

Abstract
Phosphatidylinositol 3,4,5-trisphosphate is a major intracellular messenger molecule thought to be formed almost exclusively by cytosolic, wortmannin-inhibited phosphoinositide 3-kinase family members. Inositol polyphosphate multikinase was identified as an enzyme that generates a series of water-soluble inositol phosphates. We now report the robust, physiologic, and evolutionarily conserved phosphoinositide 3-kinase activity of inositol polyphosphate multikinase, which is localized to nuclei and unaffected by wortmannin. In yeast, this inositol lipid kinase activity physiologically regulates transcription.
Keywords: lipids, nucleus, wortmannin
Water-soluble inositol phosphates comprise a family of signaling molecules with various permutations of phosphate substituents on the myo-inositol ring (1). Inositol 1,4,5-trisphosphate (IP3), the best characterized family member, which releases intracellular calcium, is generated by the hydrolysis of the lipid inositol phosphatidylinositol 4,5-bisphosphate [PI(4,5)P2] by phospholipase C (PLC) (2) and can be sequentially phosphorylated by inositol phosphate kinases (3). A recently identified family of inositol polyphosphate kinases (IPKs; PFA M accession no. PF03770) includes the inositol hexakisphosphate kinases (IP6Ks), the IP3-3 kinases, and a third member that we designated inositol polyphosphate multikinase (IPMK, also designated Ipk2) because of its ability to phosphorylate multiple sites on the inositol phosphate ring (4–6). Sequential phosphorylation of IP3 by IPMK ultimately leads to the production of inositol 1,3,4,5,6-pentakisphosphate (IP5) (Fig. 1). Yeast mutants of IMPK display transcriptional regulation defects in response to nutrients, as well as defects in sporulation, mating, and salt stress tolerance (7).
Fig. 1.

Schematic depicting known soluble inositol (Ins) kinase activities (red arrows) of IPMK previously characterized in both in vitro and in vivo assays (for review see ref. 3). Also depicted is the lipid inositol PI3K activity being described. Additional activities not shown include the phosphorylation of Ins(4,5)P2 to Ins(1,4,5)P3 and the phosphorylation of Ins(1,4,5)P3 first to Ins(1,4,5,6)P4 and subsequently to Ins(1,3,4,5,6)P5. More recently, human IPMK was shown to be capable of phosphorylating Ins(1,3,4,6)P4 to Ins(1,3,4,5,6)P5 (19).
Besides generating IP3, PI(4,5)P2 is a substrate for the predominantly cytosolic lipid inositol kinases designated phosphoinositide 3-kinases (PI3Ks) which phosphorylate the D-3 position of inositol lipids, often in response to growth factor receptor stimulation (8, 9). The formation of phosphatidylinositol 3,4,5-trisphosphate [PI(3,4,5)P3] at the plasma membrane by class I PI3Ks is the rate-limiting step in multiple pathways that regulate cell migration, growth, proliferation, and survival (8). We now report that IPMK is a robust, physiologic nuclear PI3K that regulates transcription (Fig. 1).
Materials and Methods
Lipid Kinase Assays. Lipid inositol substrates were dried under a stream of nitrogen gas and resuspended via sonication in a carrier of phosphatidylserine, 20 mM Hepes (pH 7.4), and 1 mM EDTA. Alternatively, lipid inositol substrates were resuspended in 20 mM Hepes (pH 7.4), 1 mM EDTA, and 0.5% deoxycholate. Both methods yielded similar results. Kinase reactions were performed in a total volume of 50 μl containing 10 μl of lipid resuspension providing a final concentration of 0.03 mg/ml purified/synthetic lipid inositols (Sigma, Calbiochem, Avanti Polar Lipids) or at a final concentration of 0.2 mg/ml for Folch bovine brain extracts (Sigma). Kinase reaction buffer consisted of 20 mM Hepes (pH 7.4), 6 mM MgCl2, and 10 μCi of [γ-32P]ATP (PerkinElmer–NEN, 6,000 mCi/mmol; 1 Ci = 37 GBq) in a carrier of 100 μM unlabeled ATP. Enzyme reactions were incubated either 30°C (yeast IPMK) or 37°C (rat IPMK and p110γ) for 15 min. Enzyme concentrations typically ranged between 10 and 50 ng of bacterially expressed and purified His-/GST-tagged IPMK or His-tagged human p110γ (Alexis Biochemicals). Enzymatic comparisons were made by using equal molar enzyme concentrations. Kinase reactions were stopped with 90 μl of 1 M HCl/methanol (1:1 by volume). Lipids were extracted twice with 100 μl of choloroform and resolved on silica gel 60 TLC plates in a solvent system consisting water/n-propanol/glacial acetic acid (34:65:1, by volume). Alternatively, extracted lipids were deacylated as described (10) and separated by HPLC.
