

Abstract
Both ancient and recent polyploidy, together with post-polyploidization loss of many duplicated gene copies, complicates angiosperm comparative genomics. To explore an approach by which these challenges might be mitigated, genetic maps of extant diploid and tetraploid cottons (Gossypium spp.) were used to infer the approximate order of 3016 loci along the chromosomes of their hypothetical common ancestor. The inferred Gossypium gene order corresponded more closely than the original maps did to a similarly inferred ancestral gene order predating an independent paleopolyploidization (α) in Arabidopsis. At least 59% of the cotton map and 53% of the Arabidopsis transcriptome showed correspondence in multilocus gene arrangements based on one or both of two software packages (CrimeStatII, FISH). Genomic regions in which chromosome structural rearrangement has been rapid (obscuring gene order correspondence) have also been subject to greater divergence of individual gene sequences. About 26%-44% of corresponding regions involved multiple Arabidopsis or cotton chromosomes, in some cases consistent with known, more ancient, duplications. The genomic distributions of multiple-locus probes provided early insight into the consequences for chromosome structure of an ancient large-scale duplication in cotton. Inferences that mitigate the consequences of ancient duplications improve leveraging of genomic information for model organisms in the study of more complex genomes.
The generally more-rapid evolution of gene arrangement in angiosperms (flowering plants), relative to other higher eukaryotes such as animals, appears to be at least in part due to polyploidy and its consequences (Bowers et al. 2003). Polyploidy may result from the merger of more than one genome from species whose chromosomes can pair and recombine (autopolyploidy), or from species with divergent chromosomes that normally do not pair or recombine (allopolyploidy). It has long been suspected, based on differences among taxa in chromosome number and size, that many angiosperm genomes may have undergone ancient polyploidization (Stebbins 1966). Bearing out early hints based on parallel arrangements of duplicated DNA marker loci (Kowalski et al. 1994; Paterson et al. 1996), the demonstration of large-scale duplication in the Arabidopsis genome (The Arabidopsis Genome Initiative 2000; Blanc et al. 2000; Paterson et al. 2000), its resolution into multiple events (Vision et al. 2000; Simillion et al. 2002; Bowers et al. 2003), and the dating of a subset of these events to near (β) or before (γ) the monocot-dicot divergence suggest that virtually all angiosperms are paleopolyploids (Bowers et al. 2003).
Burgeoning genomic data are revealing that not only do most angiosperms share a few ancient whole-genome duplications, but many have also undergone more recent lineage-specific duplications. For example, a duplication detected in the rice sequence (Goff et al. 2002; Paterson et al. 2003) is shared by the major cereals but not by more distant monocots such as Musa (banana) and Allium (Paterson et al. 2004). Recent investigations of large numbers of ESTs suggest genomic duplications in many additional lineages (Blanc and Wolfe 2004). The curious failure of this EST-based approach to validate the rice duplication suggests that such methods underestimate the extent of genomic duplication, for reasons that are not yet understood.
Better understanding of polyploidy and its consequences are central to comparative biology. Traditional models (Ohno 1970) suggest that polyploidy may free one duplicated gene copy to evolve new function, with the fitness of the organism buffered by the second copy. However, several recent findings are at odds with this model, in particular, the reduced species-wide polymorphism levels associated with recently duplicated genes in Arabidopsis (Moore and Purugganan 2003) and the long period for which duplicated genes retain partially redundant functions (Gu et al. 2003). Theoretical considerations suggest that the fates of duplicated genes are closely associated with effective population size (Ne) for a taxon (Lynch and Conery 2003). Thus, insights from microbes such as yeast with large Ne may not extend well to crown eukaryotes with small Ne.
The high frequency of duplications (especially in comparison to dioecious organisms such as most animals) and experimental facility of angiosperms, together with their much smaller Ne than yeast, make them attractive models for dissecting the evolutionary consequences of polyploidy in higher eukaryotes. These consequences may have many dimensions—for example, patterns of distribution of QTLs within a nucleus, across taxa, and across environments (Jiang et al. 1998; Wright et al. 1998; Ming et al. 2001), together with intricate patterns of organ-specific reciprocal expression silencing (Adams et al. 2003) all suggest that nonlinear interactions between duplicated genes and genomes may contribute novel attributes to some polyploid crops.
The complicated genome structure that results from repeated cycles of genome duplication, each followed by loss of one member of many duplicated gene pairs (“diploidization”—[Eckardt 2001b]), adds much complexity to deciphering the evolutionary history of plants. Understanding the relative relationships between taxon divergence and genome duplication is essential to making truly orthologous comparisons among angiosperm genomes (Kellogg 2003). Such information adds a powerful new tool to help resolve the fates of individual genes and gene family members by more definitive determination of orthology and more precise links between gene duplication and taxon divergence than have been realized using molecular clock-based methods.
Gossypium (cotton) is an especially appropriate system in which to explore the comparative genomics of paleopolyploids. While the cereals have often been preferred models for angiosperm comparative genomics, the relative lack of knowledge of non-Poaceae monocots is presently a hindrance in studying ancient genomic duplications. The Malvales (including cotton) are presently the nearest relative to Arabidopsis outside of the Brassicales, for which a detailed genetic map has been described. Dating of the α event to after the cotton-Arabidopsis divergence assures that the two lineages differ by at least this ancient duplication event (Bowers et al. 2003). Cultivated cottons are tetraploids of relatively recent (ca. 1 Mya) origin from A- and D-genome diploid ancestors that may themselves have shared common ancestry about 10 Mya (Cronn et al. 2002; Wendel and Cronn 2003). Colinearity among these genomes has been well characterized (Rong et al. 2004). Further, the A- and D-genomes share common gametic chromosome number (13) with all six other genome types in the genus (B, C, E, F, G, K). That several related genera have many species with n = 6 has long hinted at the possibility of ancient duplication in the cotton lineage, however, solid evidence of such an event or its consequences has been lacking. Finally, the cotton-Arabidopsis comparison offers practical benefits—research into the genetic control of the seed-borne epidermal fibers that account for most of cotton's economic importance may benefit greatly from progress in understanding the growth and development of hair-bearing epidermal cells (trichomes) in Arabidopsis (Larkin et al. 2003; Schiefelbein 2003). The recent discovery that a cotton Myb could rescue an Arabidopsis gl1 mutant, and also induce seed trichome production in Arabidopsis (Wang et al. 2004) is particularly exciting.
As a further step toward unraveling the complexities that polyploidy introduces into comparative genomics, we explore in detail the comparative chromosome structural evolution of Gossypium and Arabidopsis. Detailed genetic linkage maps of the tetraploid and D-diploid cotton genomes (Rong et al. 2004) are used to infer the probable arrangement of mapped genes in a hypothetical common ancestor, mitigating both the consequences of diploidization and the inability of genetic mapping studies to detect DNA polymorphism at all duplicated loci. Analysis of this inferred gene order substantially improves the degree of correspondence in gene arrangement detected between the hypothetical ancestors of cotton and Arabidopsis. The first insights into structural genomic consequences of an ancient whole-genome duplication in cotton are revealed. This work points the way to more effective leveraging of genomic information from botanical models in the study and improvement of major crops, and improved understanding of mechanisms underlying botanical diversity.
