

Summary
The IKKβ and NEMO/IKKγ subunits of the NF-κB activating signalsome complex are known to be essential for activating NF-κB by inflammatory and other stress-like stimuli. However, the IKKα subunit is believed to be dispensable for the latter responses and instead functions as an in vivo mediator of other novel NF-κB dependent and independent functions. In contrast to this generally accepted view of IKKα’s physiological functions, we demonstrate in mouse embryonic fibroblasts (MEFs) that, akin to IKKβ and NEMO/IKKγ, IKKα is also a global regulator of TNFα and IL-1 responsive IKK signalsome-dependent target genes including many known NF-κB targets such as Serum amyloid A3, C3, IL-6, IL-11, IL-1RA, VEGF, Ptx3, β2 microglobulin, IL-1α, Mcp-1 & 3, RANTES, Fas antigen, Jun-B, c-Fos, M/CSF and GM/CSF. Only a small number of NF-κB dependent target genes were preferentially dependent on IKKα or IKKβ. Constitutive expression of a trans-dominant IκBα super repressor (IκBαSR) in wild type MEFs confirmed that these signalsome dependent target genes were also dependent on NF-κB. A subset of NF-κB target genes were IKK dependent in the absence of exogenous stimuli suggesting that the signalsome was also required to regulate basal levels of activated NF-κB in established MEFs. Overall, a sizeable number of novel NF-κB/IKK dependent genes were identified including Secreted Frizzled, Cadherin 13, Protocadherin 7, C/EBPβ&δ, Osteoprotegerin, FOXC2&F2, BMP-2, p75 neurotrophin receptor, Caspase 11, Guanylate binding proteins 1 and 2, ApoJ/Clusterin, Interferon (α & β) receptor 2, Decorin, Osteoglycin, Epiregulin, Proliferins2&3, Stromal Cell derived factor and Cathepsins B, F and Z. SOCS-3, a negative effector of STAT3 signaling was found to be an NF-κB/IKK induced gene, suggesting that IKK mediated NF-κB activation can coordinately illicit negative effects on STAT signaling.
Introduction
The NF-κB transcription factors are pivotal regulators of gene expression programs culminating in stress-like responses and the genesis of innate and acquired immunity {reviewed in (1–4)}. A host of extracellular stimuli including inflammatory cytokines, viral and bacterial infections, oxidative and DNA damaging agents, UV light and osmotic shock can all result in NF-κB activation (1,3–5). NF-κB transcription factors bind to DNA as hetero- or homodimers that are selectively derived from five possible subunits (RelA/p65, c-Rel, RelB, p50 and p52) with each binding to half of a conserved 10 base pair consensus sequence (GGGRNWTYCC) (1,5). While the RelA/p65 and p50 subunits are ubiquitously expressed, the p52, c-Rel and RelB subunits are more functionally important in specific differentiated cell types (1,6). Cytoplasmic p50/p65 heterodimers, c-Rel homodimers and RelB are bound to IκBs (inhibitors of NF-κB) thereby sequestering them in the cytoplasm of most cells that are not experiencing a stress-like response (1,5). Activators of NF-κB mediate the site-specific phosphorylation of two amino terminal serines on each IκB which makes the nearby lysines targets for ubiquitination thereby resulting in IκB proteasomal destruction. NF-κB is then free to translocate to the nucleus and bind DNA leading to the activation of a host of inflammatory response target genes (1,5). Recent evidence has shown that NF-κB subunits dynamically shuttle between the cytoplasm and the nucleus but a dominant acting nuclear export signal in IκBα ensures their transport back to the cytoplasm. It was recently shown that nuclear retention of RelA/p65 is regulated by reversible acetylation with its acetylated form being severely compromised for interaction with IκBα (7).
In contrast to RelA/p65, c-Rel and RelB, the NF-κB p50 and p52 subunits are derived from p105 and p100 precursor proteins by removal of carboxy-terminal IκB-like domains, which possess the inhibitory properties of IκBs (8–10), with the processing of these precursor proteins being initiated by signal induced phosphorylation. Even though NF-κB is largely considered to be a transcriptional activator, under certain circumstances it can also be involved in directly repressing gene expression {reviewed in (1,5)}. In the latter scenario direct repression can result if activation domain deficient homodimers of the NF-κB p50 and p52 subunits bind to NF-κB target sequences instead of activating p50/p65 heterodimers (11–13). The IκB homologue Bcl3, an abundant nuclear IκB-like protein that is not degraded by NF-κB activating pathways, has been reported to have diverse effects on the binding of p50 or p52 homodimers to DNA depending on its state of phosphorylation, concentration and association with nuclear cofactors (14–17). Bcl-3 readily forms ternary complexes with DNA bound p50 and p52 homodimers and in that context function like a transcriptional activator, with its activation potential enhanced by interaction with the Tip60 histone acetylase (15,17–20). Complexes of Bcl3/p50 homodimers were recently shown to contribute to the transcriptional activation of the survival promoting Bcl2 NF-κB target gene (21). Bcl3-p50 complexes form with the same kinetics as p50/p65 heterodimers but are independent of p50/p65 release from IκBα, implicating a p105 proteolysis pathway in their production (10).
IκB phosphorylation is mediated by a high molecular weight signalsome complex consisting of two IκB kinases (IKKα and IKKβ, also known as IKK1/CHUK and IKK2 respectively) and a non-catalytic regulatory subunit (NEMO/NF-κB essential modulator, also known as IKKγ/IKKAP-1/FIP-3) {reviewed in (3,4,22)}. Two molecules of NEMO are believed to orchestrate the assembly of the IKKs into the high molecular weight signalsome complex partially by binding to specific carboxy-terminally conserved residues of both IKKα and IKKβ termed the NEMO binding domain (NBD) (23–26). NEMO may also facilitate the recruitment of IκBα to the IKK complex (27). The two catalytic IKK subunits differentially respond via NEMO to an array of signal induced upstream kinase activities culminating in the coordinated phosphorylation of a pair of serines in their MAPK-like T activation loops by an unknown mechanism.
The roles of the IKKs in NF-κB activation have been investigated in mice lacking IKKβ, IKKα or NEMO (28–33). Akin to mice genetically deficient for the NF-κB p65 subunit (34), murine embryos genetically null for either IKKβ or NEMO succumbed to severe liver apoptosis in utero due to a virtually complete block in NF-κB activation (28–30,35–37). These IKKβ and NEMO KO animals were severely if not completely deficient for both cytokine mediated IκB degradation and nuclear NF-κB DNA binding activity (28–30,35–37). In contrast to the IKKβ and NEMO KO mice, IKKα null animals died prenatally due to severe skin, limb and skeletal abnormalities caused by a block in the terminal differentiation of epidermal keratinocytes (31–33). Furthermore, IKKα null embryos appeared to be phenotypically normal for both cytokine induced IκBα degradation, NF-κB nuclear translocation and NF-κB DNA binding activity (32,33). Subsequent work revealed that IKKα (independent of both its kinase activity and NF-κB) controls the production of a soluble factor that induces keratinocyte differentiation (38). Activation of IKKβ depends upon signal induced phosphorylation of serines 177 and 181 in its T activation loop (39,40). However, unlike IKKβ, a catalytic inactive mutant of IKKα with alanines replacing serines 176 and 180 in its analogous T activation loop (IKKαAA) had no demonstrable affect on the activity of IKK holocomplexes to induce IκBα degradation or NF-κB DNA binding activity in response to inflammatory cytokine stimuli in either transfected cells (40) or in fibroblasts derived from IKKαAA/AA knockin mice (41). It is important to note in this context that several reports of IKKα not affecting NF-κB activation in response to inflammatory stimuli in cells derived from IKKα (−/−) or IKKαAA/AA knockin mice did not address whether nuclear NF-κB was fully transcriptionally competent in these mutant cells (32,33,41). However, there appear to be some inconsistencies with this generally accepted view, as two groups reported some deficiencies in NF-κB transcriptional competence in IKKα (−/−) embryonic fibroblasts even though IκBα degradation and NF-κB DNA binding activity appeared to be normal in these cells (31,42). Nevertheless, this body of work has led to the well accepted tenet that IKKβ alone is essential for NF-κB activation by inflammatory response mediators (22,25,43,44). More recently and in keeping with its separate and distinct functions from IKKβ, IKKα has been shown to possess at least two additional novel in vivo functions: (a) it is essential for B lymphocyte maturation (45) and Peyers patch formation via an LTβR and NIK dependent signaling pathway (46) wherein it is required to target the cytokine induced processing of the NF-κB2 (p100) precursor to produce functional NF-κB p52 subunit (47), and (b) it is required for the proliferation of mammary epithelial cells in response to RANK ligand but not TNFα signaling to activate cyclin D1 (41). Independent of these studies, IKKβ was reported to phosphorylate an IκB-like destruction motif in the p50’ p105 precursor, which produces a recognition site for βTrCP-containing SCF ubiquitin ligases with subsequent polyubiquitination of p105 causing its complete proteasomal destruction and the induced release of DNA binding p50 homodimers (10,48), providing additional support for the notion that IKKβ and IKKα have distinct roles in NF-κB activation.
In this report, we have addressed the roles of each subunit of the NF-κB activating signalsome in the global control of NF-κB target genes. By employing DNA microarrays we show that IKKα is just as critical as IKKβ and NEMO/IKKγ for the global activation of NF-κB dependent, TNFα and IL-1 responsive genes. These experiments also identified a number of novel NF-κB dependent genes including a variety of regulatory factors, growth promoting activities and inflammatory response mediators. A significant fraction of the IKK/NF-κB target genes were dependent on the IKKs for their relative levels of expression in the absence of extracellular NF-κB activating stimuli, suggesting that the signalsome is also involved in positively regulating the basal levels of NF-κB responsive genes in MEFs.
Experimental Procedures
Tissue Culture
Wild type MEFs and mutant IKKα (−/−), IKKβ (−/−) and NEMO/IKKγ (−/−) MEFs (kindly provided by Dr. Michael Karin, UC San Diego) were routinely cultured in growth media (GM) consisting of DMEM, 2 mM glutamine, 10% fetal bovine serum, 100 U/ml penicillin and 100 μg/ml streptomycin. In most experiments the endogenous IKK complex was activated by either human TNFα (10 ng/ml) (Invitrogen) or IL-1β (50 ng/ml) (Pharmingen) signaling for 2 hr with exceptions notes in the text. In some experiments de novo cellular protein synthesis was inhibited by addition of 100 μM anisomycin (SIGMA) to block translational initiation prior to the addition of TNFα followed by co-incubation with TNFα and anisomycin for up to 2 hr. A trans-dominant IκBα (SS32/36AA) super repressor (IκBαSR), with serines 32 and 36 mutated to alanines was introduced into wild type MEFs by retroviral infection as previously described (49). Following infection with a recombinant murine retrovirus harboring an IκBαSR-IRES-Neomycin expression cassette, neomycin resistant MEF populations were obtained after 6–8 days of selection in 800 μg/ml Geneticin (Invitrogen) (49).
Probe preparation, chip hybridization and data analyses
Total cellular RNAs were extracted from cell lysates with an RNeasy kit (Qiagene). Purified RNAs were converted to double-stranded cDNA with a Double Stranded cDNA synthesis kit (Invitrogen) and an oligo-dT primer containing a T7 RNA polymerase promoter (GENSET). Biotin-labeled cRNAs were generated from the cDNA samples by an in vitro transcription with T7 RNA polymerase (Enzo kit, Enzo Diagnostics). The labeled cRNAs were fragmented to an average size of 35 to 200 bases by incubation at 94°C for 35 min. Hybridization (16 hr), washing and staining protocols have been described {Affymetrix Gene ChipR Expression Analysis Technical Manual; (50)}.
We employed Affymetrix MG-U74Av2 chips which includes ~6,000 functionally characterized sequences of the murine UniGene database in addition to ~6,000 EST clusters. Chips were stained with streptavidin-phycoerythrin (Molecular Probes) and scanned with a Hewlett-Packard GeneArray Scanner. DNA microarray chip data analysis was performed using MAS4.0 software (Affymetrix). The quantitation of each gene expressed was calculated from the hybridization intensities of 16 pairs of perfectly matched (PM) and mismatched (MM) control probes (51) (Affymetrix Inc.). The average of the differences (PM minus MM) for each gene-specific probe family was calculated. The software computes a variety of different parameters to determine if an RNA molecule is present or absent (Absolute Call) and whether each transcript’s expression level has changed between the baseline and experimental samples (Difference Call). For a comparative chip file (such as TNFα stimulated Wt. MEF vs. Wt. MEF/IκBαSR), the experimental file {Wt.(S)}(S = stimulated) was compared to the baseline file {IκBαSR(S)}. To minimize false positives, the following criteria were selected for significant changes for each primary screen: (1) the change in the average difference across all probe sets was at least 2 fold; (2) for induced genes, a difference call of “increase” or “marginal increase” should be present, and an absolute call of “presence” should be associated with the experimental file; (3) for repressed genes, a difference call of “decrease” or “marginal decrease” should be present, and an absolute call of “presence” should be associated with the baseline file.
