

Abstract
We have cloned and expressed the 43 kDa N-terminal domain of Leishmania donovani topoisomerase II. This protein has an intrinsic ATPase activity and obeys Michaelis–Menten kinetics. Cross-linking studies indicate that the N-terminal domain exists as a dimer both in the presence and absence of nucleotides. Etoposide, an effective antitumour drug, traps eukaryotic DNA topoisomerase II in a covalent complex with DNA. In the present study, we report for the first time that etoposide inhibits the ATPase activity of the recombinant N-terminal domain of L. donovani topoisomerase II. We have modelled the structure of this 43 kDa protein and performed molecular docking analysis with the drug. Mutagenesis of critical amino acids in the vicinity of the ligand-binding pocket reveals less efficient inhibition of the ATPase activity of the enzyme by etoposide. Taken together, these results provide an insight for the development of newer therapeutic agents with specific selectivity.
Keywords: anti-leishmanial agent, ATPase domain, etoposide, kinetoplastid parasite, Leishmania, topoisomerase II
Abbreviations: DHBA, dihydrobetulinic acid; DMS, dimethyl suberimidate; GyrA, gyrase A; IPTG, isopropyl β-D-thiogalactoside; LdTOP2, Leishmania donovani topoisomerase II; Ni-NTA, Ni2+-nitrilotriacetate; p[NH]ppA, adenosine 5′-[β,γ-imido]triphosphate
INTRODUCTION
The protozoan parasite Leishmania donovani is the causative organism for the clinically important disease visceral leishmaniasis. The current drugs for Leishmania infections are inadequate due to low efficacy and high toxicity, and the problem is further compounded by the increasing prevalence of drug-resistant parasites. Current biomedical research always has its focus on the search for newer intervention strategies to control the public health impact of parasitic diseases. Modern development of antiparasitic drugs relies on rational design of substances directed against specific targets. In conjunction with the fact that DNA topoisomerase II is a proven target for clinically useful antitumour and antibacterial drugs [1], this enzyme has been cloned and sequenced from an array of parasitic organisms including the kinetoplastids [2]. Type II DNA topoisomerase is an enzyme that relieves the torsional stress created in DNA by transcription and replication and enables the segregation of two inter-twined DNA molecules after replication [3].
All eukaryotic type II topoisomerases are homodimers with three distinct regions/subunit. The N-terminal region has ATPase activity, the central region forms the DNA-binding and cleavage domain and the C-terminus is unconserved [4]. The eukaryotic topoisomerase II seems to have arisen by the fusion of GyrA (gyrase A) and GyrB subunits, which are prokaryotic counterparts of the eukaryotic enzyme. Homology between eukaryotic and prokaryotic enzymes is closest in the region containing the active site of the DNA cleavage (corresponding to the N-terminal domain of GyrA) and the ATPase active site (corresponding to the N-terminal domain of GyrB). The enzyme binds to a segment of DNA (G-segment) and generates a reversible double-strand break to serve as a gateway for the strand passage. The binding of ATP to the ATPase domain leads to dimerization of this domain and closure of the N-terminal protein clamp (N-gate). This movement in the N-terminal clamp can entrap another DNA segment (T-segment) and initiate a series of conformational changes, namely the passage of the T-segment through the protein-mediated DNA break, the religation of the DNA gate and release of the T-segment through the opening of its C-terminal dimer interface. Recent rapid kinetic measurements have provided further insights into this process [5,6].
The epipodophyllotoxins have been the mainstay in medical oncology for a long time, and were eventually shown to target DNA topoisomerase II by stabilizing the transient covalent DNA–topoisomerase II complexes, which results in the generation of lethal DNA double-strand breaks in cells. These events thus convert this essential enzyme into a potential cellular toxin. The type II DNA topoisomerases of the kinetoplastid protozoa are also the targets for a number of antiparasitic agents including the epipodophyllotoxins and bisdioxopiperazines [7].
Earlier reports indicate that the ATPase activity of topoisomerase II resides in its N-terminal domain. This ATPase activity is present in the 43 kDa N-terminal fragment of GyrB and the 52 kDa N-terminal fragment of human topoisomerase IIα, containing amino acids 1–435 of the intact enzyme [8–10]. Sequence analysis reveals that 1–385 amino acids of LdTOP2 (L. donovani topoisomerase II) are homologous with the N-terminal fragment of human topoisomerase IIα.
The mechanistic feature of type II DNA topoisomerase of the kinetoplastid parasites is not well studied to date in spite of this enzyme being an important drug target. In the present study, we report the ATPase activity of the 43 kDa N-terminal recombinant enzyme of LdTOP2 containing amino acid residues 1–385. Here, a biochemical approach has been undertaken to identify the interaction of ATP and etoposide with the 43 kDa protein. The presence of etoposide strongly inhibits the ATPase activity of the recombinant protein. Using the molecular modelling approach, we examined a plausible structure for the N-terminal domain of LdTOP2 on the basis of the crystal structure of GyrB from Thermus thermophillus [11] and simulated the docking of etoposide within the ATP-binding pocket. Subsequently, amino acids that were predicted to contact the drug and ATP were mutated. We detected significant changes in the ATPase activity of the mutated proteins. This study provides the first insight into the mechanistic pathway of the N-terminal domain of LdTOP2 and delineates the features differentiating the parasite enzyme from its prokaryotic and eukaryotic counterparts.