Characterization of Enzyme Kinetics. Km and Vmax determinations were performed by using a maximum of 100 μM PI(4,5)P2 (Avanti Polar Lipids) or serial dilutions of the lipid resuspended in 20 mM Hepes (pH 7.4), 1 mM EDTA, and 0.5% deoxycholate. Reactions were performed in triplicate as described above. Extracted, radiolabeled lipids were counted, and rate calculations were fitted to Michaelis–Menten equations by using sigmaplot. Competition studies using IP3 or inositol 1,3,4,6-tetrakisphosphate (A.G. Scientific) were performed in the presence of 100 μM PI(4,5)P2 and increasing concentrations of the indicated soluble inositol.
HPLC Analysis of Soluble and Lipid Inositol. Isolated glycerophosphoinositides were resuspended in 1 mM EDTA, and ∼2 × 106 cpm were resolved by using anion exchange chromatography with a Whatman PartiSphere SAX (4.6 × 250 mm) column. The column was eluted with a gradient generated by mixing 1 mM EDTA and buffer B [1 mM EDTA/1.3 M (NH4)2HPO4, pH 3.8] in one of two methods: (i) 0–5 min, 0% B; 5–105 min, 0–100% B; 105–120 min, 100% B; or (ii) 0–5 min, 0% B; 5–85 min, 0–50% B; 85–105 min, 50%–100% B; 105–120 min, 100% B. Spectrophotometric monitoring of AMP, ADP, and ATP elution times and authentic deacylated [3H]PI(4,5)P2 (PerkinElmer NEN) or p110γ-generated [32P]PI(3,4,5)P3 and [32P]PI(3,4)P2 were used as standards. Singly phosphorylated glycerophosphoinositides species eluted between AMP and ADP, whereas glycerophosphoinositide bisphosphates such as gPI(4,5)P2 eluted between ADP and ATP; gPI(3,4,5)P3 eluted after ATP. Fractions (1 ml) were collected and counted by using 4 ml of Ultima-Flo AP LCS-mixture (Packard). Extraction and analyses of soluble inositol lipids were performed as described (11).
Lipid Phosphatase Reactions. Lipid phosphatase reactions were performed as per manufacturers' recommendations using recombinant SHIP-2 (Echelon Biosciences) or recombinant PTEN (Upstate Biotechnology) and putative [32P]PI(3,4,5)P3 generated in vitro as described above. Extracted lipids were resolved by TLC and analyzed by autoradiography.
Transfections. HEK293T and Cos-7 (American Type Culture Collection) cells were transfected by using Lipofectamine 2000 (Invitrogen) as per the manufacturer's recommendation.
[3H]Inositol Labeling of Transfected Cells. Transfected HEK293T and Cos-7 cells were labeled for 48 h with 10 μCi/ml (PerkinElmer NEN, 25 Ci/mmol) in inositol-free DMEM (Specialty Media). Lipid inositols were extracted as described (10) and analyzed by HPLC as described above.
Primary Cultures. Primary cortical and hippocampal cultures were established as described (12). Immunostaining and activity assays were performed on 3- to 5-day-old cultures. Hepatocyte cultures were established by using GIBCO hepatocyte products (Invitrogen) as per the manufacturer's recommendations.
Immunostaining. Transfected cells and primary cultures were fixed in 4% paraformaldehyde for 30 min at 4°C. Cells were washed three times in Tris-buffered saline (TBS) and permeabilized in TBS containing 10% goat serum (Vector Laboratories) and 0.5% Triton X-100 for 1 h at room temperature. Primary antibodies were incubated overnight in TBS containing 10% goat serum. Primary antibodies were used at the following concentrations: anti-HA (Covance), 1:5,000; anti-βIII-tubulin (Promega), 1:2,000; anti-PI(3,4,5)P3 (13) (Echelon), 1:200; anti-neurofilament (Developmental Studies Hybridoma Bank), 1:100; anti-IPMK, 1:1,500. After primary antibody incubation, cells were washed five times in TBS containing 10% goat serum. Alexa Fluor 488 or Alexa Fluor 568 fluorescent secondaries (1:4,000) (Molecular Probes) were applied for 5 h at room temperature in TBS containing 10% goat serum. Cells were washed overnight in PBS. Identification of nuclei was performed by using Hoechst dye 33258 (Molecular Probes). Polyclonal anti-IPMK antibodies were generated in rabbits by using fulllength bacterially expressed rat IPMK; IPMK-specific antibodies were affinity purified by using Affigel-crosslinked (Bio-Rad) rat IPMK. Confocal images were obtained on a PerkinElmer UltraVIEW Spinning Disk Confocal microscope.
