

Abstract
The reaction cycle and the major structural states of the molecular chaperone GroEL and its cochaperone, GroES, are well characterized. In contrast, very little is known about the nonnative states of the substrate polypeptide acted on by the chaperonin machinery. In this study, we investigated the substrate protein human dihydrofolate reductase (hDHFR) while bound to GroEL or to a single-ring analog, SR1, by NMR spectroscopy in solution under conditions where hDHFR was efficiently recovered as a folded, enzymatically active protein from the stable complexes upon addition of ATP and GroES. By using the NMR techniques of transverse relaxation-optimized spectroscopy (TROSY), cross-correlated relaxation-induced polarization transfer (CRIPT), and cross-correlated relaxation-enhanced polarization transfer (CRINEPT), bound hDHFR could be observed directly. Measurements of the buildup of hDHFR NMR signals by different magnetization transfer mechanisms were used to characterize the dynamic properties of the NMR-observable parts of the bound substrate. The NMR data suggest that the bound state includes random coil conformations devoid of stable native-like tertiary contacts and that the bound hDHFR might best be described as a dynamic ensemble of randomly structured conformers.
Keywords: dihydrofolate reductase, transverse relaxation-optimized spectroscopy (TROSY), cross-correlated relaxation-induced polarization transfer (CRIPT), protein folding
Chaperone proteins are involved in nearly every cellular process in which proteins must become unfolded or refolded, including transport across membranes, assembly and disassembly of macromolecular complexes, and de novo folding of newly synthesized cytosolic proteins. Chaperones of the Hsp60 class, also named chaperonins, are of special interest in this regard, because folding takes place within a central, closed cavity. The chaperonin architecture consists of two homo-oligomeric rings stacked back to back. The polypeptide substrate binds in the open end of one of these rings and gets encapsulated upon subsequent binding of the cochaperonin partner protein.
The essential cellular action of chaperonin-mediated folding involves cycles of polypeptide binding and ATP/GroES-driven release, with only a small percentage of input molecules reaching the native state in any given cycle (1–5). With each round of binding, it appears that the same ensemble of chaperonin-associated nonnative conformers becomes stably associated with the chaperonin, giving the bound molecules essentially the same chance at reaching the native state with each round of release (1, 6). In the case of the Escherichia coli chaperonin GroEL, binding is mediated through a hydrophobic lining of its central cavity (7), which forms multivalent contacts (8) with exposed hydrophobic surfaces of the nonnative substrate protein (9–14). Such interaction serves to prevent substrate protein from irreversible misfolding and aggregation (15, 16).
Although the overall action of polypeptide binding by GroEL has been well described, possible correlations between the binding mode and the substrate conformation have not yet been rationalized on the molecular level. GroEL can catalyze complete unfolding of a small protein, a result obtained from hydrogen-exchange studies (17), but how is the unfolded state obtained? Is there active unfolding mediated by multivalent binding (18)? Or is the apparent unfolding action a passive result of thermodynamic partitioning, in which GroEL binds less-folded conformers with greater affinity, shifting the ensemble toward a less-folded state (19, 20)? As a basis for distinguishing between different possible mechanisms, at least the stable GroEL-bound “end-state” of a nonnative substrate bound in an open GroEL ring must be characterized.
Early studies revealed that GroEL-bound polypeptides were exquisitely protease susceptible, with their tryptophans in environments intermediate in polarity between native and fully unfolded states, suggesting an unstable tertiary structure (21). Subsequent hydrogen–deuterium exchange experiments revealed that there are only low degrees of exchange protection, suggesting the absence of stable regular secondary structures (17, 22–25). Crystallographic observations on substrates bound to GroEL have been made in two cases but involved only short peptides associating with single apical domains (26, 27). The peptides were bound in an extended state in the hydrophobic groove between two apical α-helices, but it is unclear whether such well-ordered structures inform about the behavior of full-length polypeptide substrates. Indeed, it has been suggested that these structures may more closely mimic the binding to the apical surface of the so-called “mobile loop” of the cochaperonin GroES (28). Thus, it would be desirable to directly inspect an intact nonnative polypeptide while bound to GroEL. This inspection has become possible through the development of NMR techniques that can observe very large molecules in solution (29, 30). In particular, such techniques have recently been applied to the GroEL system, examining binding of the 15N-labeled and perdeuterated 70-kDa cochaperonin, GroES, to the unlabeled 800-kDa chaperonin, GroEL. Nearly all of the 94 amide protons of the GroES subunits were observed both in the free cochaperonin and in the 870-kDa complex, and chemical shift changes were detected upon association of GroES with GroEL, localizing to the GroES mobile loop region (31). We now report NMR observations of an isotope-labeled substrate polypeptide, human dihydrofolate reductase (hDHFR), while bound to unlabeled or 15N-depleted GroEL.
Materials and Methods
hDHFR Expression. For uniform 15N-labeling, the E. coli strain BL21(DE3) harboring the T7 expression plasmid was grown on D2O-based M9 medium by using [15N]ammonium chloride as the sole nitrogen source. Samples containing an overall deuteration level of ≈85% were expressed by using unlabeled glucose as the sole carbon source, whereas samples containing >98% deuteration were expressed by using deuterated acetate as the sole carbon source (31–35), requiring preconditioning of cells, first to medium containing D2O with unlabeled glucose, followed by the full labeling medium.