MATERIALS AND METHODS
Plasmid construction and mutagenesis
The 1.1 kb N-terminal fragment and the 3.7 kb full-length LdTOP2 were cloned in NdeI/BamHI and BamHI/HindIII sites of pET16b and pET28c using sense primers 5′-GGGAATTCCATATGACAGACGCTTCCAAG-3′ and 5′-CGGGATCCTCATGACAGACGCTTCCAAG-3′, and antisense primers 5′-CGGGATCCCGCCTCCAGAAACGGCAT-3′ and 5′-CCCAAGCTTCATCAAAACATGTGGCAG-3′ respectively. The recombinant clones were transformed into BL21 (DE3) pLysS and expression from the constructs pET16b/1.1 and pET28c/3.7 yielded 43 and 137 kDa proteins respectively as visualized by SDS/PAGE. The mutants N65K (Asn65→Lys), N69K, N96K, D130G and G140D were reconstructed in the plasmid pET16b/1.1 by oligonucleotide-directed mutagenesis using Quik Change site-directed mutagenesis kit (Stratagene, La Jolla, CA, U.S.A.) following manufacturer's instructions. Primers used to construct the mutations are: N65K, 5′-GACGAAATACTGCTCAAGGCGGCGGACAACATC-3′ and 5′-GATGTTGTCCGCCGCCTTGAGCAGTATTTCGTC-3′; N69K, 5′-CTCAACGCGGCGGACAAGATCAACAACAGTAAA-3′ and 5′-TTTACTGTTGTTGATCTTGTCCGCCGCGTTGAG-3′; N96K, 5′-GAGATCACGGTCGAGAAGGACGGGGCTGGCCTC-3′ and 5′-GAGGCCAGCCCCGTCCTTCTCGACCGTGATCTC-3′; D130G, 5′-TCGAACTACAACAACGGCTCGACGTCGACGACA-3′ and 5′-TGTCGTCGACGTCGAGCCGTTGTTGTAGTTCGA-3′; G140D, 5′-ACAGCGGGTCGGCACGACTACGGCGCCAAGCTG-3′ and 5′-CAGCTTGGCGCCGTAGTCGTGCCGACCCGCTGT-3′. The constructs were sequenced to confirm the mutations.
Expression and purification of N-terminal domain of LdTOP2
Purification of the N-terminal His-tagged proteins was done as follows. Briefly, the cells containing the plasmids pET16b/1.1 and pET28c/3.7 were induced at A600 0.6 with 0.5 mM IPTG (isopropyl β-D-thiogalactoside) at 22 °C for 10 h. The cells were harvested and resuspended in phosphate buffer (pH 7.8) containing 300 mM NaCl, 200 μg/ml lysozyme, 0.1% Triton X-100, 0.25% Sarkosyl and 1 mM PMSF. Final lysis was achieved by sonication on ice and the lysate was cleared by centrifugation at 11000 g in an SS34 rotor for 20 min. The cleared lysate was passed through an Ni-NTA (Ni2+-nitrilotriacetate) agarose column (Qiagen, Chatsworth, CA, U.S.A.). After washing with phosphate buffer (pH 7.8) containing 300 mM NaCl and 30 mM imidazole, elution was done using 250 mM imidazole. For further purification, the fractions were pooled from the Ni2+ column and dialysed and loaded on to a 2 ml packed phosphocellulose column (P11 cellulose; Whatman International, Maidstone, Kent, U.K.). The column was washed with 10 ml of wash buffer (50 mM Tris/HCl, pH 7.5, 0.5 mM EDTA and 20%, v/v, glycerol) containing 600 mM KCl and finally eluted with the same buffer containing 800 mM KCl. After elution, the peak fractions were dialysed overnight against storage buffer containing 10 mM Tris/HCl (pH 7.8), 100 mM NaCl, 5 mM MgCl2, 1 mM PMSF and 15% glycerol and stored at −70 °C until further use.
To purify the proteins from polyacrylamide gels, the purified 43 kDa protein was subjected to SDS/PAGE (12% polyacrylamide). A section of the gel was stained and the corresponding band of interest was excised from the remaining (unstained) portion of the gel and the protein was electroeluted in a buffer containing 50 mM Tris/HCl (pH 7.5), 1% SDS, 5 mM DTT (dithiothreitol), 150 mM NaCl and 100 mM EDTA per 0.1 g of wet gel slice at 4 °C. The protein was precipitated by the addition of an equal volume of acetone and kept at −20 °C for 1 h. The precipitated protein was collected by centrifugation at 17000 g in an SS34 rotor for 30 min. The resultant pellet was resuspended in 50 mM Tris/HCl (pH 7.8), 100 mM NaCl and 8 M urea and refolding was achieved by overnight dialysis in the above-mentioned dialysis buffer.
ATPase assay
ATPase measurements were carried out by the pyruvate kinase/lactate dehydrogenase assay described previously [8] with the following modifications. Reaction mixture (1 ml) contained 0.1 mM NADH, 2 mM phosphoenolpyruvate, 3 units of pyruvate kinase and 4 units of lactate dehydrogenase. Reactions were initiated by mixing the reaction mixture and ATP both preequilibrated to 30 °C. Decrease in NADH concentration was monitored by measuring the absorbance at 340 nm in a UV–visible spectrophotometer (Shimadzu, Columbia, MD, U.S.A.). All assays were performed in triplicate. The Km and Vmax values were determined from the double-reciprocal plots and the Ki value was calculated from the following equation:
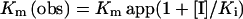 |
where Km(obs) is the Michaelis–Menten constant in the presence of the inhibitor, Kmapp is the Michaelis–Menton constant in the absence of the inhibitor, [I] is the concentration of the inhibitor and Ki is the inhibitory constant.
