

Abstract
Gene function is typically evaluated by sampling the continuum of gene expression at only a few discrete points corresponding to gene knockout or overexpression. We argue that this characterization is incomplete and present a library of engineered promoters of varying strengths obtained through mutagenesis of a constitutive promoter. A multifaceted characterization of the library, especially at the single-cell level to ensure homogeneity, permitted quantitative assessment correlating the effect of gene expression levels to improved growth and product formation phenotypes in Escherichia coli. Integration of these promoters into the chromosome can allow for a quantitative accurate assessment of genetic control. To this end, we used the characterized library of promoters to assess the impact of phosphoenolpyruvate carboxylase levels on growth yield and deoxy-xylulose-P synthase levels on lycopene production. The multifaceted characterization of promoter strength enabled identification of optimal expression levels for ppc and dxs, which maximized the desired phenotype. Additionally, in a strain preengineered to produce lycopene, the response to deoxy-xylulose-P synthase levels was linear at all levels tested, indicative of a rate-limiting step, unlike the parental strain, which exhibited an optimum expression level, illustrating that optimal gene expression levels are variable and dependent on the genetic background of the strain. This promoter library concept is illustrated as being generalizable to eukaryotic organisms (Saccharomyces cerevisiae) and thus constitutes an integral platform for functional genomics, synthetic biology, and metabolic engineering endeavors.
Keywords: functional genomics, metabolic engineering
Protein engineering via directed evolution and gene shuffling (1, 2) has been extensively applied for the systematic improvement of protein properties such as antibody-binding affinity (3), enzyme regulation (4), and increased or diverse substrate specificity (5). A similar approach whereby continuously improved mutants are generated along a selection-defined trajectory in the sequence space can also be applied for the systematic improvement or modification of other types of biological sequences, e.g., ribozymes (6, 7). We show here that promoters can also be engineered via directed evolution to achieve precise strengths and regulation and, by extension, can constitute libraries exhibiting broad ranges of genetic control.
Typically, the deletion (8) and strong overexpression (9) of genes have been the principal strategies for elucidation of gene function. These two methods sample the continuum of gene expression at only a few discrete points, determined by experimental feasibility (10) and not necessarily biological significance. Thus, the full dependency of phenotype on gene expression may not be accessible due to the limitations inherent in these methods. Gene expression is controlled by a number of factors in the cell, including promoter strength, cis- and transacting factors, cell growth stage, the expression level of various RNA polymerase-associated factors, and other gene-level regulation. Of course, gene expression may not always correspond with enzymatic activity given protein level regulation, which may also be present. Nevertheless, several groups have attempted to control gene expression through the creation of promoter libraries (11–13). In this work, we present the development of a fully characterized, homogeneous, broad-range, functional promoter library and demonstrate its applicability to the analysis of such a genetic control. By characterizing the strength of these promoters in a quantitative manner with various metrics and subsequently integrating these constructs into the genome, it is possible to deduce the precise impact of the gene dosage on the desired phenotype.
An alternative method for controlling gene expression is through the use of a single inducible promoter tested at various levels of inducer. Although inducible promoters allow for a continuous control of expression at the macroscopic level, practical applications of these systems are limited by prohibitive inducer costs, hypersensitivity to inducer concentration, and transcriptional heterogeneity at the single-cell level (14, 15). The latter factor, in particular, can limit the effect of inducers in a culture to a simple increase of the number of cells expressing the gene of interest instead of the overexpression of the gene in all cells. Inducible systems are suitable in certain applications (e.g., recombinant protein overproduction) (16); however, the elucidation of gene function and genetic control on phenotype requires well characterized promoter libraries, which behave in a similar manner at the single-cell level. As a result, the creation of a promoter library based on a constitutive promoter would eliminate the need to regulate inducer concentrations and avoid heterogeneities in cellular response.