Hierarchical clustering was performed with the Cluster program (available at http://rana.lbl.gov/) as described previously (52) and all genes showing at least 2-fold induction in the primary screen Wt. MEF vs. Wt. MEF/IκBαSR were included. The average difference values (representing the quantity of mRNA, see “above”) of the selected genes were subtracted by the average difference value of the Wt. unstimulated sample, normalized by genes to the magnitude (sum of the squares of the values) of a row vector to 1.0. The normalized data were clustered by average linkage clustering analysis of Y axis (genes) using an uncentered correlation similarity metric, as described in the program Cluster. Average difference values of 50 or less were set to 50 before Wt. unstimulated centering and normalization. The clustered data were visualized by the program TreeView (available at http://rana.lbl.gov/).
RT-PCRs and TaqMan Real-Time Quantitative PCR
RT-PCRs were performed as previously described (49,53). To establish their relative qualities, serial dilutions of cDNAs were amplified with β-actin and Gapdh specific primers for internal standardization. Similarly, linear response ranges were determined for each gene to semi-quantify their expression levels. In all cases, the sizes of PCR products corresponded to those expected for each gene. PCR primer pairs were 22–24 mers and their nucleotide sequences will be provided upon request.
TaqMan Real-time quantitative PCR was based on the fluorogenic 5’ nuclease assay (54). The same total RNA samples that were used to prepare probes for microarray hybridization were treated with DNase I followed by the RNeasy Mini protocol for RNA cleanup (Qiagen). The TaqMan probe consists of an oligonucleotide with a 5’-reporter dye (FAM) and a 3’-quencher dye (TAMRA). To measure the gene copy numbers of the target transcript, cloned plasmid DNA or mouse genomic DNA was serially diluted and used to produce a standard curve as described elsewhere (55). Data from TaqMan PCR analyses were normalized based on mRNA copy numbers of Gapdh using the TaqMan rodent Gapdh control reagents (Applied Biosystems).
Results
We previously reported that IL-1 or LPS signaling via NEMO in a differentiating pre-B cell line induced a host of novel NF-κB dependent target genes but surprisingly also coordinately down-modulated a large group of genes which were also dependent on NF-κB for their repression (49). However, in this latter cell context we were not able to determine the individual roles of the IKKα or IKKβ signalsome subunits for the stimulation or repression of these novel NF-κB/NEMO dependent genes (49). Therefore we sought an alternative biological system whereby we could determine the individual contributions of each IKK subunit for the global cellular response to NF-κB activating pro-inflammatory stimuli. Thus we began a series of DNA microarray analyses with mutant MEFs genetically null for either IKKα, IKKβ or NEMO/IKKγ and examined their genomic responses to TNFα stimulation in comparison to wild type MEFs and MEFs constitutively expressing a trans-dominant IκBα super repressor mutant.
To be certain that only signalsome target genes that were dependent on NF-κB would be evaluated, we introduced a trans-dominant IκBα super repressor (IκBαSR) by retroviral transduction into Wt. MEFs and performed two independent primary DNA microarray screens with and without 2 hr of TNFα stimulation. These primary screens reproducibly identified ~400 NF-κB dependent target genes that were stimulated 2 fold or more and which also exhibited average difference calls of increase in addition to appropriate present or absent absolute calls (see Methods). Up to 150 of the stimulated genes were effected 5 fold or more. Parallel secondary microarray screens comparing Wt. MEFs to mutant MEFs that were null for either IKKα, IKKβ or NEMO/IKKγ were subsequently performed to determine their requirements for each signalsome subunit. As with the IκBαSR screens each of the latter IKK subunit screens were performed two independent times with excellent reproducibility. The screens also identified a sizeable number of novel NF-κB and IKK dependent repressed genes, which will be described in detail in a subsequent report. The expression status of each IKK subunit in the mutant MEF lines was verified by RT-PCR with primer pairs within the targeted exons and also by western blotting. As previously reported (29,32,37) and as expected, the IKKα (−/−), IKKβ (−/−) and NEMO (−/−) MEF lines were only null for the expression of the targeted IKK subunit gene (data not shown).
Most NF-κB dependent genes require both IKKα and IKKβ for their expression
One hundred induced genes were chosen from the two primary Wt. MEF vs. Wt.MEF/IκBαSR screens and assembled into functional categories (see Table I). A significant number of the induced genes were known NF-κB positively regulated genes including: Serum Amyloid A3, IL-6, IL-11, ISG15, IL-1RA, VEGF, Ptx3, β2 microglobulin, IL-1α, Mcp-3, RANTES, Mcp-1, Fas ligand, Jun-B, c-Fos, M/CSF and GM/CSF {reviewed in (56)}. Most of these known NF-κB dependent genes required all signalsome subunits for their activity. The activity of a synthetic NF-κB promoter driven luciferase reporter gene in each IKK null line also showed low to negligible activity in response to TNFα stimulation compared to wild type MEFs (data not shown). A hierarchical clustering image of all genes induced 2 fold or more in both independent microarray screens is presented in Figure 1. Genes labeled Wt. (S) in the first two lanes of Figure 1 were induced in response to TNFα signaling, while their expressions were inhibited in the two screens of MEFs constitutively expressed IκBαSR as noted in the ninth and tenth lanes of the figure. The eleventh and last lane of the hierarchical figure is an unstimulated (US) Wt. MEF control that has been preset as described in Experimental Procedures to allow the reader to better visualize the effects on specific gene clusters. Comparisons of the first and the last lanes of Figure 1 reveal that most genes were dependent on TNFα for the relative levels of expression to varying degrees. However a subset of genes that were dependent on basal levels of NF-κB for their activity but displayed no significant TNFα induced stimulation were also present. Surprisingly, a portion of the latter TNFα independent genes were nevertheless dependent on one or more signalsome subunits for their expression (also see Table I).
Table I.
Signalsome subunit requirements of selected genes in MEFs that are dependent on NF-κB for their activity. One hundred representative genes with fold change values greater than or equal to 2, that were dependent on NF-κB for maintaining their relative expression levels, were selected from a primary Wt (+ TNFα) Vs. IκBαSR (+TNFα) screen. Genes were grouped in categories based upon their physiological functions or properties. Gene accession numbers are in the far left column adjacent their names and descriptions. In the first data column, fold changes of genes identified in two independent primary microarray screens of Wt. MEFs compared to Wt. MEFs constitutively expressing the IκBα(SS32/36AA) super repressor are provided. In data columns 2–4, the dependencies of each of these genes on IKKα, NEMO/IKKγ and IKKβ were determined in six independent microarray screens wherein Wt. MEFs were compared to mutant MEFs null for the individual IKK subunits. Fold change values from duplicate screenings were listed together. Dependence on one or more signalsome subunits was stringently evalutated by adhering to two criteria: wild type MEF absolute calls of “Present” (P) and “Increase” (I) average difference calls. Each screen was performed with cells stimulated for 2 hr with 10 ng/ml of human TNFα. Genes which were dependent on basal NF-κB for their expression were identified by performing independent microarray screens in the absence of TNFα stimulation. Redundancy hits (corresponding to different oligo regions of the same gene) are noted in the gene description column. NC denotes a “No Change” average difference call, indicating no significant dependence on that IKK subunit. Results of exposure to TNFα are presented in the indicated column as follows: - (no significant effect), +/− (~2–5 fold stimulation) and + (>10 fold stimulated). Two independent screens were conducted in all cases with similar results.
| Genes and Descriptions | Wt vs. Iκ BαSR | Wt vs. IKKα(−/−) | Wt vs. NEMO(−/−) | Wt vs. IKKβ(−/−) | TNFα | |
|---|---|---|---|---|---|---|
| Inflammation/Stress and Immune-like responses | ||||||
| X03505 | Serum amyloid A 3 | 450.7 / 471.9 | 321.0 / 269.0 | 452.0 / 269.0 | 207.0 / 489.0 | + |
| X81627 | Lipocalin 2/24p3 (2 hits) | 374.6 / 417.3 | 315 / 199.8 | 374.1 / 200.1 | 254.0 / 408.5 | − |
| X16490 | Plasminogen activator inhibitor type II/PAI-2 | 189.9 / 138.8 | 92.7 / 66.2 | 131.8 / 54.4 | 42.4 / 41.3 | + |
| X54542 | Interleukin 6 | 65.5 / 50.8 | 53.7 / 24.6 | 64.9 / 23.8 | 19.8 / 18.8 | + |
| U49513 | ScyA9/MIP-1γ | 48.4 / 35.7 | 39 / 22 | 12.5 / 7.4 | 10.3 / 12.8 | + |
| K02782 | Complement component 3 | 29.4 / 50.1 | 48.7 / 84.3 | 59.1 / 118.9 | 107.0 / 128.9 | +/− |