Results
Inferring gene order along the chromosomes of the hypothetical ancestor of the A and D genomes of Gossypium
To improve our ability to identify intragenomic duplication in cotton, existing genetic maps (Rong et al. 2004) were supplemented with 147 new DNA markers that were likely (based on Southern blot data) to detect two or more duplicated loci in a highly polymorphic cross between diploids G. trilobum and G. raimondii, adding 290 new segregating loci. This yielded a total of 1243 loci from the At subgenome (i.e., the 13 tetraploid chromosomes derived from an A-genome diploid ancestor), 1218 loci from the Dt subgenome (the 13 tetraploid chromosomes derived from a D-genome diploid ancestor), and 1014 loci from the D diploid genome (Supplemental Table 1).
A total of 781 probes were mapped to two or more loci in cotton. Alignments among homoeologous At, Dt, and D chromosomes were readily established based on 333 pairs of loci arranged in extensive blocks with corresponding order and orientation. An additional 87 pairs of loci revealed blocks of corresponding order but opposite orientation (reflecting inversions). Homoeologous reference loci were used as a framework to interpolate the probable locations of additional markers that could be mapped in only a subset of the homoeologs. In the vast majority of cases, this reflects lack of genetic polymorphism. Even in interspecific crosses, per-locus polymorphism rates in tetraploid cotton are modest (e.g., Rong et al. 2004), and the likelihood of finding polymorphisms at each of two homoeologous loci is small.
Several lines of evidence show that gene loss per se is infrequent in the At and Dt genomes. First, the vast majority of cDNA or other low-copy probes detect multiple “alloallelic” restriction fragments in tetraploid cotton, which comigrate with fragments in diploid progenitors (Reinisch et al. 1994). In a survey of 40 gene pairs based on high-resolution SSCP, 100% of the 40 genes studied were represented in tetraploid cotton by two homoeologous copies (Adams et al. 2003), suggesting a rate of gene loss that is no more than 2.5%. Lack of rearrangement following polyploid synthesis in cotton (Liu et al. 2001) and identical gene composition along corresponding At and Dt cotton BACs (Grover et al. 2004) support this view. While we cannot preclude the possibility of occasional gene loss, all available evidence points to it playing a minor role in the inability to map homoeologs in tetraploid cotton.
In the absence of appreciable gene loss, the same principles that apply to the inference of nonpolymorphic sites across the At, Dt, and D genomes are appropriate for inference of approximate gene arrangement along the chromosomes of their hypothetical common ancestor. To infer the probable gene order along the chromosomes of a hypothetical common ancestor of the At, Dt, and D genomes, the Dt genome was used as the primary map into which the other maps were merged. We chose Dt as the reference map because (1) it is virtually collinear with the D diploid map, in contrast to extensive rearrangement of the At genome relative to its A ancestor (Brubaker et al. 1999), and (2) because the Dt map had 20% more markers than the D diploid map so less inference is necessary. All common colinear loci within the same homoeologous group were used as anchor loci to start the construction of consensus maps by simple interpolation based on the relative spacings of nonanchor loci between consecutive anchors. The D map was merged into the Dt map first, followed by the At map, with the exception of chromosome ends (from the end to the first or last common marker). For regions that differed by inversion, marker locations were interpolated based on the Dt order (Fig. 1A,B; Supplemental Fig. 1; Table 1).
Figure 1.


Procedure and example of inferring approximate gene order along the chromosomes of a hypothetical common ancestor of the cotton subgenomes. (A) Illustration of consensus map assembly process. A framework of DNA markers that mapped to homoeologous sites (e.g., X1/X2 and Y1/Y2) were used to interpolate the probable locations of additional markers (e.g., M1/M2) that could be mapped in only a subset of the homoeologs (for details, see Methods). (B) Consensus map of cotton homoeologous group 3 (Chr.3, Chr14/17, and D3), as an example. Markers colored with green, blue, and red were originally from At, Dt, and diploid D chromosomes, respectively. Markers colored with brown were common to two or more homoeologous chromosomes. Vertical bars colored with yellow and green represented inverted regions on diploid D and At chromosomes, respectively, compared with the inferred map. The vertical ellipses highlighted with blue represent possible locations of centromeres, inferred as described elsewhere (Rong et al. 2004). Superscripted numbers in parentheses indicate centiMorgan locations that could not be shown between major gridlines on the map due to space, followed by markers at those locations. Markers below the maps also could not be shown at the centiMorgan locations indicated (parentheses), due to space.
Table 1.
Features of cotton consensus chromosomes
| Cotton Chra | Homoeolog (At,Dt,D) | # Ioci | Length (cM) | Largest gap (cM) | % Ioci sequenced | % Ioci with match in Arab. | % Ioci in conserved synteny with Arab.b | % Ioci with no match in Arab. |
|---|---|---|---|---|---|---|---|---|
| C01 | 7,16,1 | 245 | 195.4 | 9.9 | 91.4 | 65.2 | 38.4 | 34.8 |
| C02 | 1,15,2 | 194 | 176.4 | 6.9 | 94.8 | 52.7 | 54.6 | 47.3 |
| C03 | 2/3,17,3 | 149 | 120.7 | 14.5 | 94.0 | 66.4 | 62.4 | 33.6 |
| C04 | A02,D03,4 | 208 | 183.8 | 6.2 | 94.2 | 63.3 | 35.5 | 36.7 |
| C05 | 2/3,14,5 | 246 | 191.9 | 11.4 | 90.2 | 61.7 | 46.7 | 38.3 |
| C06 | 9,23,6 | 235 | 172.6 | 6.4 | 91.9 | 61.1 | 33.3 | 38.9 |
| C07 | A03,D02,7 | 290 | 217.2 | 7.9 | 94.1 | 61.5 | 49.4 | 38.5 |
| C08 | 12,26,8 | 248 | 159.5 | 6.4 | 91.5 | 63.3 | 52.4 | 36.7 |
| C09 | 4/5,D08,9 | 382 | 264.6 | 7.3 | 89.5 | 58.8 | 43.8 | 41.2 |
| C10 | 6,25,10 | 164 | 172.6 | 9.1 | 95.1 | 58.3 | 48.4 | 41.7 |
| C11 | 10,20,11 | 227 | 170.2 | 6.1 | 94.7 | 63.7 | 54.0 | 36.3 |
| C12 | 4/5,22,12 | 186 | 101.7 | 4.3 | 94.7 | 70.1 | 41.9 | 29.9 |
| C13 | A01,18,13 | 242 | 198.1 | 11.8 | 94.6 | 63.3 | 46.2 | 36.7 |
| Total | 3016 | 2324.7 | 14.5 | 92.8 | 62.1 | 46.1 | 37.9 |
Based on hypothetical ancestral chromosomes; does not correspond to published nomenclature for modern tetraploid chromosomes (Rong et al. 2004).
Including Ioci identified by either CS2, FISH, or both.
Differences in polymorphism associated with terminal markers may cause the respective maps to have somewhat different coverage of their respective chromosomes. As a best estimate of the coverage of the respective maps, we considered the centiMorgan length of the respective maps from the last common marker to the terminus. If the ends of the At or D maps were longer than the corresponding segments of the Dt map, the longer segment was used as the primary map for the affected end. The Dt map was the longest for only three chromosomes (2, 4, 6), thus, the maps of the other 10 chromosomes are therefore somewhat longer than the Dt map (Supplemental Fig. 1; Table 1). To deal with translocations between tetraploid (At) Chrs. 2 and 3, and 4 and 5, the affected chromosomes were split, with the appropriate regions merged into different D genome homologs (Supplemental Table 1). To implement the rules that we outline above for inferring the approximate gene arrangement along the chromosomes of a hypothetical common ancestor of the At, Dt, and D genomes, a computer program was written in PHP.