Methods
Strains and Media. Escherichia coli DH5α (Invitrogen) was used for routine transformations, as described in the protocol. E. coli K12 (MG1655) and E. coli K12 PT5-dxs, PT5-idi, and PT5-ispFD (provided by DuPont) were used for promoter engineering examples. In specified strains, lycopene expression was performed by using the pAC-LYC plasmid (17) and assayed as described (18). Assay strains were grown at 37°C with 225 rpm orbital shaking in M9-minimal media (19) containing 5 g/liter d-glucose. When necessary, the M9 media were supplemented with 0.1% casamino acids. All other strains and propagations were cultured at 37°C in LB media. Media were supplemented with 68 μg/ml chloramphenicol/20 μg/ml kanamycin/100 μg/ml ampicillin, as necessary. Glucose monitoring was conducted by using the r-Biopharm (Swansea, U.K.) kit. Cell density was monitored spectrophotometrically at 600 nm. All PCR products and restriction enzymes were purchased from New England Biolabs and used Taq polymerase. M9 minimal salts were purchased from USBiological (Swampscott, MA), and all remaining chemicals were from Sigma-Aldrich. Primers were purchased from Invitrogen, and sequence information is listed in Supporting List, which is published as supporting information on the PNAS web site.
Saccharomyces cerevisiae strain BY4741 (MATa; his3Δ1; leu2Δ0; met15Δ0; ura3Δ0) used in this study was obtained from EUROSCARF (Frankfurt). It was cultivated in yeast extract/peptone/dextrose medium (10 g of yeast extract per liter/20 g of Bacto Peptone (Becton Dickinson) per liter/20 g of glucose per liter). For yeast transformation, Frozen-EZ Yeast Transformation II (Zymo Research, Orange, CA) was used. To select and grow yeast transformants bearing plasmids with URA3 as selectable marker, a yeast synthetic complete (YSC) medium was used containing 6.7 g of yeast nitrogen base (Difco) per liter, 20 g of glucose/liter, and a mixture of appropriate nucleotides and amino acids (CSM-URA, Qbiogene, Irvine, CA), referred here as to YSC Ura–. Medium was supplemented with 1.5% agar for solid media. Yeast cells were routinely cultivated at 30°C in Erlenmeyer flasks shaken at 200 rpm. For sorting single cells (TEF promoter mutations) by FACS into microtiter plates, each well contained 200 μl of YSC Ura– supplemented with 10 mg/liter ergosterol and 420 mg/liter Tween 80 (20).
Library Construction. Nucleotide analogue mutagenesis was carried out in the presence of 20 μM 8-oxo-2′-deoxyguanosine (8-oxo-dGTP) and 6-(2-deoxy-β-d-ribofuranosyl)-3,4-dihydro-8H-pyrimido-[4,5-c][1,2]oxazin-7-one (dPTP) (TriLink BioTechnologies) (21). By using plasmid pZE-gfp(ASV) kindly provided by M. Elowitz (California Institute of Technology, Pasadena) as template (22) along with the primers PL_sense_AatII and PL_anti_EcoRI, 10 and 30 amplification cycles with the primers mentioned above were performed. The 151-bp PCR products were purified by using the GeneClean Spin Kit (Qbiogene). After digestion, the product was ligated at 16°C overnight and transformed into library efficiency E. coli DH5α (Invitrogen). Approximately 30,000 colonies were screened by eye from minimal media–casamino acid agar plates, and 200 colonies, spanning a wide range in fluorescent intensity, were picked from the plates.
To create the TEF promoter mutation library for S. cerevisiae, the plasmid p416-TEF-yECitrine was used as a template for the error-prone PCR of the TEF1 promoter by using the primers TEF_Sense and TEF_Anti. The mix of purified mutagenized PCR products was transformed into yeast together with p416-TEF, which was cut with SacI/XbaI before (in vivo cloning). The CEN/ARS plasmid P416-TEF (23), containing the native TEF1 promoter from S. cerevisiae, the CYC1 terminator, and the URA3 gene as a selectable marker, was obtained from American Type Culture Collection. The plasmid pKT140 was obtained from EUROSCARF. This plasmid contains the coding sequence of yECitrine, a yeast codon-optimized version of the yellow fluorescent protein (24), which was used as a reporter protein in this study. To clone the yECitrine gene downstream of the TEF promoter, the coding sequence of yECitrine was amplified via PCR from the plasmid pKT140 by using the primer yEC_Sense and yEC_Anti. The PCR product was cut with ClaI and XbaI and ligated to ClaI/XbaI restricted vector p416-TEF. The resulting plasmid is referred to as p416-TEF-yECitrine.