Specific labeling of the leucine residues with 15N was carried out on a transaminase-deficient strain (35). Cells were grown to mid-log in LB, then in M9 with either H2O (≈80% final deuteration) or D2O (≈90% final deuteration), then supplemented 1 h later with 1 g/liter deuterated algae extract (CELTONE-d powder, Spectra Stable Isotopes, Columbia, MD), 75 mg/liter [2H,15N]l-leucine, and 0.5 mM isopropyl β-d-thiogalactoside. Cells were harvested after 3–5 h. hDHFR was purified by using methotrexate-agarose affinity chromatography (24).
hDHFR Sample Preparation. Reference samples of folded hDHFR were dialyzed against binding buffer containing 250 μM dihydrofolate and 5 mM ATP, and 5% D2O was added before the NMR measurements. Denatured hDHFR was prepared by diluting the folded hDHFR ≈10-fold into 6.6 M guanidine hydrochloride and 50 mM Tris (pH 7.5), followed by PD10 chromatography. Samples were concentrated to ≈10 mg/ml, supplemented with 1–5 mM DTT, and frozen at –80°C until use for complex formation with GroEL or single ring variant 1 of GroEL (SR1). Denatured hDHFR reference NMR samples were prepared by dilution of the stock protein solutions into the same buffer.
Chaperonins, hDHFR–GroEL Complexes, and Refolding. GroES and SR1 were expressed as described in refs. 31 and 36. Complexes of either GroEL or SR1 with hDHFR were prepared by diluting 10 μl of 0.5 mM GuHCl-unfolded hDHFR 50-fold into binding buffer containing 15 μM chaperonin protein at 23°C. To prepare a sample, a number of such mixtures were pooled, centrifuged at 18,000 × g for 5 min to remove aggregated unbound hDHFR, then exchanged to remove residual GuHCl and concentrated to 0.3 ml and supplemented with 5% D2O. The final protein concentration was typically 60–80 mg/ml. Refolding was accomplished by addition of dihydrofolate (500 μM final), GroES (1.5:1 GroES to SR1 or GroEL), and ATP (5 mM final). After 10 min at 16°C, the sample was centrifuged, and NMR was performed.
Assay for hDHFR Enzymatic Activity. hDHFR subjected to refolding exactly as just described also was assayed for recovery of enzymatic activity (24), reflecting production of the native state. Recovery of activity was observed with single-exponential kinetics (k = 0.31 min–1), reaching nearly complete reactivation by 10 min.
Detection of Internal Dynamics of hDHFR Bound to SR1 by Using NMR. Many different experiments have been proposed to measure internal motions in proteins (37–41). However, the sensitivity of these experiments is too low for large macromolecular structures of several hundred kilodaltons in size, so we used a previously undescribed approach based on comparison of the efficiencies of 1H magnetization transfer in backbone 15N-1H moieties to generate 1H magnetization in antiphase to 15N when using either cross-correlated relaxation-induced polarization transfer (CRIPT) or insensitive nuclei enhanced by polarization transfer (INEPT) coherence transfer (42).
The transfer of 1H magnetization into 1H magnetization in antiphase to 15N, Sv, by cross-correlated relaxation between dipole–dipole (DD) coupling and chemical shift anisotropy (CSA) interactions by means of CRIPT or by scalar coupling with INEPT is described by (42, 43)
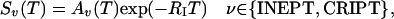 |
[1] |
with
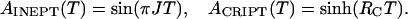 |
[2] |
RI is the transverse relaxation rate of 1HN, T is the transfer time, and RC is the relaxation rate due to cross-correlation between 1HN CSA and 1HN-15N DD coupling.
RI and RC are given by Eqs. 3 and 4 for isotropic rotational tumbling of rigid, high-molecular-weight systems at high magnetic field (44)
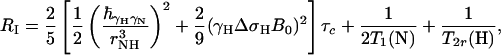 |
[3] |
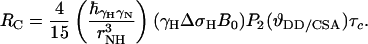 |
[4] |
τc is the isotropic rotational correlation time of an equivalent sphere representing the molecule of interest (41), rNH is the distance between the two nuclei, ΔσH is the CSA of 1HN, B0 is the static magnetic field, and γH and γN are the gyromagnetic ratios of 1H and 15N, respectively. The second-order Legendre polynome, P2(ϑDD/CSA), accounts for deviations of the angle ϑDD/CSA between the DD vector and the principal axis of the CSA tensor from 0°. The term 1/T1(N) is the longitudinal relaxation rate of 15N, and 1/T2r(H) is the contribution to the transverse relaxation rate of 1HN due to DD coupling with remote protons Hi at distances rHHi. Assuming for large structures that 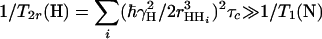 , the optimal CRIPT transfer period, TC, becomes
, the optimal CRIPT transfer period, TC, becomes
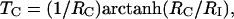 |
[5] |
and is inversely proportional to τc (Eq. 4). Measurement of CRIPT build-up curves for the determination of TC therefore enables an estimate of the size of the molecular particle studied (42), assuming that internal motions are sufficiently limited so as to have at most small effects on TC.
