

Abstract
Dopamine is a neuromodulator the functions of which in the regulation of complex behaviors such as mood, motivation, and attention are well known. Dopamine appears in the brain early in the embryonic period when none of those behaviors is robust, raising the possibility that dopamine may influence brain development. The effects of dopamine on specific developmental processes such as neurogenesis are not fully characterized. The neostriatum is a dopamine-rich region of the developing and mature brain. If dopamine influenced neurogenesis, the effects would likely be pronounced in the neostriatum. Therefore, we examined whether dopamine influenced neostriatal neurogenesis by influencing the cell cycle of progenitor cells in the lateral ganglionic eminence (LGE), the neuroepithelial precursor of the neostriatum. We show that dopamine arrives in the LGE via the nigrostriatal pathway early in the embryonic period and that neostriatal neurogenesis progresses in a dopamine-rich milieu. Dopamine D1-like receptor activation reduces entry of progenitor cells from the G1- to S-phase of the cell cycle, whereas D2-like receptor activation produces the opposite effects by promoting G1- to S-phase entry. D1-like effects are prominent in the ventricular zone, and D2-like effects are prominent in the subventricular zone. The overall effects of dopamine on the cell cycle are D1-like effects, most likely because of the preponderance of D1-like binding sites in the embryonic neostriatum. These data reveal a novel developmental role for dopamine and underscore the relevance of dopaminergic signaling in brain development.
Keywords: ganglionic eminence, dopamine, cell cycle, striatum, neurogenesis, D1 receptor
Introduction
Monoamines, such as dopamine, are among the earliest neurochemical systems to develop in the embryo. The early appearance and continued presence of dopamine throughout the prolonged sequence of CNS development suggest that an imbalance in the dopaminergic system could affect brain development. Dopaminergic imbalance in the developing brain can occur under various conditions. Drugs of abuse such as cocaine target dopaminergic systems of the developing brain and cause lasting neurological dysfunction (Hope et al., 1992; Nestler et al., 1993; Kosofsky et al., 1994; Levitt et al., 1997; Lidow et al., 2001; Stanwood et al., 2001). Cocaine penetrates the placental barrier, interferes with dopamine uptake and storage mechanisms, and disrupts dopamine receptor signaling (Kosofsky et al., 1994; Wang et al., 1995; Shearman et al., 1996; Levitt et al., 1997). Drugs such as methylphenidate that are used in the treatment of childhood behavioral afflictions also can alter dopamine levels in the brain (Moll et al., 2001). Schizophrenia also is associated with dopamine imbalance and may have a developmental origin (Bloom, 1993;Weinberger, 1995; Benes, 2000; Schwartz et al., 2000).
Although the role of dopamine in normal development of the brain and behavior is beginning to be understood, its role in modulating critical developmental processes such as neurogenesis is not fully understood. If the effects of dopamine on neurogenesis are characterized at the level of the cell cycle, it might be possible to gain further insights into the way in which chemical substances or disease processes that interfere with the dopaminergic system disrupt normal development of the brain and behavior.
Perhaps in no other telencephalic region is the role of dopamine more evident than in the neostriatum, a dopamine-rich component of the basal ganglia. Neostriatal neurons arise from the lateral ganglionic eminence (LGE). The LGE along with the medial ganglionic eminence is also a source of GABAergic neurons of the cerebral cortex, olfactory bulb, and hippocampus (Anderson et al., 1997; Tamamaki et al., 1997; Lavadas et al., 1999; Marin et al., 2000). We show that dopamine receptor activation modulates the progression of LGE progenitors from the G1-phase to the S-phase of the cell cycle.
Materials and Methods
Animals. We used CD1 mice (Charles River Laboratories, Wilmington, MA) housed in our institutional animal facility. Female mice housed with a male for the previous 15–17 hr were examined for the presence of vaginal plugs at 9:00 A.M. Presence of the plug was taken to indicate conception; the day of plug was designated embryonic day (E) 0, and the day of birth was designated postnatal day (P) 0.
The time of onset of neostriatal neurogenesis. Bromodeoxyuridine (BUdR) (Sigma, St, Louis, MO; 50 μg/gm body weight) was administered as a single intraperitoneal injection to dams carrying E11, E12, E13, or E15 mice. Offspring were anesthetized (ketamine, 50 mg/kg body weight, and xylazine, 10 mg/kg body weight, i.p.) on P30 and perfused through the heart with 70% ethanol. BUdR immunohistochemistry was performed on 4-μm-thick, paraffin-embedded coronal sections through the neostriatum (Bhide, 1996). The sections were counterstained with basic fuchsin. The distribution of BUdR-labeled cells with respect to neostriatal borders was examined (see Fig. 1A–C).
Fig. 1.
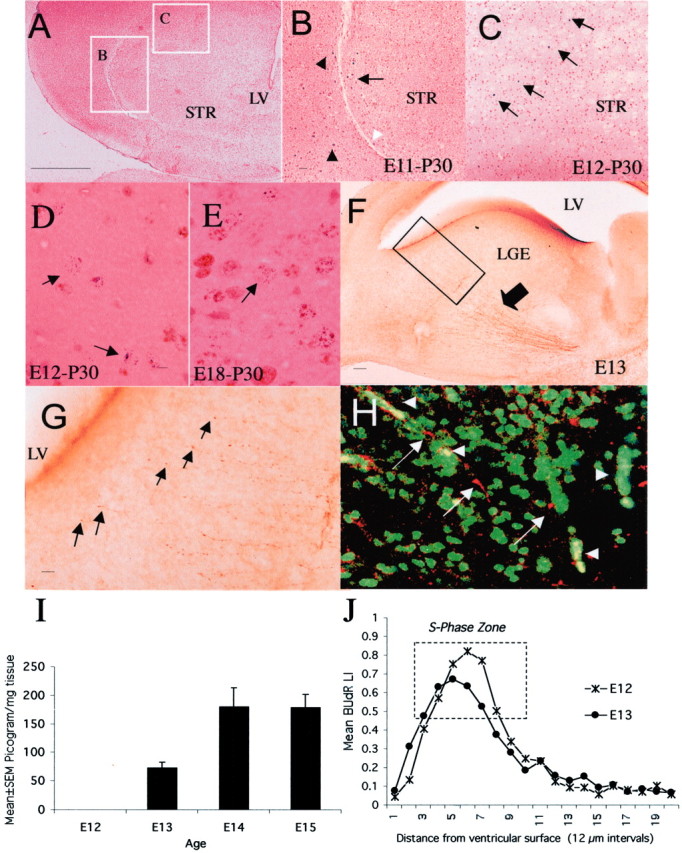
Neurogenesis in the LGE occurs in a dopamine-rich milieu. When E11 mice were exposed to BUdR, labeled cells were found at the lateral margin of the neostriatum (B,arrow) just medial to the external capsule (B, white arrowhead) as well as outside the neostriatal borders on P30 (B, black arrowheads, E11-P30). When E12 mice were exposed to BUdR, labeled cells were found throughout the neostriatum (C, arrows) as well as outside the neostriatum on P30 (E12-P30). The position of labeled cells shown in B and C is indicated bywhite rectangles in A. When a double S-phase labeling method was used, cells labeled with3H-TdR-only were present in the neostriatum on P30 if the S-phase marker injections were administered on E12 (D,arrows, E12-P30) or E18 (E, arrow, E18-P30) but not on E11, confirming that neostriatal neurogenesis began on E12.F, TH-positive axons in the neostriatum (arrow) on E13. The boxed area inF is shown at a higher magnification in Gto illustrate growing tips of TH-positive fibers (G,arrows) within 25–50 μm of the lateral ventricular border. TH-positive axons and growth cones (white arrows) are in close proximity to BUdR-labeled (green) nuclei in the LGE (H). Red blood cells that fluoresce in both the green and red filters appear yellowish orange(H, white arrowheads). Dopamine content of the forebrain was undetectable on E12 and rose dramatically between E12 and E13 (I) coincident with the arrival of TH-positive axons in the LGE (F). BUdR LI decreased between E12 and E13 in the S-phase zone of the LGE (J), coincident with the arrival of dopamine. LV, Lateral ventricle; STR, neostriatum. Scale bars:A, 250 μm; (in B) B,C, 50 μm; (in D) D,E, 5 μm; F, 50 μm; G, 10 μm; H, 20 μm.
In other experiments, BUdR and methyl tritiated thymidine (3H-TdR) were injected sequentially. Pregnant dams carrying E11, E12, E13, E15, and E18 mice were given a single injection of 3H-TdR (DuPont/New England Nuclear, Boston, MA; 5 μCi/gm body weight, i.p.). Beginning 2 hr later, BUdR was injected at 3 hr intervals for a total duration of 9 hr (E11 and E12), 15 hr (E13), or 24 hr (E15 and E18). Offspring from the3H-TdR- and BUdR-injected mothers were anesthetized on P30 and perfused through the heart with 70% ethanol. Four-micrometer-thick paraffin-embedded coronal sections through the neostriatum were processed for BUdR immunohistochemistry and autoradiography (Sheth and Bhide, 1997). Three types of labeled cells were present in the autoradiograms: BUdR-only labeled cells,3H-TdR and BUdR double-labeled cells, and3H-TdR-only labeled cells. The3H-TdR-only labeled cells left the S-phase during the 2 hr interval between the3H-TdR and the first BUdR injection and did not reenter the cell cycle (Takahashi et al., 1994). Those are the cells of interest. Their position was examined with respect to neostriatal borders.
In every type of experiment, two to three pregnant mice were used. Four to six sections each from at least three brains from each litter were analyzed for each age group.