Library Characterization
Initial Characterization. Approximately 20 μl of overnight cultures of library clones growing LB broth were used to inoculate 5 ml of M9G medium supplemented with 0.1% wt/vol casamino acid (M9G/CAA), and the cultures were grown at 37°C with orbital shaking. After 14 h, a sample of the culture was centrifuged at 18,000 × g for 2 min, and the cells were resuspended in ice-cold water. Flow cytometry was performed on a Becton Dickinson FACScan, and the geometric mean of the fluorescence distribution of each clonal population was calculated. To ensure that bulk population-averaged measurements could reflect the underlying single-cell behavior, only clones with clean monovariate distributions of fluorescence were retained for further analysis. Twenty-seven clones were isolated in this way. Sequencing revealed that these 27 clones represented 22 unique promoter sequences.
Promoter Strength Metric. Shake flasks containing 50 ml of M9G/CAA medium were inoculated with 1% vol/vol of an overnight LB culture of a library clone. The culture turbidity (A600 nm) and fluorescence (Packard Fusion microplate fluorescence reader, PerkinElmer) were monitored as a function of time. Fluorescence readings taken during the exponential growth phase were plotted as a function of turbidity. The best-fit slope to this line represents the exponential-phase steady-state concentration of GFP, fSS. Because fSS is affected by the cell growth rate oxygen-dependent maturation constant of GFP and the protease-mediated degradation of GFP as well as the promoter-driven synthesis of new GFP, it is not a suitable metric for promoter strength. Instead, we used a previously published dynamic model (25) that accounts for all of these factors. Under this model and under the assumption that the rate constant of protease-mediated degradation is the same for mature GFP as its precursor polypeptide, P, the rate of promoter-driven production of GFP can be expressed as in Eq. 1.
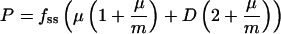 |
[1] |
In Eq. 1, μ is the growth rate, m is the maturation constant for the oxygen-dependent fluorophore activation of GFP, and D is the first-order rate constant for protease-mediated degradation. Estimates of m and D of 1.5 h–1 and 0.23 h–1, respectively (26, 27), were obtained from the literature. The parameters fSS and μ were measured separately for each member of the promoter library. P, in relative fluorescence units per absorbance unit per hour, was calculated from Eq. 1 for each clone. We performed duplicate cultures for each clone.
Transcriptional Analysis. Cultures inoculated as previously were grown for 3 h, and the total RNA was extracted from a 1.5-ml sample with a commercial kit (RNEasy, Qiagen, Valencia, CA). All samples were diluted to a final concentration of 20 μg/ml and stored at –20°C. A commercial kit for RT-PCR (iScript One-Step RT-PCR Kit with SYBR green, Bio-Rad) was used with a charge-coupled device-equipped thermal cycler (iCycler, Bio-Rad) for RT-PCR of the gfp transcript. Primers were used at a final concentration of 100 nM, and 20 ng of RNA was used as template in each 50-μl reaction. We performed duplicate cultures for each clone and duplicate extractions for each culture. The threshold cycles for each sample were calculated from the fluorescence data with proprietary software (Bio-Rad).
Chloramphenicol Resistance. pZE-promoter-cat plasmids were created by PCR of the chloramphenicol acetyltransferase (CAT) gene from pACYC184 by using primers CAT_Sense_MluI and CAT_Anti_KpnI and ligated into the proper pZE-promoter construct, which was previously digested by KpnI and MluI. Exponential-phase cultures grown in LB supplemented with kanamycin were plated onto LB agar supplemented with kanamycin and various concentrations of chloramphenicol ranging from 0 to 500 μg/ml. After overnight incubation at 37°C, the lowest concentration of chloramphenicol that inhibited the growth of a clone was recorded.
TEF Promoter Library Characterization. Measuring of specific fluorescence of TEF promoter library in S. cerevisiae was performed by using cells harvested from the logarithmic phase during growth in shake flasks. Fluorescence of yECitrine was measured by using a fluorescence spectrometer (HITACHI F-2500) with an excitation wavelength of 502 nm and an emission wavelength of 532 nm. The specific fluorescence referred to here is the ratio of fluorescence level measured and the optical density at 600 nm measured in the same cuvette.