| D14077 | Clusterin/ApoJ | 24.3 / 25.9 | 14.2 / 16.7 | 15.8 / 19.3 | 31.0 / 31.2 | + |
| U03421 | Interleukin 11 | 16.2 / 11.8 | 16.1 / 7.7 | 18.4 / 8.2 | 19.8 / 13.0 | + |
| M55544 | Guanylate binding protein 1/mGBP-1/mag-1 | 14.8 / 15.4 | 8.7 / 17.0 | 8.1 / 19.5 | 16.5 / 17.8 | + |
| X70296 | Protease-nexin I/PN-1 | 14.4 / 25.2 | 12.7 / 9.3 | 14.0 / 34.6 | NC / NC | + |
| X56602 | 15 kDa Interferon-stimulated protein/ISG15 | 13.9 / 13.1 | 13.8 / 7.0 | 16.3 / 7.3 | 13.7 / 13.8 | + |
| L32838 | Interleukin 1 receptor antagonist/IL-1RA | 13.1 / 17.4 | 236.4 / 116.8 | 284.6 / 112.5 | 139.9 / 156.6 | − |
| M95200 | Vascular endothelial growth factor/VEGF | 10.7 / 12.3 | 7.8 / 10.5 | 16.3 / 24.9 | 5.7 / 5.7 | + |
| U38261 | Superoxide dismutase 3, extracellular/EC-SOD | 8.7 / 6.0 | 10.3 / 12.0 | 3.8 / 8.0 | 3.4 / 3.5 | − |
| U29678 | CCR1 | 8.4 / 6.4 | 4.1 / 7.6 | 3.9 / 8.5 | 6.6 / 8.2 | + |
| U49430 | Ceruloplasmin/Ferroxidase | 8.1 / 8.6 | 185.5 / 99.8 | 53.2 / 36.0 | 113.6 / 124.6 | +/− |
| U43084 | Interferon-ind. Prot. with tetratricopeptide repeats | 8.0 / 3.8 | 4.9 / 8.6 | 5.9 / 10.3 | 7.3 / 9.9 | + |
| X83601 | Pentaxin related gene/ptx3 | 7.8 / 11.9 | 57.2 / 83.4 | 33.8 / 56.2 | 14.9 / 18.9 | + |
| AB031386 | Clast1/LR8 | 7.2 / 10.7 | 11.9 / 12.2 | 6.4 / 6.8 | 14.2 / 10.2 | +/− |
| M14639 | Interleukin 1 alpha | 6.8 / 7.1 | 6.0 / 4.3 | 6.3 / 4.9 | 7.0 / 7.2 | + |
| AI323667 | Immunoresponsive 2/IRG1 | 6.6 / 4.9 | 2.6 / 3.4 | 3.8 / 5.5 | 4.4 / 9.3 | + |
| AJ007970 | GTP binding protein 2/mGBP2 | 6.4 / 6.2 | 24.3 / 65.2 | 16.1 / 36.3 | 57.3 / 88.4 | + |
| X70058 | ScyA7/MARC/Mcp-3 | 6.2 / 7.2 | NC / NC | 5.3 / 5.6 | 8.9 / 9.1 | + |
| U27267 | LIX/LPS-inducible CXC chemokine | 5.7 / 8.6 | 22.7 / 21.9 | 25.5 / 32.5 | 137.1 / 198.9 | − |
| AI841894 | ERG2 homolog/p27-like | 4.6 / 6.0 | 3.7 / 2.8 | 3.9 / 5.1 | 7.9 / 8.1 | + |
| L24118 | TNF alpha induced protein 2/B94 | 4.4 / 4.3 | 4.0 / 4.2 | 34.3 / 20.2 | 73.4 / 42.2 | + |
| X81584 | Insulin-like growth factor binding protein 6 | 4.2 / 4.3 | 89.8 / 104.2 | 7.0 / 5.6 | 316 / 192.5 | + |
| AF022371 | Interferon activated gene 203 | 3.8 / 2.8 | 2.9 / 3.6 | 2.7 / 2.8 | 3.5 / 4.0 | + |
| M27034 | MHC class I D2d antigen | 3.8 / 4.0 | 10.5 / 10.9 | 4.7 / 16.1 | 16.3 / 14.0 | +/− |
| M18837 | Beta-2-microglobulin (Qb-1) gene | 3.7 / 3.9 | 8.2 / 6.3 | 5.6 / 4.0 | 17.9 / 12.1 | + |
| AF065947 | ScyA5/RANTES (2 hits) | 3.6 / 3.7 | 108.7 / 45.0 | 130.7 / 44.3 | 129.7 / 88.9 | + |
| M69069 | Histocompatibility 2, D region locus 1 | 3.4 / 3.0 | 19.9 / 15.8 | 12.8 / 17.6 | 13.9 / 17.4 | +/− |
| V00746 | MHC Class I, H2-K | 3.1 / 3.0 | 10.2 / 9.0 | 9.1 / 8.9 | 11.7 / 10.2 | +/− |
| X58609 | MHC (Qa) Q2-k Class I antigen | 3.1 / 3.2 | 13.2 / 6.1 | 6.0 / 6.8 | 9.0 / 8.7 | + |
| J04596 | GRO1 oncogene (2 hits) | 2.5 / 2.5 | 1.5 / 1.4 | 17.7 / 16.6 | 3.7 / 3.7 | − |
| M19681 | ScyA2/Mcp-1 | 2.3 / 2.8 | NC / NC | 12.6 / 12.6 | 5 / 6.8 | − |
| AW046124 | Cytochrome b-558 light chain/p22-phox | 2.2 / 2.3 | 2.2 / 2.6 | 3.0 / 3.5 | NC / NC | + |
| U92565 | Small inducible cytokine D1/Fracaltyne | 2.2 / 2.2 | 24.2 / 23.4 | 12.7 / 11.1 | 7.9 / 6.4 | + |
| M89641 | Interferon (alpha and beta) receptor 2 | 2.2 / 2.2 | 8.9 / 3.5 | 3.6 / 4.9 | 1.9 / 2.6 | +/− |
| Growth and Development | ||||||
| -Differentiation/Cell fate | ||||||
| AF099973 | Schlafen 2/Slfn2 | 22.9 / 17.2 | 8.6 / 18.0 | 8.9 / 25.1 | 16.7 / 18.8 | + |
| U88566 | Secreted frizzled-related 1/sFRP-1 | 21.6 / 47 | 66.9 / 35.9 | 70.4 / 30.2 | 12.3 / 12.3 | +/− |
| AB022100 | Cadherin 13/T-Cadherin | 13.6 / 11.9 | 14.3 / 7.6 | 3.0 / 4.5 | 1.7 / 2.1 | + |
| X61800 | CCAAT/enhancer binding protein δ (C/EBPδ) | 8.9 / 9.2 | 7.6 / 6.8 | 9.1 / 7.1 | 3.3 / 2.8 | +/− |
| AB006758 | Procadherin 7/Pcdh7/BH-pcdh | 7.6 / 7.9 | 4.8 / 4.5 | 5.8 / 14.6 | 5.0 / 5.9 | + |
| X85994 | Semaphorin E/Collapsin-like | 6.6 / 5.2 | 4.3 / 4.3 | 3.6 / 7.2 | 5.4 / 7.7 | + |
| AV252118 | Nocturnin | 6.3 / 11.8 | 11.8 / 24.3 | 10.0 / 25.1 | 5.2 / 7.4 | + |
| Y12293 | Forkhead box F2/Winged helix lun gene | 5.0 / 4.5 | 6 / 3.3 | 8.2 / 2.4 | 7.0 / 5.5 | − |
| U94331 | Osteoprotegerin/0PG | 4.9 / 5.9 | 4.8 / 4.9 | 3.1 / 3.2 | NC / NC | + |
| M61007 | CCAAT/enhancer binding protein β (C/EBPβ) | 4.4 / 4.6 | 4.5 / 4.2 | 7.4 / 7.6 | 5.6 / 6.4 | +/− |
| X13945 | Lung carcinoma myc-like oncogene/L-myc | 3.9 / 5.2 | 4.0 / 3.5 | 20.4 / 7.7 | 7.0 / 4.9 | − |
| AA611766 | Kruppel-like factor 5/IKLF | 3.6 / 3.5 | 71.2 / 36 | 81.8 / 32.7 | 9.3 / 8.9 | + |
| AF053943 | AE binding protein/ACLP | 2.5 / 2.8 | 6.9 / 6.2 | 2.7 / 2.5 | 5.8 / 4.4 | +/− |
| X74040 | Forkhead box C2/MFH-1/FoxC2 (2 hits) | 2.1 / 2.2 | 24.6 / 17 | 80.6 / 35.7 | NC / NC | +/− |
| -Growth Arrest/Apopotosis | ||||||
| AF004874 | Bone morphogenetic protein-2/BMP-2 | 46.6 / 18.1 | 17.9 / 41.6 | 15.2 / 27.9 | 13.3 / 44.7 | + |
| M83649 | Fas Antigen (2 hits) | 8.3 / 9.9 | 8.1 / 8.2 | 22.5 / 10.0 | 17.1 / 13.1 | +/− |
| U20735 | Jun-B oncogene (2 hits) | 6.8 / 7.8 | 34.5 / 4.9 | 20.6 / 6.8 | 11.1 / 12.7 | +/− |
| Y13089 | Caspase 11/ICH-3 | 5.5 / 4.5 | 4.9 / 4.1 | 3.1 / 3.8 | NC / NC | − |
| X87128 | p75 TNF receptor | 4.1 / 5.0 | 3.0 / 3.1 | 3.7 / 4.1 | 2.1 / 2.0 | + |
| -Proliferation and survival | ||||||
| D30782 | Epiregulin | 45.4 / 53.4 | 61.6 / 71.5 | 155.7 / 125.6 | 19.3 / 18.8 | + |
| D17444 | Leukemia inhibitory factor receptor/LIF(R) | 15.3 / 10.3 | 14.2 / 6.6 | 14.4 / 5.7 | 14.6 / 10.6 | +/− |
| X03020 | GM/CSF2 | 8.6 / 6.2 | 7.4 / 3.2 | 8.7 / 3.4 | 6.9 / 5.9 | + |
| M17298 | Nerve growth factor β | 8.6 / 9.7 | 27.9 / 67.0 | 17.3 / 38.2 | 7.4 / 5.5 | + |
| X16009 | Mrp1/Proliferin 3/Plf3 | 3.8 / 5.9 | 6.7 / 5.4 | NC / NC | 4.0 / 2.8 | − |
| V00727 | FBJ osteosarcoma oncogene/c-Fos | 3.6 / 5.8 | 6.2 / 3.4 | NC / 1.7 | NC / 2.9 | +/− |
| D16195 | Epithelin/Granulin/GEP | 3.4 / 3.0 | 2.8 / 2.9 | 1.9 / 1.7 | 2.7 / 2.7 | +/− |
| K03235 | Proliferin 2/Plf2 | 3.4 / 5.3 | 6.4 / 8.1 | 1.5 / NC | 2.5 / 4.1 | − |
| M21952 | M/CSF1 | 3.3 / 3.7 | 2.7 / 3.8 | 7.7 / 10.7 | 2.5 / 4.2 | + |
| AF017128 | Fos-related antigen/Fra-1 | 3.2 / 3.2 | 5.9 / 6.4 | 5.5 / 5.6 | 3.8 / 4.2 | +/− |
| D49921 | Glial cell neuroptrophic factor/GDNF | 2.9 / 3.3 | 3.5 / 6.4 | 4.4 / 6.5 | 4.5 / 4.3 | + |
| AV139913 | Stromal cell derived factor 1 (3 hits) | 2.3 / 2.6 | 137.6 / 58.5 | 22.0 / 17.3 | 2.4 / 2.8 | + |
| Y11666 | Hexokinase 2/HXK II | 2.1 / 5.5 | 8.6 / 6.9 | 54.0 / 25.0 | 1.7 / 2.4 | − |
| Signal Transduction and Cell Cycle | ||||||
| U88328 | SH2-protein 3/SOCS-3 (2 hits) | 64.9 / 40.8 | 99.8 / 51.8 | 115.5 / 50.8 | 63.4 / 80.8 | +/− |
| AF026124 | Phospholipase D3 | 36.1 / 7.3 | 3.9 / 3.4 | 40.9 / 5.3 | 3.9 / 5.5 | +/− |
| M55181 | Preproenkephalin 2 | 14.4 / 19.8 | 7.5 / 26.6 | 6.9 / 6.6 | 22.5 / 19.8 | + |
| AI845584 | Mkp-3/Dual specificity protein phosphatase 6 | 11.3 / 11.4 | 11.5 / 12.3 | 11.2 / 14.3 | 5.5 / 6.1 | + |
| U94828 | Regulator of G-protein signaling 16/Rgs16 (2 hits) | 9.3 / 5.7 | NC / NC | 3.8 / 4.2 | 4.1 / 4.5 | + |
| AI060729 | Transmembrane 7 | 5.3 / 5.4 | 4.0 / 4.5 | 2.8 / 2.6 | 5.0 / 4.3 | +/− |
| AI846152 | MC1P1/Calcineurin interacting protein | 3.7 / 4.2 | 3.5 / 3.3 | 1.9 / NC | 2.5 / 2.2 | + |
| AF000236 | G protein coupled orphan receptor/RDC1 | 3.7 / 3.9 | 3.5 / 6.1 | NC / NC | NC / NC | − |
| AW046181 | Serum/glucocorticoid regulated kinase/Sgk | 2.2 / 2.8 | 10.9 / 9.3 | 3.1 / 2.4 | 2.9 / 2.1 | + |
| Adhesion/Extracellular Matrix | ||||||
| X53929 | Decorin | 27.1 / 46.1 | 26.0 / 29.0 | 3.0 / 3.7 | 229.8 / 258.1 | + |
| AA647799 | Osteoglycin (2 hits) | 21.6 / 20.8 | 17.6 / 26.9 | 5.9 / 8.3 | 51.4 / 80.6 | + |
| D00613 | Matrix gamma-carboxyglutamate (gla) protein | 11.0 / 30.6 | 46.5 / 21.7 | 65.7 / 24.3 | 8.6 / NC | + |
| L17324 | Nidogen/Entactin | 5.3 / 7.7 | 6.8 / 5.1 | 2.2 / NC | 8.5 / 6.6 | − |
| Metabolic pathways | ||||||
| AW122933 | Ecto-nucleotidase 2 | 29.0 / 31.9 | 21.4 / 42.2 | 21.4 / 51.5 | 35.8 / 50.3 | − |
| AJ131851 | Cathepsin F | 18.5 / 26.0 | 7.7 / 8.9 | 11.2 / 6.2 | 5.5 / 6.0 | + |
| X98055 | Glutathione S-transferase/GSTT1 | 11.6 / 21.2 | 16.6 / 32.9 | 14.2 / 38.8 | 13.3 / 20.7 | + |
| L09737 | GTP cyclohydrolase 1 | 10.9 / 40.9 | 17.5 / 35.0 | 17.0 / 40.8 | 26.0 / 41.5 | + |
| X60367 | Cellular Retinol binding protein/CRBP1 | 10.9 / 10.7 | 16.8 / 36.9 | NC / NC | 7.0 / 6.6 | − |
| U09816 | GM2a ganglioside activator | 10.4 / 10.2 | 9.3 / 10.7 | 9.8 / 24.0 | 6.0 / 5.7 | + |
| K02236 | Metallothionein 2 | 8.7 / 13.8 | 9.5 / 17.5 | 8.9 / 19.3 | 9.4 / 17.1 | +/− |
| V00835 | Metallothionein 1 | 7.7 / 16.7 | 291.3 / 204.4 | 8.0 / 11.8 | 15.7 / 46.9 | + |
| AI851255 | Cathepsin B | 6.5 / 8.0 | 2.7 / 3.0 | 3.1 / 2.2 | 5.6 / 5.1 | + |
| AF059213 | Cholesterol 25-hydroxlase | 5.7 / 6.4 | 6.0 / 7.6 | 8.3 / 10.7 | 18.9 / 17.1 | + |
| AF045692 | Solute carrier protein/Xpct | 5.4 / 3.6 | 5.5 / 10.0 | 4.1 / 9.0 | 6.7 / 7.7 | − |
| D88994 | AMP deaminase 3 | 4.2 / 5.8 | 3.5 / 5.9 | 4.0 / 6.7 | 3.4 / 2.1 | + |
| U85247 | α-Nacetylglucosaminidase/NAGLU | 2.9 / 3.3 | 3.5 / 6.4 | 4.4 / 6.5 | 4.5 / 4.3 | + |
| U36993 | Cyp7b P450 | 2.7 / 3.2 | 23.0 / 12.0 | 28.1 / 11.9 | 26.6 / 17.4 | − |
| AJ242663 | Cathepsin Z | 2.5 / 3.0 | 2.6 / 2.4 | 2.7 / 2.5 | 3.9 / 3.5 | +/− |
| AI845514 | ABC transporter | 2.1 / 2.3 | 3.3 / 9.3 | 2.3 / 3.0 | 3.1 / 4.4 | + |
Figure 1.