Protein cross-linking
Protein cross-linking was performed in a buffer containing 10 mM Tris/HCl (pH 8.0), 100 mM KCl, 4 mM DTT, 4 mM MgCl2 and 1.15 μM of the purified 43 kDa protein. Where indicated, samples were incubated with 1 mM ATP, ADP and p[NH]ppA (adenosine 5′-[β,γ-imido]triphosphate) for 1 h at 25 °C. DMS (dimethyl suberimidate; 4 μg/ml) was added and the incubation was continued for 4 h at 30 °C. Samples were subjected to SDS/PAGE (10% polyacrylamide) and analysed by Western blotting using a rabbit polyclonal antibody raised against the 43 kDa protein [4].
Molecular modelling
The model of the N-terminal 43 kDa fragment of LdTOP2 was based on the available crystal structure of the ATP-binding domain of GyrB from Thermus thermophilus [PDB (Protein Data Bank) accession no. 1kij]. Prediction of the three-dimensional structure of the N-terminal domain of LdTOP2 and its binary complex with ATP was done on the basis of knowledge-based homology modelling using Insight II 2000 of Accelrys (San Diego, CA, U.S.A.) and ABGEN [12] and our in-house package of MODELYN and ANALYN (MODELYN, a molecular modelling program; version PC-1.0; Indian Copyright no. 9/98, 1998, developed by C. Mandal) in UNIX as well as MS Windows environment. The structure of etoposide was built using the program ‘Builder’ module of Insight II followed by energy minimization and molecular dynamics using ‘Discover’ module and Cff91 force field on a Silicon Graphics® OCTANE workstation. The initial docking of etoposide into the ATP-binding pocket of LdTOP2 was made manually by superimposing the ribose moiety of ATP with the benzene ring of the drug. Energy minimizations and molecular dynamics were performed to get the most stable structure. For all energy minimizations, a combination of steepest and conjugate gradient methods (100 steps each) was used with a convergence criterion of 0.001 kcal/mol (1 cal=4.184 J). These steps were repeated until satisfactory conformational parameters were obtained. While carrying out energy minimization and molecular dynamics of complexes, positional constraint was applied to all residues which were more than 10 Å (1 Å=0.1 nm) away from the ligand. Secondary structures were predicted from sequences using SPOM and analysed using MODELYN and RIBBONS.
RESULTS
Cloning and purification of the N-terminal fragment of LdTOP2
ATP binding and hydrolysis by topoisomerase II is known to mediate sequential conformational changes in the enzyme, reaching from the N-terminal clamp to the C-terminal dimerization region [14]. To investigate the biochemical properties associated with the GyrB homologue of LdTOP2 (Figure 1A), we cloned the N-terminal 1–385 amino acids of LdTOP2 in pET16b, and for comparison with the full-length enzyme, the wild-type LdTOP2 (1236 amino acids) was cloned in pET28c. The resultant clones pET16b/1.1 and pET 28c/3.7 were transformed in BL21 (DE3) plysS and induced with 0.5 mM IPTG. Purification of the 43 kDa protein is shown in Figure 1(B).
Figure 1. Schematic representation of LdTOP2 and purification of the N-terminal domain of the protein.

(A) Schematic diagram. A co-ordinate of full-length LdTop 2: (A) wild-type LdTOP2 containing amino acid residues 1–1236, showing regions of homology with the GyrB and GyrA subunits of bacterial gyrase, and (B) N-terminal LdTOP2 protein. (B) SDS/10% polyacrylamide gel. Lanes 1 and 2, 2 μg cell extracts of E. coli BL21 (DE3) pLysS harbouring the pET16b/1.1 kb plasmid in the absence and presence of 0.5 mM IPTG respectively; lane 3, 2 μg 43 kDa protein purified through an Ni-NTA and phosphocellulose column. The gel was stained by silver stain. Sizes of molecular mass standards (MBI Fermentas, Flamborough, ON, Canada) in kDa are shown.
Properties of the 43 kDa protein
The ATPase activity of the 43 and 137 kDa proteins were quantified by PK/LDH-linked enzyme assay (Figure 2A) at a fixed substrate concentration. The ATPase activity of the two proteins was investigated as a function of enzyme concentration and found to exhibit a linear dependence of rate on enzyme concentrations in the range 5.8–140 nM. At higher enzyme concentrations, the rate of ATP hydrolysis reaches a plateau because of the unavailability of the substrate (ATP). Typically, 10 nM of the 43 kDa protein was found to have an activity of 50 nM/s (Figure 2A). This behaviour is in marked contrast with that of the 43 kDa N-terminal domain of GyrB, which exhibits a nonlinear dependence of the ATPase rate on enzyme concentration [8], whereas the N-terminal domain of human topoisomerase II also exhibits a linear dependence on enzyme concentration [10]. Interestingly, the ATPase activity of the 43 kDa protein remains unchanged in the presence of DNA, whereas, in contrast, the activity of the 137 kDa protein is enhanced by 2-fold in the presence of DNA (Figure 2B).
Figure 2. Kinetics of ATPase activity of the 43 kDa protein.

(A) ATPase activity of the 43 kDa (▲) and 137 kDa (●) proteins as a function of protein concentration. Rates are initial velocities; the substrate (ATP) concentration was 2 mM. (B) The ATPase activity of the 43 and 137 kDa proteins at 2 mM ATP and 100 nM protein concentration, in the absence and presence of DNA respectively. (C) Dependence of ATPase rate of the 43 kDa protein on ATP concentration at constant enzyme concentration. ○, phosphocellulosepurified protein; ●, refolded protein. The Km was calculated by the Lineweaver–Burk plot (inset plot). The data shown are means±S.D. for three independent experiments.