Promoter Delivery Construction. Promoter replacements were conducted by using PCR product recombination (28) with using the pKD46 plasmid expressing the λ red recombination system and pKD13 as the template for PCR. Promoter replacements were verified through colony PCR by using the k1, k2, and kt primers along with the verification primers listed below. To create the cassette for promoter replacement, two fragments were amplified via PCR. Fragment 1 contained the promoter with primer homology to the upstream region of the endogenous promoter. Fragment 2 contained the kanamycin maker from pKD13 and had homology to an area downstream of the endogenous promoter or gene. These two fragments had an internal homology to each other of 25 bp to allow for self annealing and subsequent amplification of a single cassette, which was used (≈100 ng) for the transformation. For the case of deoxy-xylulose-P synthase (dxs), the entire gene was amplified and used as a third fragment, which was annealed with the previous two. This provided higher recombination efficiency due to the longer homology region. A complete list of primers is provided in Supporting List.
Results
Characterization of the Promoter Library. A derivative of the constitutive bacteriophage PL-λ promoter (29) was mutated through error-prone PCR (30), cloned into a reporter plasmid upstream of a low-stability GFP gene (26), and screened in E. coli based on the fluorescence signal in a glucose minimal medium, supplemented with 0.1% casamino acids to attenuate GFP toxicity. Nearly 200 promoter mutants, spanning a wide range of GFP fluorescence, were selected. Many of these initially screened promoters exhibited large variations in fluorescence between several trials or did not have an acceptable single-cell-level homogeneity. Twenty-two mutants were finally chosen to form a functional promoter library based on reproducible and homogeneous single-cell fluorescence distributions, as measured by flow cytometry (Figs. 5–7, Table 1, and Supporting Text, which are published as supporting information on the PNAS web site). Fig. 1 illustrates the process of creating and subsequently selecting these promoters.
Fig. 1.

Generation of the functional promoter library. A variant of the constitutive bacteriophage PL-λ promoter was mutated through error-prone PCR, used in a plasmid construct to drive the expression of gfp, then screened based on fluorescence of colonies. The chosen constructs have a wide range of fluorescence both on a culture-wide and on a single-cell level, as illustrated by representative flow cytometry histograms at the bottom. All of the selected promoters have a uniform expression level on a single-cell level, as measured by GFP signal.
In light of the uncertainty surrounding the concept of promoter strength (31) and the poor reliability of single reporter-gene-based systems, we performed a multifaceted characterization of each library member. We first determined the promoter strength in the library strains (in units of GFP fluorescence per cell per hour) by measuring culture fluorescence and by using a dynamic equation balancing GFP production and degradation (25). Through replicate culturing, the promoter strength of the library members was found to span a 196-fold range with a mean spacing of 29% between adjacent members (Fig. 2).
Fig. 2.

Comprehensive characterization of the promoter library. Several orthogonal metrics were used to characterize the promoter library and ensure the consistent behavior of all its members for various genes and culturing conditions. We show here three metrics that were chosen for quantifying transcriptional of the promoters: (i) The dynamics of GFP production based on fluorescence, (ii) measurement of the relative mRNA transcript levels in the cultures, and (iii) testing of the MIC for chloramphenicol in an additional library of constructs where the promoter drove the expression of chloramphenicol acetyltransferase. The overall strong correlation between the various metrics suggests a broad-range utility of the promoter library for a variety of genes and conditions.
Next, to characterize the promoter library directly at the transcriptional level, we measured the relative mRNA levels of gfp transcripts in the above cultures by quantitative RT-PCR. The high correlation between fluorescence and mRNA level (Fig. 2) confirmed that expression was transcriptionally controlled. The mRNA level spanned a 325-fold range with a mean spacing of 32% between adjacent members. We then formed an “average promoter strength metric” for each promoter by averaging the scaled mRNA and fluorescence data.
Finally, to verify the constitutive nature of all of the promoters, each was redeployed into a new construct driving the reporter gene cat. Cultures bearing these constructs were assayed for resistance to chloramphenicol on a rich solid-phase medium. The minimum inhibitory concentration (MIC) spanned a 26-fold range with a mean spacing between MIC values of 17% (which is biased because of the discrete levels of chloramphenicol tested).