Tyrosine hydroxylase immunohistochemistry. Brains from E11, E12, E13, E14, E15, and E17 mice were immersion fixed in 4% paraformaldehyde in 0.1 m phosphate buffer. Immunohistochemistry was performed using tyrosine hydroxylase (TH) antibody (rabbit polyclonal antiserum; Eugene Tech, Eugene, OR; diluted 1:1000) and DAB as the chromagen on 50-μm-thick cryostat or vibratome sections of the brain. In other experiments, E13 mice were exposed continuously to BUdR for 6.5 hr before they were killed (a total of three BUdR injections at 3 hr intervals, and then they were killed 0.5 hr after the third BUdR injection). Brains were removed and fixed in paraformaldehyde. Double-labeling immunohistochemistry was performed on vibratome sections of the brains using fluorescent secondary antibodies against TH (Cy-3-conjugated donkey anti-rabbit IgG;Jackson ImmunoResearch, West Grove, PA) and BUdR (Cy-2-conjugated donkey anti-mouse IgG; Jackson ImmunoResearch). The sections were examined in a laser confocal microscope.
Estimation of dopamine content. Dopamine content was estimated by HPLC (LC-4B; Bioanalytical Systems, West Lafayette, IN) in the forebrains of E12, E13, E14, and E15 mice. In separate experiments, dopamine content was analyzed only in the basal forebrain from E13 mice whose mothers had received ascorbic acid orl-DOPA plus ascorbic acid in drinking water from E10 to E13. The measurements were standardized on a per milligram of tissue basis. Although samples of the basal forebrain from E13 mice could be collected, the small size of the brain on E11 and E12 made it difficult to collect and analyze such samples reliably. Therefore, to maintain uniformity of sampling across the age groups, in the developmental study, forebrain samples were used in all cases. Because only the basal forebrains were analyzed in thel-DOPA administration study, the dopamine content in the two series of experiments are different. Six to eight brains from each of two to three litters for each age were analyzed.
Explant cultures. Timed-pregnant CD1 mice carrying E13 or E14 embryos were anesthetized, and the embryos were removed one at a time. Embryos (or entire litters) that did not fall within the recommended range for E13 or E14 (Theiler, 1972) were discarded. From each embryo, explants of the dorsal wall of the cerebral hemispheres were prepared as described previously (Takahashi et al., 1999). The explants were placed in a 50 μl droplet of rat tail collagen held on ice in a 35 mm Petri dish. The dish was transferred to an incubator (37°C, 95% air/5% CO2). Approximately 30 min after explantation, when collagen had gelled, 2.5 ml of the culture medium (DMEM/F12 with l-glutamine; Invitrogen, Grand Island, NY; final concentration 1×) was added to each Petri dish, and BUdR (10 μm) was added to the medium.
Explants were fixed with 70% ethanol at 2 hr intervals from the time of addition of the medium and BUdR, for a total duration of 12 hr. Four-micrometer-thick, paraffin-embedded coronal sections from the mid portion of each explant were selected and processed for BUdR immunohistochemistry. The BUdR labeling index (LI; BUdR-labeled cells divided by all cells within a defined area) was calculated within a 120 × 240 μm2 sector of the LGE. The sector was further subdivided into 20 bins (each bin was 12 × 120 μm2). BUdR LI was calculated for each bin.
D1-like receptor agonists SKF 38393 or SKF 81297 (1 or 10 μm; RBI, Natick, MA) or D2-like agonists quinpirole hydrochloride or PD128907 hydrochloride (1 or 10 μm; RBI, Natick, MA) were added to the culture medium at the same time as BUdR. In other experiments, D1 receptor antagonist Schering 23390 or D2 receptor antagonist eticlopride (RBI, Natick, MA) were used to block the specific receptors before the addition of the agonists. Dopamine (Sigma) was added to the culture medium at the same time as BUdR, at a final concentration of 1, 10, 50, or 100 μm. Glutathione (10 μm; Sigma) or ascorbic acid (0.01%; Sigma) was added to the culture medium to retard oxidation of dopamine. Control cultures were incubated with no drugs, with the receptor antagonist alone, or with the antioxidant alone. BUdR was added to the medium in all cultures.
The following method was used to measure cell output.3H-TdR and BUdR injections were administered 2 hr apart to pregnant mice carrying E13 embryos. Explants were prepared after the BUdR injection, and BUdR was added to the culture medium (Takahashi et al., 1999). After 12 hr exposure to dopamine plus antioxidants or antioxidant alone (control), cultures were fixed. Coronal sections of the explants were stained immunohistochemically for BUdR and processed for autoradiography.3H-TdR-only labeled cells were identified in the sections and counted within a 120 × 240 μm2 sector of the LGE.
In each experiment, we used 18–24 explants taken from 9–12 embryos (CD1 mice have large litters; 10–13 embryos per litter is common). The explants were assigned randomly to different experimental groups. One group was always the control group (no additives, receptor antagonist only or antioxidant only). We calculated BUdR LI for each explant (four sections per explant) and calculated mean and SEM values for a given experimental group (consisting of four to six explants). We performed 10 experiments using E13 explants and 4 using E14 explants to test the effects of dopamine on BUdR LI. We performed five experiments each to examine the effects of D1- and D2-agonists and to examine specificity of the effects using the antagonists.
Dopamine receptor binding and G-protein coupling. Embryonic heads (E13) or brains (E14 onward) were snap frozen in isopentane cooled in liquid nitrogen and sectioned in the coronal plane at 12 μm thickness on the cryostat. The sections were mounted on glass slides and incubated with tritiated D1-like (Schering 23390, 1 nm) or D2-like (raclopride, 3 nm) antagonists. Nonspecific binding was assessed with 10 μm dopamine. The optical density of the autoradiograms was measured over the LGE and the neostriatum (including the area corresponding to the 20 bins in which the BUdR LI was analyzed) using a computerized image analysis system (MCID, Ontario, Canada). Optical density values were converted to “nanocurie per milligram of tissue equivalent” after calibration with Amersham tritiated polymer standards (Arlington Heights, IL). The nanocurie per milligram of tissue equivalent values were converted to femtomoles of bound ligand per milligram of tissue on the basis of the specific activity of the antagonists used. The experiments were performed on two to three embryos (or newborn mice) for each age group taken from each of three litters.
For G-protein coupling assays, 12-μm-thick cryostat sections of the whole embryonic heads were used from E15 and E16 mice. An iodinated D2-like antagonist, [125I]iodosulpiride, was used because it provides a more intense signal than the tritiated antagonist. Consecutive sections were incubated with [125I]iodosulpiride (0.3 nm), with 2 μm dopamine and [125I]iodosulpiride, or with the nonhydrolyzable GTP analog GTPγS (10 μm), [125I]iodosulpiride, and dopamine. After the incubations and washes, the sections were exposed to film.
In vivo administration of dopaminergic drugs. We administered two intraperitoneal injections of the D1 receptor agonist SKF 81297 (10 or 20 mg/kg), or saline (control groups) to pregnant mice carrying E13 embryos. The injections were spaced 3 hr apart. BUdR (50 mg/kg, i.p.) was administered 1 hr after the second SKF 81297 injection. The mice were killed 2 hr after the BUdR injection, and the embryonic heads were processed for paraffin-wax histology. Thus, the embryos were exposed to SKF 81297 for 6 hr and to BUdR for the final 2 hr. We used three litters per each dose of SKF 81297 and two litters in the control (saline) group. BUdR LI was calculated in four sections each from four to five embryonic brains from each litter.
The dopamine precursor l-DOPA was administered to pregnant mice in drinking water from E10 to E13. l-DOPA (2 mg/ml; Sigma) was dissolved in 0.025% ascorbic acid (Sigma; an antioxidant that protects l-DOPA). Control groups received 0.025% ascorbic acid alone. BUdR was administered to the pregnant mice on E13. The embryos were killed 2 hr after the BUdR injection, and their heads were processed for paraffin-wax histology. We used three litters per each dose ofl-DOPA and three litters in the control (saline) group. BUdR LI was calculated in four sections from each of four embryonic brains from each litter.
The D1-like antagonist Schering 23390 was injected into the forebrain ventricles of E15 mice in utero. Pregnant mice were anesthetized, and a laparotomy was performed to expose the gravid uterine horns. The orientation of the embryonic head inside the intact amniotic sac was established without incising the uterine wall. One microliter solution of Schering 23390 (RBI/Sigma; 100 μm in PBS) or PBS was injected into the brain using a Hamilton syringe fitted with a glass electrode tip. The solutions contained 0.025% fast green. In every successful injection, the lateral ventricles turned dark green as the fast green filled the ventricles. Every embryo in which the anatomical landmarks could be established unequivocally was injected. Only those embryos in which the injection site could be verified by visual inspection of the green color in the ventricles were used for further analyses. Once the injections were completed, the abdominal incision was closed, and the mother was allowed to recovered from the effects of anesthesia. BUdR was injected into the mother 2 hr after the completion of the last intraventricular injection. The mother was anesthetized again 2 hr after the BUdR injection, and the embryos were removed and processed for histology. We used five litters for Schering 23390 injections and four litters for saline injections. Two to six embryos per litter were injected. BUdR LI was calculated in four sections from each embryonic brain.
BUdR LI calculation in vivo. Embryonic heads (E13) or brains (E15) were immersed in 70% ethanol and processed for paraffin-wax histology and BUdR immunohistochemistry (Bhide, 1996). BUdR LI was calculated within a 120 × 200 μm2sector of the LGE. The sector was divided into 20 bins, 12 × 10 μm2, and the LI was also calculated separately for each bin.
In all of the in vivo and in vitro experiments, BUdR LI calculation was performed in sections chosen from the midportion of the rostrocaudal extent of the LGE to ascertain uniform sampling across the different experimental groups.