Nuclear Extraction and Immunoprecipitations. IPMK immunprecipitations were performed by using 1 mg of cell extract from isolated nuclei prepared as described (14). Nuclei were lysed in 20 mM Hepes, pH 7.4/1.5 mM MgCl/400 mM NaCl/1% Nonidet P-40/0.2 mM EDTA, and protease inhibitor mixture (Sigma). Alternatively, nuclear lysates were prepared by using NE-PER Nuclear extraction reagent (Pierce). Lysates were incubated with 2 μg of anti-IPMK antibody and 30 μl of protein G Plus/protein A-agarose suspension (EMD Biosciences) for 4 h at 4°C. Immunoprecipitates were washed five times with a wash buffer consisting of 20 mM Hepes (pH 7.4), 500 mM NaCl, and 1% Nonidet P-40 followed by two washes in 1× kinase reaction buffer. Reactions were performed on immunoprecipitates as described above, but incubated for 1 h as IPMK-specific antibodies significantly inhibit enzymatic activity. PI3K p85 immunoprecipitation was performed as per manufacturer's protocol (Upstate Biotechnology), typically from 1 mg of cell lysate. Control immunoprecipitates were performed with equal concentrations of purified rabbit IgG (Vector Laboratories).
Akt Actvation Assays. Transfected HEK293T cells were treated with IGF-1 (50 ng/ml) for 5 or 30 min in the presence or absence of wortmannin (500 nM). Cells were briefly rinsed with ice-cold PBS and lysed in 100°C 2× Nupage LDS sample loading buffer (Invitrogen). Proteins were separated by Nupage Bis-Tris electrophoresis (Invitrogen), transferred onto poly(vinylidene difluoride) membranes, and analyzed by Western blotting techniques using anti-Akt, anti-phospho-Akt (Ser-473) (Cell Signaling Technologies), and anti-HA (Covance) antibodies per manufacturers' recommendations.
Western Analysis. Western analysis of nuclear extracts was performed with standard techniques. Proteins (30 μg per lane) were separated by using Nupage Bis-Tris electrophoresis (Invitrogen) and transferred onto poly(vinylidene difluoride) membranes.
Yeast Strains. Wild-type (strain BY4741) and all yeast mutants, with the exception of plc1ΔipmkΔ double mutant yeast, were obtained from Research Genetics/Invitrogen (15). plc1ΔipmkΔ double mutant yeast were generated by using standard homologous recombination techniques (16). The entire PLC1 gene in the ipmkΔ strain was replaced by LEU2 generated by PCR amplification of the marker gene from the PRS405 plasmid.
Yeast Growth Conditions, Transformations, and Drug Treatments. Yeast were grown in standard yeast extract/peptone/dextrose (YPD) or synthetic medium supplemented with a complete or appropriate amino acid mixture of the Synthetic Complete supplement purchased from Qbiogene. Yeast transformations were performed by using the Alkali-Cation Yeast Transformation kit (Qbiogene). Where indicated, heterologous expression of proteins was achieved by using either pRS415 or p426-GPD yeast expression vectors (17).
Northern Analysis. For Northern analysis, yeast were grown in standard yeast extract/peptone/dextrose (YPD) or in synthetic defined (SD) drop-out medium (Qbiogene). Yeast was grown to early logarithmic phase and total RNA was extracted by standard phenol-chloroform techniques. Probes to ARG8, GAPDH, and ACT1 were made by random priming from a PCR product of each full-length gene.
Results and Discussion
To explore the lipid kinase activities of IPK family members, we incubated mammalian and yeast IPMKs and IP6Ks with bovine brain lipid extracts or PI(4,5)P2 in the presence of [γ-32P]ATP (Fig. 2 A–C). No lipid kinase activity is detectable with any of the IP6Ks tested (data not shown). In contrast, with three distinct bovine brain lipid extracts as well as with purified PI(4,5)P2, both yeast and mammalian IPMKs generate a lipid inositol product comigrating on TLC with PI(3,4,5)P3 produced by recombinant p110γ, a PI3K catalytic subunit. Whereas yeast and mammalian IPMKs form only a single product, PI3K also forms PI(3,4)P2 and PI (3)P in addition to PI(3,4,5)P3 when incubated with lipid extracts, consistent with previous reports (9). Thus, IPMK is more selective than class I PI3Ks in its lipid catalytic activity.
Fig. 2.