In partial summary, we assembled 13 maps representing the inferred gene orders along the chromosomes of the hypothetical common ancestor of the A and D genomes of Gossypium. Each consensus map was named according to the name of the diploid D genome chromosome, but with the prefix C (“consensus”). The inferred map of the hypothetical ancestral cotton chromosomes included 3016 loci, spanning 2324.7 cM, with the largest gap being 14.5 cM (Table 1).
Cotton sequences having Arabidopsis homologs
A total of 2162 (92.5%) of probes detecting 2800 (92.8%) of loci in the inferred ancestral cotton map could be sequenced (Table 1; Supplemental Table 1). Among these, 1738 (62.1%) of the sequenced loci had one or more unambiguous homologs in the Arabidopsis genome based on 7437 BLAST matches that met a threshold of E < 10-10 (listed in Supplemental Table 2). A total of 1005 cotton loci matched 3106 Arabidopsis genes that fell within 30 genes of one another along the inferred pre-α gene orders, and thus, were considered tandem or proximal duplicates likely to have arisen by illegitimate recombination and/or transposition. Similarly, 386 Arabidopsis genes matched pairs of cotton loci <5 cM apart, which were also considered proximal duplicates (the rationale for setting these proximity thresholds are in Methods). To avoid the detection of false correspondence based on multiple matches to proximal duplications, only one member of these groups of genes was kept for further analysis. Removal of 2305 proximal duplicates from the match file left 5132 matching pairs that were considered “potential orthologs” and used in comparative analyses.
Distribution of Arabidopsis putative orthologs along inferred ancestral cotton chromosomes
The distribution of best-matching Arabidopsis sequences on inferred ancestral cotton chromosomes was not random. Among 1720 pairs of neighboring cotton loci, 220 (12.8%) corresponded to genes from the same Arabidopsis α-duplicated segment (Supplemental Table 2), significantly (P = 3.02 × 10-63) more than the ∼75 explicable by chance, if cotton genes were randomly distributed across the 34 α-duplicated segments that collectively comprise 89% of the Arabidopsis genes. In addition, 143 (8.3%) pairs of loci corresponding to genes from the same Arabidopsis-duplicated segment(s) were separated by only one conflicting locus (corresponding to a different Arabidopsis duplicated segment), significantly higher than the 71 explicable by chance (P = 1.14 × 10-17).
Inferred ancestral Arabidopsis gene arrangement shows more conserved synteny with cotton than modern Arabidopsis gene order
To explore the extent and distribution of conserved synteny between cotton and Arabidopsis, two software packages, CS2 and FISH (each described in the Methods), were each used to analyze the 5132 pairs of “potential orthologs.” CS2 and FISH, respectively, detected 187 clusters, including 800 pairs of matching loci and 310 blocks, including 715 pairs of matching loci. Details of corresponding regions are summarized in Supplemental Tables 3 and 4 and presented in full in Supplemental Table 2.
To explore the consequences of duplication/diploidization in Arabidopsis for genomic comparisons, we compared the inferred ancestral cotton map with both the modern Arabidopsis genome and with inferred pre-α-duplication Arabidopsis gene orders (Bowers et al. 2003). When CS2 was used for comparison of cotton to the modern Arabidopsis genome, 270 (33.8%) of the 800 pairs of matching loci no longer occurred in clusters, reflecting gene loss in modern Arabidopsis chromosomes following the α event. As an example (Fig. 2), a region of 31.1 cM with 67 loci was selected from C06. Among 39 cotton loci that had homologs in Arabidopsis, 24 showed conserved synteny with pre-α duplications, but only 17 with modern Arabidopsis. Using the inferred ancestral Arabidopsis gene order increases the detection of conserved synteny, so it was used in further studies.
Figure 2.

Conserved synteny between a segment of C06 and Arabidopsis α duplicates α11 and α14. Loci with “*” are those that could not be sequenced. Loci with “%” were sequenced, but no orthologs were found in Arabidopsis. Loci highlighted in blue showed conserved synteny with α11 and in green with α14. The remaining loci had orthologs in Arabidopsis, either showing conserved synteny with other duplications, or no synteny. Duplicated Arabidopsis genes contributing to assembly of inferred gene orders were linked with dashed lines.
Patterns of conserved synteny between cotton and Arabidopsis
While both CS2 and FISH revealed cotton-Arabidopsis correspondence between many common regions, there were also noteworthy differences in reciprocal comparisons that used cotton (Supplemental Table 3) or Arabidopsis (Supplemental Table 4) as the foundation. Based on evaluation of matching loci in their distribution along the cotton chromosomes, CS2 detected correspondence over nearly twice as much of the genome as FISH, both in total and along each chromosome. Most regions detected by CS2 were larger than, and inclusive of the regions detected by FISH, excepting regions determined with only two pairs of matching loci (CS2 requires at least three matching pairs for significance). Overall, a total of 1372.1 cM, or 59.0% of the cotton consensus map, showed nonrandom correspondence, putatively synteny, with at least one Arabidopsis α-duplicated segment based on CS2. Different cotton chromosomes varied in the portion over which correspondence could be inferred, from 81.1% for C05 to 40.4% for C10 (Supplemental Table 3).
In most cases (81.1% with CS2, 96.8% with FISH), a single genomic region in the hypothetical ancestral cotton map corresponded to only one to two Arabidopsis-duplicated segments. These percentages varied among chromosomes—for example, over 80% of correspondence on C01 and C10 involved only one Arabidopsis α region, while on C03, nearly 70% of correspondence involved two or more Arabidopsis regions. Evaluation of putative orthologs (as described above) in their distribution along the Arabidopsis genome (measured in the number of genes along the segments) also showed much correspondence with cotton (detailed in Supplemental Table 4). Segments of Arabidopsis pre-α gene orders covering 12,402 genes (53.5%) showed correspondence with the cotton consensus map using CS2. Again, FISH detected lower correspondence (27.8% of the Arabidopsis transcriptome) across the whole genome, and on all but three individual pre-α segments (α01, α17, αS07).
Ancient duplication in the cotton genome since its divergence from a common ancestor shared with Arabidopsis
Previously (Rong et al. 2004), we noted some hints of segmental duplication within individual cotton subgenomes. We further investigated this in two ways. First, 39,079 ESTs for G. arboreum (a diploid A-genome genotype) were assembled into unigene sets using Phrap with minmatch 20, minscore 100, and repeat stringency 0.95. Next, genes (ESTs) were blasted against one another, and the synonymous substitution rate for the best-matching sequence was calculated using standard methods. This yielded a marked peak at a Ks value of about 0.45 (Fig. 3), suggestive of a large-scale duplication about 15-30 Mya, depending on the neutral substitution rate used (Gaut et al. 1996; Koch et al. 2000). In either case, this is too recent to be shared with Arabidopsis, in that the cotton and Arabidopsis lineages appear to have diverged no later than 83-86 Mya based on evidence from fossilized pollen (Benton 1993). A second peak at about Ks 1.5 may perhaps reflect the Arabidopsis β event, but needs further investigation. Similar patterns of distribution of Ks values were recently reported by others (Blanc and Wolfe 2004).
Figure 3.