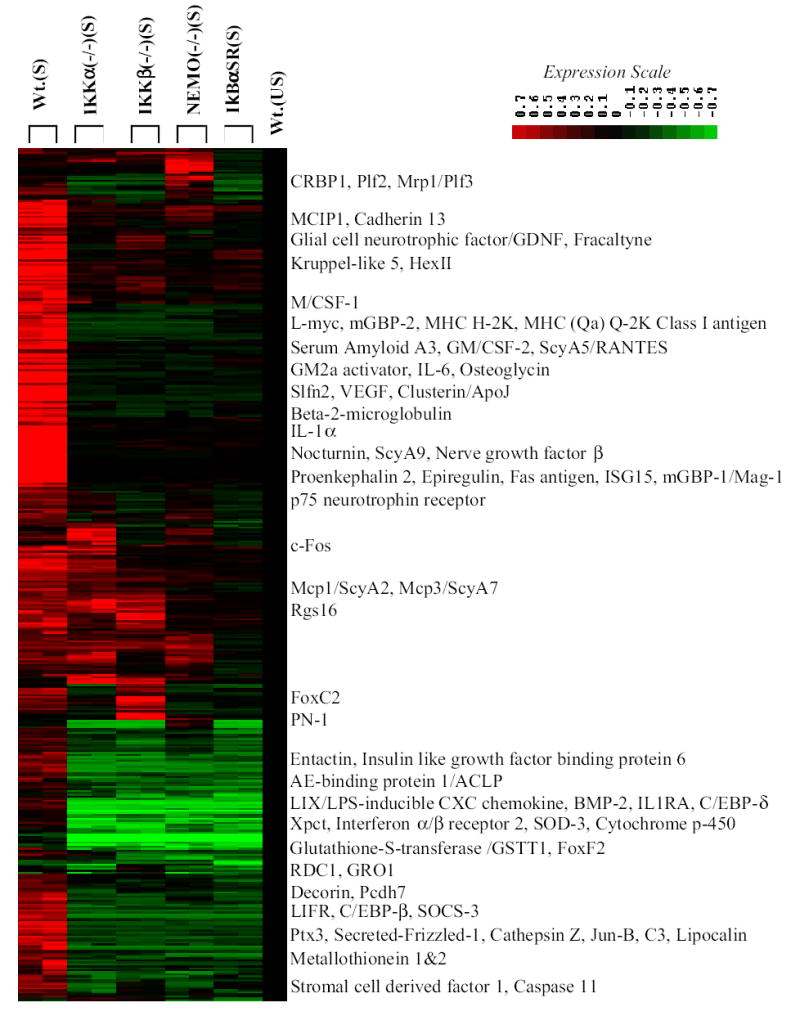
Hierarchical cluster image of gene expression patterns of NF-κB dependent/TNFα stimulated target genes in MEFs in the presence and absence of individual signalsome subunits. Expression profiles of all genes displaying induction of 2 fold or more and average difference calls of increase (as described in Experimental Procedures) in primary microarray screens of 2 hr TNFα stimulated Wt. MEFs (lanes 1&2) vs. Wt. MEFs constitutively expressing an IκBα(SS32/36AA) super repressor (lanes 9&10) were submitted to hierarchial clustering in comparison to TNFα stimulated (S) IKKα(−/−) (lanes 3&4), IKKβ(−/−) (lane 5&6) and NEMO/IKKγ(−/−) (lanes 7&8) and unstimulated (US) Wt. MEFs (lane 11). The locations of selected genes are indicated and their fold change values are presented in Table I.
Of great significance, the vast majority of the NF-κB induced genes were co-dependent on IKKα, IKKβ and NEMO/IKKγ for their expression with exceptional classes of genes that either required IKKα without a significant need for IKKβ and vice versa (see Fig. 1 and Table I). Similar results were obtained with an independent source of wild type MEFs (data not shown). Genes whose expression levels were not significantly altered by the loss of IKKα, IKKβ or NEMO/IKKγ are listed as no change (NC) calls in Table I. A small group of probable IKKα independent/IKKβ dependent genes clustered together in Figure 1 (see Rgs16 and Mcp-1/ScyA2 and Mcp-3/ScyA7). Similarly, a small number of NF-κB targets with the apparent properties of IKKβ independent/IKKα dependent genes also clustered together (exemplified by FoxC2 and PN-1 in Figure 1). A small subset of probable NEMO independent genes were also present (see CRBP1, Plf2, Mrp1/Plf3, RDC1 in Table I) and three of these genes clustered together in Figure 1 as well (see CRBP1, Plf2 and Mrp1/Plf3). This latter class of NEMO independent genes were also part of the group of TNFα independent NF-κB targets. As shown in Table II, three additional microarray screens revealed that 44 out of the 100 selected genes in Table I were also dependent on each IKK subunit for their response to IL-1. Sixteen of the eighteen genes in Table I which were not dependent on TNFα were also not stimulated by IL-1. Mcp-1 and HexII, two IKK dependent genes which were not affected by TNFα, were stimulated by IL-1. In keeping with the TNFα results (see Table I), the induction of Mcp-1 & 3 by IL-1 were more dependent on IKKβ and NEMO/IKKγ than IKKα (see Table II). In addition, Decorin was less dependent on NEMO in the response to both TNFα and IL-1. To further assess the importance of IKKα for the stimulation of NF-κB target genes by TNFα, we also performed TNFα stimulations for 4, 8 and 12 hours. As shown in Table III, 39 of the 82 genes in Table I, which showed evidence of TNFα inducibility, remained dependent on IKKα for their TNFα induction. It is also important to note that Wt. (S) vs. Wt (US) comparisons showed that the 39 genes in Table III represent all of the genes in Table I which remained significantly responsive to TNFα for more than 2 hours. So in actuality none of the genes in Table I selectively lose their IKKα dependence during prolonged exposures to TNFα. Given that the NF-κB pathway is known to attenuate its own activity by inducing the expression IκBs, a fall off in the ability of TNFα to persistently maintain the induced expression levels of a number of IKK/NF-κB target genes is not that surprising (1,5).
Table II.
Signalsome subunit requirements of the selected genes in Table I for IL-1 dependent signaling. Forty-four of the one hundred selected genes in Table I were found to be responsive to IL-1 signaling with similar dependencies on all signalsome subunits. Fold change values from DNA microarray screenings of IL-1 stimulated Wt. MEF cells compared to IL-1 stimulated IKK subunit knock-out MEF mutant cells are listed. Fold change values from the original TNFα chip screenings (Table I) are included for comparison.
| TNFalpha
|
Interleukin 1
|
|||||
|---|---|---|---|---|---|---|
| Genes and Descriptions | Wt vs. I κ BαSR | Wt vs. IKKα(−/−) | Wt vs. IKKα(−/−) | Wt vs. NEMO(−/−) | Wt vs. IKKβ(−/−) | |
| Inflammation/Stress and Immune-like responses | ||||||
| X03505 | Serum amyloid A 3 | 450.7 / 471.9 | 321.0 / 269.0 | 214.8 | 215.8 | 35.3 |
| U49513 | ScyA9/MIP-1γ | 48.4 / 35.7 | 39 / 22 | 57.3 | 294 | 50.3 |
| K02782 | Complement component 3 | 29.4 / 50.1 | 48.7 / 84.3 | 38.1 | 11.6 | 17.3 |
| M55544 | Guanylate binding protein 1/GBP-1 | 14.8 / 15.4 | 8.7 / 17.0 | 15 | NC | 6.9 |
| X56602 | 15 kDa Interferon-stimulated protein/ISG15 | 13.9 / 13.1 | 13.8 / 7.0 | 35.9 | 4.7 | 13.1 |
| U29678 | CCR1 | 8.4 / 6.4 | 4.1 / 7.6 | 6.9 | 8.4 | 1.5 |
| U49430 | Ceruloplasmin/Ferroxidase | 8.1 / 8.6 | 185.5 / 99.8 | 138.8 | 4 | 23.9 |
| U43084 | Interferon-ind. Prot. with tetratricopeptide repeats | 8.0 / 3.8 | 4.9 / 8.6 | 13.8 | 1.7 | 3.9 |
| X83601 | Pentaxin related gene/ptx3 | 7.8 / 11.9 | 57.2 / 83.4 | 12.8 | 9.7 | 3.1 |
| AB031386 | Clast1/LR8 | 7.2 / 10.7 | 11.9 / 12.2 | 3.7 | 2.2 | 1.9 |
| AI323667 | Immunoresponsive 2/IRG1 | 6.6 / 4.9 | 2.6 / 3.4 | 51.5 | 111.9 | 22.7 |
| X70058 | ScyA7/MARC/Mcp-3 | 6.2 / 7.2 | NC / NC | 2 | 5.3 | 16.9 |
| AI841894 | ERG2 homolog/p27-like | 4.6 / 6.0 | 3.7 / 2.8 | 3.4 | 2.5 | 26.7 |
| AF022371 | Interferon activated gene 203 | 3.8 / 2.8 | 2.9 / 3.6 | 13.3 | 3.1 | 3.7 |
| M27034 | MHC class I D2d antigen | 3.8 / 4.0 | 10.5 / 10.9 | 88.9 | 5.4 | NC |
| M18837 | Beta-2-microglobulin (Qb-1) gene | 3.7 / 3.9 | 8.2 / 6.3 | 6.9 | 4.4 | NC |
| AF065947 | ScyA5/RANTES (2 hits) | 3.6 / 3.7 | 108.7 / 45.0 | 150.3 | 148.3 | 27.1 |
| M69069 | Histocompatibility 2, D region locus 1 | 3.4 / 3.0 | 19.9 / 15.8 | 16 | 9.4 | 7.1 |
| V00746 | MHC Class I, H2-K | 3.1 / 3.0 | 10.2 / 9.0 | 8.5 | 4.6 | 3.9 |
| X58609 | MHC (Qa) Q2-k Class I antigen | 3.1 / 3.2 | 13.2 / 6.1 | 9.8 | 4.8 | NC |
| J04596 | GRO1 oncogene (2 hits) | 2.5 / 2.5 | 1.5 / 1.4 | 2.8 | 10.4 | 5.5 |
| M19681 | ScyA2/Mcp-1 | 2.3 / 2.8 | NC / NC | 2 | 8.9 | 59.9 |
| AW046124 | Cytochrome b-558 light chain/p22-phox | 2.2 / 2.3 | 2.2 / 2.6 | 6 | 4.3 | 24 |
| M89641 | Interferon (alpha and beta) receptor 2 | 2.2 / 2.2 | 8.9 / 3.5 | 2.4 | 1.8 | 11.3 |
| Growth and Development | ||||||
| -Differentiation/Cell fate | ||||||
| AF099973 | Schlafen 2/Slfn2 | 22.9 / 17.2 | 8.6 / 18.0 | 56.6 | 56.7 | 9.1 |
| AB006758 | Procadherin 7/Pcdh7/BH-pcdh | 7.6 / 7.9 | 4.8 / 4.5 | 5.1 | 31.4 | 8.3 |
| -Growth Arrest/Apopotosis | ||||||
| AF004874 | Bone morphogenetic protein-2/BMP-2 | 46.6 / 18.1 | 17.9 / 41.6 | 11.2 | 43 | 1.9 |
| M83649 | Fas Antigen (2 hits) | 8.3 / 9.9 | 8.1 / 8.2 | 5.7 | 5 | 3.4 |
| Y13089 | Caspase 11/CASP 4 homolog | 5.5 / 4.5 | 4.9 / 4.1 | 22.7 | 6.8 | 3.5 |
| X87128 | p75 TNF receptor | 4.1 / 5.0 | 3.0 / 3.1 | 4 | 2.6 | NC |
| -Proliferation and survival | ||||||
| V00727 | FBJ osteosarcoma oncogene/c-Fos | 3.6 / 5.8 | 6.2 / 3.4 | 3.5 | 6.8 | 1.6 |
| D16195 | Epithelin/Granulin/GEP | 3.4 / 3.0 | 2.8 / 2.9 | 5.5 | NC | 11.6 |
| AV139913 | Stromal cell derived factor 1 (3 hits) | 2.3 / 2.6 | 137.6 / 58.5 | 25.7 | 2.1 | 9.3 |
| Y11666 | Hexokinase 2/HXK II | 2.1 / 5.5 | 8.6 / 6.9 | 2.5 | 41.5 | 6.9 |
| Signal Transduction and Cell Cycle | ||||||
| AF026124 | Phospholipase D3 | 36.1 / 7.3 | 3.9 / 3.4 | 2.7 | NC | 11.9 |
| AI846152 | MC1P1/Calcineurin interacting protein | 3.7 / 4.2 | 3.5 / 3.3 | 2.1 | 1.6 | 4.3 |
| Adhesion/Extracellular Matrix | ||||||
| X53929 | Decorin | 27.1 / 46.1 | 26.0 / 29.0 | 11.6 | NC | 13.6 |
| Metabolic pathways | ||||||
| AW122933 | Ecto-nucleotidase 2 | 29.0 / 31.9 | 21.4 / 42.2 | 113.7 | 10.8 | 16 |
| AJ131851 | Cathepsin F | 18.5 / 26.0 | 7.7 / 8.9 | 4.3 | NC | 2 |
| L09737 | GTP cyclohydrolase 1 | 10.9 / 40.9 | 17.5 / 35.0 | 30.6 | 15.8 | 4.3 |
| U09816 | GM2a ganglioside activator | 10.4 / 10.2 | 9.3 / 10.7 | 33 | 4 | 15.4 |
| AI851255 | Cathepsin B | 6.5 / 8.0 | 2.7 / 3.0 | 10.1 | 1.8 | 10.4 |
| D88994 | AMP deaminase 3 | 4.2 / 5.8 | 3.5 / 5.9 | 9.6 | 7.7 | 2.7 |
| AJ242663 | Cathepsin Z | 2.5 / 3.0 | 2.6 / 2.4 | 3.8 | 2.6 | 27.9 |
Table III.