Usually, three distinct regimes are distinguished when assessing the influence of internal motions (characterized by the correlation time τm) on the overall relaxation rates (45). For slow internal motions with τm » τc (τm in the microsecond to millisecond time range), the most evident effect is resonance line broadening (39, 41, 46). For high-molecular-weight systems, such additional line broadening may be difficult to quantitate, because it is added to the inherently large line width because of transverse relaxation (30, 47, 48). Internal motions with correlation times τm comparable with τc, with 1 ≤ τm/τc ≤ 10 (τm in the nanosecond to microsecond time range) affect directly the effective correlation time for transverse relaxation (41). Finally, very rapid internal motions, with τm « τc (τm in the picosecond to nanosecond time range) reduce the efficiency of relaxation pathways, but the respective correlation times cannot be measured directly by NMR methods (41, 49, 50). Internal motions with τm/τc ≤ 10 that are not correlated with the overall rotational tumbling always reduce RC and RI (41). Slow internal motions (τm » τc), conversely, do not influence RC but can increase the RI value due to chemical shift modulation (39, 41). Here, we make use of this situation to assess the internal dynamics of substrate proteins bound to GroEL/GroES by estimating the effective RC and RI values by using a combination of INEPT and CRIPT build-up curves.
For the determination of RC and RI from build-up curves, the optimal INEPT transfer delay TI and the ratio between the optimal CRIPT and INEPT transfer efficiencies, AC/I, are needed. These parameters are defined by
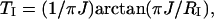 |
[6] |
and
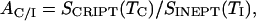 |
[7] |
where J = 93 Hz is the 1J15N1H coupling constant in the amide moiety. RI can be estimated from experimental TI values (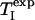 ) by using Eq. 6
) by using Eq. 6
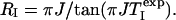 |
[8] |
With the experimental value for AC/I, 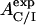 , one can then estimate RC by numerical evaluation of the roots of the function F(RC)
, one can then estimate RC by numerical evaluation of the roots of the function F(RC)
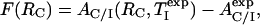 |
[9] |
where AC/I(RC, 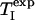 ) is given by
) is given by
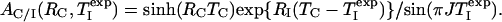 |
[10] |
TC and RI are functions of RC and 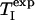 (Eqs. 5 and 8). The roots of Eq. 9 were determined numerically by applying the Newton–Raphson method (51), using the maple 9 software package (Maplesoft, Waterloo, ON, Canada).
(Eqs. 5 and 8). The roots of Eq. 9 were determined numerically by applying the Newton–Raphson method (51), using the maple 9 software package (Maplesoft, Waterloo, ON, Canada).
NMR Spectroscopy. All NMR spectra were recorded on a DRX 750-MHz spectrometer (Bruker, Fällanden, Switzerland) equipped with a triple-resonance probehead and a shielded z-gradient coil. NMR spectra were collected at 25°C. Two-dimensional [15N,1H] transverse relaxation-optimized spectroscopy (TROSY), 2D [15N,1H]-cross-correlated relaxation-enhanced polarization transfer (CRINEPT)–heteronuclear multiple-quantum correlation (HMQC)–[1H]-TROSY, and 2D [15N,1H]-CRIPT–TROSY experiments were recorded as described in refs. 31 and 44. Details of the parameter settings are given in the figure legends. The spectra were processed with the program prosa (52) and analyzed with the program xeasy (53).
Results
NMR Observation of Substrate Binding to Chaperonins. Our experimental setup (Fig. 1) includes that hDHFR was assessed in the folded state by using NMR spectroscopy. The 2D [15N,1H]-TROSY spectrum of [u-15N,u∼80% 2H]hDHFR (Fig. 2a) contains ≈190 resonances from backbone amide groups, which show large chemical shift dispersion in the proton and nitrogen dimensions, as is characteristic of a folded protein. There is evidence for two conformations of the protein, which differ in the occupation of the NADPH binding site of hDHFR (54–56). The 15N-labeled, deuterated protein was then denatured in 6.6 M guanidinium hydrochloride. This solution was diluted with “binding buffer” at pH 6.1 that contained either GroEL or its single-ring variant SR1 (Fig. 1). At pH 6.1 and without folate ligands, hDHFR diluted from denaturant is efficiently bound by the chaperonin (24). The resulting binary complexes with SR1 or GroEL are large structures of 420 or 820 kDa, respectively.
Fig. 1.

Flow chart of the experimental setup used for the formation and analysis of complexes of hDHFR (red) with SR1 or GroEL (blue). The three states assessed by NMR are indicated by horizontal arrows. Uniformly 15N-labeled and deuterated hDHFR was denatured in 6.6 M aqueous guanidine hydrochloride solution and then diluted 50-fold into buffer containing either of the chaperonin proteins SR1 or GroEL. Unbound aggregated protein was removed by centrifugation. The substrate–chaperonin complex used for the NMR studies was collected from the supernatant. After the NMR measurements, hDHFR was refolded from the complex by addition of a mixture of GroES (green), ATP, and dihydrofolate (DHF). Two sets of NMR spectra that were recorded, respectively, with uniform or residue-specific 15N-labeling of hDHFR, are shown in Fig. 2, as indicated.
Fig. 2.