Patterns of DNA sequence similarity mong G. arboreum ESTs. By analyses described in the text, similarity among best-matching sequences shows a marked peak at a Ks value of about 0.45, suggestive of a large-scale duplication about 15-30 Mya, depending on the neutral substitution rate used (Gaut et al. 1996; Koch et al. 2000).
To explore the consequences of possible paleopolyploidy of cotton at the level of chromosome structure, loci that were duplicated on nonhomoeologous chromosomes and/or within the same subgenome were mapped to each of their multiple locations on the inferred ancestral map and identified by the addition of a lower-case letter at the end of the probe name (Fig. 1; Supplemental Fig. 1; Supplemental Table 1). FISH and CS2 were used to analyze the distribution of these multiple loci. CS2 showed correspondence over 1200.1 cM, or 51.8% of the genome, more than twice the correspondence detected with FISH (564.1 cM, 24.3%: Supplemental Table 5; Fig. 4). Individual corresponding regions detected with CS2 were generally larger and often included the regions detected by FISH (Supplemental Table 5; Fig. 4). Individual chromosomes varied widely in the portion for which correspondence to other parts of the genome could be identified, from 7.4% (C10 with FISH) to 68% (C02 with CS2).
Figure 4.


Duplication in hypothetical ancestral cotton chromosomes. This map includes only those sequence-tagged probes mapped on two or more places and showing the duplications. The vertical bars highlighted with blue on the map represent the possible locations of centromeres (see Fig. 1B). Solid and broken vertical lines beside the markers represented the duplicated regions detected with FISH and CS2, respectively. The 13 colors identified the chromosomes containing duplicated regions, as shown in the legend.
Both FISH and CS2 suggested occasional correspondence of individual regions of one subgenome to multiple regions of the other, an observation that on the surface may indicate still more ancient duplication events in the cotton lineage. However, such associations are suspect in that they accounted for a disproportionately large share of the incongruities between inferences made using the two packages. Overall, the lengths of corresponding regions on individual chromosomes as estimated by CS2 versus FISH were significantly correlated with one another, but only at a modest level (r = 0.28). There was very strong agreement (r = 0.51) in the portions of individual chromosomes that matched only one other region of the genome. However, there was virtually no relationship between the two packages (r = 0.017) in portions matching two other regions. Higher-order matches lacked adequate data for meaningful comparison. A one-to-one relationship was indicated for 50% and 74% of the corresponding regions by CS2 and FISH, respectively. As illustrated in Figure 4, the number of corresponding regions, in particular those detected by CS2, appears to be related to marker density of the consensus map. Postulated centromeric regions appear to be especially high in both marker density and CS2-inferred correspondence among chromosomal segments. However, we question whether the multiple associations among chromosomes in these regions truly represent conserved synteny—other work in our group (Bowers et al. 2005) has suggested that intercentromeric gene movement may be associated with evolution of bivalent chromosome pairing in recently formed polyploids. We speculate that such gene movement may cause CS2 to falsely infer conserved syntenies. More information is needed to carefully evaluate this hypothesis, in particular, the exact locations of all cotton centromeres (which are presently approximated). However, at present, we remain cautious of these multichromosomal associations, and suggest that the more stringent FISH algorithm may better represent true synteny (or lack thereof) in these regions.
Further improving the detection of conserved synteny between cotton and Arabidopsis
The combination of cotton segments showing paleohomoeology with one another appears likely to further improve the identification of conserved synteny between cotton and Arabidopsis. Two corresponding chromosomal segments from C05 and C09, respectively (Fig. 5), show six corresponding loci in collinear order (Gate2BC05, pAR0050, Gate4CD03, Unig26C03, pAR0945, and Gate2DH05), except for an inversion between pAR0945 and Gate2DH05 at the ends of two chromosome segments. If a consensus gene arrangement is constructed from the two segments, additional conserved syntenies are identified. For example, when each cotton segment was considered separately, no one showed conserved synteny with α03 on Arabidopsis chromosome 1. But, when the two cotton homoeologous segments are merged, correspondences to two new portions of α03 are identified (Fig. 5). Based on a consensus arrangement inferred from interleaving the two segments, a total of 11 additional Arabidopsis segments showed correspondence, with a maximum of two corresponding to the same cotton region. Sufficient data to apply this approach on a genome-wide scale in cotton may greatly extend the length of chromosomal segments over which conserved synteny can be inferred.
Figure 5.

Additional conserved synteny between Cotton and Arabidopsis α duplicates detected by merger of ancient duplicated cotton chromosomal segments of C05 and C09. Common markers between C05 and C09 were highlighted in red. The number in parenthesis after gene 1683 is the Arabidopsis chromosome. The additional conserved syntenic blocks from Arabidopsis were presented as vertical dashed bars in between segments of consensus cotton chromosomes C05 and C09. The corresponding loci (named per Fig. 2) were linked with lines, blue from C05 and green from C09. Solid and broken vertical lines beside the maps represent conserved syntenic regions detected with FISH and CS2, respectively, when C05 and C09 were individually analyzed for conserved synteny with Arabidopsis.
Discussion
The relatively close relationship of cotton and Arabidopsis, detailed genetic map for cotton, and potential importance of using functional genomic information and tools from Arabidopsis to aid in dissecting economically important pathways in cotton make this system an excellent case study for exploring comparisons of gene order among divergent taxonomic families. The inferred map of 3016 loci spanning 2324.7 cM in the genome of a hypothetical common ancestor of the A and D Gossypium genomes, by itself, is a valuable tool for a wide range of applications. Due to the modest levels of DNA polymorphism among modern (AD) polyploid cottons, we can genetically map both members of a homoeologous gene set in only about 20% of cases, even using interspecific crosses. The inferred map predicts the locations of the remaining 80% of homoeologs that cannot be mapped in any one cross, resolving many incongruities between maps of different tetraploid species (which often segregate for polymorphic alleles at different homoeologs). The inferred map is an excellent resource from which markers can be selected for marker assisted selection, identification of introgression lines, QTL mapping, and SNP discovery using sensitive new techniques that permit identification of informative DNA marker alleles in sequence-tagged sites such as these that had previously been mapped as RFLPs. A growing number of such studies have been done or are in progress (Jiang et al. 1998, 2000a,b; Wright et al. 1998, 1999; Saranga et al. 2004).
The inferred ancestral map is especially important for linking the tetraploid cotton genetic map to emerging BAC-based physical maps, in that it mitigates not only the omission of monomorphic loci, but also any gene loss associated with “diploidization” subsequent to AD polyploid formation. Most of the mapped sequences have been used to anchor high-coverage bacterial artificial chromosome (BAC) libraries of G. hirsutum, G. barbadense, and G. raimondii (see http://www.plantgenome.uga.edu/cotton/CottonDBFrames.htm), the latter of which is also being completely fingerprinted (http://www.plantgenome.uga.edu/projects.htm#Cotton). This will permit us to resolve more precisely the true arrangements of loci that are too closely linked to order with confidence based on genetic recombination.