NF-κB target genes which retain their dependence on IKKα upon prolonged exposure to TNFα. Fold change values of Wt. MEF cells comapred to IKKα (−/−) MEFs at different time points of TNFα stimulation are listed. Thiry-nine of the eighty-two NF-κB/IKK/TNFα dependent selected genes in Table I remained dependent on IKKα after exposure to TNFα for 4, 8 and 12 hours. As discussed in the text, these 39 genes represented all of the TNFα dependent genes in Table I, which retained their TNFα dependencies after prolonged exposure to the cytokine.
| Genes and Descriptions | 4h TNFα Wt vs. IKKα(−/−) | 8h TNFα Wt vs. IKKα(−/−) | 12h TNFα Wt vs. IKKα(−/−) | |
|---|---|---|---|---|
| Inflammation/Stress and Immune-like responses | ||||
| X83601 | Pentaxin related gene/ptx3 | 29.3 | 21 | 20.6 |
| AJ007970 | GTP binding protein 2/mGBP2 | 17.3 | 13.6 | 17 |
| U49430 | Ceruloplasmin/Ferroxidase | 33.5 | 35.2 | 61.3 |
| AI841894 | ERG2 homolog/p27-like | 8 | 9.8 | 9 |
| L24118 | TNF alpha induced protein 2/B94 | 17.2 | 21 | 4.2 |
| X81584 | Insulin-like growth factor binding protein 6 | 27.8 | 9.6 | 7.4 |
| M18837 | Beta-2-microglobulin (Qb-1) gene | 5.1 | 6 | 5.1 |
| AF065947 | ScyA5/RANTES (2 hits) | 85.8 | 166.4 | 226.4 |
| M69069 | Histocompatibility 2, D region locus 1 | 9.1 | 9.3 | 9.7 |
| V00746 | MHC Class I, H2-K | 5.2 | 5.8 | 9.3 |
| X58609 | MHC (Qa) Q2-k Class I antigen | 10.1 | 5.6 | 5.5 |
| J04596 | GRO1 oncogene (2 hits) | 14.2 | 12.8 | 10 |
| U92565 | Small inducible cytokine D1/Fracaltyne | 72.6 | 38.5 | 8.7 |
| M89641 | Interferon (alpha and beta) receptor 2 | 4.7 | 4.8 | 3.3 |
| Growth and Development | ||||
| -Differentiation/Cell fate | ||||
| AB022100 | Cadherin 13 | 3.1 | 3.7 | 6.4 |
| X61800 | CCAAT/enhancer binding protein δ (C/EBPδ) | 1.5 | 1.7 | 2.1 |
| X85994 | Semaphorin E/Collapsin-like | 2 | 4.1 | 3.7 |
| AV252118 | Nocturnin | 4.4 | 3 | 4.2 |
| U94331 | Osteoprotegerin/0PG | 3 | 2.1 | 2 |
| AF053943 | AE binding protein/ACLP | 4.9 | 4.4 | 3.4 |
| X74040 | Forkhead box C2/MFH-1/FoxC2 (2 hits) | 1.9 | 2 | 2.3 |
| -Growth Arrest/Apopotosis | ||||
| AF004874 | Bone morphogenetic protein-2/BMP-2 | 4.2 | 1.7 | 3.2 |
| M83649 | Fas Antigen (2 hits) | 5.1 | 3.5 | 3.9 |
| U20735 | Jun-B oncogene (2 hits) | 19.4 | 6.8 | 16.2 |
| -Proliferation and survival | ||||
| M17298 | Nerve growth factor β | 6.2 | 6.7 | 3.9 |
| D49921 | Glial cell neuroptrophic factor/GDNF | 5.7 | 11.2 | 11.3 |
| AV139913 | Stromal cell derived factor 1 (3 hits) | 28.1 | 47.2 | 37.4 |
| Signal Transduction and Cell Cycle | ||||
| M55544 | Guanylate binding protein 1/GBP-1 | 4.9 | 3.8 | 4.2 |
| AI060729 | Transmembrane 7 | 2.5 | 3 | 4.7 |
| AW046181 | Serum/glucocorticoid regulated kinase/Sgk | 3.2 | 2.9 | 4.5 |
| Adhesion/Extracellular Matrix | ||||
| X53929 | Decorin | 2.2 | 10.8 | 29.3 |
| AA647799 | Osteoglycin (2 hits) | 3.3 | 3.5 | 3.3 |
| Metabolic pathways | ||||
| X98055 | Glutathione S-transferase/GSTT1 | 3.9 | 5.5 | 8.8 |
| U09816 | GM2a ganglioside activator | 4.1 | 5.6 | 2.7 |
| V00835 | Metallothionein 1 | 4.5 | 5 | 5.7 |
| AF059213 | Cholesterol 25-hydroxlase | 2.5 | 2.8 | 6.3 |
| D88994 | AMP deaminase 3 | 2.2 | 3.5 | 5.9 |
| U85247 | α-Nacetylglucosaminidase/NAGLU | 3 | 4.4 | 6.5 |
| AI845514 | ABC transporter | 2.3 | 2 | 3 |
We chose several examples of genes that were co-dependent on NF-κB and the signalsome for their induced expression for re-examination by semi-quantitative RT-PCR or quantitative TaqMan real time PCR. As shown in Figure 2, TaqMan PCRs were performed for ISG15 and RANTES with and without 2 hours of TNFα or IL-1 stimulation. RANTES and ISG15 were strongly stimulated by either TNFα or IL-1 in wild type MEFs. However, their expression was reduced to negligible levels if not strongly inhibited in the IKKα, IKKβ or NEMO/IKKγ null cells. In keeping with the TaqMan results RT-PCRs conducted with primer pairs specific for IL-6, C3, SOCS-3, IL-1RA and ISG15 show that they are critically dependent on IKKα and IKKβ for their expression (see Figure 3). Performing TNFα stimulations along with anisomycin to block translation initiation revealed that up to 50% of the TNFα dependent IKK and NF-κB dependent genes were likely to be direct targets of the NF-κB/IKK signaling pathway (data not shown).
Figure 2.
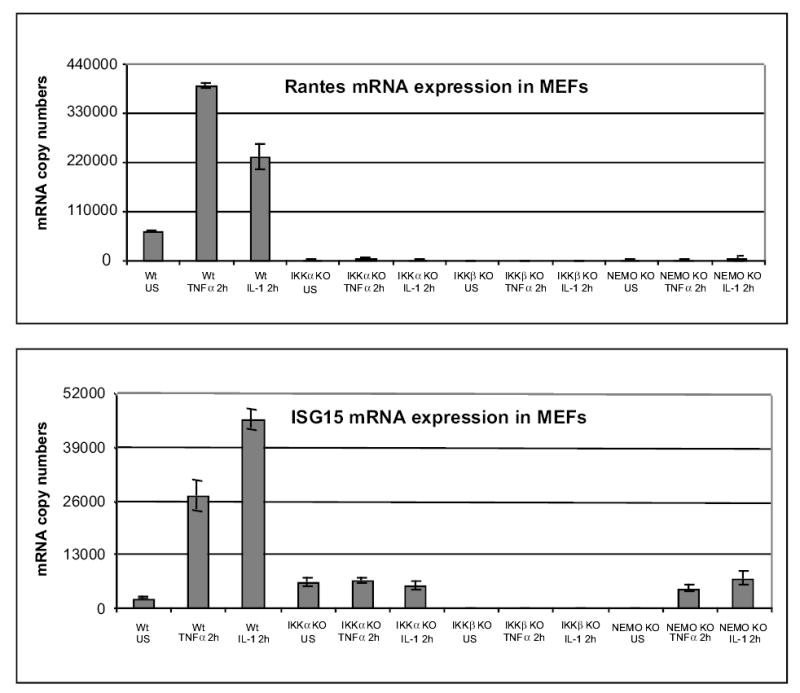
TaqMan real-time PCR validations of selected induced hits from gene chip screenings. Total cellular RNAs were isolated from wild type and mutant MEFs with and without stimulation by TNFα and or IL-1 for 2 hours. RT and PCR was carried out using TaqMan quantitation (showing mRNA copy numbers detected in 40 ng total RNA). The copy numbers of gene transcripts were determined according to DNA standard and normalized with Gapdh (see Experimental Procedures). All TaqMan PCR reactions of each individual sample were performed in triplicate, then the copy numbers and standard error were determined.
Figure 3.
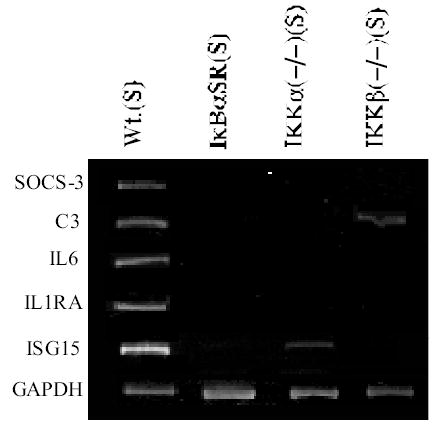
Semi-quantitative RT-PCRs reveal the IKKα and IKKβ requirements of selected MEF genes within 2 hours of TNFα stimulation. All RT-PCRs were performed in the linear response range for each transcript (in comparison to a GAPDH reference control) and products were resolved on 6% PAGE and revealed by ethidium bromide staining. RT-PCRs were performed at least three independent times with similar results as described in Experimental Procedures.
A significant portion of the NF-κB target genes in MEFs were surprisingly dependent on the IKK subunits in the absence of extracellular NF-κB activating stimuli. Examples of the latter class of genes included: PLF2&3, L-Myc, Caspase 11, FOXF2, RDC-1, Lipocalin, IL-1RA, Mcp-1, CRBP1, Entactin and P450 (see Table I and Figure 1). A larger subset of genes exhibited partial dependence on TNFα signaling for their relative levels of expression but nevertheless remained extremely dependent on the IKKs in the absence of a stimulus. These latter results imply that the IKKs are required to maintain basal NF-κB activity to ensure the differential expression of specific subsets of NF-κB target genes. It is important to note in this context that constitutively activated IKKs have been observed in specific types of human lymphoid malignancies (57–59). Thus it will be important to determine in subsequent work if these IKK dependent/signal independent genes are also present in primary MEFs or whether this is a physiological property of immortalized, established cells.
Discussion
IKKα plays general role in the global induction of NF-κB dependent inflammatory response genes
In this study, we specifically sought to address the contributions of each component of the signalsome in NF-κB regulated gene expression by examining their individual effects on a host of specific NF-κB chromosomal target genes in mouse embryo fibroblasts in response to TNFα stimulation. We found that IKKα is equally important as IKKβ and NEMO/IKKγ for the expression of NF-κB dependent, induced genes in these cells. Indeed many known NF-κB target genes such as IL-6, RANTES, Fas antigen, C3, Mcp-3, Ptx3, MIP-1γ, c-Fos, Serum amyloid A3, ISG-15, VEGF, IL-11, IL-1α, GM/CSF2, M/CSF1, Proenkephalin, GRO1, β2 Microglobulin and several other MHC molecules were not stimulated by TNFα nor IL-1 in the absence of IKKα. This is the first study revealing an essential role for IKKα in the global control of NF-κB-dependent gene expression in response to two major inflammatory response cytokines. Indeed, the largest subset of NF-κB dependent genes were those involved in inflammatory, stress or immune-like responses which exhibited a strong co-dependency on IKKα and IKKβ with few exceptions (see Table I and Figure 1). A small number of the NF-κB dependent chromosomal targets also revealed preferential dependencies on IKKα or IKKβ, indicating that their roles in NF-κB activation appear to be target gene dependent in the same cellular background and in response to the same extracellular signal. In addition, the unexpected presence of NF-κB/IKK dependent, signal independent genes suggests that the signalsome may also play a role in maintaining the activities of genes regulated by basal levels of activated NF-κB at least in immortalized, established MEFs. Since IKKα has been reported to be dispensable for the liberation of NF-κB DNA binding activity, it is important to interpret our findings in the context of a number of other reports revealing important IκB independent aspects of NF-κB activation, which lend support to our findings that IKKα (in addition to IKKβ) plays a pleotropic role in NF-κB mediated inflammatory responses.