Two-dimensional [15N,1H]-correlation spectra of deuterated hDHFR either uniformly labeled with 15N or with specific 15N-labeling of the 19 Leu residues. (a) Two-dimensional [15N,1H]-TROSY spectrum of [u-15N; u∼85%2H]hDHFR. (b) Two-dimensional [15N,1H]-CRINEPT–HMQC spectrum of [u-15N; u∼85% 2H]hDHFR bound to unlabeled SR1. The sample contained ≈50 μM [15N,2H]-hDHFR and ≈240 μM unlabeled SR1 and was prepared by dilution of hDHFR from denaturant into a buffer containing SR1 (Fig. 1). The peak clusters for the side-chain 15N-1H2 groups of Gln and Asn are indicated by I and II. (c) 2D [15N,1H]-TROSY spectrum of [u-15N; u∼85% 2H]hDHFR refolded from the complex with SR1 in b; the refolded protein was obtained after incubation of the sample with 5 mM ATP, ≈320 mM GroES, and 0.5 mM DHF at 15°C. (d–f) The same experiments as presented in a–c, except that [15N,2H-Leu;u∼80%2H]DHFR was used instead of the uniformly 15N-labeled hDHFR. The spectra were collected with the following parameters. (a and d) Acquired data size 75 × 1,024 complex points, t1,max = 18.75 ms; t2,max = 97.6 ms; INEPT transfer period = 4.8 ms; 48 scans were recorded per t1-increment. (b and e) Acquired data size 50 × 1,024 complex points; t1,max = 12.5 ms; t2,max = 97.6 ms; CRINEPT transfer period = 2.5 ms; 512 (b) or 3,072 (e) scans per t1-increment. Before Fourier transformation, the data were multiplied along the t1 dimension with a sine function shifted by 10° and in the t2 dimension with an empirically optimized exponential function. (c and f) Acquired data size 110 × 1,024 (c) and 20 × 1,024 (f) complex points; t1,max = 27.5 ms (c) and t1,max = 5.0 ms (f); t2,max = 97.6 ms; INEPT transfer period = 5.4 ms. For all experiments, the recycle delay was 1 s.
For the NMR characterization of the binary complexes of hDHFR bound to GroEL or SR1, we measured 2D [15N,1H]-TROSY, 2D [15N,1H]-CRIPT–TROSY and 2D [15N-1H]-CRINEPT–HMQC–[1H]-TROSY spectra (31, 44). In these experiments, the chaperonin proteins were either at natural isotope distribution or uniformly 14N,2H-labeled, such that they do not give rise to 15N NMR signals, and the observed spectra arise solely from [u-15N,u∼80% 2H]hDHFR. We obtained the highest sensitivity with the 2D [15N,1H]-CRINEPT–HMQC–[1H]-TROSY scheme (42). The peak patterns obtained for the complexes of hDHFR with GroEL and SR1 were very similar (data not shown), with higher signal intensities in the SR1 complexes. The spectra of hDHFR bound to unlabeled or 14N,2H-labeled SR1 were identical. Therefore, we used unlabeled SR1 for further studies. A sample of [u-15N; u∼85% 2H]hDHFR diluted from denaturant in the absence of chaperonin yielded no NMR signal after centrifugation at 18,000 × g to remove aggregated hDHFR, ruling out the possibility that the observed signals arise from unbound hDHFR.
NMR Characterization of Bound hDHFR. The distribution of resonances in 2D [15N,1H]-CRINEPT–HMQC–[1H]-TROSY spectra of the stable binary complex between unlabeled SR1 and [u-15N; u∼85%2H]hDHFR (Fig. 2b) has a small chemical shift dispersion, indicating that the observed species does not adopt stable regular secondary or tertiary structure. The resonances in Fig. 2b also show large line widths in both the 15N and 1H dimensions. This line broadening could simply be caused by the slow overall tumbling of the large complex, for which efficient transverse relaxation will broaden the resonance lines, and/or by millisecond timescale internal motions (39, 40, 46). Further, the substrate might be bound in different conformations, so that the NMR spectrum would consist of the sum of the spectra of the individual species, leading to inhomogeneous line broadening.
To gain further insight into the dynamic properties of hDHFR bound to SR1, we recorded 15N-selected 1D 1H NMR spectra of [u-15N; u∼85% 2H]hDHFR bound to unlabeled SR1, after a CRIPT or an INEPT magnetization transfer step (Fig. 3a). The signal of the 1D spectrum using the INEPT element shows nearly three times higher signal intensity than that using the CRIPT element, and the resonances in both spectra have antiphase character. For a substrate rigidly bound to the 400-kDa SR1, one would expect that the CRIPT and INEPT transfer have similar transfer efficiencies at 750 MHz and that one component of the doublet would vanish because of fast transverse relaxation (42) and thereby remove the antiphase character of the signal. Therefore, the data of Fig. 3a are an indication of internal mobility of hDHFR relative to SR1 (see Discussion).
Fig. 3.