Fig. 2 displays the high correlation among these three metrics of promoter performance. These data indicate that the library exhibits a high dynamic range, which behaves similarly regardless of the gene being regulated. Moreover, these conditions test the promoter library in contrasting medium and growth environments (liquid minimal vs. solid complex medium), further underscoring the constitutive nature of the library promoters. The disparity between the number of initially and finally selected promoters illustrates the need for a comprehensive analysis of the promoters. Although many subsets of mutations can elicit a change in promoter strength, not all are guaranteed to lead to a reproducible, homogenous, and linear relationship between promoter strength and reporter. Relying solely on bulk culture-based measurements can lead to misclassification of the behavior of the promoter at the single-cell level and thus complicate quantitative gene expression studies, such as those performed in this study.
Application of the Promoter Library. We applied the functional promoter library to introduce precise transcriptional control in the investigation of specific genetic effects on a cellular phenotype. We performed chromosomal promoter delivery into the region upstream of the targeted gene, replacing the native promoter and its inherent regulation modality.
The utility of the promoter library was tested by investigating the effect of two endogenous genes [phosphoenolpyruvate carboxylase (ppc) and dxs) on two divergent phenotypes, growth yield and lycopene production. First, we investigated the growth yield from glucose as a function of the expression level of the ppc gene in E. coli. E. coli's native ppc promoter was replaced with varying-strength promoter–ppc constructs, and these mutants were cultured while biomass and glucose concentrations were periodically monitored. Fig. 3a presents the exponential-phase biomass yields as a function of the average promoter strength metric. Increasing ppc levels have a positive effect on the biomass yield only to a certain point. This increase reaches a plateau, and further increases in the ppc level have a negative effect on the biomass yield. These results illustrate an optimum in the expression level of ppc that is above that found from endogenous expression.
Fig. 3.

Implementation of the promoter library for introducing genetic control. The phenotypes associated with integrating the promoters into the chromosome are tested by using three genes. (a) Selected promoters were integrated into the promoter region of ppc, and strains were cultured in M9-minimal media with only glucose as the carbon source. Although the knockout of ppc is lethal in glucose media, there is a clear maximum yield from glucose and thus an optimal expression level of ppc. (b) Selected promoters were integrated in front of the dxs gene in a recombinant wild-type strain of E. coli, and strains were later assayed for the production of lycopene. A clear maximum in lycopene production was obtained. From the wild-type production level, the native dxs promoter strength can be inferred to be ≈0.26, according to our metric. (c) Selected promoters were integrated in front of the dxs gene in a recombinant strain also overexpressing ispFD and idi. In this case, the linear response of lycopene yield to the promoter strength illustrates a rate-limiting behavior of dxs across all tested promoter strengths.
In this second case, volumetric productivity of lycopene accumulation in glucose medium was investigated as a function of the expression levels of the dxs gene in two different E. coli strains: the wild-type K12 strain and a previously engineered strain, which already produces lycopene in high titers (18). Fig. 3b shows the lycopene production in these dxs constructs in a wild-type (K12) background. Elevating dxs expression increases lycopene accumulation only until a certain point. Beyond this optimum, increased dxs expression is detrimental for lycopene production. Finally, the strength of the native dxs promoter can be inferred from this analysis, as is illustrated on the graph (Fig. 3b).
In contrast to the above results, a linear relationship was obtained when similar promoter–dxs constructs were placed in an engineered strain (18) overexpressing downstream genes in the isoprenoid pathway (ispFD and idi). Fig. 3c illustrates a nearly linear response of lycopene production to varying levels of dxs expression, suggesting that in the new genetic background, dxs has become rate-limiting.
Extension of the Promoter Library. We applied the promoter engineering concept to S. cerevisiae as well. By screening a library of TEF1 promoter mutants, also created by error-prone PCR, a promoter collection was obtained that drove a wide dynamic range of YFP production in S. cerevisiae (Fig. 4). Thus, the promoter engineering paradigm can yield libraries of promoter for precise genetic control despite the profound differences in bacterial and eukaryotic transcription mechanisms (32, 33).
Fig. 4.