The inferred ancestral map of cotton, together with similar pre-α Arabidopsis gene orders, far surpassed the usefulness of comparisons made between extant Arabidopsis and cotton gene orders to reveal conserved synteny. Cotton and Arabidopsis may have shared a common ancestor about 83-86 Mya (Bowers et al. 2003), only about 20-30 Myr before the divergence of the cereals that are often considered models for comparative genomics. Instances of synteny or collinearity have been identified (Eckardt 2001a) even for species that are thought to have diverged from Arabidopsis as much as 100 Mya or more (Paterson et al. 1996; Ku et al. 2000; Mayer et al. 2001; Rossberg et al. 2001; Salse et al. 2002) based largely on sequenced BACs or other large DNA fragments. However, to the degree that gene loss followed polyploidization events in these lineages, these studies chronically underestimated the degree of conserved synteny that existed (Bowers et al. 2003; Kellogg 2003). These issues are at least partly mitigated by the comparison of the inferred ancestral cotton map and the pre-α Arabidopsis gene order, which permits us to identify conserved syntenies between individual cotton chromosomes and typically three to five Arabidopsis α-duplicated segments (Fig. 5).
Herein, we begin the process of unraveling the consequences of an ancient duplication so far only known in the Gossypium genus, beginning to reveal the sizes and locations of duplicated regions. Growing evidence suggests that historical estimates of the role of paleopolyploidy in angiosperm evolution (Stebbins 1966) were underestimates, and that the evolutionary history of all angiosperm genomes includes one or more cycles of polyploidization (Bowers et al. 2003; Blanc and Wolfe 2004). Our findings in cotton were foreshadowed by classical cytogenetic studies (Muravenko et al. 1998) hinting that the ancestors of cultivated allotetraploid cotton experienced polyploidization events that far predate the formation of modern allotetraploids in the Pleistocene. Wendel and Cronn (2003) systematically reviewed the research results in this area and proposed that present AD-genome allotetraploids are likely to be at least paleooctaploid.
Much remains to be done. While we have found tentative correspondence of segments covering about half of the genome, we must assume that many small rearrangements within these segments have escaped detection by the coarse resolution afforded by the (albeit relatively large) number of genetically mapped DNA markers in hand. Additional genetic information and eventually genomic sequence will further clarify the probable patterns of gene arrangement that predated this duplication in the cotton lineage.
Although we have concentrated in this work on duplication events that occurred more recently than the divergence of the cotton and Arabidopsis lineages from a common ancestor, the effects of still more ancient duplications shared by the lineages are also evident. In several cases, different Arabidopsis α-duplicated segments matching the same cotton region corresponded to one another as a result of more ancient β or γ duplications found by Bowers et al. (2003) (Supplemental Table 2). For example, the cotton chromosomal region shown in Figure 2 mainly corresponded with α11 and α14, except for two loci having conserved synteny with α20. The loci showing conserved synteny between C06 and four members of these two α duplications were largely collinear on both cotton and Arabidopsis chromosomes. From Bowers et al. (2003), the homoeologous regions of α11 and α14 are both thought to derive from the more ancient gene order called β4.
The true properties of the “other half” of the genome, in which we have not yet found even early evidence of ancient duplication, remain to be clarified. For many of these we may simply have too little information to discern a “signal” of structural conservation from among the many factors that may contribute noise to this data set. However, genomic regions in which chromosome structural rearrangement has been rapid (leaving no conserved synteny), appear also to have been subject to rapid divergence of individual gene sequences. In analysis of potential orthologs with FISH and CS2, 802 (46.1%) detected nonrandom correspondence, putatively conserved synteny between cotton and Arabidopsis (Table 1; Supplemental Table 2). To evaluate their patterns of distribution across the genome, the consensus maps were subdivided into bins of 10 cM in length. While all chromosomes (ranging from 89.5% to 95.1%: Table 1) and most bins (Fig. 6) contained very similar proportions of sequenced loci, the percentage of loci showing putative synteny with Arabidopsis was highly variable. We further plotted the percentage of loci for which an Arabidopsis homolog could not be identified (Table 1). There was a strong negative correlation (r = -0.57) between the percentage of loci (per bin) showing conserved synteny with Arabidopsis, and the percentage of loci showing no match in Arabidopsis. This indicates that different genomic regions may tolerate rearrangement at different rates, a hypothesis that has previously been suggested based on the failure to identify ancient duplications in centromeric regions of both Arabidopsis and rice. A fascinating question for further study is whether such rapidly evolving regions contain a disproportionate share of genes that account for morphological or physiological divergence between taxa, including reproductive isolation.
Figure 6.

(A) Percentage of sequenced loci, loci showing conserved synteny between cotton and Arabidopsis α duplicates, and loci showing no orthologs in Arabidopsis. (B) Total mapped locus number in 10-cM bins along cotton consensus map C02 (a typical example). Ellipses represent possible location of centromeres, inferred as described elsewhere (Rong et al. 2004).
Ongoing improvement of analytical tools may also help to better resolve long-range comparative data. The main difference between CS2 and FISH is the method by which individual points in the data matrix are taken as evidence for conserved synteny. In CS2, Euclidean distance was used and if points are spatially close enough, they will be flagged as correspondence. This algorithm may thus be especially prone to false positives in regions of the genome in which low recombination per unit physical distance results in high marker cosegregation and inability to resolve true orders of genes along the chromosome. As we noted, CS2 tended to detect more correspondence and longer segments in marker-rich genomic regions, but multiple associations in these regions appear likely to include some false positives. In contrast, in FISH, Manhattan distance was used and only the points close enough to meet likelihood thresholds and also in a roughly diagonal line are taken as evidence of correspondence. However, deviations from such a diagonal may be caused by a number of factors. For example, localized inversions (Grant et al. 2000; Vision et al. 2000; Salse et al. 2002) are relatively frequent and often cause recently arisen deviations from an overall pattern of ancient correspondence. Further, in comparisons of genetic maps to sequences (or transcript maps as done herein), differences between recombinational and physical distances will again cause deviations from the diagonal. Finally, with relatively sparse data from genetic maps, many corresponding segments may only be comprised of the minimal set of three (CS2) or even two (Brownstein et al. 2003) data points. These factors motivated our decision to compare the two different algorithms. While the ultimate solution to these limitations will be to sequence the underlying genomes, in the meantime, many investigators may reap much benefit from botanical (or other) models in advancing the study of a wide range of additional genomes by careful consideration of the consequences of paleopolyploidy, using these or other approaches.
Methods
Assembling an inferred map of the hypothetical cotton ancient genome
Genetic linkage maps of the At (tetraploid A sub genome), Dt (tetraploid D sub genome), and D (diploid D genome) chromosomes, reported by Rong et al. (2004), were used in the construction of the inferred ancestral cotton map. A total of 290 new loci were added to the D genome map since publication of the prior map. The probes, procedures, and population used for mapping these new loci were as reported by Rong et al. (2004).