Characterization of the dynamic properties of the hDHFR/SR1 complex by using CRIPT and INEPT magnetization transfer. (a) 15N-selected 1D 1H NMR spectra of [u-15N, u∼85% 2H]hDHFR bound to unlabeled SR1, by using a CRIPT (lower trace) or an INEPT (upper trace) magnetization transfer step. In both experiments, the transfer period was set to 2.5 ms. The asterisks indicate the antiphase components of the observed signals. The vertical line at 8.15 ppm identifies the proton frequency at which the intensities for the CRIPT/INEPT buildup measurements shown in b were measured. (b) Plots of the experimental magnetization transfer efficiencies for CRIPT (circles) and INEPT (squares) vs. the length of the transfer period T for a 1H-15N cross peak of hDHFR in the 1:1 complex with unlabeled SR1. The dashed black line indicates an INEPT buildup curve fitted to the experimental data by using Eqs. 1 and 2. The spectra were recorded on a Bruker DRX 750 spectrometer. The number of transients per T value in the CRIPT and INEPT experiments was 512.
Refolding of hDHFR. For an assessment of whether or not the bound state observed by NMR is a true folding intermediate, we added GroES, ATP, and dihydrofolate to the hDHFR/SR1 complex (24) (Fig. 1). Such incubation produced efficient refolding of hDHFR to an enzymatically active form within 10 min. In the 2D [15N,1H]-TROSY spectrum of the recovered substrate (Fig. 2c), the broad peaks in the random coil region of the spectrum in Fig. 2b have completely disappeared, and a widely dispersed spectrum characteristic of a globular protein is present, which contains a large proportion of the 15N-1H cross peaks of folded hDHFR (Fig. 2a). We do not presently know why only part of the hDHFR spectrum is seen after dissociation from SR1; quite likely this result is because of the lower concentration of the refolded protein. Nonetheless, recovering a globular protein with hDHFR enzymatic activity and hDHFR-like NMR chemical shifts supports the hypothesis that the NMR-observable GroEL-bound hDHFR represents a bona fide folding intermediate in the chaperonin cycle. More direct evidence for this conclusion comes from the experiments with residue-selective labeling in the following section.
Studies with [15N,2H]Leu-hDHFR. The large line widths and the severe overlap of the resonances in the spectrum of the binary complex make it difficult to estimate the number of resonances observed in Fig. 2b. Assuming that all of the side-chain NH2 groups of Asn and Gln contribute to the peak clusters indicated by I and II in Fig. 2b, we estimate that the intensities of the other peaks in Fig. 2b correspond to ≈25% of all backbone 15N-1H moieties of hDHFR. This apparent low intensity could be due either to more extensive line broadening of the backbone resonances or to their representing only a subset of signals, corresponding to discrete parts of the molecule.
For a further investigation of this issue, we chose to simplify the spectrum by labeling only the Leu residues with 15N. A 2D [15N,1H]-HSQC spectrum of the denatured [15N,2H-Leu;U∼80% 2H]hDHFR contains the expected 19 resonances (see Fig. 5, which is published as supporting information on the PNAS web site), and folded hDHFR shows the typical wide distribution of the Leu resonances (Fig. 2d). The spectrum of the complex with SR1 prepared with this material, however, contains only one continuous area of signal intensity (Fig. 2e), and a precise count of the number of cross-peaks is not possible. The NMR spectrum of the enzymatically active hDHFR recovered from the binary complex as described in the preceding section (Fig. 2f) shows the same chemical shift dispersion and closely similar peak patterns as native hDHFR (Fig. 2d), but as with uniformly 15N-labeled hDHFR (Fig. 2c), only an incomplete set of the resonance lines is seen (see the preceding section).
Discussion
The qualitative evaluation in the preceding section of solution NMR data on the complexes between hDHFR and either SR1 or GroEL indicates that the NMR-observable parts of hDHFR do not adopt a defined 3D structure and have high internal mobility. This conclusion is in line with previously reported small amide proton protection factors for hDHFR (6, 24) and other substrate proteins (17) in complex with GroEL. Here, we elaborate further on the analysis of the signal build-up data of Fig. 3 and on the biological implications of the present observations.
Protein Dynamics from NMR Signal Build-Up Measurements. To test the significance of the data in Fig. 3, we performed model calculations of buildup curves over a wide range of different relaxation parameters. Fig. 4 shows pairs of TI,AC/I values calculated for different RC values by applying Eq. 10, where the thick solid lines indicate trajectories with constant RC in the TI,AC/I plane. TI,AC/I combinations for an isolated 15N-1H two-spin system lie on the dotted curve, and those for 15N-1H moieties in perdeuterated regular antiparallel β-sheets and α-helices, respectively, lie on the broken curves. Experimental TI,AC/I combinations for the complexes of GroES with SR1 and of hDHFR with SR1 are indicated as black diamonds. The GroES/SR1 value is compatible with a β-sheet in a rigid protein structure with the correlation time for GroES/SRI of τc = 175 ns (open diamond in Fig. 4); the respective estimated RC values (Eq. 9) are 200 and 220 Hz. In contrast, the hDHFR/SR1 complex has a much lower RC value of ≈100 Hz, which is incompatible with a rigid structure of the size of SR1. From the TI value of 2.5 ms for hDHFR/SR1, Eq. 10 shows that the RI value is ≈350 Hz, as compared with the RI value for the GroES/SR1 complex of ≈250 Hz. The increased RI value is an indication for slow internal motions, which also would explain the line broadening observed in the 2D [15N,1H]-HMQC spectrum.