As a function of the substrate (ATP) concentration, the rate of ATP hydrolysis by the 43 kDa protein shows a hyperbolic dependence (Figure 2C), indicative of Michaelis–Menten kinetics. The values of Kmapp (0.33 mM) and Vmaxapp (833 nM/s) were determined from these data (Figure 2C, inset). To address the issue whether the ATPase activity was intrinsic to the N-terminal domain of LdTOP2 and was not attributable to some contaminating protein, the purified 43 kDa protein was applied to an SDS/polyacrylamide gel and the protein band was excised and refolded as described in the Materials and methods section. The ATPase activity of this refolded protein was essentially unchanged (Figure 2C). In addition, the induced bacterial cell extract harbouring pET16b when passed through an Ni-NTA agarose and phosphocellulose column, as described for the overexpressed 43 kDa protein, failed to show any ATPase activity (results not shown).
The optimal conditions for the ATPase reactions were explored. Over a range of pH values (6.8–8.8), we found the optimum pH to be 7.5. The ATPase activity of the 43 kDa protein was Mg2+-dependent and the optimum Mg2+ concentration was approx. 5 mM. At high salt concentrations (100 mM NaCl and 100 mM KCl), the ATPase activity was abolished. So, the following ATPase reaction conditions were used in the experiments described in this paper: 10 mM Tris/HCl (pH 7.5), 50 mM NaCl, 50 mM KCl and 5 mM MgCl2.
Dimerization
A salient feature of the ATPase activity of the N-terminal domain of GyrB is its nonlinear dependence on enzyme concentration, an observation that, when coupled with binding and structural data, strongly supports the idea that this domain dimerizes in the presence of nucleoside triphosphate [8,9,18]. The observation of a linear dependence of the ATPase rate on enzyme concentration for the 43 kDa protein raised the possibility that dimerization was either not occurring with this fragment or was not the rate-limiting step of the ATPase reaction. To address this issue, we performed cross-linking experiments with this fragment. Figure 3 shows that DMS cross-links higher molecular mass forms of this protein both in the absence and presence of p[NH]ppA, ADP and ATP. There are two visible cross-linked bands in these lanes, having sizes consistent with the dimeric forms of the enzyme. The untreated 43 kDa protein (lane 1) contains a small amount of dimers, which can be attributed to the fact that treatment with SDS does not completely dissociate the dimeric forms. From these data, we suggest that the dimeric form of the N-terminal domain of LdTOP2 is stable both in the absence and presence of nucleotides.
Figure 3. Cross-linking of the 43 kDa protein.

Western blot (with antibody specific to the N-terminal domain of LdTOP2) showing cross-linking of the 43 kDa protein by the DMS in the absence and presence of 1 mM nucleotides as indicated. Sizes of molecular mass standards in kDa are shown. AMP-PNP, p[NH]ppA.
Effect of drugs on ATPase activity of LdTOP2
Recently, it has been shown that in addition to cleavable complex formation, etoposide inhibits the ATPase activity and competes with ATP for interaction with wild-type and truncated human TOP2α [15,16]. To address the possibility of an additional drug-binding site on the N-terminal domain of LdTOP2, we have studied the effect of etoposide on the ATPase activity of the truncated LdTOP2. Etoposide is an effective inhibitor of the ATPase activity of the 43 kDa protein (Figure 4A). At a concentration of 10 μM, etoposide inhibited the rate of enzyme-catalysed ATP hydrolysis by approx. 73% with a Ki of 1.97 μM, but complete inhibition of ATPase activity was not apparent even at 50 μM drug concentration (results not shown). Figure 4(B) shows the double-reciprocal plot of this inhibition. Luteolin, a naturally occurring flavonoid, and DHBA (dihydrobetulinic acid), a derivative of pentacyclic triterpenoid, have been reported to show anti-leishmanial activity mediating its effect through topoisomerases of the parasite [17]. The effect of these drugs on the ATPase activity of the N-terminal domain of LdTOP2 was investigated. It was observed that DHBA does not interfere with ATP hydrolysis even at 400 μM concentration, whereas luteolin at 20 μM concentration was found to inhibit the rate of enzyme-catalysed ATP hydrolysis by 57% (Figure 4C).
Figure 4. Effects of topoisomerase II-targeted compounds on enzyme-catalysed ATP hydrolysis.

(A) ATPase activity in the absence (○) or presence of 5 μM (■) and 10 μM (▲) etoposide respectively. (B) Lineweaver–Burk representation of the inhibition of ATPase activity in the absence (○) and in the presence of 5 μM (■) and 10 μM (◆) etoposide. (C) ATPase activity of the 43 kDa protein at 2 mM ATP concentration in the absence or presence of 20 μM luteolin, 200 μM DHBA and 400 μM DHBA. The data shown are means±S.D. for three independent experiments.