Results
Neostriatal neurogenesis occurs in a dopamine-rich milieu
We examined the temporal relationship between the onset of neostriatal neurogenesis and the arrival of dopamine in the developing neostriatum. A single injection of BUdR on E12, E13, or E15 but not E11 produced BUdR-labeled cells in the neostriatum on P30 (Fig.1). The E11 injections resulted in labeled cells at the lateral border of the neostriatum (Fig.1B, arrow) as well as outside neostriatal borders on P30 (Fig. 1B, black arrowheads). When we used a double-S-phase labeling method, which permits delineation of a cohort of cells that underwent their last S-phase over a precisely defined 2 hr interval (Takahashi et al., 1994), 3H-TdR-only labeled cells were present in the P30 neostriatum after injections on E12 (Fig.1D, arrows), E13, or E18 (Fig.1E, arrow) but not E11. Therefore, we conclude that neostriatal neurons are produced in mice on and after E12.
TH-positive axons were present in the developing neostriatum on E13 (Fig. 1F). A few TH-positive axons left the main bundle and approached the lateral ventricular border (Fig.1G). TH-positive axons and growth cones were in close proximity to BUdR-labeled, proliferating progenitor cells in the LGE on E13 (Fig. 1H). Thus, presumptive dopaminergic axons enter the LGE and are in close proximity to dividing LGE progenitor cells on E13. Substantial numbers of TH-positive axons entered the LGE and the neostriatum on E14 and later (data not shown).
TH immunoreactivity in the E13 LGE/neostriatum is indicative of but does not confirm the presence of dopamine. Therefore, dopamine content was estimated by HPLC in the forebrains of E12, E13, E14, and E15 mice. Dopamine content of the forebrain was undetectable on E12 and rose substantially to 73.5 ± 10.1 pg/mg tissue (mean ± SEM) on E13 and remained high thereafter (Fig. 1I).
Onset of dopaminergic innervation coincides with changes in cell proliferation in the LGE
We estimated the proportion of cells in S-phase in the LGE on E12 and E13, before and after dopamine arrived, respectively, by calculating the BUdR LI after a single “pulse” of BUdR. We found a 15.7% reduction in the BUdR LI in the S-phase zone, which was identified (Fig. 1J) on the basis of criteria established previously (Bhide, 1996), between E12 and E13 (mean ± SEM values: E12, 0.53 ± 0.02; E13, 0.44 ± 0.02;t test; p = 0.02). The reduction indicates that fewer cells entered S-phase on E13 compared with E12.
If temporal coincidence alone were sufficient, the reduction in the BUdR LI could be interpreted as a consequence of the arrival of dopamine. However, other factors might have contributed to the LI reduction. We developed an explant culture system in which we could use dopamine or its receptor agonists/antagonists as the only variable and determine unequivocally whether dopamine influenced LGE cell proliferation.
An explant culture system to study cell cycle parameters in the LGEin vitro
Explants of the entire ganglionic region, containing the LGE and the medial ganglionic eminence (Fig.2A), were cultured, and BUdR LI was calculated in a 120 × 240 μm2 sector of the LGE (Fig.2B). This sector corresponded to the region in which cell cycle kinetics were analyzed in vivo (Bhide, 1996;Sheth and Bhide, 1997). The sector was divided into 20 bins. Each bin was 12 × 120 μm2 wide. When the LI was analyzed for the ventricular zone (VZ) only (bins 1–7), it steadily increased during the 0.5–12.0 hr culture period (Fig.2C), indicating that new cells were entering S-phase and that the G1–S-phase checkpoint was cleared. In the next series of analyses, the LI was calculated for each of the 20 bins and for each of the six 2 hr labeling intervals (from 0.5 to 12.0 hr). The marked rise in the LI in bin 1 between 0.5 and 12.0 hr (Fig.2D) shows that nuclei of cells in S-phase at the time of explantation and of other cells entering S-phase later (therefore labeled with BUdR) moved through G2-phase and into mitotic phase (M-phase or prophase (Bhide, 1996; Takahashi et al., 1999). Thus, the S–G2–M checkpoint was also cleared successfully. The interkinetic nuclear migration was preserved in the explants.
Fig. 2.
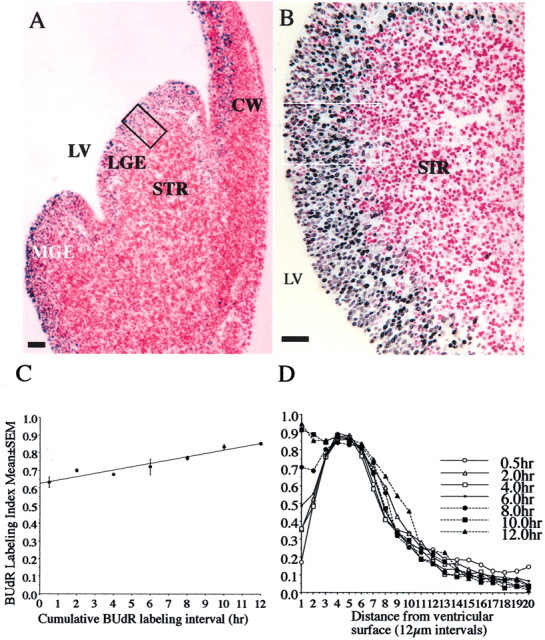
Photomicrographs of a 4-μm-thick, paraffin wax-embedded section through an E13 explant that was cultured for 12 hr with continuous exposure to BUdR (A). The section was processed for BUdR immunohistochemistry and stained with basic fuchsin. BUdR-labeled and non-BUdR-labeled (i.e., basic fuchsin-only labeled) cells are shown at higher magnification in B. BUdR LI (BUdR-labeled cells divided by all cells) was calculated within a 120 × 240 μm wide sector of the LGE (A,B, boxed region). The sector was subdivided into 20 bins (12 × 120 μm) using a microscope ocular grid. Initially, the BUdR LI was calculated for the VZ (corresponding to bins 1–7) and plotted against labeling interval (C). VZ consists predominantly of progenitor cells (and only very few postmitotic cells), fulfilling the criteria for a cell population the cell cycle kinetics of which can be analyzed by cumulative S-phase labeling methods (Nowakowski et al., 1989). The BUdR LI increased linearly in the VZ from 0.5 to 12 hr (C), suggesting that the progenitor cells entered S-phase successfully clearing G1- to S-phase transition. When the BUdR LI was calculated for each of the 20 bins for each labeling interval (0.5 to 12 hr) and plotted against distance from the ventricular surface, the LI profiles changed in step with the interkinetic nuclear migration of the progenitor cells (D). The BUdR LI in bin 1 increased progressively, showing that BUdR-labeled cells left the S-phase and arrived at the ventricular surface for mitosis. Thus, G2- to M-phase transition occurred successfully in the explants.LV, Lateral ventricle; LGE, lateral ganglionic eminence; MGE, medial ganglionic eminence;STR, neostriatum; CW, cerebral wall. Scale bars: A, 150 μm; B, 75 μm.
The incidence of cell death was analyzed in the explants using a modified terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) procedure (Gavrieli et al., 1992; Verney et al., 2000). The percentage of TUNEL-labeled nuclei (compared with all nuclei, on a per 100 μm2 basis) was ∼0.1%. Cell death was also examined by counting pyknotic nuclei in basic fuchsin-stained sections (Takahashi et al., 1999; Verney et al., 2000). By this method also, the incidence of pyknotic profiles was <0.1%. There were virtually no TUNEL-positive or pyknotic profiles in the VZ/subventricular zone (SVZ) region. The few profiles that were seen were in the differentiating striatal fields. There was no histological evidence of necrosis (e.g., swollen nuclei or cells, vacuoles, or large extracellular spaces) in the explants (Fig.2A,B).
Dopamine D1 and D2 receptor binding sites in the embryonic neostriatum
Both D1-like and D2-like binding sites were present in the neostriatum on E13, but their density increased markedly during development (e.g., 15- and 23-fold increase in D2- and D1-like binding sites, respectively, from E13 to E14) (Table1). Examples of D1-like binding are shown in Figure 3. We were unable to acquire good quality photographs of the autoradiographic films from the D2-like receptor binding experiments, because the binding intensities were very low. We performed additional experiments to illustrate D2-like receptor binding in the embryonic neostriatum (see Fig. 7), which we describe in a later section.
Table 1.
Dopamine D1- and D2-like receptor binding in the developing neostriatum
| Age | E13 | E14 | E15 | E17 | P0 |
|---|---|---|---|---|---|
| nCi/mg tissue equivalent (mean ± SEM) | |||||
| D1-like binding | 0.065 ± 0.023 | 1.552 ± 0.103 | 2.09 ± 0.217 | 4.068 ± 0.575 | 5.466 ± 0.569 |
| D2-like binding | 0.017 ± 0.014 | 0.262 ± 0.01 | 0.224 ± 0.037 | 0.893 ± 0.181 | 1.06 ± 0.213 |
| Femtomoles of bound ligand per milligram of tissue | |||||
| D1-like binding | 0.927 | 22.038 | 29.680 | 57.769 | 77.6223 |
| D2-like binding | 0.219 | 3.295 | 2.823 | 11.253 | 13.360 |
The binding intensities were standardized to nanocuries per milligram (nCi/mg) of tissue equivalent across the different replications by calibration against tritiated polymer standards. Then, the nCi/mg of tissue equivalent values were converted to femtomoles of bound ligand per milligram of tissue on the basis of the specific activity of the antagonists used. Both D1- and D2-like binding increases during development.
Fig. 3.