Extension of promoter engineering to other systems. The basic concepts in this paper are further extended to a eukaryotic system (S. cerevisiae) by using the TEF1 promoter. A similar wide range of yECitrine fluorescence is obtained from selected clones of the original promoter library. These results, along with other current work, indicate the ability to select for promoters responsible for tuning precise genetic control.
Discussion
The nearly 200 random promoter mutants we screened varied widely in their expression strength and clonal expression heterogeneity. Screening for only those promoters that drive stable monovariate expression in culture by flow cytometry was critical for deployment of our promoter constructs in pathway analysis and expression optimization. Isolating only the homogeneous expressers allowed us to establish a well defined metric of promoter strength, which combined data from several experimental assessments of gene expression levels. Using only a single technique to assess promoter strength often resulted in a scattering of the data, confounding the analysis of gene expression studies. The reliance on bulk averages would obscure the underlying relationship between expression and phenotype. The use of an integrated system allowed us to bypass the instabilities and inherent mutation rates associated with the overexpression of endogenous genes by using plasmid-based systems (34). Furthermore, this and other promoter libraries appear to have a broad host range (11), perhaps due to construction based on a heterologous constitutive promoter and reliance on the general polymerase machinery in the cell. This is exemplified through the three different strain backgrounds used in this study.
Enabled by a fully characterized library, we tested the promoter engineering concept for the analysis of two different phenotypes in E. coli. In the first, the expression of ppc was modulated to effect biomass yield from glucose. This gene expresses phosphoenol pyruvate (PEP) carboxylase, a key anaplerotic enzyme. A ppc knockout is lethal for E. coli in glucose minimal medium (35). Furthermore, overexpression of this gene has been shown to improve the growth yield on glucose (36). These data imply two possibilities: either biomass yield is a monotonically increasing function of ppc expression, or there exists a particular ppc expression level that maximizes yield. Our data show that the latter is the case. Possible reasons why ever-increasing ppc levels lead eventually to a decrease in yield include the metabolic burden of severe overexpression of ppc or, more likely, the creation of a futile ATP-wasting cycle in metabolism, where PEP is converted to oxaloacetate by ppc and back again by pck, the gene for PEP carboxykinase.
In addition to the global pleiotropic phenotype of growth yield, we also used promoter engineering in the study of a single metabolic pathway, by modulating dxs expression and measuring lycopene biosynthesis. Kinetic control of metabolic pathways is often distributed and depends on the expression level of several genes within the pathway (37). The gene dxs represents the first committed step in isoprenoid synthesis in E. coli and has been implicated in control of lycopene production (38); however, the quantitative nature of this control was unclear, and promoter delivery experiments also allowed us to quantify this control in multiple backgrounds (Fig. 3 b and c). In the case of wild-type E. coli, an optimal dxs expression was again apparent. Past the optimum, increasing dxs expression lowers lycopene yield, presumably because of the inadequate activity of downstream enzymes in the isoprenoid pathway and resulting toxic buildup of DXP. In contrast, in a strain already engineered to overexpress idi, ispF, and ispD, downstream genes in lycopene biosynthesis, no maximum is apparent. A linear response to an enzyme concentration is expected for rate-controlling genes exhibiting a high flux control coefficient for a given pathway (39), suggesting that even at the highest expression levels examined in this study, the dxs-catalyzed reaction is rate-limiting for lycopene biosynthesis. We also note that cell density in both strains was greatly reduced in the constructs harboring low-strength promoters, which was expected, because dxs is an essential gene. A significant step in performing these quantitative functional genomics studies is creating a reliable characterized promoter library for which confidence in the cellular gene expression level may be placed. When this initial step is established, it is possible to quantitatively analyze the control a single enzyme exerts in a given pathway of interest, exemplified by the dxs example.
The creation of a library of promoter mutants in yeast illustrates the applicability of this approach in both prokaryotic and eukaryotic contexts. As with E. coli, flow cytometry allowed isolation of only those promoters with relatively homogeneous reporter gene expression. It is possible to further extend and refine the selection process to create libraries of conditional promoters, active only under specified conditions. We have recently applied this selection methodology to create conditional genetic control elements that are responsive to environmental perturbations (e.g., oxygen concentration) (data not shown).