Modelling of the ATP-binding pocket of LdTOP2
In order to study the N-terminal fragment of LdTOP2, the amino acid sequence of the 43 kDa protein was compared with that of Homo sapiens, Escherichia coli and T. thermophilus using CLUSTAL W. Figure 5 shows the regions of alignment of the N-terminal fragment of LdTOP2 with that of the above three species. The N-terminal 43 kDa protein of LdTOP2 shares a similarity of 48, 53 and 55% with the N-terminal domain of human topoisomerase IIα and GyrB from E. coli and T. thermophilus respectively. The model of the N-terminal 43 kDa protein of LdTOP2 was constructed using the crystal structure of the entire N-terminal domain of GyrB (43 kDa) from the thermophilic bacteria T. thermophilus (PDB accession no. 1KIJ) with p[NH]ppA and novobiocin [11]. The most reliable homology model generated for the parasite protein comprises 357 amino acids (starting from 29 to 385 amino acids) and shows root-mean-square deviation from standard bond length of 0.0269 Å and from standard bond angle of 3.36°. The structure reveals that the parasite protein can be divided into an N-terminal and a C-terminal domain separated by a hinge. The N-terminus contains a seven-stranded β-sheet and two long and three short α-helices. The C-terminus comprises one four-stranded β-sheet and two long and one short α-helices. The strand connectivity is also rather unusual. The whole structure is tightly folded with short connecting regions between the secondary structural elements (Figure 6A).
Figure 5. Alignment of the N-terminal domain of LdTOP2 with the N-terminal fragment of human topoisomerase IIα and the N-terminal fragment of gyrase of E. coli and T. thermophilus.

Multiple alignment of sequences from E. coli (GenBank® accession no. BAB38057), T. thermophilus (PDB accession no. 1KIJ), L. donovani (GenBank® accession no. AF150876) and H. sapiens (accession no. NM_001067) using CLUSTAL W. Asterisk and dot indicate identity and strong similarity respectively. Dashes indicate the gaps used to optimize alignment.
Figure 6. Three-dimensional structure of the N-terminal 43 kDa protein of LdTOP2.

Ribbon representation of the 43 kDa protein with only ATP (A), depicting its secondary structures: α-helices rendered in blue, β-sheets in yellow and random coils in orange. View of the modelled LdTOP2 with ATP (B) and with etoposide (C). The amino acid side chains of the protein are depicted in ball and stick representation the ATP and etoposide in solid sticks (carbon, green; nitrogen, blue; oxygen, red; hydrogen, white; and phosphorus, pink). The hydrogen bonds in dotted lines and bond distances in Å are shown.
There are several hydrophobic amino acids that line the ATP-binding site. The hydrogen bonds that stabilize ATP within the ATP-binding pocket primarily involve the adenine group and the amino acid residues Val190 and Asn96. Asp68 and Asn69 form hydrogen bonds with the sugar moiety and Ala143 forms a hydrogen bond with the PO3− group of the ligand (Figure 6B). Asn65, although it does not form any hydrogen bond with ATP, is found to be present in close proximity of the molecule. To test the relevance of the modelling data, we have introduced point mutations in the ATP-binding domain of LdTOP2, thereby changing residues that were predicted to be involved in ligand binding. Three point mutations (N65K, N69K and N96K) were introduced in the ATP-binding pocket in which the neutral amino acids were replaced with basic ones, the prediction being that both positive charge and bulkier side chain of the latter would interfere with the binding of ATP to these residues. Based on the unexpected antagonism between ATP and etoposide for binding with the N-terminus of topoisomerase II, we examined how etoposide might fit into this small, well-structured domain, by molecular docking analysis. First, etoposide was built using INSIGHT and its most stable conformation was analysed using molecular dynamics simulation and energy minimization. Due to covalent and steric constraints, etoposide preserves a cross-shaped structure with one axis made of sugar and 4-hydroxy 3,5-dimethoxy phenyl groups and the other axis formed by the 4-ringed moiety (results not shown). The planar shape of the ringed moiety is conserved among many topoisomerase II inhibitors. Docking of etoposide in the ATP-binding pocket revealed a good fit. The amino acids putatively involved in the binding of etoposide are Asn65, Val77, Tyr127, Asn128 and Asn96 (Figure 6C), which collectively contribute to four stabilizing hydrogen bonds to the drug. Point mutations were introduced in the amino acids (N65K and D130G) of the ATP-binding pocket involved in interaction with etoposide. Asp130, although not visible in Figure 6(C), is present in close proximity to the drug. Finally, as a general control for the validity of the molecular substitution technique and the predicted structure, Gly140 of the walker motif was replaced with Asp. If our modelling of the protein is correct, then G140D should affect the binding of ATP.
All the mutant proteins were expressed in E. coli and purified to homogeneity just like the wild-type protein (Figure 7A, inset). To compare their activities, assays were performed as described above. As for the mutations N69K, D130G and G140D, substantial reduction in the ATPase activity were found, but no reduction in the ATPase activity was detected for the other two mutations N65K and N96K (Figure 7A). Moreover, the activity of the protein containing N65K and N96K mutations were not inhibited by etoposide, reflecting their inability to interact with the drug (Figure 7B) in the same way as mutation of E155F and K123A alters the etoposide binding ability of human topo-isomerase II but does not change ATP binding [15].
Figure 7. ATPase activity of the mutant proteins.

(A) ATPase activity of the mutant proteins and the wild-type 43 kDa protein at 2 mM ATP concentration. The inset shows the purification of the mutant proteins. Lanes 1–5 show purification of N65K, N69K, N96K, D130G and G140D respectively. (B) ATPase activity of the 43 kDa protein, N65K and N96K in the absence and presence of 50 μM etoposide respectively. The data shown are means±S.D. for three independent experiments.