Fig. 4.

Dependence of the 1HN-15N DD/CSA cross-relaxation rate RC on the optimal INEPT transfer delay TI and on the magnetization transfer efficiency ratio, AC/I, as defined in Eqs. 9 and 10. The thick lines are trajectories with constant value of RC in the TI,AC/I plane, as indicated on the left above each curve. On the right the curves terminate at RC = RI. The dotted line represents combinations of TI,AC/I values calculated for an isolated 15N-1H moiety in structures devoid of internal motions, by using Eqs. 1–7. The broken curves are for 15N-1H moieties located in a uniformly 15N,2H-labeled antiparallel β-sheet [β(2H)] and a uniformly 15N-2H-labeled α-helix [α(2H)], respectively, and take into account the effects of DD coupling with remote protons. Clearly, the most favorable case is the isolated 15N,1H group, and the accessible area of the TI,AC/I plane in real systems extends from the dotted curve to the left. Within this area, the filled diamonds indicate the experimental values for hDHFR bound to SR1 (Fig. 3b) and for GroES in a 1:1 complex with SR1 (44), and the open diamond indicates the value calculated for the 15N-1H moieties in an otherwise perdeuterated antiparallel β-sheet reorienting with the rotational correlation time for the GroES/SR1 complex, i.e., τc = 175 ns.
Overall, the analysis of the CRIPT and INEPT build-up curves shows that hDHFR is subject to internal mobility in the SR1 complex, which includes components with correlation times in the microsecond to millisecond time range, as well as components with frequencies in the picosecond to nanosecond time range. Such wide distribution of the frequencies of internal motions has previously been suggested for “collapsed conformations” of polypeptide chains, which would not have well-defined tertiary structure packing (57, 58). For the hDHFR/GroEL system, the interpretation of this analysis is limited, because we have not yet succeeded in determining whether or not the entire polypeptide chain of the bound hDHFR is NMR-observable or in identifying which parts of the bound hDHFR are observed (see Results). Quite generally, however, the approach used here opens an avenue for assessing the dynamics of distinct polypeptide segments in very large structures.
Implications for the Mechanism of GroEL Action. The NMR data presented in this work support a general model of GroEL action in which the chaperonin maintains proteins in an unfolded state while they are bound. The initial act of binding of nonnative proteins in an open ring may be associated with an unfolding action on the part of the chaperonin, but that is not resolvable by the present analysis. In particular, in the present study, hDHFR was diluted from guanidine denaturant into an aqueous mixture containing GroEL, where early folding intermediates are formed in <20 ms and are efficiently recognized by GroEL (ref. 24 and M. Goldberg, J. Beechem, and A.L.H., unpublished data). Thereby it is difficult to assess whether partially structured states become less structured upon binding to GroEL. However, GroEL also efficiently binds later-folding intermediates of hDHFR, which are formed many seconds or even several minutes after dilution from denaturant (24). It thus may become informative, as further understanding of the NMR-observable features of GroEL-bound hDHFR is attained, to assess whether these more structured states will produce similar or different spectroscopic behavior when they become associated with GroEL.
The apparent dynamic nature of GroEL-bound hDHFR also invites the question of whether there is ongoing binding and release of distinct segments of hDHFR from the apical domains of GroEL within the binary complex. Although we do not know as yet with certainty whether apical-associated stretches of polypeptide are NMR-observable (see Results), it is noteworthy that the observed hDHFR resonances were not visibly affected by deuteration of SR1.
In sum, here we have provided direct observations of a substrate protein while bound to the chaperonin GroEL in solution by using advanced NMR methods. The present work makes clear that we are confronting the problem of one smaller, relatively unstructured substrate polypeptide, moving with a distinct set of motional regimes inside the far larger chaperonin “container,” which presents a previously undescribed set of physical problems whose resolution will provide important biological insights. Further techniques of both biology and physics may be needed to resolve this system.
Supplementary Material
Acknowledgments
We thank Krystyna Furtak for help in constructing the plasmids used for the protein expression. This work was supported by the Howard Hughes Medical Institute, the National Institutes of Health, and the Schweizerischer Nationalfonds and the Eidgenössische Technische Hochschule Zürich through the National Center for Competence in Research (NCCR) Structural Biology.
Author contributions: R.H., E.B.B., A.L.H., and K.W. designed research; R.H., E.B.B., J.F., and G.W. performed research; R.H., E.B.B., and J.F. contributed new reagents/analytic tools; R.H., E.B.B., J.F., G.W., and K.W. analyzed data; and R.H., G.W., A.L.H., and K.W. wrote the paper.
Abbreviations: CRIPT, cross-correlated relaxation-induced polarization transfer; CRINEPT, cross-correlated relaxation-enhanced polarization transfer; CSA, chemical shift anisotropy; DD, dipole–dipole; HMQC, heteronuclear multiple-quantum correlation; hDHFR, human dihydrofolate reductase; INEPT, insensitive nuclei enhanced by polarization transfer; SR1, single ring variant 1 of GroEL; TROSY, transverse relaxation-optimized spectroscopy.