Even though it is well established that IKKβ is essential for the release of NF-κB from IκB and the subsequent acquisition of NF-κB DNA binding activity (22,28–30,40,43,60), a number of studies have revealed additional levels of control in the cytokine and IKK mediated control of NF-κB activation that are independent of its liberation from IκB. Inhibitors of phosphatidylcholine specific phospholipase C and protein kinase C were initially reported to block the activation of NF-κB by TNFα and IL-1 signaling without effecting IκBα degradation or NF-κB DNA binding activity (61). Subsequently, similar results were reported for the mechanism of phosphatidylinositol-3-OH kinase (PI3K) and PI3K-activated kinase B/Akt dependent NF-κB activation by IL-1 signaling, which was shown to involve the phosphorylation of the RelA/p65 activation domain (62). Several other studies suggesting that NF-κB transcriptional competence was regulated independent of IκB revealed that the catalytic subunit of protein kinase A (PKAc) phosphorylated RelA/p65 thereby facilitating its binding to the transcriptional co-activators CREB binding protein (CBP) and its p300 homolog (63–66). TNFα induced p65/RelA transactivation was blocked by specific inhibitors of the p38 stress and mitogen-activated protein kinases (MAPK) (67) and TNFα mediated p65 phosphorylation was localized to serine 529 within the p65 transcriptional activation domain (TAD) (68). Multiple serines within c-Rel’s carboxy-proximal TAD were subsequently shown to be necessary for TNFα-induced c-Rel activation; and PI3K and ζPKC were also identified as two putative downstream effectors, whose activities were both necessary for c-Rel transactivation activity (69,70). Continuing along the same theme, PI3K- and Akt- dependent signaling pathways were reported to stimulate the p65 TAD via IKKβ and were also functionally and mechanistically correlated with Akt’s anti-apoptotic activity (71). RelA/p65 serine 536 was subsequently shown to be phosphorylated by IKKβ in vitro and in vivo (39,72). The molecular requirements for Akt mediated activation of the p65 TAD were furthered dissected to reveal that: (a) p65 TAD serines 529 and 536 were both required by Akt signaling, which operated at least in part via IKKβ; and (b) Akt and IL-1 signaling also activated p38 in an undefined IKKα dependent pathway, which appeared in part to facilitate p65 engagement with the CBP/p300 co-activator (73). Akt activation in vivo requires PIP3 (phosphatidylinositol 3,4,5-triphosphate), a natural product of PI3K activity; and PIP3 is down-regulated by PTEN, a lipid phosphatase and tumor suppressor (74). PTEN was initially reported to inhibit TNFα induced NF-κB transactivation and DNA binding activity (75,76). However, a more recent study showed that PTEN only inhibited p65 transactivation and not NF-κB DNA binding, which was rescued by over-expression of activated forms of PI3K, Akt or Akt and IKK providing additional support for the controversial role of the PI3K-Akt pathway in uniquely controlling NF-κB transactivation potential (77). Interestingly, more recent efforts have shown that efficient IL-1 and Akt mediated NF-κB transactivation appears to require both IKKα and IKKβ, which are also co-dependent on each other for p65 TAD phosphorylation (42). Taken together, the above findings reveal that important gaps remain in our knowledge regarding the physiological significance and controversial mechanisms of action of the IKK complex for establishing the transcriptional competence of DNA bound NF-κB. Furthermore, our findings, that IKKα plays an unexpectedly general role in the global competence of NF-κB, indicate that IKKα is likely to be a direct or indirect contributor to a number of these phenomenon.
Novel NF-κB dependent genes encoding regulators of gene expression, differentiation and cellular fate
In addition to revealing an unappreciated critical role for IKKα in the expression of NF-κB dependent genes associated with inflammatory/stress like responses, we also identified novel cellular regulator that were subject to NF-κB control. FoxF2 (78) and FoxC2 (79), members to the Forkhead/Winged Helix family of DNA binding transcription factors, were both dependent on NF-κB for their levels of expression. The highly related C/EBPβ and C/EBP/δ leucine zipper class transcription factors, which contribute to the activation of inflammatory responses (80), were coordinately dependent on NF-κB and the IKKs. Two members of the cadherin superfamily of glycoproteins, (non-classical proto-cadherin 7/Pcdh7 and Cadherin 13/T-Cadherin) (81,82), were TNFα dependent targets of the IKKs in MEFs. Cadherins contain multiple extracellular cadherin/EC domains that promote homophilic adhesive interactions which are believed to play critical roles in embryonic morphogenesis and in neuronal circuitry of the brain (83). Osteoprotegerin (84,85) and BMP-2 (86), mediators of bone resorption and formation respectively were both NF-κB targets with BMP-2 being dependent on IKKα and IKKβ, while Osteoprotegerin did not appear to be significantly dependent on IKKβ in response to TNFα signaling. Nocturin, a probably circadian clock effector in mammalian cells (87), was strongly dependent on each subunit of the signalsome for its induction by TNFα induction in MEFs. Secreted frizzled, a Wnt binding secreted protein bearing homology to the ligand binding domain of the frizzled family of transmembrane receptors (88), was highly dependent on each signalosome subunit for its expression which was enhanced by TNFα. Expression of L-myc, a member of the c-Myc family of oncoproteins (89), was surprisingly dependent on the IKKs and NF-κB in the absence of a stimulus.
Positive and negative effectors of cellular proliferation and mortality were amongst the genes dependent on IKK/NF-κB signaling
Genes encoding proteins that directly influence cellular growth required the IKKs and NF-κB for their expression. Epiregulin (an EGF-like autocrine growth factor for kerotinocytes) (90), Granulin/epithelin precursor/GEP (a potent MEF specific growth factor that functions independent of insulin-like growth factor receptor) (91), Stromal cell derived growth factor (a potent lymphocyte chemotactic chemokine activity produced by stromal cells) (92) and Leukemia inhibitory factor receptors (93) were amongst the TNFa responsive IKK dependent genes. However, Proliferin 2/PLF2 and Proliferin 3/PLF3/Mrp1 (94,95), prolactin related hormones with angiogenic properties which also stimulate endothelial cell chemotaxis, belonged to a subset of genes which were dependent on IKKα and IKKβ in the absence of TNFα stimulation in MEFs. Several neurotrophic activities including the p75 neurotrophin receptor, nerve growth factor β and glial cell-derived neurotrophic factor (GDNF) were also dependent on the IKKs and TNFα signaling for their levels of expression and p75 was also responsive to IL-1 signaling.
Several negative effectors of cellular growth, cell cycle progression, cellular viability or inflammatory reactions were also surprisingly dependent on IKKα and IKKβ for their TNF stimulation including Clusterin/ApoJ, BMP-2 and Schlafen 2 and the p75 neurotrophin receptor, while Caspase 11 appeared to be dependent on IKKα and NEMO in the absence of extracellular NF-κB stimuli. BMP-2 has been reported to promote apoptosis in a SMAD independent, protein kinase C dependent pathway by increasing the Bax/Bcl2 ratio and increasing the release of cytochrome C from mitochondria. Enforced expression of Schlafen 2 in transgenic mice has been reported to block double positive thymocyte maturation and to retard fibroblast cell growth in vitro (96). Nerve growth factor has been reported to illicit pro-apoptotic effects on neuroblastoma cells via the p75 neurotrophin receptor, while it can also promote a survival response upon signaling via the homologous TrkA neurotrophin receptor (97). The p75NTR has also been shown to promote apoptosis by binding to beta amyloid peptide, an effect which is enhanced by IL-1 signaling (98). Because IKK mediated NF-κB activation by TNFα has been shown to promote neuronal cell survival (99), subsequent activation of p75 by NF-κB could also be a double edged sword, contributing under some physiological situations to apoptotic responses. Caspase 11/Ich-3 is a member of the ice/ced family of death promoting proteins (100). It is dramatically induced by mediators of septic shock and promotes apoptosis, which can be abrogated by the Bcl-2 survival factor (100). Therefore, under some physiological circumstances IKK mediated NF-κB activation can have unexpected inhibitory effects on cellular growth, cell cycle progression and cellular viability.
Clusterin/ApoJ, a molecular chaperone-like glycoprotein, has been well documented to accumulate at the sites of tissue remodeling and degeneration in various disease states (101,102). ApoJ was also repressed in proliferating cells and its over-expression was recently shown to impede cell cycle progression of transformed cells in vitro (103). Clusterin/ApoJ was also recently reported to act in an anti-inflammatory capacity in vivo by regulating immune complex metabolism and clearance with ApoJ deficient mice exhibiting enhanced kidney aging due to immune complex deposition (104). Therefore, induction of Cluterin/ApoJ by NF-κB could conceivably protect against immune complex mediated inflammatory reactions in vivo.
Important regulators of signal transduction and metabolic pathways requiring NF-κB/IKK signaling for their expression
Components of NF-κB independent signal transduction and metabolic pathways were also amongst the novel target genes downstream of IKK mediated NF-κB activation. SOCS-3, a negative regulator of STAT3 signaling (105), was a TNFα dependent NF-κB/IKK target, revealing a novel type of regulatory cross talk wherein NF-κB has the potential to simultaneously inhibit STAT signaling pathways. SOCS-3 was also recently shown to be an intracellular effector of IL-10 induced anti-inflammatory responses in macrophages, where it was capable of blocking the LPS induced expression of a number of NF-κB target genes including IL-6, TNFα and GM-CSF (106). Consequently, the activation of SOCS-3 by NF-κB/IKK could conceiveable represent a novel mechanism to attentuate NF-κB induced inflammatory responses. In addition to SOC-3, intracellular effectors of other NF-κB independent signaling pathways were also found to be NF-κB/IKK dependent. GBP1/Mag-1 and mGBP2, 65-kDa GTPases which were known to be amongst the genes activated in the cellular response to IFN-γ (107,108), were both found to be strongly dependent on NF-κB and each IKK for its induction by TNFα and mGBP1/Mag-1 was also dependent on IKKα for its stimulation by IL-1. Interestingly, Interferon (alpha and beta) receptor 2 (the murine homolog of the human interferon alpha receptor) was also found to be dependent on IKKα and IKKβ for its TNFα stimulation (109). Rgs16, a negative regulator of G- protein-coupled receptor (GPCR) signaling induced in response to bacterial infection (110,111) that we had previously shown was dependent on NEMO and NF-κB in a differentiating pre-B cell line (49) was dependent on NEMO but not significantly on either IKKα or IKKβ in MEFs. RDC1, an orphan G-protein coupled receptor and a novel HIV/SIV co-receptor (112), was dependent on NF-κB and IKKα for its TNFα independent expression but did not appear to be independent of NEMO nor IKKβ. MCIP1, (myocyte-enriched calcineurin interacting protein) which is located in the Down syndrome critical region and can act as a blocker of calcineurin signaling (113), was dependent on IKKα and IKKβ for its TNF induced expression. The 22-kDa subunit of Cytochrome b-558/p22-Phox, an essential component of the phagocytic NADPH-oxidase responsible for superoxide generation and absent in inherited chronic granulomatours disease (CGD) (114,115), was dependent on IKKα but not IKKβ for its stimulation by TNFα while it was dependent on both catalytic IKKs for its activation by IL-1 signaling. Ceruloplasmin/Ferroxidase, a copper and iron binding oxidoreductase which is upregulated in acute phase inflammatory responses (116,117), was highly dependent on the IKKs for its expression and stimulation by TNFα and IL-1. NAGLU (α-Nacetylglucosaminidase), which is required for heparin sulfate degradation and known to be responsible for the rare autosomal recessive disorder Sanfilippo syndrome (118), required IKKα and IKKβ for its response to TNFα. In addition, the GM2 activator, which plays an essential role in the lysosomal degradation of GM2 gangliosides and is the causal deficiency of neurodegenerative Tay-Sachs and Sandhoff diseases (119), required each IKK for its response to TNFα and IL-1. Cholesterol 25-hydroxylase, which synthesizes 25-hyroxycholesterol (a co-repressor that reduces cholesterol biosynthesis and blocks sterol regulatory element binding protein processing) (120), was amongst the IKK/NF-κB/TNFα dependent genes. Co-ordinate TNFα induction of Decorin and Osteoglycin, two members of the lecuine rich repeat family of proteoglycans (121), was also dependent on each IKK subunit; and Decorin was also responsive to IL-1 signaling. Finally, three Cathepsin cysteine proteinases (Cathepsins B, F and Z) (122–124) were also co-ordinately dependent on IKKα and IKKβ for their stimulation by TNFα and IL-1.
To summarize we have shown that akin to IKKβ and NEMO/IKKγ, IKKα is also a critically important regulator of inflammatory response gene targets. Future work will in part be directed towards identifying the IKKα protein targets that are responsible for its essential role in the regulation of NF-κB dependent, inflammatory response genes. Given these observations, it will also be important to identify the IKKα independent, IKKβ and NEMO/IKKγ specific target genes that are responsible for their unique roles in maintaining cellular survival in vivo. In light of our findings, IKKα could represent a more efficacious target for pharmaceutical intervention than either IKKβ or NEMO/IKKγ to block the unwanted, deleterious side effects of chronic and acute inflammatory reactions without compromising cell viability.
Acknowledgments
The assistance of Ms. Darlene Balzarano with a portion of the RT-PCRs assays is greatly appreciated. We also thank Dr. Michael Karin for providing us with the IKKα, IKKβ and NEMO/IKKγ null MEFs. This research was supported in part by an NIH grant awarded to KBM.