DISCUSSION
A key question in understanding the mechanism of action of topoisomerase II is to elucidate the role of ATP binding and hydrolysis in the overall process of DNA-strand passage reaction. In the present study, we report for the first time the kinetics of ATP hydrolysis by the N-terminal domain of LdTOP2. We have constructed N-terminal truncation mutant of LdTOP2 and studied the ATPase activity associated with this fragment. The data presented in this paper show conclusively that the ATPase activity resides within the 43 kDa N-terminal fragment of the LdTOP2 protein. We have found that ATP hydrolysis by the parasite protein follows Michaelis–Menten kinetics having a Km of 0.33 mM. This result is consistent with that obtained from the 52 kDa N-terminal fragment of human TOP2α [10] but is in contrast with the kinetics of ATP hydrolysis by the N-terminal ATPase domain of GyrB (43 kDa) that is distinctly non-Michaelian [8]. Thus the Km value of the ATPase domain of the parasite protein is in keeping with the Km values of 0.3 and 0.47 mM calculated for the ATPase activity of yeast and human topoisomerase II in the absence of DNA [10,19]. This suggests that substrate binding may remain unaffected whether the ATPase domain is a part of the holoenzyme or exists by itself.
The most striking difference between the ATPase activities of the N-terminal domain of L. donovani and human topoisomerase II is that the activity of the parasite protein does not get stimulated in the presence of DNA [10]. No direct evidence for the interaction of the ATPase domain of GyrB with DNA exists. It is predicted that stable binding of DNA by the 43 kDa domain of GyrB occurs only in the context of A2B2 tetramer [8], where the principal interaction between the A and B proteins is via the C-terminal 47 kDa fragment of B protein, which in the presence of the A subdomain forms a complex capable of relaxing DNA [20,21]. For the 43 kDa N-terminal fragment of LdTOP2, we did not detect any DNA dependence of ATPase activity under the given assay conditions. In the case of amino acid residues 1–409 of yeast topoisomerase II, it has been observed that there is a lack of ATPase stimulation by DNA in 100 mM salt concentration, which led to the interpretation that direct interaction between DNA and ATPase sites of a type II DNA topoisomerase is not important in the catalysis of ATP hydrolysis [22]. Studies on human topoIIα also suggest that the magnitude of DNA stimulation of ATPase activity is dependent on the size of the N-terminal fragment [22–24]. In keeping with this, we have also observed that the N-terminal domain of LdTOP2 is DNA-independent, whereas the full-length enzyme exhibits DNA-dependent ATPase activity.
Another property of the N-terminal domain of LdTOP2 is that it exists as a dimer, like the N-terminal fragment of human topoisomerase IIα [10,25]. This property is strikingly different from that observed in the case of gyrase where dimerization occurs only in the presence of nucleoside triphosphate [8–10]. In Leishmania, the existence of the protein as a dimer is consistent with the linear dependence of rate on enzyme concentration, i.e. no dimerization step is detectable in the kinetics of ATP hydrolysis. Moreover, it has been proposed that the eukaryotic domain forms a stable dimer through its interaction at its C-terminus, such that it does not dissociate during the ATP binding and hydrolysis cycles that result in clamp opening and closing. As a consequence, the N-terminal fragment behaves as a permanent dimer and shows ATPase kinetics that is not dependent on a dimerization step [10].
Much evidence has led to the notion that the catalytic site of topoisomerase II plays a key role in drug protein interaction in which the central catalytic core of topoisomerase II is sufficient to form a stable complex with a DNA double-strand break in response to bisdioxopiperazines and epipodophyllotoxins [26]. Very little is known about the effects of DNA cleavage-enhancing drugs on any step of topoisomerase II catalytic cycle other than cleavage–religation [27,28]. Genistein and other flavone-based compounds, which are inhibitors of the cleavage activity of the topoisomerase II, also interact with the ATP-binding site of tyrosine kinases [29–32]. The ATPase activity and double-stranded DNA unwinding activities of replicative helicase Rep A has been found to be inhibited by luteolin [33]. Moreover, eukaryotic topoisomerase II as well as B subunit of gyrase share the same consensus of ATP-binding motif with protein kinases [32]. It is possible that these and other topoisomerase II-targeted agents might alter the interaction of the enzyme with its high-energy cofactor. Therefore the effects of DNA cleavage-enhancing enzymes on the ATPase activity of topoisomerase II were examined. Consistent with these facts, the ATPase activity of the 43 kDa protein was inhibited by luteolin.
Etoposide and teniposide are successful anticancer agents that target human topoisomerase II. In Trypanosoma equiperdum, it has been reported that etoposide induces topoisomerase II-mediated cleavable complex formation [7]. There is evidence that etoposide binds competitively to the ATP-binding site of human toposiomerase II [15,16]. Several point mutations have been identified within the ATP-binding pocket of human topoIIα that lower the affinity of etoposide in vitro [15]. Another study has also shown that expression of the wild-type and N-terminal human topoisomerase IIα in yeast enhances drug resistance in vivo [16]. Consistent with this, we have found that etoposide inhibits the hydrolysis of ATP by the N-terminal domain of LdTOP2 (Figure 4C).
Recent evidence suggests that there may be sufficient conservation in the ATP-binding domain of topoisomerase across different kingdoms [34]. So, to understand better the interaction of the N-terminal domain with etoposide, we have performed molecular modelling of this domain. The modelling studies predicted that specific amino acid residues like Asn65, Asn69, Asn96 and Asp130 are involved in the interaction with ATP and etoposide. Point mutations were introduced into the ligand-binding pocket in the amino acids involved in interaction with the ligand and the drug. The results suggest that these mutants confer etoposide resistance in part because of the failure of the mutant proteins to interact with the drug. Moreover, these results support the hypothesis that residues Asn65 and Asn96 help to stabilize etoposide without being directly implicated in the correct co-ordination of ATP. Consistent with the prediction that Gly140 will contact ATP and not the drug, we found that substitution of Asp for Gly140 interferes with the interaction with ATP.
