3820. [DOI] [PubMed] [Google Scholar]
- 57.Carson, M. (1991) J. Appl. Crystallogr. 24, 958–961. [Google Scholar]
- 58.Jones, T. A., Zou, J. Y., Cowan, S. W. & Kjeldgaard, M. (1991) Acta Crystallogr. A 47, 110–119. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}