Yeast and rat IPMKs produce a single lipid product comigrating with PI(3,4,5)P3 when incubated with a number of lipid mixture extracts or purified lipid inositols. Yeast IPMK (A), rat IPMK (B), or recombinant human PI3K p110γ (C) were incubated in the presence of [γ-32P]ATP with type III, type I, or phosphoinositide-enriched type I bovine Folch extracts as well as purified PI(4,5)P2. (D) IPMK displays substrate specificity producing phosphorylated reaction products only in the presence of PI(4,5)P2. Lipid reaction products were separated by TLC and analyzed by autoradiography. (E) Definitive identification of IPMK lipid kinase reaction products was obtained by HPLC analysis and comparisons of deacylated lipids (glycerophosphoinositides) isolated from in vitro reactions with yeast IPMK, rat IPMK, and PI3K p110γ using PI(4,5)P2 as substrate. More polar peaks eluting after PI(3,4,5)P3 are likely artifacts of chemical deacylation and appear to comigrate with IP4.
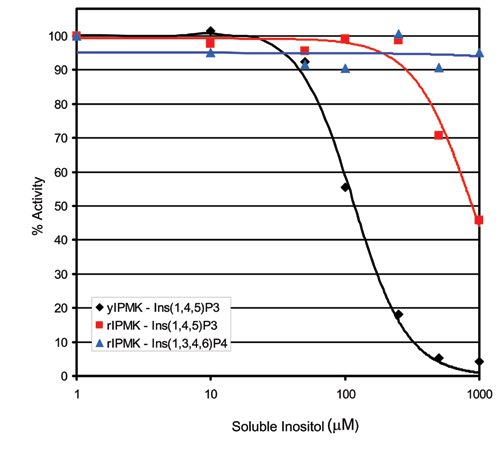
To delineate the specificity of IPMK, we incubated it with [γ-32P]ATP and a variety of synthetic lipid inositols (Fig. 2D). Catalytic activity is evident only with PI(4,5)P2, further emphasizing the selectivity of IPMK. Yeast and rat IPMK display similar kinetic parameters to those observed for canonical PI3Ks (18) with apparent Km values for PI(4,5)P2 of 30 μM and 6 μM, respectively (Fig. 7, which is published as supporting information on the PNAS web site). Maximal specific activities for yeast and rat IPMKs are ∼0.5 μmol/min/mg and ∼0.7 μmol/min/mg, respectively (Fig. 7). Competition assays in which yeast and rat IPMK lipid kinase reactions are performed in the presence of known soluble inositol substrates suggest that PI(4,5)P2 successfully competes with either IP3 or inositol 1,3,4,6-tetrakisphosphate, a recently identified substrate for IPMK's 5-kinase activity (19) (Fig. 8, which is published as supporting information on the PNAS web site).
To obtain definitive evidence for the catalytic step, we monitored the formation of PI(3,4,5)P3 by HPLC, analyzing the purified, deacylated reaction products. With both yeast and mammalian IPMKs, we observe almost exclusive formation of PI(3,4,5)P3 whose migratory properties are identical to those observed for PI3K reaction products (Fig. 2E). Further confirmation of the reaction products as the physiologically relevant PI(3,4,5)P3 isomer was obtained by incubation of the reaction products of rat and yeast IPMKs with either PTEN (Phosphatase and TENsin homologue deleted on chromosome 10), a PI(3,4,5)P3-3 phosphatase, or SHIP-2 (Src Homology 2 domain-containing Inositol Phosphatase), a PI(3,4,5)P3-5 phosphatase. PTEN phosphatase treatment of the reaction products results in the loss of the radiolabeled phosphate consistent with PTEN′s 3-phosphatase activity and the presumed D-3 position labeling by [γ-32P]ATP. SHIP-2 phosphatase treatment results in a radiolabeled product comigrating with a PI(3,4)P2 standard fitting with its 5-phosphatase specificity (Fig. 9, which is published as supporting information on the PNAS web site).
A hallmark of PI3K family members is their potent inhibition by wortmannin, a naturally occurring fungal metabolite that covalently modifies and inactivates PI3K (8). Wortmannin inhibits p110γ PI3K at concentrations as low as 5 nM, but fails to influence mammalian or yeast IPMK lipid kinase activity at concentrations as high as 10 μM (Fig. 3). Similarly, IPMK is unaffected by LY294002, a second selective PI3K inhibitor, at concentrations as high as 500 μM (data not shown).
Fig. 3.

Yeast and mammalian IPMKs are wortmannin-insensitive PI3Ks. Human PI3K p110γ (A), rat IPMK (B), and yeast IPMK (C) were incubated with PI(4,5)P2 and [γ-32P]ATP in the presence of increasing concentrations of wortmannin. Reaction products were separated by TLC and analyzed by autoradiography.