It was previously noted that many probes detected multiple polymorphic loci (Rong et al. 2004). Many of the duplicates were distributed at collinear locations on the At, Dt, and D chromosomes, respectively, permitting us to identify homoeologous groups of At, Dt, and D chromosomes. Inferred ancestral maps were assembled from the chromosomes of a homoeologous group by using common colinear loci for alignment. The Dt genome was used as the primary map to which the other maps were merged into. The D map was merged first, followed by the At map, with the exception of chromosome ends (from the end to the first common marker). If the ends of the At or D maps were longer than the corresponding fragments of the Dt map, the longer fragment was used as the primary map for the mapping of this end (Fig. 1A). While this imposes modest bias in the recombinational lengths of terminal regions, it was necessary to accommodate the likelihood that the longest terminal interval covers more chromatin than the others. If the markers are in a collinear order, but in the opposite direction, the order on the Dt chromosome was used. Loci that are duplicated on the homoeologous chromosomes but not in collinear order, or on nonhomoeologous chromosomes, are identified by the addition of a lower-case letter at the end of the probe name in the consensus map. Relative spacing along the consensus map of the loci in regions between two consecutive markers shared by At or D and Dt are calculated as follows:
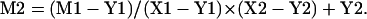 |
Here, M is the marker located on At or D and intended to be merged into the inferred map. M2 is the relative location (distance from the top of the inferred maps, cM) of the marker (M) in the inferred map (Fig. 1A). M1 is the original location of marker M on At or D. Y1 and Y2 are the location (distance from the top of the each homoeologous chromosome, cM) of the first common marker (Y) on the At (or D) and inferred map, respectively. X1 and X2 are the location of the second common marker (Y) on the At (or D) and inferred map, respectively. To efficiently build the inferred map according to the rules and formula mentioned above, a Web-based computer program was compiled using PHP. The code of the program is available at http://www.plantgenome.uga.edu/MapMerger/.
Computer software
The FISH (Calabrese et al. 2003) and CrimeStatII (Levine 2002) software packages were both used to identify putatively duplicated genomic regions within the hypothetical ancestral cotton genome, as well as putatively corresponding regions between cotton and Arabidopsis. Both packages treat single locus matches between a pair of genomes as point features that occur in a two-dimensional grid. The problem of identifying significant regions of colinearity between two genomes is thus reduced to the problem of finding significant clusters of points in Euclidean space.
FISH was designed for Fast Identification of Segmental Homologs (Calabrese et al. 2003). FISH preprocesses the matched locus data between two genomes to enforce symmetry and remove noise from the data set, and then identifies sets of neighbors. A dynamic programming algorithm is then used to follow a trace-back path that picks the string of neighbors that will yield the maximally extended block. To be considered neighbors by FISH, the Manhattan distance
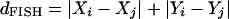 |
between a pair of points i and j must be less than the threshold distance
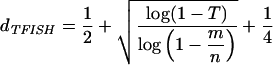 |
where m = number of points, n = number of cells, and T = the probability of having one or more neighbors within a distance less than dTFISH under the assumption that each cell contains a point with probability m/n.
The CS2 package was developed by Ned Levine and Associates (Levine 2002) under funding from the National Institute of Justice and has been made freely available for educational and research purposes (http://www.icpsr.umich.edu/NACJD/crimestat.html). Although initially developed for the analysis of crime-occurrence data, the package performs measurements of central tendency, spatial autocorrelation, and hot-spot analysis that can be applied to any spatial data set. Interoperability with existing GIS programs such as ArcView (http://www.esri.com) and integrated Dynamic Data Exchange protocols make CS2 a flexible tool for both analyzing and visualizing spatial datasets. This package has proven to be useful to scientists studying spatial patterns in ecology (Worrall et al. 2004) epidemiology (Brownstein et al. 2003), and cellular biology (Fuss and Linn 2002) and could prove to be a powerful tool for assessing spatial patterns in genomic data sets.
One CS2 tool useful to genomics is the nearest neighbor hierarchical clustering analysis, which provides a means of delineating significant clusters of points in Euclidean space. This tool identifies clusters by finding nearest neighbors that are separated by a Euclidean distance
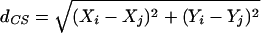 |
that is less than a minimum threshold distance. This threshold distance is defined to be a one-tailed confidence interval around the expected random values for distance to nearest neighbors such that,
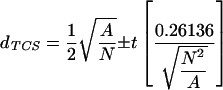 |
where A is the Area being surveyed, N is the number of sampled incidents, and t is the t-value associated with a probability level from the Student's t-distribution. These nearest neighbor sets are joined into hierarchical clusters with a minimum size and probability level set by the user. CS2 will accept cluster sizes as low as two points, and can join points with probability values ranging from P = 0.00001 up to P = 0.999. The output from this analysis is a text file identifying points and their cluster association, as well as an ellipsoid shape file that may be exported to ArcView for visualization of the distribution of clusters.
Identification of ancient duplications in the hypothetical cotton ancestor
Excluding the collinear anchor loci used in the construction of consensus maps, many duplicated loci were found at nonhomoeologous locations. To evaluate whether these reflected ancient whole-genome or large-scale duplication in the hypothetical ancestor, these loci were analyzed with FISH and CS2. For FISH, all markers were arranged in their recombinational order along the map, with co-segregating markers arranged alphabetically (i.e., arbitrarily) at a locus. A total of 13 map files were established, with one file per chromosome. Match files are composed of pairs of loci from the same probe (named as matching loci). A total of 169 match files (13 × 13) were created for FISH analysis, using the following parameters: minimum block size, 2 (default is 3); minimum bit score, 200 (as default); T, 0.05 (as default).
For CS2, the same set of match data as in FISH was used. Instead of the marker name used in the above match files, the accumulated distances from Chromosome 1 (C01) to C13 were calculated and used as the locus name for all chromosomes. Only one file was therefore created for all 13 chromosomes and is composed of two columns, numbers in each column representing one of the matching loci. The first column was displayed as the x-axis and the second as the y-axis. CS2 analysis used the following parameters: minimum points (pairs of matching loci) per cluster, three; number of standard deviations for ellipses, one; simulation runs, 0. The likelihood was set to 1%.
The lengths of conserved syntenic blocks were estimated by calculating the distance between the delimiting markers in the block detected by FISH or in the cluster detected by CS2.
Arabidopsis gene duplication database
Assembly of inferred ancestral gene orders along duplicated segments in the Arabidopsis genome is described elsewhere (Bowers et al. 2003). Briefly, a total of 26,028 Arabidopsis gene sequences were downloaded from NCBI, encoded by their chromosomal order and transcriptional orientation, and compared with each other using BLASTP. A total of 23,172 genes were shown to be in duplicated regions and grouped into 34 nonoverlapping chromosomal segments. Name and linear order of these 23,172 genes in the assembled duplications was explained by Bowers et al. (2003) and can be found at http://www.nature.com/nature or http://www.plantgenome.uga.edu/ploid.html.
Cotton probe DNA sequencing and BLAST search against Arabidopsis sequence database
Mapped cotton probes were sequenced as reported by Rong et al. (2004) and listed in Supplemental Table 1. Cotton sequences as query were compared with Arabidopsis gene sequences by searching all 26,028 genes in the Arabidopsis sequence database using BLASTX. Here, cotton query sequences are DNA sequences and Arabidopsis are translated protein sequences. The top 10 matches that met a threshold of E < 10-10 were used for further analysis.
Evaluation of conserved synteny between cotton and Arabidopsis
Both FISH and CS2 were also applied in the analysis of conserved synteny between cotton and Arabidopsis, and the same parameters used as in the identification of ancient duplication in the hypothetical cotton ancestor. Cotton-mapping data used here was the same as that in the identification of cotton ancient duplication by CS2 with the following modifications. The location of a given marker was determined by the accumulated locus number along chromosomes instead of the accumulated genetic distance. The marker location for a given marker was standardized by multiplying the marker location of a given locus by the ratio of the total Arabidopsis gene number to cumulative cotton gene number. This was based on the assumption that cotton and Arabidopsis have a similar gene number. This was done so that the two genomes could be aligned on x- and y-axes and direct comparisons made in CS2 and have the same chance to be compared in FISH.