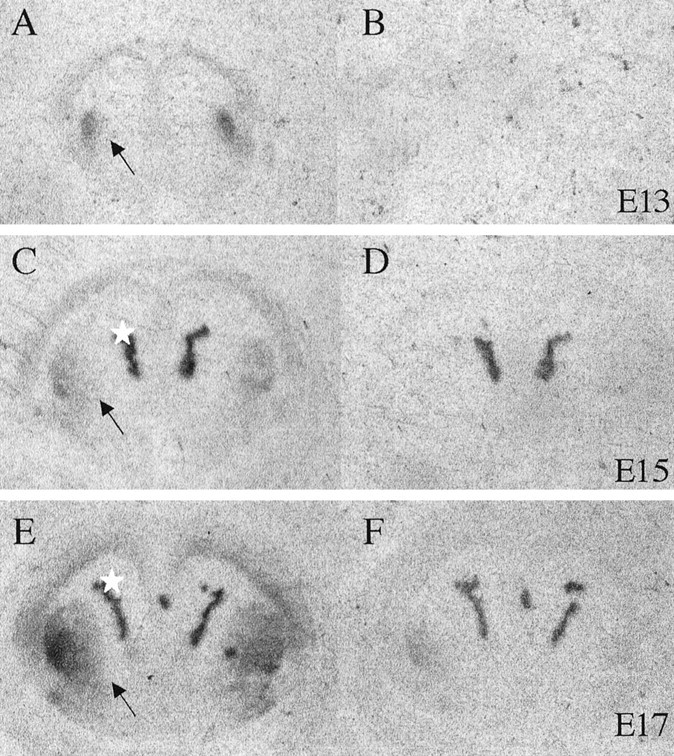
Dopamine D1 receptor binding in the neostriatum of E13 (A, B), E15 (C, D), and E17 (E, F) mice. Sections were incubated with tritiated D1 antagonist Schering 23390 (1 nm;A, C, E) or dopamine (10 μm) plus the antagonist to reveal nonspecific or total binding (B, D, F) and exposed to autoradiographic film. Binding is pronounced in the neostriatum (arrows), and nonspecific binding is seen in the choroid plexus (asterisks). The nonspecific binding was seen also in dopamine-exposed sections (D, F) and was accounted for when calculating the binding intensities shown in Table 1.
Fig. 7.

D2 receptor coupling to G-proteins in sections of E15 (A–C) and E16 (D–F) mouse brains. Shown are autoradiograms of consecutive 12-μm-thick coronal sections through the entire heads of E15 (A–C) and E16 (D–F) mice incubated with [125I]iodosulpiride (D2-Antagonist) alone (A,D), [125I]iodosulpiride plus 2 μm dopamine (B, E), or [125I]iodosulpiride plus 2 μmdopamine plus 10 μm GTPγS (C,F). Outlines of the brain are shown inA and D. The labeling is evident at this coronal plane only in the neostriatum (arrows).
On the basis of the receptor binding data alone, we hypothesized that the effects of dopamine on LGE cell proliferation would be D1-like effects because of the seeming preponderance of D1-like binding sites, provided the effects of activation of each receptor subtype were different. However, we did not know whether D1-like and D2-like receptor activation produced the same or different effects on LGE cell cycle kinetics. We addressed that issue next.
D1-like and D2-like receptor agonists produce opposite effects on BUdR LI
We exposed E13 LGE explants to the D1-like receptor agonist SKF 38393 (1 or 10 μm) or SKF 81297 (1 or 10 μm) for 12 hr in the presence of BUdR and calculated the BUdR LI. Both agonists activate D1 and D5 receptors. SKF 38393 (10 μm) produced ∼22% reduction in the BUdR LI, and SKF 81297 (1 μm) produced ∼16% reduction (Fig.4A,B). In both cases, the decrease was statistically significant (mean ± SEM values: SKF 38393, 0.38 ± 0.03; SKF 81297, 0.41 ± 0.04; control, 0.49 ± 0.02; t test; p = 0.001 for SKF 38393 and p = 0.02 for SKF 81297). SKF 38393 did not produce significant effects at 1 μm concentration. The reduction in the BUdR LI indicates that during the 12 hr labeling period, fewer cells entered S-phase from G1-phase.
Fig. 4.

Two dopamine D1-like receptor agonists SKF 81297 (A) and SKF 38393 (B) reduced the BUdR LI in LGE explants from E13 mice. Two D2-like agonists, quinpirole and PD 128907, produced the opposite effects: they increased the BUdR LI (C, D). The explants were cultured with the agonists or without any drug (Control) for 12 hr with continuous exposure to BUdR. The BUdR LI was calculated for the entire LGE spanning 20 bins, to include the VZ as well as the SVZ. The BUdR LI in Figure 2C is for the VZ only (i.e., for bins 1–7) and is higher than the values here because the SVZ region (included in the analysis here) also contains nonproliferating (postmitotic) cells that do not become BUdR labeled. The incidence of cell death was <0.1% under all conditions. Four to six explants were analyzed in each experiment group. The mean and SEM values are based on the five replications.
We examined the effects of two D2-like receptor agonists, quinpirole hydrochloride and PD128907 hydrochloride. Quinpirole hydrochloride is a D2 receptor agonist with some selectivity to D3 receptors, whereas PD128907 is a selective D3 receptor agonist. Both D2-like agonists produced effects that were opposite of those produced by the D1-like agonists. BUdR LI increased by ∼43% in explants exposed to 10 μm quinpirole hydrochloride and by ∼20% in explants exposed to 10 μm PD128907, compared with control explants (Fig. 4C,D) (mean ± SEM: control, 0.49 ± 0.01; quinpirole, 0.70 ± 0.01; PD128907, 0.59 ± 0.02; t test; p = 0.0001 for quinpirole and p = 0.001 for PD 128907). The two agonists did not produce significant effects at 1 μm concentration. The increase in the BUdR LI suggests that a higher proportion of LGE cells entered S-phase during the 12 hr period.
Next, we examined the specificity of the D1-like and D2-like effects. D1-like receptor antagonist Schering 23390 was used to block D1-like receptors before the addition of SKF 38393. Schering 23390 (10 μm) virtually completely eliminated the effects of SKF 38393 [mean ± SEM: SKF 38393 = 0.38 ± 0.03; SKF 38393 + Schering 23390 = 0.46 ± 0.03; control (no drugs) = 0.45 ± 0.01]. We used SKF 38393 in these experiments because it was effective at higher concentrations (10 μm) compared with SKF 81297. The antagonist blocked the effects of SKF 38393, ruling out the possibility of nonspecific effects even in the case of the agonist that was used at higher concentrations.
The D2-like receptor antagonist eticlopride (10 μm) eliminated the effects of quinpirole hydrochloride (10 μm) (quinpirole HCl = 0.70 ± 0.01; eticlopride + quinpirole HCl, 0.46 ± 0.02; control, 0.45 ± 0.01). When the explants were exposed to Schering 23390 (10 μm) alone or eticlopride (10 μm) alone, the LI was not significantly different from that in the control (no drug) explants (mean ± SEM values: Schering 23390 alone, 0.45 ± 0.02; eticlopride alone, 0.45 ± 0.01). The incidence of TUNEL-positive or pyknotic profiles was under 0.1% in the controls and in all of the experimental conditions.
The LGE consists of two progenitor populations, a pseudostratified ventricular epithelium in the VZ and a secondary proliferative population (SPP) in the SVZ (Bhide, 1996; Sheth and Bhide, 1997). We examined whether the D1-like and D2-like receptor activation influenced the two progenitor populations differently. The VZ and SVZ border was determined to be at bin 7 (Fig.5A), on the basis of criteria defined previously (Bhide, 1996; Sheth and Bhide, 1997), and the BUdR LI was calculated for the VZ (bins 1–7) and SVZ (bins 8–20) separately.
Fig. 5.

Effects of D1-like and D2-like agonists on BUdR LI were examined separately for the VZ and the SVZ in the E13 LGE. The BUdR LI was plotted as a function of distance from the ventricular surface by calculating the LI for each of 20 bins (A). The border between the VZ and the SVZ was set at bin 7 (A). The D1-like agonist SKF 81297 reduced the LI in the VZ (B) but did not produce significant effects in the SVZ (B). The D2-like agonist quinpirole increased the LI in both the VZ (D) and the SVZ (E), with a larger increase in the latter.
SKF 81297 produced a 14% reduction in the BUdR LI in the VZ (Fig.5B) (mean ± SEM values: SKF 81297, 0.73 ± 0.03; control, 0.85 ± 0.01; t test; p = 0.003) but no statistically significant change in the LI in the SVZ (Fig. 5C) (mean ± SEM values: SKF 81297, 0.21 ± 0.06; control, 0.27 ± 0.02; t test; p= 0.29). We used the D1-like agonist SKF 81297 rather than SKF 38393 in these experiments because it was effective at a lower concentration (1 μm) than SKF 38393 (Fig.4A,B). Generally, lower concentrations of agonists are less likely to produce nonspecific effects. The D2-like agonist quinpirole hydrochloride increased the LI in both VZ (by ∼10%) and SVZ (∼63%) (Fig.5D,E). The mean ± SEM values for the VZ are as follows: quinpirole hydrochloride, 0.94 ± 0.01; control, 0.85 ± 0.01 (t test; p = 0.005). The mean ± SEM values for the SVZ are as follows: quinpirole hydrochloride, 0.44 ± 0.06; control, 0.27 ± 0.02 (t test, p = 0.007). Thus, the effects of the D2-like agonist were more pronounced in the SVZ than in the VZ.
Dopamine reduces BUdR LI in explants of the LGE in the same manner as the D1 receptor agonists
We cultured explants of E13 LGE for 10–12 hr in the presence of 1, 10, 50, or 100 μm dopamine, BUdR (10 μm), glutathione (10 μm), or ascorbic acid (0.01%). The latter are antioxidants that retard oxidation of dopamine in the culture medium (Marien et al., 1984; Schmidt et al., 1996;Porter et al., 1999). Control explants were cultured, in parallel, with BUdR and the antioxidant. Dopamine, 50 μm and 100 μm, produced ∼20% reduction in the BUdR LI (mean ± SEM values: 50 μm dopamine, 0.34 ± 0.04; 100 μm dopamine, 0.35 ± 0.03; control, 0.45 ± 0.01; t test; p = 0.03). Dopamine, 1 μm or 10 μm, did not cause statistically significant changes (mean ± SEM values: 1 μm dopamine, 0.45 ± 0.01; 10 μm dopamine, 0.44 ± 0.01). The incidence of TUNEL-positive or pyknotic profiles was not altered by dopamine even at 100 μm concentrations (<0.1% incidence of cell death in control and dopamine-exposed explants). The effects of dopamine on BUdR LI are similar to those produced by the D1-like agonists and opposite those produced by the D2-like agonists.