In summary, our results provide the first information of its kind about the N-terminal domain of topoisomerase II from a kinetoplastid parasite. We also provide information about the interaction of the N-terminal domain of LdTOP2 with etoposide, the catalytic inhibitor of topoisomerase II. We have identified a drug-binding site in the N-terminal domain of LdTOP2 in addition to the high-affinity drug-binding site in the core domain. The complexes of the N-terminal domain of LdTOP2 with ATP and etoposide reveal the structural background of competitive inhibition with ATP. This knowledge will allow the structure-based designing of novel inhibitors in the foreseeable future.
Acknowledgments
This work is dedicated to the memory of Professor A. N. Bhaduri. We are grateful to Dr D. Bhattacharya (Indian Institute of Chemical Biology) for his helpful comments and valuable discussions. This work was supported by a grant (BT/PR2643/BRB/10/250/2001) from the Department of Biotechnology, Government of India. T.S. and R.D. were supported by a fellowship from Council of Scientific and Industrial Research, India.
References
- 1.Kellener U., Sehested M., Jensen P. B., Gieseler F., Rudolph P. Culprit and victim-DNA topoisomerase II. Lancet Oncol. 2002;3:235–243. doi: 10.1016/s1470-2045(02)00715-5. [DOI] [PubMed] [Google Scholar]
- 2.Das A., Dasgupta A., Sengupta T., Majumder H. K. Topoisomerase of kinetoplastid parasite as potential chemotherapeutic targets. Trends Parasitol. 2004;20:381–387. doi: 10.1016/j.pt.2004.06.005. [DOI] [PubMed] [Google Scholar]
- 3.Wang J. C. Cellular roles of DNA topoisomerases: a molecular perspective. Nat. Rev. Mol. Cell Biol. 2002;3:430–440. doi: 10.1038/nrm831. [DOI] [PubMed] [Google Scholar]
- 4.Sengupta T., Mukherjee M., Mandal C. N., Das A., Majumder H. K. Functional dissection of the C-terminal domain of type II DNA topoisomerase from the kinetoplastid hemoflagellate Leishmania donovani. Nucleic Acids Res. 2003;31:5305–5316. doi: 10.1093/nar/gkg727. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 5.Harkins T. T., Lindsley J. E. Pre-steady-state analysis of ATP hydrolysis by Saccharomyces cerevisiae DNA topoisomerase II. 1. A DNA-dependent burst in ATP hydrolysis. Biochemistry. 1998;37:7292–7298. doi: 10.1021/bi9729099. [DOI] [PubMed] [Google Scholar]
- 6.Baird C. L., Harkins T. T., Morris S. K., Lindsley J. E. Topoisomerase II drives DNA transport by hydrolysing one ATP. Proc. Natl. Acad. Sci. U.S.A. 1999;96:13685–13690. doi: 10.1073/pnas.96.24.13685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nenortas E. C., Bodley A. L., Shapiro T. A. DNA topoisomerases: a new twist for antiparasitic chemotherapy? Biochim. Biophys. Acta. 1998;1400:349–354. doi: 10.1016/s0167-4781(98)00146-8. [DOI] [PubMed] [Google Scholar]
- 8.Ali J. A., Jackson A. P., Howells A. J., Maxwell A. The 43-kilodalton N-terminal fragment of DNA gyrase B protein hydrolyses ATP and binds coumarin drugs. Biochemistry. 1993;32:2717–2724. doi: 10.1021/bi00061a033. [DOI] [PubMed] [Google Scholar]
- 9.Ali J. A., Orphanides G., Maxwell A. Nucleotide binding to the 43-kilodalton N-terminal fragment of the DNA gyrase B protein. Biochemistry. 1995;34:9801–9808. doi: 10.1021/bi00030a018. [DOI] [PubMed] [Google Scholar]
- 10.Gardiner L. P., Roper D. I., Hammonds T. R., Maxwell A. The N-terminal domain of human topoisomerase IIα is a DNA-dependent ATPase. Biochemistry. 1998;37:16997–17004. doi: 10.1021/bi9818321. [DOI] [PubMed] [Google Scholar]
- 11.Lamour V., Hoermann L., Jeltsch J. M., Oudet P., Moras D. An open conformation of Thermus thermophilus gyrase B ATP-binding domain. J. Biol. Chem. 2002;277:18947–18953. doi: 10.1074/jbc.M111740200. [DOI] [PubMed] [Google Scholar]
- 12.Mandal C., Kingery B. D., Anchin J. M., Subramanium S., Linthicum D. S. ABGEN: a knowledge based automated approach for antibody structure modeling. Nat. Biotechnol. 1996;14:323–328. doi: 10.1038/nbt0396-323. [DOI] [PubMed] [Google Scholar]
- 13. Reference deleted.