Additionally, the analysis of libraries of promoters may be studied to deduce a linkage between sequence and phenotype. To this end, it would be possible to create correlations between mutation sites and promoter metrics such as strengths or variability in gene expression (40). Further application and study of this promoter library can greatly facilitate efforts in synthetic biology aiming to create synthetic genetic operons. The cataloging of promoter sequences along with their behavior can help in the selection of components to be used in synthetic gene networks such as toggle switches (41) and for creating polygenic operons with prescribed ratios of gene expression.
Conclusion
We have created a general framework for the precise quantitative control of gene expression in vivo. Our strategy allows (i) achievement of any desired expression level for a specific gene, (ii) optimization of gene expression for maximal (or minimal) pathway function, and (iii) a means for the analysis of the distribution of genetic control on pathway behavior. In two disparate examples, we have shown that pathway function can exhibit well-defined extrema with respect to levels of gene expression. The existence of these extrema evinces the need for precise gene-dosage studies for the full understanding of pathway behavior. The creation and detailed characterization of a promoter library, as described here, are a facile and robust means to such an end.
Supplementary Material
Acknowledgments
We thank Michael Elowitz, Kyle Jensen, Juan Pedraza, and Alexander van Oudenaarden for assistance with this work. We acknowledge financial support by the DuPont–MIT Alliance and National Science Foundation Grant no. BES-0331364. E.N. thanks the Berliner Chancengleichheitsprogramm für Frauen in Forschung und Lehre for financial support.
Author contributions: H.A. designed research; H.A., C.F., and E.N. performed research; H.A. and C.F. analyzed data; and H.A., C.F., and E.N. wrote the paper.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: dxs, deoxy-xylulose-P synthase; ppc, phosphoenolpyruvate carboxylase.
References
- 1.Stemmer, W. P. (1994) Nature 370, 389–391. [DOI] [PubMed] [Google Scholar]
- 2.Glieder, A., Farinas, E. T. & Arnold, F. H. (2002) Nat. Biotechnol. 20, 1135–1139. [DOI] [PubMed] [Google Scholar]
- 3.Boder, E. T., Midelfort, K. S. & Wittrup, K. D. (2000) Proc. Natl. Acad. Sci. USA 97, 10701–10705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nelms, J., Edwards, R. M., Warwick, J. & Fotheringham, I. (1992) Appl. Environ. Microbiol. 58, 2592–2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fa, M., Radeghieri, A., Henry, A. A. & Romesberg, F. E. (2004) J. Am. Chem. Soc. 126, 1748–1754. [DOI] [PubMed] [Google Scholar]
- 6.Ferguson, A., Boomer, R. M., Kurz, M., Keene, S. C., Diener, J. L., Keefe, A. D., Wilson, C. & Cload, S. T. (2004) Nucleic Acids Res. 32, 1756–1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tao, L., Jackson, R. E. & Cheng, Q. (2005) Metab. Eng. 7, 10–17. [DOI] [PubMed] [Google Scholar]
- 8.Zhou, L., Lei, X. H., Bochner, B. R. & Wanner, B. L. (2003) J. Bacteriol. 185, 4956–4972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nishino, K., Inazumi, Y. & Yamaguchi, A. (2003) J. Bacteriol. 185, 2667–2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jana, S. & Deb, J. K. (2005) Appl. Microbiol. Biotechnol. 67, 289–298. [DOI] [PubMed] [Google Scholar]
- 11.Jensen, P. R. & Hammer, K. (1998) Appl. Environ Microbiol. 64, 82–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jorgensen, C. M., Hammer, K., Jensen, P. R. & Martinussen, J. (2004) Eur. J. Biochem. 271, 2438–2445. [DOI] [PubMed] [Google Scholar]
- 13.Khlebnikov, A., Risa, O., Skaug, T., Carrier, T. A. & Keasling, J. D. (2000) J. Bacteriol. 182, 7029–7034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Siegele, D. A. & Hu, J. C. (1997) Proc. Natl. Acad. Sci. USA 94, 8168–8172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mnaimneh, S., Davierwala, A. P., Haynes, J., Moffat, J., Peng, W. T., Zhang, W., Yang, X., Pootoolal, J., Chua, G., Lopez, et al. (2004) Cell 118, 31–44. [DOI] [PubMed] [Google Scholar]
- 16.San, K. Y., Bennett, G. N., Chou, C. H. & Aristidou, A. A. (1994) Ann. N.Y. Acad. Sci. 721, 268–276. [DOI] [PubMed] [Google Scholar]
- 17.Cunningham, F. X., Jr., Sun, Z., Chamovitz, D., Hirschberg, J. & Gantt, E. (1994) Plant Cell 6, 1107–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Alper, H., Jin, Y.-S., Moxley, J. & Stephanopoulos, G. (2005) Metab. Eng. 7, 155–164. [DOI] [PubMed] [Google Scholar]
- 19.Maniatis, T., Fritsch, E. F. & Sambrook, J. (1982) Molecular Cloning: A Laboratory Manual (Cold Spring Harbor Lab. Press, Plainview, NY).