We found that 10 μm dopamine reduced the BUdR LI by ∼23% in the E14 LGE over the 12 hr period (mean ± SEM values: 10 μm dopamine, 0.42 ± 0.02; control, 0.55 ± 0.03; t test; p = 0.01). Dopamine, 1 μm, did not produce statistically significant differences (mean ± SEM values: 0.52 ± 0.01). Thus, a lower concentration of dopamine was effective in the E14 explants compared with the E13 explants.
Dopamine increases cell output in E13 LGE explants
Cell output, i.e., cells leaving the cell cycle, was increased by ∼45% in cultures exposed to 100 μm dopamine compared with control cultures (mean ± SEM: dopamine, 26.9 ± 3.6; control, 18.5 ± 2.3; t test; p = 0.004). This is consistent with the finding that dopamine reduced G1- to S-phase entry of LGE progenitors.
Effects of dopamine and its receptor activation in the LGEin vivo
We administered dopaminergic drugs to mice to examine the effects on the BUdR LI in the intact brain in vivo. We used a 2 hr BUdR labeling paradigm rather than the more laborious 12 hr cumulative labeling paradigm in these experiments. The 2 hr BUdR exposure labels cells in S-, G2-, and M-phases (Bhide, 1996;Mitsuhashi et al., 2001). Therefore, changes in the 2.0 hr BUdR LI produced by the dopaminergic drugs reflect changes in the number of cells in S-, G2-, and M-phases, and also, in turn, the corresponding changes in the number of cells in G1-phase or cells exiting the cell cycle (the latter will be non-BUdR labeled).
We administered the D1 receptor agonist SKF 81297 by intraperitoneal injections to pregnant mice carrying E13 embryos. Previous studies showed that D1-like agonists crossed the placental and blood–brain barriers and activated D1 receptors in the embryonic brain (Shearman et al., 1997). We observed a dose-dependent effect of SKF 81297 on BUdR LI (Fig.6A). SKF 81297, 20 mg/kg, reduced the BUdR labeling index in the LGE by ∼15% compared with saline-injected controls (mean ± SEM values: control, 0.53 ± 0.01; 20 mg SKF 81297, 0.45 ± 0.03;p = 0.02; t test), whereas 10 mg/kg did not produce significant changes (10 mg SKF 81297: 0.52 ± 0.01;p = 0.07). The reduction in the BUdR LI indicates that fewer cells entered S-phase from G1-phase (Bhide, 1996; Mitsuhashi et al., 2001).
Fig. 6.

The effects of D1-like receptor agonist SKF 81297 (A, B) and l-DOPA (C, D) on BUdR LI in the LGE of E13 mice in vivo and the effects of blocking the D1-like receptors on BUdR LI in the LGE of E15 micein vivo (E, F). BUdR exposure was for 2 hr, and the LI was calculated for the entire LGE sector (A, C, E) as well as for each of 20 bins to analyze interkinetic nuclear migration (B, D, F). SKF 81297 was injected intraperitoneally into pregnant mice in two doses to produce a 6 hr exposure of the E13 embryos to the drug. Control groups received saline injections. BUdR was administered to the mother 2 hr before it was killed. SKF 81297 reduced the BUdR LI in the LGE at 20 mg/kg but had no effect at 10 mg/kg (A). The dopamine precursor, l-DOPA, and the antioxidant ascorbic acid were administered to pregnant mice in drinking water from E10 to E13. Control groups received ascorbic acid only. BUdR was administered to the mothers 2 hr before they were killed. l-DOPA also reduced the BUdR LI in the LGE (C). Thus, as in the explants, D1-agonist and dopamine reduced the BUdR LI. The D1-like receptor antagonist Schering 23390 was injected into the forebrain ventricles of E15 mice in utero. Two hours later, BUdR was administered to the mother carrying the injected embryos. The mice were killed 2 hr after the BUdR administration. Schering 23390 increased the BUdR LI (E). The increase in the LI indicates that augmenting D2-like receptor activation by endogenous dopamine as a result of blocking the endogenous D1-like receptors promoted G1- to S-phase entry. The BUdR LI is lower at E15 (E) than at E13 (A, C) because of the normal developmental decline in cell proliferation. The distribution of BUdR LI in the 20 bins in the drug-administered groups was similar to that in the control groups in all three experiments (Fig.6B,D,F). Therefore, the interkinetic nuclear migration was preserved after the drug administration, indicating that there was no gross perturbation of cell cycle progression. The marked rise in the LI in bin 1 in the E13 plots (B, D, arrows) is not evident in the E15 plots (F). The size of the SVZ increases significantly from E13 to E15. The SVZ progenitors do not migrate to the ventricular border (bin 1) for M-phase. Therefore, there is a lower increase in the BUdR LI in that region on E15 compared with E13.
We administered the dopamine precursor l-DOPA to pregnant mothers in drinking water. Previous studies indicated thatl-DOPA administered to pregnant mothers in drinking water caused a significant increase in the dopamine content of the embryonic brain (Zhou and Palmiter, 1995; Rios et al., 1999). We found that there was an increase of ∼50% in the dopamine content of the basal forebrain of embryos receiving l-DOPA (mean ± SEM values, picogram per milligram of tissue: ascorbic acid alone, 104 ± 24; l-DOPA + ascorbic acid, 157 ± 9.9;t test; p = 0.03).
l-DOPA reduced the BUdR LI in the LGE by ∼11% (Fig.6C) (mean ± SEM values: control, 0.53 ± 0.01;l-DOPA, 0.47 ± 0.02; p = 0.03; t test). As discussed above for the SKF 81297 data, these reductions suggest that l-DOPA reduced the number of cells entering S-phase. The reduction in the BUdR LI after l-DOPA administration was smaller than the reduction after SKF 81297 administration, probably because the higher dopamine levels activated both the D1-like and D2-like receptors.
D2 receptors are coupled to Gi proteins. We examined whether the D2 receptor–G-protein coupling occurred in the embryonic neostriatum using sections of the brain from E15 and E16 mice. We chose E15 and E16 because D2-like binding sites are at a higher density (Table 1) at these ages than at E13 or E14. The experiment consisted of three steps; the data are shown in Figure7. We used an iodinated ligand in these experiments rather than the tritiated ligand used in the receptor binding assays (Table 1), because the iodinated ligand produces a more intense signal in the autoradiograms, although it increases the background and reduces resolution in the photographs. In the first step, we showed that the D2 antagonist radioligand [125I]iodosulpiride binds to D2 receptors in neostriatum (Fig.7A,D). It reconfirmed the existence of D2-like binding sites demonstrated by using the tritiated ligand (shown in Table 1 for E15). In the second step, we showed that 2 μm dopamine binds to the receptors and displaces [125I]iodosulpiride (virtually no label in Fig. 7B,E). In the third step, we added the nonhydrolyzable GTP analog GTPγS to the incubation medium along with [125I]iodosulpiride and dopamine (Fig.7C,F). The labeling in the presence of all three chemicals was comparable with that in the presence of [125I]iodosulpiride alone (Fig. 7, compare A, D with C,F).
In the third step (Fig. 7C,F), the excess GTPγS bound to the α subunit of the G-proteins that were coupled to D2-like receptors (Gi proteins). That binding triggered their dissociation from the receptors (“uncoupling” the receptor from the G-protein) and hence lowered the affinity of the receptors for the agonist dopamine. Therefore, dopamine did not bind to the receptors in the presence of GTPγS. The affinity of the antagonist, [125I]iodosulpiride, does not depend on the coupling state of the receptor: it will bind to coupled as well as uncoupled receptors. Therefore, even in the presence of GTPγS and dopamine, [125I]iodosulpiride bound to D2 receptors (Fig. 7C,F). Although the displacement of [125I]iodosulpiride by a relatively low concentration of the endogenous agonist dopamine in the first experiment (Fig. 7B,E) is an indication that D2 receptors are coupled, it is possible that dopamine displaced [125I]iodosulpiride from uncoupled D2 receptors as well. To rule out that possibility rigorously, the third series of experiments using GTPγS, dopamine, and [125I]iodosulpiride was performed. These data show that the D2-like receptors are G-protein coupled in the embryonic striatum and in that sense “function” in a manner similar to that in the mature brain.
In the final series of in vivo experiments, we injected a D1-like receptor antagonist into the embryonic brain to block D1-like receptor activation and reveal the effects of D2-like receptor activation on BUdR LI by endogenous dopamine. Although our goal was to illustrate the effects of D2-like receptor activation on the BUdR LI, we did not inject a D2-like agonist. We reasoned that activation of D2-like receptors by exogenous D2-like agonist might not be sufficient to override the effects of activation of the abundant D1-like receptors by the endogenous dopamine.
We injected the D1-like receptor antagonist Schering 23390 or PBS (control) into the forebrain ventricles of E15 mice in utero (1 μl of a 100 μm solution in PBS; an estimated maximum final concentration of 5–10 μm, assuming a ventricular volume of 10–20 μl and allowing for diffusion of the drug into the brain). We chose E15 mice rather than E13 mice because intrauterine intraventricular injections were difficult to perform on E13 and our success rate was low. The mothers were administered BUdR 2 hr after the Schering 23390 or PBS injections, and the embryos were removed for histological analyses 2 hr after the BUdR injection. The BUdR LI in the LGE was increased by ∼40% in the embryos receiving Schering 23390 compared with the vehicle-injected embryos (Fig. 6E) (mean ± SEM values: Schering 23390, 0.31 ± 0.02; control, 0.22 ± 0.01; t test; p = 0.004).
The 2 hr BUdR LI on E15 is lower than that on E13 in both the control and experimental groups (Fig. 6, compare E withA, C). The reason for the lower LI is that there is a normal decline in proliferative activity and lengthening of the cell cycle during the interval between E13 and E15 (Caviness et al., 1995; Takahashi et al., 1997).