To ascertain whether IPMK possesses lipid kinase activity in intact cells, we transfected Cos-7 cells with HA-IPMK (Fig. 4). As described for yeast (20) and human (21) IPMK, we detected HA-IPMK almost exclusively in nuclei. Using PI(3,4,5)P3-specific antibodies, we observed pronounced nuclear staining for PI(3,4,5)P3 in IPMK-transfected cells, whereas in mock-transfected cells, PI(3,4,5)P3 is almost entirely cytosolic (Fig. 4 A–F). Isolated, intact nuclei from transfected cells also display wortmannin-insensitive PI3K activity in vitro when incubated with [γ-32P]ATP and PI(4,5)P2 (Fig. 10, which is published as supporting information on the PNAS web site). Biochemical analysis further confirms that IPMK forms PI(3,4,5)P3 in intact cells. HPLC analysis of [3H]inositol-labeled cells transfected with HA-IPMK reveals a significant increase in PI(3,4,5)P3 levels when compared to mock-transfected cells (Fig. 4G).
Fig. 4.

In vivo production of nuclear PI(3,4,5)P3 by IPMK. Cos-7 cells transfected with HA-IPMK (A–C) or empty vector (D–F) were fixed and stained via immunofluorescence with anti-HA (A and D, green) and anti-PI(3,4,5)P3 (B and E, red) antibodies. Merged images of cells transfected with HA-IPMK (C) or empty vector (F) reveal an increase in nuclear PI(3,4,5)P3 staining in HA-IPMK-transfected cells that colocalize with nuclear HA-IPMK (overlap shown in yellow). Images were acquired by confocal microscopy. Biochemical identification of increases in PI(3,4,5)P3 in HA-IPMK-transfected cells was accomplished by HPLC analysis (G) of glycerophosphoinositides prepared from [3H]inositol-labeled cells overexpressing HA-IPMK or GFP.
To assess the disposition and lipid kinase activity of endogenous IPMK, we used antibodies raised against rat IPMK. Western analysis of isolated nuclei from primary rat brain cultures and liver lysates reveals a single band of ∼44 kDa, corresponding to the predicted molecular size of IPMK (Fig. 5A). In cerebral cortical, hippocampal, and hepatocyte cultures, IPMK immunoreactivity is predominantly nuclear and occurs in a punctuate fashion (Fig. 5 B–D).
Fig. 5.

Endogenous IPMK localizes to the nucleus with a punctuate disposition. (A) In both cortical and hepatocyte primary cultures, the IPMK antibody recognizes a single band of the predicted molecular size. Immunolabeling of E17–18 rat embryo primary cortical (B) and hippocampal (C) cultures was performed by using anti-IPMK (green) and either anti-βIII-tubulin or anti-neurofilament (cytoplasmic neuronal markers) (red) antibodies. (D) Similar punctuate, nuclear localization of IPMK (green) is evident in adult rat hepatocytes. Hoechst staining (not shown) was used to verify nuclear localization. Images were acquired by confocal microscopy. (E) Immunoprecipitation of endogenous IPMK from nuclear lysates of adult rat liver or cortical cultures reveals wortmannin-resistant PI3K activity. Immunoprecipitates were incubated in the presence of [γ-32P]ATP and PI(4,5)P2; reaction products were separated by TLC and analyzed by autoradiography. Control reactions performed with PI3K p85 immunoprecipitates of whole-cell, cortical culture lysates display wortmannin-sensitive PI3K activities (E). Rabbit IgG fails to immunoprecipitate any PI3K activity.
We examined the lipid kinase activity of endogenous IPMK in rat liver and cerebral cortical cultures after immunoprecipitation of IPMK (Fig. 5E). In immunoprecipitates from isolated nuclear lysates, we observed formation of PI(3,4,5)P3, which is unaffected by 500 nM wortmannin. The pattern of lipid kinase activity observed is essentially the same as that observed with immunoprecipitated PI3K, although in the latter case enzyme activity is abolished by wortmannin.
To ascertain whether the nuclear PI(3,4,5)P3 produced by IPMK influences the canonical PI3K signaling cascade, we transfected IPMK or kinase-dead IPMK into HEK293T cells and monitored levels of phospho-Akt, reflecting activation of the PI3K pathway (Fig. 11, which is published as supporting information on the PNAS web site) (8). Phospho-Akt levels are unaffected by overexpression of wild-type or kinase-dead IPMK under a variety of serum starvation or stimulation paradigms, consistent with IPMK's lack of involvement in the canonical PI3K signaling pathway.