- 20.Moller, K., Olsson, L. & Piskur, J. (2001) J. Bacteriol. 183, 2485–2489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zaccolo, M. & Gherardi, E. (1999) J. Mol. Biol. 285, 775–783. [DOI] [PubMed] [Google Scholar]
- 22.Elowitz, M. B. & Leibler, S. (2000) Nature 403, 335–338. [DOI] [PubMed] [Google Scholar]
- 23.Mumberg, D., Muller, R. & Funk, M. (1995) Gene 156, 119–122. [DOI] [PubMed] [Google Scholar]
- 24.Sheff, M. A. & Thorn, K. S. (2004) Yeast 21, 661–670. [DOI] [PubMed] [Google Scholar]
- 25.Leveau, J. H. & Lindow, S. E. (2001) J. Bacteriol. 183, 6752–6762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Andersen, J. B., Sternberg, C., Poulsen, L. K., Bjorn, S. P., Givskov, M. & Molin, S. (1998) Appl. Environ. Microbiol. 64, 2240–2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cormack, B. P., Valdivia, R. H. & Falkow, S. (1996) Gene 173, 33–38. [DOI] [PubMed] [Google Scholar]
- 28.Datsenko, K. A. & Wanner, B. L. (2000) Proc. Natl. Acad. Sci. USA 97, 6640–6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lutz, R. & Bujard, H. (1997) Nucleic Acids Res. 25, 1203–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zaccolo, M., Williams, D. M., Brown, D. M. & Gherardi, E. (1996) J. Mol. Biol. 255, 589–603. [DOI] [PubMed] [Google Scholar]
- 31.Horn, G. T. & Wells, R. D. (1981) J. Biol. Chem. 256, 2003–2009. [PubMed] [Google Scholar]
- 32.Lee, T. I. & Young, R. A. (2000) Annu. Rev. Genet 34, 77–137. [DOI] [PubMed] [Google Scholar]
- 33.Browning, D. F. & Busby, S. J. (2004) Nat. Rev. Microbiol. 2, 57–65. [DOI] [PubMed] [Google Scholar]
- 34.Zaslaver, A., Mayo, A. E., Rosenberg, R., Bashkin, P., Sberro, H., Tsalyuk, M., Surette, M. G. & Alon, U. (2004) Nat. Genet. 36, 486–491. [DOI] [PubMed] [Google Scholar]
- 35.McAlister, L. E., Evans, E. L. & Smith, T. E. (1981) J. Bacteriol. 146, 200–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liao, J. C., Chao, Y. P. & Patnaik, R. (1994) Ann. N.Y. Acad. Sci. 745, 21–34. [DOI] [PubMed] [Google Scholar]
- 37.Stephanopoulos, G. & Vallino, J. J. (1991) Science 252, 1675–1681. [DOI] [PubMed] [Google Scholar]
- 38.Kim, S. W. & Keasling, J. D. (2001) Biotechnol. Bioeng. 72, 408–415. [DOI] [PubMed] [Google Scholar]
- 39.Kacser, H. & Acerenza, L. (1993) Eur. J. Biochem. 216, 361–367. [DOI] [PubMed] [Google Scholar]
- 40.Blake, W. J., Kaern, M., Cantor, C. R. & Collins, J. J. (2003) Nature 422, 633–637. [DOI] [PubMed] [Google Scholar]
- 41.Gardner, T. S., Cantor, C. R. & Collins, J. J. (2000) Nature 403, 339–342. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.