- 14.Bjergbaek L., Kingma P., Nielsen I. S., Wang Y., Westergaard O., Osheroff N., Andersen A. Communication between the ATPase and cleavage/religation domains of human topoisomerase IIα. J. Biol. Chem. 2000;275:13041–13048. doi: 10.1074/jbc.275.17.13041. [DOI] [PubMed] [Google Scholar]
- 15.Leroy D., Kajava A. V., Frei C., Gasser S. M. Analysis of etoposide binding to subdomains of human DNA topoisomerase IIα in the absence of DNA. Biochemistry. 2001;40:1624–1634. doi: 10.1021/bi0019141. [DOI] [PubMed] [Google Scholar]
- 16.Vilian N., Tsai-Pflugfelder M., Benoit A., Gasser S. M., Leroy D. Modulation of drug sensitivity in yeast cells by the ATP-binding domain of human DNA topoisomerase IIα. Nucleic Acids Res. 2003;31:5714–5722. doi: 10.1093/nar/gkg737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chowdhury A. R., Majumder H. K. DNA topoisomerases in life and death: implications in kinetoplastid protozoa. Curr. Mol. Med. 2004;4:711–722. doi: 10.2174/1566524043360302. [DOI] [PubMed] [Google Scholar]
- 18.Wigley D. B., Davies G. J., Dodson E. J., Maxwell A., Dodson G. Crystal structure of an N-terminal fragment of the DNA gyrase B protein. Nature (London) 1991;351:624–629. doi: 10.1038/351624a0. [DOI] [PubMed] [Google Scholar]
- 19.Lindsley J. E., Wang J. C. On the coupling of ATP usage and DNA transport by yeast DNA topoisomerase II. J. Biol. Chem. 1993;268:8096–8104. [PubMed] [Google Scholar]
- 20.Brown P. O., Peebles C. L., Cozeralli N. R. A topoisomerase from Escherichia coli related to DNA gyrase. Proc. Natl. Acad. Sci. U.S.A. 1979;76:6110–6114. doi: 10.1073/pnas.76.12.6110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gellert M., Fisher L. M., O'Dea M. H. DNA gyrase: purification and catalytic properties of a fragment of gyrase B protein. Proc. Natl. Acad. Sci. U.S.A. 1979;76:6289–6293. doi: 10.1073/pnas.76.12.6289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Olland S., Wang J. C. Catalysis of ATP hydrolysis by two NH(2)-terminal fragments of yeast DNA topoisomerase II. J. Biol. Chem. 1999;274:21688–21694. doi: 10.1074/jbc.274.31.21688. [DOI] [PubMed] [Google Scholar]
- 23.Campbell S., Maxwell A. The ATP operated clamp of human DNA topoisomerase II alpha: hyperstimulation of ATPase by ‘piggy-back’ binding. J. Mol. Biol. 2002;320:171–188. doi: 10.1016/S0022-2836(02)00461-8. [DOI] [PubMed] [Google Scholar]
- 24.Hammond T. R., Maxwell A. The DNA dependence of the ATPase activity of human DNA topoisomerase IIalpha. J. Biol. Chem. 1997;272:32696–32703. doi: 10.1074/jbc.272.51.32696. [DOI] [PubMed] [Google Scholar]
- 25.Hu T., Sage H., Hsieh T. ATPase domain of eukaryotic topoisomerase II. J. Biol. Chem. 2002;274:5944–5951. doi: 10.1074/jbc.M111394200. [DOI] [PubMed] [Google Scholar]
- 26.Chang S., Hu T., Hsieh T. Analysis of a core domain in Drosophila DNA topoisomerase II. Targeting of an antitumor agent ICRF-159. J. Biol. Chem. 1998;273:19822–19828. doi: 10.1074/jbc.273.31.19822. [DOI] [PubMed] [Google Scholar]
- 27.Hsiang Y. H., Jiang J. B., Liu L. F. Topoisomerase II-mediated DNA cleavage by amonafide and its structural analogs. Mol. Pharmacol. 1989;36:371–376. [PubMed] [Google Scholar]
- 28.Schneider E., Lawson P. A., Ralph R. K. Inhibition of protein synthesis reduces the cytotoxicity of 4′-(9-acridinylamino)methanesulfon-m-anisidide without affecting DNA breakage and DNA topoisomerase II in a murine mastocytoma cell line. Biochem. Pharmacol. 1989;38:263–269. doi: 10.1016/0006-2952(89)90036-1. [DOI] [PubMed] [Google Scholar]
- 29.Akiyama T., Ishida J., Nakagawa S., Ogawara H., Watanabe S., Itoh N., Shibuya M., Fukami Y. Genistein, a specific inhibitor of tyrosine-specific protein kinases. J. Biol. Chem. 1987;262:5592–5595. [PubMed] [Google Scholar]
- 30.Graziani Y., Erikson E., Erikson R. L. The effect of quercetin on the phosphorylation activity of the Rous sarcoma virus transforming gene product in vitro and in vivo. Eur. J. Biochem. 1983;135:583–589. doi: 10.1111/j.1432-1033.1983.tb07692.x. [DOI] [PubMed] [Google Scholar]
- 31.Srivastava A. K. Inhibition of phosphorylase kinase, and tyrosine protein kinase activities by quercetin. Biochem. Biophys. Res. Commun. 1985;131:1–5. doi: 10.1016/0006-291x(85)91761-9. [DOI] [PubMed] [Google Scholar]
- 32.Markovits J., Linassier C., Fosse P., Couprie J., Pierre J., Jacquemin-Sablon A., Saucier J. M., Le Pecq J. B., Larsen A. K. Inhibitory effects of tyrosine kinase inhibitor genistein on mammalian DNA topoisomerase II. Cancer Res. 1989;49:5111–5117. [PubMed] [Google Scholar]
- 33.Xu H., Ziegelin G., Schroder W., Frank J., Ayora S., Alonso J. C., Lanka E., Saenger W. Flavones inhibit the hexameric replicative helicase RepA. Nucleic Acids Res. 2001;29:5058–5066. doi: 10.1093/nar/29.24.5058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Corbett K. D., Berger J. M. Structure of topoisomerase VI-B subunit: implication for type II topoisomerase mechanism and evolution. EMBO J. 2003;22:151–163. doi: 10.1093/emboj/cdg008. [DOI] [PMC free article] [PubMed] [Google Scholar]