Although a physiologic function for IPMK has not been delineated in mammalian tissues, reports (22, 23) have suggested that yeast IPMK (also known as ArgRIII, Arg82, or Ipk2) functions as a regulatory component of the arginine-sensitive ArgR-Mcm1 transcriptional complex required for the suppression or induction of several gene products mediating arginine catabolism and synthesis. We focused on ARG8 (acetylornithine aminotransferase), whose mRNA levels are markedly augmented in ipmkΔ yeast. Using Northern analysis, we confirm the pronounced up-regulation of ARG8 mRNA in ipmkΔ yeast (Fig. 6 A and B). We show that this up-regulation is caused by the loss of IPMK, as transformation of the deficient yeast with yeast IPMK abolishes the up-regulation of ARG8 mRNA. Moreover, deletion of IPK1 or IP6K, the upstream inositol kinases necessary for the production of IP6 and IP7, does not affect ARG8 mRNA levels.
Fig. 6.

Northern analyses of ARG8 mRNA levels in yeast reveal lipid-kinase-dependent transcriptional regulation by IPMK. (A) Wild-type, ipk1Δ, and ip6kΔ yeast transformed with empty vector (p415, LEU2) show low levels of ARG8 expression when grown in synthetic drop-out medium. In contrast, ipmkΔ yeast show robust derepression of ARG8. The ARG8 phenotype of ipmkΔ is rescued by transformation with IPMK but not by transformation with kinase-dead (K133/A) IPMK. A deletion of an aspartate-rich region of IPMK (amino acids 285–300) partially impairs the ability of IPMK to rescue the ARG8 phenotype of ipmkΔ yeast. plc1Δ yeast, which lack detectable soluble inositol polyphosphates (C), display wild-type, repressed levels of ARG8 (B), whereas plc1ΔipmkΔ yeast display elevated ARG8 mRNA levels (B) despite possessing a nearly identical absence of inositol polyphosphates (C).
The kinase activity of IPMK is required for the regulation of ARG8, because transformation of the deficient yeast with kinase-dead IPMK (K133A) fails to rescue up-regulated ARG8 mRNA levels (Fig. 6A). We wondered whether regulation of ARG8 involves the inositol kinase or the lipid kinase activities of IPMK. Because yeast possess only one PLC isoform, phenotypic comparisons of plc1Δ yeast with yeast lacking downstream inositol polyphosphosphate kinases has clarified functional roles for inositol polyphosphate metabolism (4, 24–26). Despite having no detectable inositol polyphosphates (Fig. 6C), plc1Δ yeast display wild-type levels of ARG8 mRNA (Fig. 5B), suggesting that inositol polyphosphate synthesis is not necessary for ARG8 regulation. However, ipmkΔ yeast possess aberrantly high levels of IP2 and IP3 (Fig. 6C), which may function as negative regulators of ARG8 repression. To exclude a role for water-soluble inositol polyphosphates in ARG8 disregulation in ipmkΔ yeast, we generated plc1ΔipmkΔ double mutant yeast that, like plc1Δ yeast, display a lack of higher inositol polyphosphates (Fig. 6C). Like ipmkΔ yeast, plc1Δ ipmkΔ yeast display elevated ARG8 mRNA levels (Fig. 6B) that can only be rescued with enzymatically active yeast IPMK (data not shown). Thus, the ARG8 transcription regulatory activity of IPMK appears to reflect its lipid inositol kinase rather than its water-soluble inositol kinase activity.
Previous reports have identified a necessary role for IPMK in stabilizing the ArgR-Mcm1 transcriptional complex (composed of yeast IPMK, Arg80, Arg81, and Mcm1) via direct interactions with Mcm1 and Arg80 (20). With the exception of the catalytic domains, rat IPMK shares little homology to yeast IPMK and lacks conserved regulatory domains necessary for mediating IPMK's protein–protein interactions in yeast (20). Consistent with a requirement for appropriate recruitment or localization of IPMK in mediating ARG8 regulation, overexpression of rat IPMK in ipmkΔ or plc1ΔipmkΔ yeast fails to rescue the ARG8 phenotype (data not shown).
Our findings reveal IPMK to be a robust evolutionarily conserved inositol lipid kinase with PI3K activity. IPMK demonstrates greater substrate specificity than class I PI3Ks, as IPMK phosphorylates only PI(4,5)P2. Also, whereas class I PI3Ks are predominantly cytosolic and are potently inhibited by wortmannin, IPMK is almost exclusively nuclear and is unaffected by wortmannin or other PI3K selective inhibitors.
Very recently (27, 28), the first crystal structures were solved for an IPK family member, the exclusively metazoan IP3-3 kinase, providing a framework for understanding the mechanism of catalysis and constraints on substrate specificity for the entire IPK family. Structure database comparisons revealed that IP3-3 kinases possess significant structural similarity to type IIβ phosphatidylinositol phosphate kinases, suggesting a direct relationship between the evolutionary development of lipid inositol kinase activities and soluble inositol kinase activities. However, structural constraints unique to the evolutionarily recent IP3-3 kinases preclude IP3-3 kinases from functioning as lipid kinases (27, 28). These constraints are absent in yeast and mammalian IPMKs, allowing for both lipid and soluble inositol kinase activities.