We analyzed the BUdR LI on the basis of “bins” in all threein vivo experiments. The overall pattern of distribution of BUdR LI in the 20 bins in the drug-administered group was similar to that in the control group in all three experiments (Fig.6B,D,F). Thus, although there were significant differences between the control and experimental groups in the BUdR LI, the interkinetic nuclear migration was preserved after the drug administration, indicating that there was no gross perturbation of cell cycle progression.
The BUdR LI plots are different between E15 (Fig. 6F) and E13 (Fig. 6B,D) mice at the level of bin 1. The marked rise in the LI in this bin in the E13 LGE (Fig. 6B,D, arrows) is not evident in the E15 LGE. A large SVZ develops in the LGE by E15 (P. G. Bhide, unpublished observations). The SVZ progenitors do not undergo interkinetic nuclear migration (Bhide, 1996; Sheth and Bhide, 1997). Therefore, a large number of BUdR-labeled LGE progenitors do not move toward the ventricular surface (bin 1) for M-phase on E15 but undergo M-phase away from the ventricular surface, thereby causing a smaller increase in the BUdR LI in bin 1.
Discussion
Our data show that dopamine arrives in the LGE via the nigrostriatal pathway on E13 and that the dopamine content of the forebrain remains high from E13 onward throughout the period of neostriatal neurogenesis. Dopamine receptor activation modulates the cell cycle of LGE progenitors. Specifically, dopamine D1-like receptor activation reduces G1- to S-phase entry, whereas D2-like receptor activation promotes G1- to S-phase entry. The D1-like effects are prominent in the VZ, and the D2-like effects are prominent in the SVZ. D1-like receptor binding sites appear to predominate over the D2-like binding sites in the embryonic neostriatum. However, the D2-like receptors are coupled to Gi proteins in the embryonic neostriatum. The effects of dopamine on the BUdR LI are D1-like effects, most likely because of the apparent abundance of D1-like binding sites.
The changes produced by dopamine and its receptor agonists on BUdR LI in the present study are generally ∼15–20%, although D2-like agonists produced larger increases (40–60%) in the BUdR LI, and dopamine produced a larger increase (45%) in cell output. The 15–20% changes in BUdR LI might appear relatively modest. However, we point out that our analyses were performed in explants of the LGE where neuroepithelial integrity is maintained and the progenitor cells are not “liberated” from their communal constraints. In our explant system, even basic fibroblast growth factor, one of the most potent mitogenic factors for CNS progenitor cells, produced only 10–20% increases in the BUdR LI (Goto et al., 2002), although it increased proliferation to a much higher degree in dissociated cell preparations (Ghosh and Greenberg, 1995). Therefore, a comparison between the magnitude of the effects on cell proliferation in explants or in vivo and the effects in dissociated cells should take into account the differences between the experimental systems.
We suggest that dopamine receptors exert a tonic influence on cell cycle rather than radically altering the course of neurogenesis. In other words, dopamine receptor activation modulates the normal course of neurogenesis by altering the existing molecular machinery of the cell cycle, without completely overriding the control mechanisms. In this scenario, dopamine receptors act in concert with each other and with other mitogenic and anti-mitogenic signals to influence proliferative tone. The significance of tonic effects should not be underestimated. Loss of proliferative tone, that is, relatively small and short-lived fluctuation in neuroepithelial cell cycle kinetics and cell output, can profoundly alter the total number of cells produced by the neuroepithelium over many cell cycles (Caviness et al., 1995;Takahashi et al., 1997). If the normal pattern of cell output is altered “slightly” and transiently, by only ∼10% over an 8–10 hr period in the course of 2 of 11 rounds of cell divisions, the total number of cells produced can increase by up to 550% (Takahashi et al., 1997).
Previous reports indicated that dopamine influenced cell proliferation in vascular smooth muscle cells (Yasunari et al., 1997), in cultured human meningioma cells (Schrell et al., 1990), in Chinese hamster ovary cells (Lajiness et al., 1993; Scarselli et al., 1999), in olfactory neuronal cell lines (Coronas et al., 1997), and in the pituitary (Saiardi et al., 1997). Dopamine was found to influence differentiation of dissociated striatal neurons (Schmidt et al., 1996). Thus, our findings fit within the larger context of the action of dopamine on the molecular machinery of the cell cycle.
Other neurotransmitters and neuroactive substances also influence neurogenesis. The excitatory neurotransmitter glutamate and the inhibitory neurotransmitter GABA decrease G1- to S-phase entry in the neocortical neuroepithelium (LaMantia, 1995;LoTurco et al., 1995; Gallo et al., 1996; Ma et al., 1998; Owens et al., 1999; Haydar et al., 2000). Prenatal cocaine exposure, which affects dopamine, serotonin, and norepinephrine levels, alters neurogenesis in the primate neocortex (Lidow and Song, 2001, Lidow et al., 2001). Pituitary adenylate cyclase activating polypeptide and vasoactive intestinal polypeptide also have potent effects on cell proliferation in the telencephalic neuroepithelium (Pincus et al., 1994; Lu and DiCicco-Bloom, 1997; DiCicco-Bloom et al., 1998; Carey et al., 2002).
We show that D1-like and D2-like receptor activation produce mutually opposing effects on BUdR LI. The balance between the level of activation of the two receptors ultimately must set the threshold for overall dopaminergic effects on neurogenesis. The diversity in receptor distribution among the different brain regions and in different cell types confers an enormous level of selectivity to the effects of dopamine on neurogenesis. Progenitor subpopulations, for example, progenitors in the VZ versus SVZ, glial versus neuronal progenitors, oligodendrocyte progenitors versus others, may express exclusively or predominantly D1-like or D2-like receptors (Bongarzone et al., 1998). Depending on the size of each subpopulation, a predominantly D1-like or D2-like overall effect may be elicited at a given developmental stage. Moreover, synergistic interactions may exist between the two receptors (Spealman et al., 1992) or interactions between dopaminergic and other neurotransmitter receptors (Cepeda and Levine, 1998) or growth factors (Guillin et al., 2001; Kuppers and Beyer, 2001).
Our finding that D2-like agonists increase the BUdR LI in both the VZ and the SVZ and that the effects are more pronounced in the SVZ suggests the possibility that D2-like receptor stimulation is involved in the development of the SPP in the SVZ. In support of this, D3 receptor mRNA is expressed selectively in the ganglionic neuroepithelium early in embryonic development, earlier than in the dorsal forebrain (Diaz et al., 1997). An SPP emerges in the LGE earlier than in the dorsal forebrain (Bhide, 1996; Sheth and Bhide, 1997). Therefore, D2-like receptor activation may promote the establishment and maintenance of the SPP. These findings may be relevant to understanding a role for dopamine in the regulation of cell proliferation in the SVZ of the mature brain.
The relevance of the role of dopamine in cell cycle regulation is unlikely to be limited to neostriatal histogenesis. Neurons produced in the ganglionic region migrate long distances to establish interneuronal circuitry throughout the forebrain (Anderson et al., 1997; Tamamaki et al., 1997; Lavadas et al., 1999; Marin et al., 2000). Any diminution of GABAergic neuronal deployment from the ganglionic region may cause general (vs focal or localized) deficits or disturbances in GABAergic circuitry such as those associated with epilepsy and schizophrenia (Akbarian et al., 1995; Benes et al., 1996; Flint and Kriegstein, 1997;Woo et al., 1998). Interestingly, some of those illnesses, especially schizophrenia, also show altered dopaminergic function (Benes, 2000;Tallman, 2000). Therefore, understanding dopaminergic influences on neurogenesis is critical not only for understanding mechanisms regulating neostriatal development but also development of inhibitory interneuron circuits throughout the telencephalon.
Our data reveal a novel role for dopamine in brain development, namely a role in cell cycle regulation. Understanding the effects of dopamine on the cell cycle may facilitate interpretation of changes in the cytoarchitecture of the mature brain produced by dopamine imbalance in developing brain. Prenatal cocaine exposure (Lidow, 1995), loss of dopamine receptor (Xu et al., 1994, 1997; Drago et al., 1998), or deficiencies in dopamine biosynthetic machinery (Rios et al., 1999) alter the cytoarchitecture of the brain permanently. Thus, our data underscore the significance of taking into account the effects of dopamine on neurogenesis when interpreting changes in CNS cytoarchitecture caused by a disruption of the dopaminergic system in the developing brain.
Footnotes
This work was supported by United States Public Health Service Grants HD 05515, NS 43426, and NS 62005, a grant from the National Alliance for Autism Research, and funds from the Massachusetts General Hospital. N.O. was supported by a fellowship from the Japanese Society for the Promotion of Science awarded to her and Dr. Koujiro Tohyama at Iwate Medical University, Morioka, Japan. We thank Aparna M. Das for assistance with histology and Drs. Barry E. Kosofsky, James E. Crandall, Verne S. Caviness Jr, Michael A. Schwarzschild, and Takayuki Mitsuhashi for advice and support throughout the course of this research.
Correspondence should be addressed to Dr. Pradeep G. Bhide, Developmental Neurobiology, Massachusetts General Hospital, 149 Thirteenth Street, Charlestown, MA 02129. E-mail:Bhide@helix.mgh.harvard.edu.
T. Goto's present address: Neurology, Tokyo Metropolitan Kiyose Children's Hospital, 1-3-1 Umezono, Kiyose-shi, Tokyo 204-8567, Japan.