References
- 1.Baldwin A., Jr Annu Rev Immunol. 1996;14:649–683. doi: 10.1146/annurev.immunol.14.1.649. [DOI] [PubMed] [Google Scholar]
- 2.May MM, Ghosh S. ImmunolToday. 1998;19:80–88. [Google Scholar]
- 3.Mercurio F, Manning AM. Oncogene. 1999;18:6163–6171. doi: 10.1038/sj.onc.1203174. [DOI] [PubMed] [Google Scholar]
- 4.Barkett M, Gilmore TD. Oncogene. 1999;18:6910–6924. doi: 10.1038/sj.onc.1203238. [DOI] [PubMed] [Google Scholar]
- 5.Ghosh S, May MJ, Kopp EB. Annu Rev Immunol. 1998;16:225–260. doi: 10.1146/annurev.immunol.16.1.225. [DOI] [PubMed] [Google Scholar]
- 6.Liou HC, Sha WC, Scott ML, Baltimore D. Mol Cell Biol. 1994;14:5349–5359. doi: 10.1128/mcb.14.8.5349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen LF, Fischle W, Verdin E, Greene WC. Science. 2001;293:1653–1657. doi: 10.1126/science.1062374. [DOI] [PubMed] [Google Scholar]
- 8.Liou HC, Nolan GP, Ghosh S, Fujita T, Baltimore D. EMBO J. 1992;11:3003–3009. doi: 10.1002/j.1460-2075.1992.tb05370.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hatada EN, Naumann M, Scheidereit C. Embo J. 1993;12:2781–2788. doi: 10.1002/j.1460-2075.1993.tb05939.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Heissmeyer V, Krappmann D, Wulczyn FG, Scheidereit C. Embo J. 1999;18:4766–4778. doi: 10.1093/emboj/18.17.4766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kang SM, Tran AC, Grilli M, Lenardo MJ. Science. 1992;256:1452–1456. doi: 10.1126/science.1604322. [DOI] [PubMed] [Google Scholar]
- 12.Plaksin D, Baeuerle PA, Eisenbach L. J Exp Med. 1993;177:1651–1662. doi: 10.1084/jem.177.6.1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brown AM, Linhoff MW, Stein B, Wright KL, Baldwin AS, Jr, Basta PV, Ting JP. Mol Cell Biol. 1994;14:2926–2935. doi: 10.1128/mcb.14.5.2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wulczyn FG, Naumann M, Scheidereit C. Nature. 1992;358:597–599. doi: 10.1038/358597a0. [DOI] [PubMed] [Google Scholar]
- 15.Bours V, Franzoso G, Azarenko V, Park S, Kanno T, Brown K, Siebenlist U. Cell. 1993;72:729–739. doi: 10.1016/0092-8674(93)90401-b. [DOI] [PubMed] [Google Scholar]
- 16.Nolan GP, Fujita T, Bhatia K, Huppi C, Liou HC, Scott ML, Baltimore D. Mol Cell Biol. 1993;13:3557–3566. doi: 10.1128/mcb.13.6.3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dechend R, Hirano F, Lehmann K, Heissmeyer V, Ansieau S, Wulczyn FG, Scheidereit C, Leutz A. Oncogene. 1999;18:3316–3323. doi: 10.1038/sj.onc.1202717. [DOI] [PubMed] [Google Scholar]
- 18.Fujita T, Nolan GP, Liou HC, Scott ML, Baltimore D. Genes Dev. 1993;7:1354–1363. doi: 10.1101/gad.7.7b.1354. [DOI] [PubMed] [Google Scholar]
- 19.Pan J, McEver RP. J Biol Chem. 1995;270:23077–23083. doi: 10.1074/jbc.270.39.23077. [DOI] [PubMed] [Google Scholar]
- 20.Hirano F, Tanaka H, Hirano Y, Hiramoto M, Handa H, Makino I, Scheidereit C. Mol Cell Biol. 1998;18:1266–1274. doi: 10.1128/mcb.18.3.1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kurland JF, Kodym R, Story MD, Spurgers KB, McDonnell TJ, Meyn RE. J Biol Chem. 2001;276:45380–45386. doi: 10.1074/jbc.M108294200. [DOI] [PubMed] [Google Scholar]
- 22.Karin M. Oncogene. 1999;18:6867–6874. doi: 10.1038/sj.onc.1203219. [DOI] [PubMed] [Google Scholar]
- 23.Krappmann D, Hatada EN, Tegethoff S, Li J, Klippel A, Giese K, Baeuerle PA, Scheidereit C. J Biol Chem. 2000;275:29779–29787. doi: 10.1074/jbc.M003902200. [DOI] [PubMed] [Google Scholar]
- 24.Li XH, Fang X, Gaynor RB. J Biol Chem. 2001;276:4494–4500. doi: 10.1074/jbc.M008353200. [DOI] [PubMed] [Google Scholar]
- 25.Hatada EN, Krappmann D, Scheidereit C. Current Opinion in Immunology. 2000;12:52–58. doi: 10.1016/s0952-7915(99)00050-3. [DOI] [PubMed] [Google Scholar]
- 26.May MJ, D’Acquisto F, Madge LA, Glockner J, Pober JS, Ghosh S. Science. 2000;289:1550–1554. doi: 10.1126/science.289.5484.1550. [DOI] [PubMed] [Google Scholar]
- 27.Yamamoto Y, Kim DW, Kwak YT, Prajapati S, Verma U, Gaynor RB. J Biol Chem. 2001;276:36327–36336. doi: 10.1074/jbc.M104090200. [DOI] [PubMed] [Google Scholar]
- 28.Li Q, Van Antwerp D, Mercurio F, Lee KF, Verma IM. Science. 1999;284:321–325. doi: 10.1126/science.284.5412.321. [DOI] [PubMed] [Google Scholar]
- 29.Li ZW, Chu W, Hu Y, Delhase M, Deerinck T, Ellisman M, Johnson R, Karin M. J Exp Med. 1999;189:1839–1845. doi: 10.1084/jem.189.11.1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tanaka M, Fuentes ME, Yamaguchi K, Durnin MH, Dalrymple SA, Hardy KL, Goeddel DV. Immunity. 1999;10:421–429. doi: 10.1016/s1074-7613(00)80042-4. [DOI] [PubMed] [Google Scholar]
- 31.Li Q, Lu Q, Hwang JY, Buscher D, Lee KF, zpisua-Belmonte JC, Verma IM. Genes Dev. 1999;13:1322–1328. doi: 10.1101/gad.13.10.1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hu Y, Baud V, Delhase M, Zhang P, Deerinck T, Ellisman M, Johnson R, Karin M. Science. 1999;284:316–320. doi: 10.1126/science.284.5412.316. [DOI] [PubMed] [Google Scholar]
- 33.Takeda K, Takeuchi O, Tsujimura T, tami S, Adachi O, Kawai T, Sanjo H, Yoshikawa K, Terada N, Akira S. Science. 1999;284:313–316. doi: 10.1126/science.284.5412.313. [DOI] [PubMed] [Google Scholar]
- 34.Beg AA, Sha WC, Bronson RT, Ghosh S, Baltimore D. Nature. 1995;376:167–170. doi: 10.1038/376167a0. [DOI] [PubMed] [Google Scholar]
- 35.Rudolph D, Wen-Chen Y, Wakeham A, Rudolph B, Nallainathan D, Potter J, Elia AJ, Mak TW. Genes and Dev. 2000;14:854–862. [PMC free article] [PubMed] [Google Scholar]
- 36.Schmidt-Supprian M, Bloch W, Courtois G, Addicks K, Israel A, Rajewsky K, Pasparakis M. Mol Cell. 2000;5:981–992. doi: 10.1016/s1097-2765(00)80263-4. [DOI] [PubMed] [Google Scholar]
- 37.Makris C, Godfrey VL, Krahn-Senftleben G, Takahashi T, Roberts JL, Schwarz T, Feng L, Johnson RS, Karin M. Mol Cell. 2000;5:969–979. doi: 10.1016/s1097-2765(00)80262-2. [DOI] [PubMed] [Google Scholar]
- 38.Hu Y, Baud V, Oga T, Kim KI, Yoshida K, Karin M. Nature. 2001;410:710–714. doi: 10.1038/35070605. [DOI] [PubMed] [Google Scholar]
- 39.Mercurio F, Zhu H, Murray BW, Shevchenko A, Bennett BL, Li J, Young DB, Barbosa M, Mann M, Manning A, Rao A. Science. 1997;278:860–866. doi: 10.1126/science.278.5339.860. [DOI] [PubMed] [Google Scholar]
- 40.Delhase M, Hayakawa M, Chen Y, Karin M. Science. 1999;284:309–313. doi: 10.1126/science.284.5412.309. [DOI] [PubMed] [Google Scholar]
- 41.Cao Y, Bonizzi G, Seagroves TN, Greten FR, Johnson R, Schmidt EV, Karin M. Cell. 2001;107:763–775. doi: 10.1016/s0092-8674(01)00599-2. [DOI] [PubMed] [Google Scholar]
- 42.Sizemore N, Lerner N, Dombrowski N, Sakurai H, Stark GR. J Biol Chem. 2002;277:3863–3869. doi: 10.1074/jbc.M110572200. [DOI] [PubMed] [Google Scholar]
- 43.Karin M, Ben-Neriah Y. Annu Rev Immunol. 2000;18:621–663. doi: 10.1146/annurev.immunol.18.1.621. [DOI] [PubMed] [Google Scholar]
- 44.Ghosh, S., and Karin, M. (2002) Cell 109 Suppl, S81–96 [DOI] [PubMed]
- 45.Kaisho T, Takeda K, Tsujimura T, Kawai T, Nomura F, Terada N, Akira S. J Exp Med. 2001;193:417–426. doi: 10.1084/jem.193.4.417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Matsushima A, Kaisho T, Rennert PD, Nakano H, Kurosawa K, Uchida D, Takeda K, Akira S, Matsumoto M. J Exp Med. 2001;193:631–636. doi: 10.1084/jem.193.5.631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Senftleben U, Cao Y, Xiao G, Greten FR, Krahn G, Bonizzi G, Chen Y, Hu Y, Fong A, Sun SC, Karin M. Science. 2001;293:1495–1499. doi: 10.1126/science.1062677. [DOI] [PubMed] [Google Scholar]
- 48.Heissmeyer V, Krappmann D, Hatada EN, Scheidereit C. Mol Cell Biol. 2001;21:1024–1035. doi: 10.1128/MCB.21.4.1024-1035.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li J, Peet GW, Balzarano D, Li X, Massa P, Barton RW, Marcu KB. J Biol Chem. 2001;276:18579–18590. doi: 10.1074/jbc.M100846200. [DOI] [PubMed] [Google Scholar]
- 50.Mahadevappa M, Warrington JA. Nat Biotechnol. 1999;17:1134–1136. doi: 10.1038/15124. [DOI] [PubMed] [Google Scholar]
- 51.Lockhart DJ, Dong H, Byrne MC, Follettie MT, Gallo MV, Chee MS, Mittmann M, Wang C, Kobayashi M, Horton H, Brown EL. Nat Biotechnol. 1996;14:1675–1680. doi: 10.1038/nbt1296-1675. [DOI] [PubMed] [Google Scholar]
- 52.Eisen MB, Spellman PT, Brown PO, Botstein D. Proc Natl Acad Sci U S A. 1998;95:14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McKenzie FR, Connelly MA, Balzarano D, Muller JR, Geleziunas R, Marcu KB. Mol Cell Biol. 2000;20:2635–2649. doi: 10.1128/mcb.20.8.2635-2649.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Livak KJ, Flood SJ, Marmaro J, Giusti W, Deetz K. PCR Methods Appl. 1995;4:357–362. doi: 10.1101/gr.4.6.357. [DOI] [PubMed] [Google Scholar]
- 55.Li X, Wang X. Brain Res Brain Res Protoc. 2000;5:211–217. doi: 10.1016/s1385-299x(00)00015-5. [DOI] [PubMed] [Google Scholar]
- 56.Pahl HL. Oncogene. 1999;18:6853–6866. doi: 10.1038/sj.onc.1203239. [DOI] [PubMed] [Google Scholar]
- 57.Kordes U, Krappmann D, Heissmeyer V, Ludwig WD, Scheidereit C. Leukemia. 2000;14:399–402. doi: 10.1038/sj.leu.2401705. [DOI] [PubMed] [Google Scholar]
- 58.Davis RE, Brown KD, Siebenlist U, Staudt LM. J Exp Med. 2001;194:1861–1874. doi: 10.1084/jem.194.12.1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hinz M, Loser P, Mathas S, Krappmann D, Dorken B, Scheidereit C. Blood. 2001;97:2798–2807. doi: 10.1182/blood.v97.9.2798. [DOI] [PubMed] [Google Scholar]
- 60.Baud V, Liu ZG, Bennett B, Suzuki N, Xia Y, Karin M. Genes Dev. 1999;13:1297–1308. doi: 10.1101/gad.13.10.1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bergmann M, Hart L, Lindsay M, Barnes PJ, Newton R. J Biol Chem. 1998;273:6607–6610. doi: 10.1074/jbc.273.12.6607. [DOI] [PubMed] [Google Scholar]
- 62.Sizemore N, Leung S, Stark GR. Mol Cell Biol. 1999;19:4798–4805. doi: 10.1128/mcb.19.7.4798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gerristen ME, Williams AJ, Neish AS, Moore S, Shi Y, Collins T. Proc Natl Acad Sci U S A. 1997;94:2927–2932. doi: 10.1073/pnas.94.7.2927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Perkins ND, Felzien LK, Betts JC, Leung K, Beach DH, Nabel GJ. Science. 1997;275:523–527. doi: 10.1126/science.275.5299.523. [DOI] [PubMed] [Google Scholar]