Although exact functions of IPMK-mediated nuclear PI(3,4,5)P3 production in mammalian tissues are unclear, in yeast, IPMK PI3K activity appears to regulate transcription. Functional roles for soluble inositol polyphosphate kinase activity in transcriptional regulation and chromatin remodeling have been proposed (4, 25), and kinase-independent transcriptional regulation has also been suggested (24). Our identification of IPMK's lipid kinase activity provides an alternative context for the analysis of transcriptional regulation permitting clarification of previously conflicting interpretations.
Most studies of signaling pathways regulated by PI3K have focused on tyrosine kinase-linked receptors and their associated cytosolic signaling cascades. However, selective nuclear lipid inositol pathways have been extensively described (29, 30). Critical elements of the inositol lipid kinase pathways occur in nuclei, including enzymes that generate PI(4,5)P2, PLC isoforms, and the lipid inositol phosphatases, PTEN and SHIP, that dephosphorylate PI(3,4,5)P3 (1, 30). Nuclear, transcriptional regulatory proteins that act as receptors for lipid inositols, including PI(3,4,5)P3, have also been identified (31). Whereas cytosolic lipids often comprise classic lipid bilayers, nuclear inositol lipids are detergent-resistant and not associated with nuclear membranes (29). Thus, alternative techniques may be necessary to elucidate inositol lipid dynamics and regulation in nuclei. The absence in yeast of receptor-tyrosine kinase pathways and associated class I PI3Ks, as well as the inability to biochemically detect PI(3,4,5)P3 in yeast extracts had implied that PI(3,4,5)P3 signaling is relegated to higher organisms, although in animal tissues, PI(3,4,5)P3 is thought to comprise <0.005% of total inositol lipids (32). However, the recent discovery of PI(3,4,5)P3 in fission yeast lacking a PTEN homologue (33) confirms the existence of more primitive biosynthetic pathways for the lipid inositol. Our demonstration that IPMK is a robust physiologic nuclear PI3K that regulates transcription implies functional importance for PI(3,4,5)P3 in the nuclei of both mammals and yeast.
Supplementary Material
Acknowledgments
We sincerely thank Lynda Hester for her technical expertise and assistance. This work was supported by United States Public Health Service Grant DA-000266 (to S.H.S.).
Author contributions: A.C.R., B.N.K., S.H.S., and A.S. designed research; A.C.R., A.M.S., B.N.K., K.J.H., and A.S. performed research; A.C.R., B.N.K., S.H.S., and A.S. analyzed data; and A.C.R., S.H.S., and A.S. wrote the paper.
Abbreviations: IP3, inositol 1,4,5-trisphosphate; PI(4,5)P2, inositol phosphatidylinositol 4,5-bisphosphate; PLC, phospholipase C; IPK, inositol polyphosphate kinase; IPMK, inositol polyphosphate multikinase; PI3K, phosphoinositide 3-kinase; PI(3,4,5)P3, phosphatidylinositol 3,4,5-trisphosphate.
References
- 1.Irvine, R. F. & Schell, M. J. (2001) Nat. Rev. Mol. Cell Biol. 2, 327–338. [DOI] [PubMed] [Google Scholar]
- 2.Berridge, M. J. (1993) Nature 361, 315–325. [DOI] [PubMed] [Google Scholar]
- 3.Shears, S. B. (2004) Biochem. J. 377, 265–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Odom, A. R., Stahlberg, A., Wente, S. R. & York, J. D. (2000) Science 287, 2026–2029. [DOI] [PubMed] [Google Scholar]
- 5.Saiardi, A., Nagata, E., Luo, H. R., Sawa, A., Luo, X., Snowman, A. M. & Snyder, S. H. (2001) Proc. Natl. Acad. Sci. USA 98, 2306–2311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Saiardi, A., Erdjument-Bromage, H., Snowman, A. M., Tempst, P. & Snyder, S. H. (1999) Curr. Biol. 9, 1323–1326. [DOI] [PubMed] [Google Scholar]
- 7.Dubois, E. & Messenguy, F. (1994) Mol. Gen. Genet. 243, 315–324. [DOI] [PubMed] [Google Scholar]
- 8.Cantley, L. C. (2002) Science 296, 1655–1657. [DOI] [PubMed] [Google Scholar]
- 9.Vanhaesebroeck, B., Leevers, S. J., Ahmadi, K., Timms, J., Katso, R., Driscoll, P. C., Woscholski, R., Parker, P. J. & Waterfield, M. D. (2001) Annu. Rev. Biochem. 70, 535–602. [DOI] [PubMed] [Google Scholar]
- 10.Kurt, R., Serunian, L. A., and Cantley, L. C. (1990) Methods in Inositide Research (Raven, New York).