References
- 1.Akbarian S, Kim JJ, Potkin SG, Hagman JO, Tafazzoli A, Bunney WE, Jr, Jones EG. Gene expression for glutamic acid decarboxylase is reduced without loss of neurons in prefrontal cortex of schizophrenics. Arch Gen Psychiatry. 1995;52:258–266. doi: 10.1001/archpsyc.1995.03950160008002. [DOI] [PubMed] [Google Scholar]
- 2.Anderson SA, Eisenstat DD, Shi L, Rubenstein JLR. Interneuron migration from basal forebrain to neocortex: dependence on Dlx genes. Science. 1997;278:474–476. doi: 10.1126/science.278.5337.474. [DOI] [PubMed] [Google Scholar]
- 3.Benes FM. Emerging principles of altered neural circuitry in schizophrenia. Brain Res Brain Res Rev. 2000;31:251–269. doi: 10.1016/s0165-0173(99)00041-7. [DOI] [PubMed] [Google Scholar]
- 4.Benes FM, Vincent SL, Marie A, Kahn Y. Up-regulation of GABA receptor binding on neurons of the prefrontal cortex in schizophrenic subjects. Neuroscience. 1996;75:1021–1031. doi: 10.1016/0306-4522(96)00328-4. [DOI] [PubMed] [Google Scholar]
- 5.Bhide PG. Cell cycle kinetics in the embryonic mouse corpus striatum. J Comp Neurol. 1996;374:506–522. doi: 10.1002/(SICI)1096-9861(19961028)374:4<506::AID-CNE3>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 6.Bloom FE. Advancing a neurodevelopmental origin for schizophrenia. Arch Gen Psychiatry. 1993;50:224–227. doi: 10.1001/archpsyc.1993.01820150074008. [DOI] [PubMed] [Google Scholar]
- 7.Bongarzone ER, Howard SG, Schonmann V, Campagnoni AT. Identification of the dopamine D3 receptor in oligodendrocyte precursors: potential role in regulating differentiation and myelin formation. J Neurosci. 1998;18:5344–5353. doi: 10.1523/JNEUROSCI.18-14-05344.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carey RG, Li B, DiCicco-Bloom E. Pituitary adenylate cyclase activating polypeptide anti-mitogenic signaling in cerebral cortical progenitors is regulated by p57Kip2-dependent CDK2 activity. J Neurosci. 2002;22:1583–1591. doi: 10.1523/JNEUROSCI.22-05-01583.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Caviness VS, Jr, Takahashi T, Nowakowski RS. Numbers, time and neocortical neuronogenesis: a general developmental and evolutionary model. Trends Neurosci. 1995;18:379–383. doi: 10.1016/0166-2236(95)93933-o. [DOI] [PubMed] [Google Scholar]
- 10.Cepeda C, Levine MS. Dopamine and N-methyl-d-aspartate receptor interactions in the neostriatum. Dev Neurosci. 1998;20:1–18. doi: 10.1159/000017294. [DOI] [PubMed] [Google Scholar]
- 11.Coronas V, Feron F, Hen R, Sicard G, Jourdan F, Moyse E. In vitro induction of apoptosis or differentiation by dopamine in an immortalized olfactory neuronal cell line. J Neurochem. 1997;69:1870–1881. doi: 10.1046/j.1471-4159.1997.69051870.x. [DOI] [PubMed] [Google Scholar]
- 12.Diaz J, Ridray S, Mignon V, Griffon N, Schwartz JC, Sokoloff P. Selective expression of dopamine D3 receptor mRNA in proliferative zones during embryonic development of the rat brain. J Neurosci. 1997;17:4282–4292. doi: 10.1523/JNEUROSCI.17-11-04282.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.DiCicco-Bloom E, Lu N, Pintar JE, Zhang J. The PACAP ligand/receptor system regulates cerebral cortical neurogenesis. Ann NY Acad Sci. 1998;865:274–289. doi: 10.1111/j.1749-6632.1998.tb11188.x. [DOI] [PubMed] [Google Scholar]
- 14.Drago J, Padungchaichot P, Accili D, Fuchs S. Dopamine receptors and dopamine transporter in brain function and addictive behaviors: insights from targeted mouse mutants. Dev Neurosci. 1998;20:188–203. doi: 10.1159/000017313. [DOI] [PubMed] [Google Scholar]
- 15.Flint AC, Kriegstein AR. Mechanisms underlying neuronal migration disorders and epilepsy. Curr Opin Neurol. 1997;10:92–97. doi: 10.1097/00019052-199704000-00004. [DOI] [PubMed] [Google Scholar]
- 16.Gallo V, Zhou JM, McBain CJ, Wright P, Knutson PL, Armstrong RC. Oligodendrocyte progenitor cell proliferation and lineage progression are regulated by glutamate receptor-mediated K+ channel block. J Neurosci. 1996;16:2659–2670. doi: 10.1523/JNEUROSCI.16-08-02659.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gavrieli Y, Sherman Y, ben-Sasson S. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J Cell Biol. 1992;119:493–501. doi: 10.1083/jcb.119.3.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ghosh A, Greenberg ME. Distinct roles for bFGF and NT-3 in the regulation of cortical neurogenesis. Neuron. 1995;15:89–103. doi: 10.1016/0896-6273(95)90067-5. [DOI] [PubMed] [Google Scholar]
- 19.Goto T, Takahashi T, Miyama S, Nowakowski RS, Bhide PG, Caviness VS., Jr Developmental regulation of the effects of fibroblast growth factor-2 and 1-octanol on neuronogenesis: implications for a hypothesis relating to mitogen-antimitogen opposition. J Neurosci Res. 2002;69:714–722. doi: 10.1002/jnr.10361. [DOI] [PubMed] [Google Scholar]
- 20.Guillin O, Diaz J, Carroll P, Griffon N, Schwartz JC, Sokoloff P. BDNF controls dopamine D3 receptor expression and triggers behavioural sensitization. Nature. 2001;411:86–89. doi: 10.1038/35075076. [DOI] [PubMed] [Google Scholar]
- 21.Haydar TF, Wang F, Schwartz ML, Rakic P. Differential modulation of proliferation in the neocortical ventricular and subventricular zones. J Neurosci. 2000;20:5764–5774. doi: 10.1523/JNEUROSCI.20-15-05764.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hope B, Kosofsky B, Hyman SE, Nestler EJ. Regulation of immediate early gene expression and AP-1 binding in the rat nucleus accumbens by chronic cocaine. Proc Natl Acad Sci USA. 1992;89:5764–5768. doi: 10.1073/pnas.89.13.5764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kosofsky BE, Wilkins AS, Gressens P, Evrard P. Transplacental cocaine exposure: a mouse model demonstrating neuroanatomic and behavioral abnormalities. J Child Neurol. 1994;9:234–241. doi: 10.1177/088307389400900303. [DOI] [PubMed] [Google Scholar]
- 24.Kuppers E, Beyer C. Dopamine regulates brain-derived neurotrophic factor (BDNF) expression in cultured embryonic mouse striatal cells. NeuroReport. 2001;12:1175–1179. doi: 10.1097/00001756-200105080-00025. [DOI] [PubMed] [Google Scholar]
- 25.Lajiness ME, Chio CL, Huff RM. D2 dopamine receptor stimulation of mitogenesis in transfected Chinese hamster ovary cells: relationship to dopamine stimulation of tyrosine phosphorylations. J Pharmacol Exp Ther. 1993;267:1573–1581. [PubMed] [Google Scholar]
- 26.LaMantia AS. The usual suspects: GABA and glutamate may regulate proliferation in the neocortex. Neuron. 1995;15:1223–1225. doi: 10.1016/0896-6273(95)90002-0. [DOI] [PubMed] [Google Scholar]
- 27.Lavadas AA, Grigoriou M, Pachnis V, Parnavelas JG. The medial ganglionic eminence gives rise to a population of early neurons in the developing cerebral cortex. J Neurosci. 1999;99:7881–7888. doi: 10.1523/JNEUROSCI.19-18-07881.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Levitt P, Harvey JA, Friedman E, Simansky K, Murphy EH. New evidence for neurotransmitter influences on brain development. Trends Neurosci. 1997;20:269–274. doi: 10.1016/s0166-2236(96)01028-4. [DOI] [PubMed] [Google Scholar]
- 29.Lidow MS. Prenatal cocaine exposure adversely affects development of the primate cerebral cortex. Synapse. 1995;21:332–341. doi: 10.1002/syn.890210408. [DOI] [PubMed] [Google Scholar]
- 30.Lidow MS, Song ZM. Effect of cocaine on cell proliferation in the cerebral wall of monkey fetuses. Cereb Cortex. 2001;11:545–551. doi: 10.1093/cercor/11.6.545. [DOI] [PubMed] [Google Scholar]
- 31.Lidow MS, Bozian D, Song Z. Cocaine affects cerebral neocortical cytoarchitecture in primates only if administered during neocortical neuronogenesis. Brain Res Dev Brain Res. 2001;128:45–52. doi: 10.1016/s0165-3806(01)00139-0. [DOI] [PubMed] [Google Scholar]
- 32.LoTurco JJ, Owens DF, Heath MJS, Davis MBE, Kriegstein AR. GABA and glutamate depolarize cortical progenitor cells and inhibit DNA synthesis. Neuron. 1995;15:1287–1298. doi: 10.1016/0896-6273(95)90008-x. [DOI] [PubMed] [Google Scholar]
- 33.Lu NR, DiCicco-Bloom E. Pituitary adenylate cyclase-activating polypeptide is an autocrine inhibitor of mitosis in cultured cortical precursor cells. Proc Natl Acad Sci USA. 1997;94:3357–3362. doi: 10.1073/pnas.94.7.3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ma W, Liu QY, Maric D, Sathanoori RS, Chang YH, Barker JL. Basic FGF-responsive telencephalic precursor cells express functional GABAA receptor Cl− channels in vitro. J Neurobiol. 1998;35:277–286. doi: 10.1002/(sici)1097-4695(19980605)35:3<277::aid-neu5>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 35.Marien MR, Shin SH, Cooper GR, Jhamandas K. Stability of dopamine in an in vitro incubation system using Medium 199. Can J Physiol Pharmacol. 1984;62:600–603. doi: 10.1139/y84-096. [DOI] [PubMed] [Google Scholar]