Similarly, all 34 Arabidopsis α duplications were combined consecutively into a single unit in ascending order of the duplicate segment numbers (in the nomenclature of Bowers et al. 2003). The location of a given Arabidopsis gene was assigned as the accumulated gene number. In order to avoid bias from proximal duplicated genes on the estimation of conserved synteny between cotton and Arabidopsis, the distance (gene number) among the top 10 Arabidopsis matches to each cotton query sequence were calculated before running FISH and CS2. If the best matches were <30 genes apart along the Arabidopsis duplicated segment, we tentatively considered the genes to be proximal duplicates, and only one gene from such groups was kept for further analysis. The middle gene in the proximal duplicates was kept because it was the best representative of all proximal genes. If there was an even number of genes in the proximal duplication, the first of the middle two genes was kept. We used the number 30 because it was statistically unlikely (5%) that more than one match fell in an interval of this size by chance, even allowing consideration of the top 10 matches. Similarly, if a single Arabidopsis gene matched multiple cotton loci <5 cM apart, we counted only a single match.
Supplementary Material
Acknowledgments
This work has been supported in part by grants from the USDA National Research Initiative (97-35300-5305), and National Science Foundation Plant Genome Research Program (DBI-9872630, DBI-0211700).
Footnotes
[Supplemental material is available online at www.genome.org.]
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.3907305. Article published online before print in August 2005.
References
- Adams, K.L., Cronn, R., Percifield, R., and Wendel, J.F. 2003. Genes duplicated by polyploidy show unequal contributions to the transcriptome and organ-specific reciprocal silencing. Proc. Natl. Acad. Sci. 100: 4649-4654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Arabidopsis Genome Initiative 2000. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 408: 796-815. [DOI] [PubMed] [Google Scholar]
- Benton, M.J. 1993. The fossil record 2. Chapman and Hall, New York.
- Blanc, G. and Wolfe, K.H. 2004. Widespread paleopolyploidy in model plant species inferred from age distributions of duplicate genes. Plant Cell 16: 1667-1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanc, G., Barakat, A., Guyot, R., Cooke, R., and Delseny, I. 2000. Extensive duplication and reshuffling in the Arabidopsis genome. Plant Cell 12: 1093-1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowers, J.E., Chapman, B.A., Rong, J., and Paterson, A.H. 2003. Unravelling angiosperm genome evolution by phylogenetic analysis of chromosomal duplication events. Nature 422: 433-438. [DOI] [PubMed] [Google Scholar]
- Bowers, J.E., Arias, M.A., Asher, R., Avise, J.A., Ball, R.T., Brewer, G.A., Buss, R.W., Chen, A.H., Edwards, T.M., Estill, J.C., et al. 2005. Comparative physical mapping links retention of microsynteny to chromosome structure and recombination in grasses. Proc. Natl. Acad. Sci. (in press). [DOI] [PMC free article] [PubMed]
- Brownstein, J.S., Holford, T.R., and Fish, D. 2003. A climate-based model predicts the spatial distribution of the lyme disease vector ixodes scapularis in the United States. Environ. Health Perspect. 111: 1152-1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brubaker, C.L., Paterson, A.H., and Wendel, J.F. 1999. Comparative genetic mapping of allotetraploid cotton and its diploid progenitors. Genome 42: 184-203. [Google Scholar]
- Calabrese, P.P., Chakravarty, S., and Vision, T.J. 2003. Fast identification and statistical evaluation of segmental homologies in comparative maps. Bioinformatics 19: i74-i80. [DOI] [PubMed] [Google Scholar]
- Cronn, R.C., Small, R.L., Haselkorn, T., and Wendel, J.F. 2002. Rapid diversification of the cotton genus (Gossypium: Malvaceae) revealed by analysis of sixteen nuclear and chloroplast genes. Amer. J. Bot. 89: 707-725. [DOI] [PubMed] [Google Scholar]
- Eckardt, N.A. 2001a. Everything in its place: Conservation of gene order among distantly related plant species. Plant Cell 13: 723-725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ____. 2001b. A sense of self: The role of DNA sequence elimination in allopolyploidization. Plant Cell 13: 1699-1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuss, J. and Linn, S. 2002. Human DNA polymerase ε colocalizes with proliferating cell nuclear antigen and DNA replication late, but not early, in S phase. J. Biol. Chem. 277: 8658-8666. [DOI] [PubMed] [Google Scholar]
- Gaut, B.S., Morton, B.R., McCaig, B.C., and Clegg, M.T. 1996. Substitution rate comparisons between grasses and palms: Synonymous rate differences at the nuclear gene Adh parallel rate differences at the plastid gene rbcL. Proc. Natl. Acad. Sci. 93: 10274-10279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goff, S.A., Ricke, D., Lan, T.H., Presting, G., Wang, R.L., Dunn, M., Glazebrook, J., Sessions, A., Oeller, P., Varma, H., et al. 2002. A draft sequence of the rice genome (Oryza sativa L. ssp japonica). Science 296: 92-100. [DOI] [PubMed] [Google Scholar]
- Grant, D., Cregan, P., and Shoemaker, R.C. 2000. Genome organization in dicots: Genome duplication in Arabidopsis and synteny between soybean and Arabidopsis. Proc. Natl. Acad. Sci. 97: 4168-4173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grover, C.E., Kim, H., Wing, R.A., Paterson, A.H., and Wendel, J.F. 2004. Incongruent patterns of local and global genome size evolution in cotton. Genome Res. 14: 1474-1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu, Z., Steinmetz, L.M., Gu, X., Scharfe, C., Davis, R.W., and Li, W-H. 2003. Role of duplicate genes in genetic robustness against null mutations. Nature 421: 63-66. [DOI] [PubMed] [Google Scholar]
- Jiang, C., Wright, R., El-Zik, K., and Paterson, A.H. 1998. Polyploid formation created unique avenues for response to selection in Gossypium (cotton). Proc. Natl. Acad. Sci. 95: 4419-4424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, C., Wright, R.J., Woo, S.S., DelMonte, T.A., and Paterson, A.H. 2000a. QTL analysis of leaf morphology in tetraploid Gossypium (cotton). Theor. Appl. Genet. 100: 409-418. [Google Scholar]
- Jiang, C.X., Chee, P.W., Draye, X., Morrell, P.L., Smith, C.W., and Paterson, A.H. 2000b. Multilocus interactions restrict gene introgression in interspecific populations of polyploid Gossypium (cotton). Evolution 54: 798-814. [DOI] [PubMed] [Google Scholar]
- Kellogg, E.A. 2003. It's all relative. Nature 422: 383-384. [DOI] [PubMed] [Google Scholar]
- Koch, M.A., Haubold, B., and Mitchell-Olds, T. 2000. Comparative evolutionary analysis of chalcone synthase and alcohol dehydrogenase loci in Arabidopsis, Arabis, and related genera (Brassicaceae). Mol. Biol. Evol. 17: 1483-1498. [DOI] [PubMed] [Google Scholar]
- Kowalski, S., Lan, T-H., Feldmann, K., and Paterson, A.H. 1994. Comparative mapping of Arabidopsis thaliana and Brassica oleracea chromosomes reveals islands of conserved gene order. Genetics 138: 499-510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ku, H.M., Vision, T., Liu, J.P., and Tanksley, S.D. 2000. Comparing sequenced segments of the tomato and Arabidopsis genomes: Large-scale duplication followed by selective gene loss creates a network of synteny. Proc. Natl. Acad. Sci. 97: 9121-9126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkin, J.C., Brown, M.L., and Schiefelbein, J. 2003. How do cells know what they want to be when they grow up? Lessons from epidermal patterning in Arabidopsis. Annu. Rev. Plant Biol. 54: 403-430. [DOI] [PubMed] [Google Scholar]
- Levine, N. 2002. CrimeStat: A spatial statistics program for the analysis of crime incident locations (v 2.0). Ned Levine & Associates, Houston, TX, and the National Institute of Justice, Washington, DC.