- 11.Saiardi, A., Sciambi, C., McCaffery, J. M., Wendland, B. & Snyder, S. H. (2002) Proc. Natl. Acad. Sci. USA 99, 14206–14211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dawson, V. L., Dawson, T. M., London, E. D., Bredt, D. S. & Snyder, S. H. (1991) Proc. Natl. Acad. Sci. USA 88, 6368–6371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen, R., Kang, V. H., Chen, J., Shope, J. C., Torabinejad, J., DeWald, D. B. & Prestwich, G. D. (2002) J. Histochem. Cytochem. 50, 697–708. [DOI] [PubMed] [Google Scholar]
- 14.Vann, L. R., Wooding, F. B., Irvine, R. F. & Divecha, N. (1997) Biochem. J. 327, 569–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Winzeler, E. A., Shoemaker, D. D., Astromoff, A., Liang, H., Anderson, K., Andre, B., Bangham, R., Benito, R., Boeke, J. D., Bussey, H., et al. (1999) Science 285, 901–906. [DOI] [PubMed] [Google Scholar]
- 16.Baudin, A., Ozier-Kalogeropoulos, O., Denouel, A., Lacroute, F. & Cullin, C. (1993) Nucleic Acids Res. 21, 3329–3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mumberg, D., Muller, R. & Funk, M. (1995) Gene 156, 119–122. [DOI] [PubMed] [Google Scholar]
- 18.Carpenter, C. L., Duckworth, B. C., Auger, K. R., Cohen, B., Schaffhausen, B. S. & Cantley, L. C. (1990) J. Biol. Chem. 265, 19704–19711. [PubMed] [Google Scholar]
- 19.Chang, S. C., Miller, A. L., Feng, Y., Wente, S. R. & Majerus, P. W. (2002) J. Biol. Chem. 277, 43836–43843. [DOI] [PubMed] [Google Scholar]
- 20.El Bakkoury, M., Dubois, E. & Messenguy, F. (2000) Mol. Microbiol. 35, 15–31. [DOI] [PubMed] [Google Scholar]
- 21.Nalaskowski, M. M., Deschermeier, C., Fanick, W. & Mayr, G. W. (2002) Biochem. J. 366, 549–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dubois, E., Bercy, J. & Messenguy, F. (1987) Mol. Gen. Genet. 207, 142–148. [DOI] [PubMed] [Google Scholar]
- 23.Bechet, J., Greenson, M. & Wiame, J. M. (1970) Eur. J. Biochem. 12, 31–39. [DOI] [PubMed] [Google Scholar]
- 24.Dubois, E., Dewaste, V., Erneux, C. & Messenguy, F. (2000) FEBS Lett. 486, 300–304. [DOI] [PubMed] [Google Scholar]
- 25.Steger, D. J., Haswell, E. S., Miller, A. L., Wente, S. R. & O'Shea, E. K. (2003) Science 299, 114–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.York, J. D., Odom, A. R., Murphy, R., Ives, E. B. & Wente, S. R. (1999) Science 285, 96–100. [DOI] [PubMed] [Google Scholar]
- 27.Gonzalez, B., Schell, M. J., Letcher, A. J., Veprintsev, D. B., Irvine, R. F. & Williams, R. L. (2004) Mol. Cell 15, 689–701. [DOI] [PubMed] [Google Scholar]
- 28.Miller, G. J. & Hurley, J. H. (2004) Mol. Cell 15, 703–711. [DOI] [PubMed] [Google Scholar]
- 29.Irvine, R. F. (2003) Nat. Rev. Mol. Cell Biol. 4, 349–360. [DOI] [PubMed] [Google Scholar]
- 30.Martelli, A. M., Tabellini, G., Borgatti, P., Bortul, R., Capitani, S. & Neri, L. M. (2003) J. Cell Biochem. 88, 455–461. [DOI] [PubMed] [Google Scholar]
- 31.Jones, D. R. & Divecha, N. (2004) Curr. Opin. Genet. Dev. 14, 196–202. [DOI] [PubMed] [Google Scholar]
- 32.Leslie, N. R. & Downes, C. P. (2002) Cell. Signalling 14, 285–295. [DOI] [PubMed] [Google Scholar]
- 33.Mitra, P., Zhang, Y., Rameh, L. E., Ivshina, M. P., McCollum, D., Nunnari, J. J., Hendricks, G. M., Kerr, M. L., Field, S. J., Cantley, L. C. & Ross, A. H. (2004) J. Cell Biol. 166, 205–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}