References
- 1.Weissman, J. S., Kashi, Y., Fenton, W. A. & Horwich, A. L. (1994) Cell 78, 693–702. [DOI] [PubMed] [Google Scholar]
- 2.Todd, M. J., Viitanen, P. V. & Lorimer, G. H. (1994) Science 265, 659–666. [DOI] [PubMed] [Google Scholar]
- 3.Fenton, W. A. & Horwich, A. L. (2003) Q. Rev. Biophys. 36, 229–256. [DOI] [PubMed] [Google Scholar]
- 4.Thirumalai, D. & Lorimer, G. H. (2001) Annu. Rev. Biophys. Biomol. Struct. 30, 245–269. [DOI] [PubMed] [Google Scholar]
- 5.Hartl, F. U. & Hayer-Hartl, M. (2002) Science 295, 1852–1858. [DOI] [PubMed] [Google Scholar]
- 6.Gross, M., Robinson, C. V., Mayhew, M., Hartl, F. U. & Radford, S. E. (1996) Protein Sci. 5, 2506–2513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Braig, K., Otwinowski, Z., Hegde, R., Boisvert, D. C., Joachimiak, A., Horwich, A. L. & Sigler, P. B. (1994) Nature 371, 578–586. [DOI] [PubMed] [Google Scholar]
- 8.Farr, G. W., Furtak, K., Rowland, M. B., Ranson, N. A., Saibil, H. R., Kirchhausen, T. & Horwich, A. L. (2000) Cell 100, 561–573. [DOI] [PubMed] [Google Scholar]
- 9.Fenton, W. A., Kashi, Y., Furtak, K. & Horwich, A. L. (1994) Nature 371, 614–619. [DOI] [PubMed] [Google Scholar]
- 10.Mendoza, J. A., Rogers, E., Lorimer, G. H. & Horowitz, P. M. (1991) J. Biol. Chem. 266, 13044–13049. [PubMed] [Google Scholar]
- 11.Landry, S. J. & Gierasch, L. M. (1991) Biochemistry 30, 7359–7362. [DOI] [PubMed] [Google Scholar]
- 12.Itzhaki, L. S., Otzen, D. E. & Fersht, A. R. (1995) Biochemistry 34, 14581–14587. [DOI] [PubMed] [Google Scholar]
- 13.Lin, Z. L., Schwarz, F. P. & Eisenstein, E. (1995) J. Biol. Chem. 270, 1011–1014. [DOI] [PubMed] [Google Scholar]
- 14.Thiyagarajan, P., Henderson, S. J. & Joachimiak, A. (1996) Structure (London) 4, 79–88. [DOI] [PubMed] [Google Scholar]
- 15.Goloubinoff, P., Gatenby, A. A. & Lorimer, G. H. (1989) Nature 337, 44–47. [DOI] [PubMed] [Google Scholar]
- 16.Ranson, N. A., Dunster, N. J., Burston, S. G. & Clarke, A. R. (1995) J. Mol. Biol. 250, 581–586. [DOI] [PubMed] [Google Scholar]
- 17.Zahn, R., Spitzfaden, C., Ottiger, M., Wüthrich, K. & Plückthun, A. (1994) Nature 368, 261–265. [DOI] [PubMed] [Google Scholar]
- 18.Reid, B. G. & Flynn, G. C. (1996) J. Biol. Chem. 271, 7212–7217. [DOI] [PubMed] [Google Scholar]
- 19.Zahn, R. & Plückthun, A. (1994) J. Mol. Biol. 242, 165–174. [DOI] [PubMed] [Google Scholar]
- 20.Walter, S., Lorimer, G. H. & Schmid, F. X. (1996) Proc. Natl. Acad. Sci. USA 93, 9425–9430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Martin, J., Langer, T., Boteva, R., Schramel, A., Horwich, A. L. & Hartl, F. U. (1991) Nature 352, 36–42. [DOI] [PubMed] [Google Scholar]
- 22.Robinson, C. V., Gross, M., Eyles, S. J., Ewbank, J. J., Mayhew, M., Hartl, F. U., Dobson, C. M. & Radford, S. E. (1994) Nature 372, 646–651. [DOI] [PubMed] [Google Scholar]
- 23.Zahn, R., Perrett, S. & Fersht, A. R. (1996) J. Mol. Biol. 261, 43–61. [DOI] [PubMed] [Google Scholar]
- 24.Goldberg, M. S., Zhang, J., Sondek, S., Matthews, C. R., Fox, R. O. & Horwich, A. L. (1997) Proc. Natl. Acad. Sci. USA 94, 1080–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen, J. W., Walter, S., Horwich, A. L. & Smith, D. L. (2001) Nat. Struct. Biol. 8, 721–728. [DOI] [PubMed] [Google Scholar]
- 26.Buckle, A. M., Zahn, R. & Fersht, A. R. (1997) Proc. Natl. Acad. Sci. USA 94, 3571–3575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen, L. L. & Sigler, P. B. (1999) Cell 99, 757–768. [DOI] [PubMed] [Google Scholar]
- 28.Shewmaker, F., Maskos, K., Simmerling, C. & Landry, S. J. (2001) J. Biol. Chem. 276, 31257–31264. [DOI] [PubMed] [Google Scholar]
- 29.Pervushin, K., Riek, R., Wider, G. & Wüthrich, K. (1997) Proc. Natl. Acad. Sci. USA 94, 12366–12371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Riek, R., Pervushin, K. & Wüthrich, K. (2000) Trends Biochem. Sci. 25, 462–468. [DOI] [PubMed] [Google Scholar]
- 31.Fiaux, J., Bertelsen, E. B., Horwich, A. L. & Wüthrich, K. (2002) Nature 418, 207–211. [DOI] [PubMed] [Google Scholar]
- 32.Sambrook, J., Fritsch, E. F. & Maniatis, T. (1989) Molecular Cloning: A Laboratory Manual (Cold Spring Harbor Lab. Press, Woodbury, NY).