- Liu, B., Brubaker, C.L., Mergeai, G., Cronn, R.C., and Wendel, J.F. 2001. Polyploid formation in cotton is not accompanied by rapid genomic changes. Genome 44: 321-330. [PubMed] [Google Scholar]
- Lynch, M. and Conery, J.S. 2003. The origins of genome complexity. Science 302: 1401-1404. [DOI] [PubMed] [Google Scholar]
- Mayer, K., Murphy, G., Tarchini, R., Wambutt, R., Volckaert, G., Pohl, T., Dusterhoft, A., Stiekema, W., Entian, K-D., Terryn, N., et al. 2001. Conservation of microstructure between a sequenced region of the genome of rice and multiple segments of the genome of Arabidopsis thaliana. Genome Res. 11: 1167-1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ming, R., Liu, S-C., Irvine, J.E., and Paterson, A.H. 2001. Comparative QTL analysis in a complex autopolyploid: Candidate genes for determinants of sugar content in Sugarcane. Genome Res. 11: 2075-2084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore, R.C. and Purugganan, M.D. 2003. The early stages of duplicate gene evolution. Proc. Natl. Acad. Sci. 100: 15682-15687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muravenko, O.V., Fedotov, A.R., Punina, E.O., Fedorova, L.I., Grif, V.G., and Zelenin, A.V. 1998. Comparison of chromosome BrdU-Hoechst-Giemsa banding patterns of the A1 and (AD) (2) genomes of cotton. Genome 41: 616-625. [Google Scholar]
- Ohno, S. 1970. Evolution by gene duplication. Springer, Berlin, Germany.
- Paterson, A.H., Lan, T.H., Reischmann, K.P., Chang, C., Lin, Y.R., Liu, S.C., Burow, M.D., Kowalski, S.P., Katsar, C.S., DelMonte, T.A., et al. 1996. Toward a unified genetic map of higher plants, transcending the monocot-dicot divergence. Nat. Genet. 14: 380-382. [DOI] [PubMed] [Google Scholar]
- Paterson, A.H., Bowers, J., Burow, M., Draye, X., Elsik, C., Jiang, C., Katsar, C., Lan, T., Lin, Y., Ming, R., et al. 2000. Comparative genomics of plant chromosomes. Plant Cell 12: 1523-1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paterson, A., Bowers, J., Peterson, D., Estill, J., and Chapman, B. 2003. Structure and evolution of cereal genomes. Curr. Opin. Genet. Dev. 13: 644-650. [DOI] [PubMed] [Google Scholar]
- Paterson, A.H., Bowers, J.E., and Chapman, B.A. 2004. Ancient polyploidization predating divergence of the cereals, and its consequences for comparative genomics. Proc. Natl. Acad. Sci. 101: 9903-9908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinisch, A.J., Dong, J.M., Brubaker, C.L., Stelly, D.M., Wendel, J.F., and Paterson, A.H. 1994. A Detailed RFLP map of cotton, Gossypium hirsutum X Gossypium barbadense: Chromosome organization and evolution in a disomic polyploid genome. Genetics 138: 829-847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rong, J-K., Abbey, C., Bowers, J.E., Brubaker, C.L., Chang, C., Chee, P.W., Delmonte, T.A., Ding, X.L., Garza, J.J., Marler, B.S., et al. 2004. A 3347-locus genetic recombination map of sequence-tagged sites reveals features of genome organization, transmission and evolution of cotton (Gossypium). Genetics 166: 389-417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossberg, M., Theres, K., Acarkan, A., Herrero, R., Schmitt, T., Schumacher, K., Schmitz, G., and Schmidt, R. 2001. Comparative sequence analysis reveals extensive microcolinearity in the lateral suppressor regions of the tomato, Arabidopsis, and Capsella genomes. Plant Cell 13: 979-988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salse, J., Piegu, B., Cooke, R., and Delseny, M. 2002. Synteny between Arabidopsis thaliana and rice at the genome level: A tool to identify conservation in the ongoing rice genome sequencing project. Nucleic Acids Res. 30: 2316-2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saranga, Y., Menz, M., Jiang, C., Wright, R., Yakir, D., and Paterson, A.H. 2004. Genetic and physiological dissection of adaptations associated with cotton productivity under arid conditions. Plant, Cell Environ. 27: 263-277. [Google Scholar]
- Schiefelbein, J. 2003. Cell-fate specification in the epidermis: A common patterning mechanism in the root and shoot. Curr. Opin. Plant Biol. 6: 74-78. [DOI] [PubMed] [Google Scholar]
- Simillion, C., Vandepoele, K., Van Montagu, M.C.E., Zabeau, M., and Van de Peer, Y. 2002. The hidden duplication past of Arabidopsis thaliana. Proc. Natl. Acad. Sci. 99: 13627-13632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stebbins, G. 1966. Chromosomal variation and evolution; polyploidy and chromosome size and number shed light on evolutionary processes in higher plants. Science 152: 1463-1469. [DOI] [PubMed] [Google Scholar]
- Vision, T., Brown, D., and Tanksley, S. 2000. The origins of genomic duplications in Arabidopsis. Science 290: 2114-2117. [DOI] [PubMed] [Google Scholar]
- Wang, S., Wang, J-W., Yu, N., Li, C-H., Luo, B., Gou, J-Y., Wang, L-J., and Chen, X-Y. 2004. Control of plant trichome development by a cotton fiber MYB gene. Plant Cell 16: 2323-2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendel, J.F. and Cronn, R.C. 2003. Polyploidy and the evolutionary history of cotton. Adv. Agron. 78: 139-186. [Google Scholar]
- Worrall, J.J., Sullivan, K.F., Harrington, T.C., and Steimel, J.P. 2004. Incidence, host relations and population structure of Armillaria ostoyae in Colorado campgrounds. Forest Ecol. Management 192: 191-206. [Google Scholar]
- Wright, R., Thaxton, P., El-Zik, K., and Paterson, A.H. 1998. D-subgenome bias of Xcm resistance genes in tetraploid Gossypium (Cotton) suggests that polyploid formation has created novel avenues for evolution. Genetics 149: 1987-1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright, R., Thaxton, P., Paterson, A.H., and El-Zik, K. 1999. Molecular mapping of genes affecting pubescence of cotton. J. Heredity 90: 215-219. [Google Scholar]
WEB SITE REFERENCES
- http://www.plantgenome.uga.edu/cotton/CottonDBFrames.htm, http://www.plantgenome.uga.edu/projects.htm#Cotton, http://www.plantgenome.uga.edu/MapMerger/, and http://www.plantgenome.uga.edu/ploid.html; Plant Genome mapping laboratory, University of Georgia.
- http://www.icpsr.umich.edu/NACJD/crimestat.html; The National Archive of Criminal Justice Data (NACJD).
- http://www.esri.com; Geographic Information Systems software (some also applicable to genomics).
- http://www.nature.com/nature; Nature journal.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.