- 36.Marin O, Anderson SA, Rubenstein JL. Origin and molecular specification of striatal interneurons. J Neurosci. 2000;20:6063–6076. doi: 10.1523/JNEUROSCI.20-16-06063.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mitsuhashi T, Aoki Y, Eksioglu YZ, Takahashi T, Bhide PG, Reeves SA, Caviness VS., Jr Overexpression of p27Kip1 lengthens G1-phase in a mouse model that targets inducible gene expression to CNS progenitor cells. Proc Natl Acad Sci USA. 2001;98:6435–6440. doi: 10.1073/pnas.111051398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Moll GH, Hause S, Ruther E, Rothenberger A, Huether G. Early methylphenidate administration to young rats causes a persistent reduction in the density of striatal dopamine transporters. J Child Adolesc Psychopharmacol. 2001;11:15–24. doi: 10.1089/104454601750143366. [DOI] [PubMed] [Google Scholar]
- 39.Nestler EJ, Hope BT, Widnell KL. Drug addiction: a model for the molecular basis of neural plasticity. Neuron. 1993;11:995–1006. doi: 10.1016/0896-6273(93)90213-b. [DOI] [PubMed] [Google Scholar]
- 40.Nowakowski RS, Lewin SB, Miller MW. Bromodeoxyuridine immunohistochemical determination of the lengths of the cell cycle and the DNA-synthetic phases for an anatomically defined population. J Neurocytol. 1989;18:311–318. doi: 10.1007/BF01190834. [DOI] [PubMed] [Google Scholar]
- 41.Owens DF, Liu X, Kriegstein AR. Changing properties of GABA(A) receptor-mediated signaling during early neocortical development. J Neurophysiol. 1999;82:570–583. doi: 10.1152/jn.1999.82.2.570. [DOI] [PubMed] [Google Scholar]
- 42.Pincus DW, DiCicco-Bloom E, Black IB. Trophic mechanisms regulate mitotic neuronal precursors: role of vasoactive intestinal peptide (VIP). Brain Res. 1994;663:51–60. doi: 10.1016/0006-8993(94)90461-8. [DOI] [PubMed] [Google Scholar]
- 43.Porter LL, Rizzo E, Hornung JP. Dopamine affects parvalbumin expression during cortical development in vitro. J Neurosci. 1999;19:8990–9003. doi: 10.1523/JNEUROSCI.19-20-08990.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rios M, Habecker B, Sasaoka T, Eisenhofer G, Tian H, Landis S, Chikaraishi D, Roffler-Tarlov S. Catecholamine synthesis is mediated by tyrosinase in the absence of tyrosine hydroxylase. J Neurosci. 1999;19:3519–3526. doi: 10.1523/JNEUROSCI.19-09-03519.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Saiardi A, Bozzi Y, Baik JH, Borrelli E. Antiproliferative role of dopamine: loss of D2 receptors causes hormonal dysfunction and pituitary hyperplasia. Neuron. 1997;19:115–126. doi: 10.1016/s0896-6273(00)80352-9. [DOI] [PubMed] [Google Scholar]
- 46.Scarselli M, Barbier P, Salvadori F, Armogida M, Collecchi P, Pardini C, Vaglini F, Maggio R, Corsini GU. Apomorphine has a potent antiproliferative effect on Chinese hamster ovary cells. J Neural Trans. 1999;55:47–55. doi: 10.1007/978-3-7091-6369-6_5. [DOI] [PubMed] [Google Scholar]
- 47.Schmidt U, Beyer C, Oestreicher AB, Reisert I, Schilling K, Pilgrim C. Activation of dopaminergic D1 receptors promotes morphogenesis of developing striatal neurons. Neuroscience. 1996;74:453–460. doi: 10.1016/0306-4522(96)00201-1. [DOI] [PubMed] [Google Scholar]
- 48.Schrell UM, Fahlbusch R, Adams EF, Nomikos P, Reif M. Growth of cultured human cerebral meningiomas is inhibited by dopaminergic agents: presence of high affinity dopamine-D1 receptors. J Clin Endocrinol Metab. 1990;71:1669–1671. doi: 10.1210/jcem-71-6-1669. [DOI] [PubMed] [Google Scholar]
- 49.Schwartz JC, Diaz J, Pilon C, Sokoloff P. Possible implications of the dopamine D(3) receptor in schizophrenia and in antipsychotic drug actions. Brain Res Brain Res Rev. 2000;31:277–287. doi: 10.1016/s0165-0173(99)00043-0. [DOI] [PubMed] [Google Scholar]
- 50.Shearman LP, Collins LM, Meyer JS. Characterization and localization of [125I]RTI-55-labeled cocaine binding sites in fetal and adult rat brain. J Pharmacol Exp Ther. 1996;277:1770–1783. [PubMed] [Google Scholar]
- 51.Shearman LP, Zeitzer J, Weaver DR. Widespread expression of functional D1-dopamine receptors in fetal rat brain. Brain Res Dev Brain Res. 1997;102:105–115. doi: 10.1016/s0165-3806(97)00091-6. [DOI] [PubMed] [Google Scholar]
- 52.Sheth AN, Bhide PG. Concurrent cellular output from two proliferative populations in the early embryonic mouse corpus striatum. J Comp Neurol. 1997;383:220–230. [PubMed] [Google Scholar]
- 53.Spealman RD, Bergman J, Madras BK, Kamien JB, Melia KF. Role of D1 and D2 dopamine receptors in the behavioral effects of cocaine. Neurochem Int. 1992;20[Suppl]:147S–152S. doi: 10.1016/0197-0186(92)90228-j. [DOI] [PubMed] [Google Scholar]
- 54.Stanwood GD, Washington RA, Levitt P. Identification of a sensitive period of prenatal cocaine exposure that alters the development of the anterior cingulate cortex. Cereb Cortex. 2001;11:430–440. doi: 10.1093/cercor/11.5.430. [DOI] [PubMed] [Google Scholar]
- 55.Takahashi T, Nowakowski RS, Caviness VS., Jr Mode of cell proliferation in the developing mouse neocortex. Proc Natl Acad Sci USA. 1994;91:375–379. doi: 10.1073/pnas.91.1.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Takahashi T, Nowakowski RS, Caviness VS., Jr The mathematics of neocortical neuronogenesis. Dev Neurosci. 1997;19:17–22. doi: 10.1159/000111179. [DOI] [PubMed] [Google Scholar]
- 57.Takahashi T, Bhide PG, Goto T, Miyama S, Caviness VS., Jr Proliferative behavior of the murine cerebral wall in tissue culture: cell cycle kinetics and checkpoints. Exp Neurol. 1999;156:407–417. doi: 10.1006/exnr.1999.7023. [DOI] [PubMed] [Google Scholar]
- 58.Tallman JF. Development of novel antipsychotic drugs. Brain Res Brain Res Rev. 2000;31:385–390. doi: 10.1016/s0165-0173(99)00055-7. [DOI] [PubMed] [Google Scholar]
- 59.Tamamaki N, Fujimori KE, Takauji R. Origin and route of tangentially migrating neurons in the developing neocortical intermediate zone. J Neurosci. 1997;17:8313–8323. doi: 10.1523/JNEUROSCI.17-21-08313.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Theiler K. The house mouse: development and normal stages from fertilization to 4 weeks of age. Springer; Berlin: 1972. [Google Scholar]
- 61.Verney C, Takahashi T, Bhide PG, Nowakowski RS, Caviness VS., Jr Independent controls for neocortical neuron production and histogenetic cell death. Dev Neurosci. 2000;22:125–138. doi: 10.1159/000017434. [DOI] [PubMed] [Google Scholar]
- 62.Wang X-H, Levitt P, Grayson DR, Murphy EH. Intrauterine cocaine exposure of rabbits: persistent elevation of GABA-immunoreactive neurons in anterior cingulate cortex but not visual cortex. Brain Res. 1995;689:32–46. doi: 10.1016/0006-8993(95)00528-x. [DOI] [PubMed] [Google Scholar]
- 63.Weinberger DR. From neuropathology to neurodevelopment. Lancet. 1995;346:552–557. doi: 10.1016/s0140-6736(95)91386-6. [DOI] [PubMed] [Google Scholar]
- 64.Woo TU, Whitehead RE, Melchitzki DS, Lewis DA. A subclass of prefrontal gamma-aminobutyric acid axon terminals are selectively altered in schizophrenia. Proc Natl Acad Sci USA. 1998;95:5341–5346. doi: 10.1073/pnas.95.9.5341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Xu M, Moratalla R, Gold LH, Hiroi N, Koob GF, Graybiel AM, Tonegawa S. Dopamine D1 receptor mutant mice are deficient in striatal expression of dynorphin and in dopamine-mediated behavioral responses. Cell. 1994;79:729–742. doi: 10.1016/0092-8674(94)90557-6. [DOI] [PubMed] [Google Scholar]
- 66.Xu M, Koeltzow TE, Santiago GT, Moratalla R, Cooper DC, Hu XT, White NM, Graybiel AM, White FJ, Tonegawa S. Dopamine D3 receptor mutant mice exhibit increased behavioral sensitivity to concurrent stimulation of D1 and D2 receptors. Neuron. 1997;19:837–848. doi: 10.1016/s0896-6273(00)80965-4. [DOI] [PubMed] [Google Scholar]
- 67.Yasunari K, Kohno M, Hasuma T, Horio T, Kano H, Yokokawa K, Minami M, Yoshikawa J. Dopamine as a novel antimigration and antiproliferative factor of vascular smooth muscle cells through dopamine D1-like receptors. Arterioscler Thromb Vasc Biol. 1997;17:3164–3173. doi: 10.1161/01.atv.17.11.3164. [DOI] [PubMed] [Google Scholar]
- 68.Zhou QY, Palmiter RD. Dopamine-deficient mice are severely hypoactive, adipsic, and aphagic. Cell. 1995;83:1197–1209. doi: 10.1016/0092-8674(95)90145-0. [DOI] [PubMed] [Google Scholar]