- 33.Shekhtman, A., Ghose, R., Goger, M. & Cowburn, D. (2002) FEBS Lett. 524, 177–182. [DOI] [PubMed] [Google Scholar]
- 34.Venters, R. A., Huang, C. C., Farmer, B. T., Trolard, R., Spicer, L. D. & Fierke, C. A. (1995) J. Biomol. NMR 5, 339–344. [DOI] [PubMed] [Google Scholar]
- 35.Fiaux, J., Bertelsen, E. B., Horwich, A. L. & Wüthrich, K. (2004) J. Biomol. NMR 29, 289–297. [DOI] [PubMed] [Google Scholar]
- 36.Horwich, A. L., Burston, S. G., Rye, H. S., Weissman, J. S. & Fenton, W. A. (1998) Methods Enzymol. 290, 141–146. [DOI] [PubMed] [Google Scholar]
- 37.Peng, J. W. & Wagner, G. (1994) Methods Enzymol. 239, 563–596. [DOI] [PubMed] [Google Scholar]
- 38.Kay, L. E. (1998) Nat. Struct. Biol. 5, 513–517. [DOI] [PubMed] [Google Scholar]
- 39.Palmer, A. G., Kroenke, C. D. & Loria, J. P. (2001) Methods Enzymol. 339, 204–238. [DOI] [PubMed] [Google Scholar]
- 40.Korzhnev, D. M., Billeter, M., Arseniev, A. S. & Orekhov, V. Y. (2001) Prog. Nucl. Magn. Reson. Spectrosc. 38, 197–266. [Google Scholar]
- 41.Luginbühl, P. & Wüthrich, K. (2002) Prog. Nucl. Magn. Reson. Spectrosc. 40, 199–247. [Google Scholar]
- 42.Riek, R., Wider, G., Pervushin, K. & Wüthrich, K. (1999) Proc. Natl. Acad. Sci. USA 96, 4918–4923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wider, G. (1998) Prog. Nucl. Magn. Reson. Spectrosc. 32, 193–275. [Google Scholar]
- 44.Riek, R., Fiaux, J., Bertelsen, E. B., Horwich, A. L. & Wüthrich, K. (2002) J. Am. Chem. Soc. 124, 12144–12153. [DOI] [PubMed] [Google Scholar]
- 45.Brüschweiler, R. & Ernst, R. R. (1992) J. Chem. Phys. 96, 1758–1766. [Google Scholar]
- 46.Sandström, J. (1982) Dynamic NMR Spectroscopy (Academic, London).
- 47.Fernandez, C. & Wider, G. (2003) Curr. Opin. Struct. Biol. 13, 570–580. [DOI] [PubMed] [Google Scholar]
- 48.Tugarinov, V., Hwang, P. M. & Kay, L. E. (2004) Annu. Rev. Biochem. 73, 107–146. [DOI] [PubMed] [Google Scholar]
- 49.Lipari, G. & Szabo, A. (1982) J. Am. Chem. Soc. 104, 4546–4559. [Google Scholar]
- 50.Lipari, G. & Szabo, A. (1982) J. Am. Chem. Soc. 104, 4559–4570. [Google Scholar]
- 51.Press, W. H., Teukolsky, S. A., Vetterling, W. T. & Flannery, B. P. (1988) Numerical Recipes in C: The Art of Scientific Computing (Cambridge Univ. Press, Cambridge, U.K.).
- 52.Güntert, P., Dötsch, V., Wider, G. & Wüthrich, K. (1992) J. Biomol. NMR 2, 619–629. [Google Scholar]
- 53.Bartels, C., Xia, T. H., Billeter, M., Güntert, P. & Wüthrich, K. (1995) J. Biomol. NMR 6, 1–10. [DOI] [PubMed] [Google Scholar]
- 54.Falzone, C. J., Wright, P. E. & Benkovic, S. J. (1991) Biochemistry 30, 2184–2191. [DOI] [PubMed] [Google Scholar]
- 55.Johnson, J. M., Meiering, E. M., Wright, J. E., Pardo, J., Rosowsky, A. & Wagner, G. (1997) Biochemistry 36, 4399–4411. [DOI] [PubMed] [Google Scholar]
- 56.Meiering, E. M. & Wagner, G. (1995) J. Mol. Biol. 247, 294–308. [DOI] [PubMed] [Google Scholar]
- 57.Baum, J., Dobson, C. M., Evans, P. A. & Hanley, C. (1989) Biochemistry 28, 7–13. [DOI] [PubMed] [Google Scholar]
- 58.Kim, S., Bracken, C. & Baum, J. (1999) J. Mol. Biol. 294, 551–560. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.