- 65.Zhong H, Suyang H, Erdjument-Bromage P, Tempst P, Ghosh S. Cell. 1997;89:413–424. doi: 10.1016/s0092-8674(00)80222-6. [DOI] [PubMed] [Google Scholar]
- 66.Zhong H, Voll RE, Ghosh S. Mol Cell. 1998;1:661–671. doi: 10.1016/s1097-2765(00)80066-0. [DOI] [PubMed] [Google Scholar]
- 67.Vanden Berghe W, Plaisance S, Boone E, De Bosscher K, Schmitz ML, Fiers W, Haegeman G. J Biol Chem. 1998;273:3285–3290. doi: 10.1074/jbc.273.6.3285. [DOI] [PubMed] [Google Scholar]
- 68.Wang D, Baldwin AS., Jr J Biol Chem. 1998;273:29411–29416. doi: 10.1074/jbc.273.45.29411. [DOI] [PubMed] [Google Scholar]
- 69.Martin AG, Fresno M. J Biol Chem. 2000;275:24383–24391. doi: 10.1074/jbc.M909396199. [DOI] [PubMed] [Google Scholar]
- 70.Martin AG, San-Antonio B, Fresno M. J Biol Chem. 2001;276:15840–15849. doi: 10.1074/jbc.M011313200. [DOI] [PubMed] [Google Scholar]
- 71.Madrid LV, Wang CY, Guttridge DC, Schottelius AJ, Baldwin AS, Jr, Mayo MW. Mol Cell Biol. 2000;20:1626–1638. doi: 10.1128/mcb.20.5.1626-1638.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sakurai H, Chiba H, Miyoshi H, Sugita T, Toriumi W. J Biol Chem. 1999;274:30353–30356. doi: 10.1074/jbc.274.43.30353. [DOI] [PubMed] [Google Scholar]
- 73.Madrid LV, Mayo MW, Reuther JY, Baldwin AS., Jr J Biol Chem. 2001;276:18934–18940. doi: 10.1074/jbc.M101103200. [DOI] [PubMed] [Google Scholar]
- 74.Cantley LC, Neel BG. Proc Natl Acad Sci U S A. 1991;96:4240–4245. doi: 10.1073/pnas.96.8.4240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Koul D, Yao Y, Abbruzzese JL, Yung WK, Reddy SA. J Biol Chem. 2001;276:11402–11408. doi: 10.1074/jbc.M007806200. [DOI] [PubMed] [Google Scholar]
- 76.Gustin JA, Maehama T, Dixon JE, Donner DB. J Biol Chem. 2001;276:27740–27744. doi: 10.1074/jbc.M102559200. [DOI] [PubMed] [Google Scholar]
- 77.Mayo MW, Madrid LV, Westerheide SD, Jones DR, Yuan XJ, Baldwin AS, Jr, Whang YE. J Biol Chem. 2002;277:11116–11125. doi: 10.1074/jbc.M108670200. [DOI] [PubMed] [Google Scholar]
- 78.Miura N, Kakinuma H, Sato M, Aiba N, Terada K, Sugiyama T. Genomics. 1998;50:346–356. doi: 10.1006/geno.1998.5288. [DOI] [PubMed] [Google Scholar]
- 79.Fang J, Dagenais SL, Erickson RP, Arlt MF, Glynn MW, Gorski JL, Seaver LH, Glover TW. Am J Hum Genet. 2000;67:1382–1388. doi: 10.1086/316915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Caivano, M., Gorgoni, B., Cohen, P., and Poli, V. (2001) J Biol Chem [DOI] [PubMed]
- 81.Yoshida K, Yoshitomo-Nakagawa K, Seki N, Sasaki M, Sugano S. Genomics. 1998;49:458–461. doi: 10.1006/geno.1998.5271. [DOI] [PubMed] [Google Scholar]
- 82.Takeuchi T, Misaki A, Liang SB, Tachibana A, Hayashi N, Sonobe H, Ohtsuki Y. J Neurochem. 2000;74:1489–1497. doi: 10.1046/j.1471-4159.2000.0741489.x. [DOI] [PubMed] [Google Scholar]
- 83.Yagi T, Takeichi M. Genes Dev. 2000;14:1169–1180. [PubMed] [Google Scholar]
- 84.Simonet WS, Lacey DL, Dunstan CR, Kelley M, Chang MS, Luthy R, Nguyen HQ, Wooden S, Bennett L, Boone T, Shimamoto G, DeRose M, Elliott R, Colombero A, Tan HL, Trail G, Sullivan J, Davy E, Bucay N, Renshaw-Gegg L, Hughes TM, Hill D, Pattison W, Campbell P, Boyle WJ, et al. Cell. 1997;89:309–319. doi: 10.1016/s0092-8674(00)80209-3. [DOI] [PubMed] [Google Scholar]
- 85.Lomaga MA, Yeh WC, Sarosi I, Duncan GS, Furlonger C, Ho A, Morony S, Capparelli C, Van G, Kaufman S, van der Heiden A, tie A, Wakeham A, Khoo W, Sasaki T, Cao Z, Penninger JM, Paige CJ, Lacey DL, Dunstan CR, Boyle WJ, Goeddel DV, Mak TW. Genes Dev. 1999;13:1015–1024. doi: 10.1101/gad.13.8.1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hay E, Lemonnier J, Fromigue O, Marie PJ. J Biol Chem. 2001;276:29028–29036. doi: 10.1074/jbc.M011265200. [DOI] [PubMed] [Google Scholar]
- 87.Wang Y, Osterbur DL, Megaw PL, Tosini G, Fukuhara C, Green CB, Besharse JC. BMC Dev Biol. 2001;1:9. doi: 10.1186/1471-213X-1-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Rattner A, Hsieh JC, Smallwood PM, Gilbert DJ, Copeland NG, Jenkins NA, Nathans J. Proc Natl Acad Sci U S A. 1997;94:2859–2863. doi: 10.1073/pnas.94.7.2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Legouy E, DePinho R, Zimmerman K, Collum R, Yancopoulos G, Mitsock L, Kriz R, Alt FW. Embo J. 1987;6:3359–3366. doi: 10.1002/j.1460-2075.1987.tb02657.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Shirakata Y, Komurasaki T, Toyoda H, Hanakawa Y, Yamasaki K, Tokumaru S, Sayama K, Hashimoto K. J Biol Chem. 2000;275:5748–5753. doi: 10.1074/jbc.275.8.5748. [DOI] [PubMed] [Google Scholar]
- 91.Zanocco-Marani T, Bateman A, Romano G, Valentinis B, He ZH, Baserga R. Cancer Res. 1999;59:5331–5340. [PubMed] [Google Scholar]
- 92.Bleul CC, Fuhlbrigge RC, Casasnovas JM, Aiuti A, Springer TA. J Exp Med. 1996;184:1101–1109. doi: 10.1084/jem.184.3.1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Tanaka M, Hara T, Copeland NG, Gilbert DJ, Jenkins NA, Miyajima A. Blood. 1999;93:804–815. [PubMed] [Google Scholar]
- 94.Groskopf JC, Syu LJ, Saltiel AR, Linzer DI. Endocrinology. 1997;138:2835–2840. doi: 10.1210/endo.138.7.5276. [DOI] [PubMed] [Google Scholar]
- 95.Toft DJ, Rosenberg SB, Bergers G, Volpert O, Linzer DI. Proc Natl Acad Sci U S A. 2001;98:13055–13059. doi: 10.1073/pnas.231364798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Schwarz DA, Katayama CD, Hedrick SM. Immunity. 1998;9:657–668. doi: 10.1016/s1074-7613(00)80663-9. [DOI] [PubMed] [Google Scholar]
- 97.Bono F, Lamarche I, Bornia J, Savi P, Della Valle G, Herbert JM. FEBS Lett. 1999;457:93–97. doi: 10.1016/s0014-5793(99)01006-6. [DOI] [PubMed] [Google Scholar]
- 98.Perini G, Della-Bianca V, Politi V, Della Valle G, Dal-Pra I, Rossi F, Armato U. J Exp Med. 2002;195:907–918. doi: 10.1084/jem.20011797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Mattson MP, Culmsee C, Yu Z, Camandola S. J Neurochem. 2000;74:443–456. doi: 10.1046/j.1471-4159.2000.740443.x. [DOI] [PubMed] [Google Scholar]
- 100.Wang S, Miura M, Jung Y, Zhu H, Gagliardini V, Shi L, Greenberg AH, Yuan J. J Biol Chem. 1996;271:20580–20587. doi: 10.1074/jbc.271.34.20580. [DOI] [PubMed] [Google Scholar]
- 101.Silkensen JR, Schwochau GB, Rosenberg ME. Biochem Cell Biol. 1994;72:483–488. doi: 10.1139/o94-065. [DOI] [PubMed] [Google Scholar]
- 102.Rosenberg ME, Silkensen J. Int J Biochem Cell Biol. 1995;27:633–645. doi: 10.1016/1357-2725(95)00027-m. [DOI] [PubMed] [Google Scholar]
- 103.Bettuzzi S, Scorcioni F, Astancolle S, Davalli P, Scaltriti M, Corti A. Oncogene. 2002;21:4328–4334. doi: 10.1038/sj.onc.1205594. [DOI] [PubMed] [Google Scholar]
- 104.Rosenberg ME, Girton R, Finkel D, Chmielewski D, Barrie IA, Witte DP, Zhu G, Bissler JJ, Harmony JAK, Aronow BJ. Mol Cell Biol. 2002;22:1893–1902. doi: 10.1128/MCB.22.6.1893-1902.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Starr R, Willson TA, Viney EM, Murray LJ, Rayner JR, Jenkins BJ, Gonda TJ, Alexander WS, Metcalf D, Nicola NA, Hilton DJ. Nature. 1997;387:917–921. doi: 10.1038/43206. [DOI] [PubMed] [Google Scholar]
- 106.Berlato C, Cassatella MA, Kinjyo I, Gatto L, Yoshimura A, Bazzoni F. J Immunol. 2002;168:6404–6411. doi: 10.4049/jimmunol.168.12.6404. [DOI] [PubMed] [Google Scholar]
- 107.Wynn TA, Nicolet CM, Paulnock DM. J Immunol. 1991;147:4384–4392. [PubMed] [Google Scholar]
- 108.Boehm U, Guethlein L, Klamp T, Ozbek K, Schaub A, Futterer A, Pfeffer K, Howard JC. J Immunol. 1998;161:6715–6723. [PubMed] [Google Scholar]
- 109.Uze G, Lutfalla G, Bandu MT, Proudhon D, Mogensen KE. Proc Natl Acad Sci U S A. 1992;89:4774–4778. doi: 10.1073/pnas.89.10.4774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Beadling C, Druey KM, Richter G, Kehrl JH, Smith KA. J Immunol. 1999;162:2677–2682. [PubMed] [Google Scholar]
- 111.Panetta R, Guo Y, Magder S, Greenwood MT. Biochem Biophys Res Commun. 1999;259:550–556. doi: 10.1006/bbrc.1999.0817. [DOI] [PubMed] [Google Scholar]
- 112.Shimizu N, Soda Y, Kanbe K, Liu HY, Mukai R, Kitamura T, Hoshino H. J Virol. 2000;74:619–626. doi: 10.1128/jvi.74.2.619-626.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Rothermel B, Vega RB, Yang J, Wu H, Bassel-Duby R, Williams RS. J Biol Chem. 2000;275:8719–8725. doi: 10.1074/jbc.275.12.8719. [DOI] [PubMed] [Google Scholar]
- 114.Parkos CA, Dinauer MC, Walker LE, Allen RA, Jesaitis AJ, Orkin SH. Proc Natl Acad Sci U S A. 1988;85:3319–3323. doi: 10.1073/pnas.85.10.3319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Dinauer MC, Pierce EA, Bruns GA, Curnutte JT, Orkin SH. J Clin Invest. 1990;86:1729–1737. doi: 10.1172/JCI114898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Aldred AR, Grimes A, Schreiber G, Mercer JF. J Biol Chem. 1987;262:2875–2878. [PubMed] [Google Scholar]
- 117.Klomp LW, Farhangrazi ZS, Dugan LL, Gitlin JD. J Clin Invest. 1996;98:207–215. doi: 10.1172/JCI118768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Tessitore A, Villani GR, Di Domenico C, Filocamo M, Gatti R, Di Natale P. Hum Genet. 2000;107:568–576. doi: 10.1007/s004390000429. [DOI] [PubMed] [Google Scholar]
- 119.Liu Y, Hoffmann A, Grinberg A, Westphal H, McDonald MP, Miller KM, Crawley JN, Sandhoff K, Suzuki K, Proia RL. Proc Natl Acad Sci U S A. 1997;94:8138–8143. doi: 10.1073/pnas.94.15.8138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Lund EG, Kerr TA, Sakai J, Li WP, Russell DW. J Biol Chem. 1998;273:34316–34327. doi: 10.1074/jbc.273.51.34316. [DOI] [PubMed] [Google Scholar]
- 121.Matsushima N, Ohyanagi T, Tanaka T, Kretsinger RH. Proteins. 2000;38:210–225. doi: 10.1002/(sici)1097-0134(20000201)38:2<210::aid-prot9>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 122.Qian F, Frankfater A, Chan SJ, Steiner DF. DNA Cell Biol. 1991;10:159–168. doi: 10.1089/dna.1991.10.159. [DOI] [PubMed] [Google Scholar]
- 123.Santamaria I, Velasco G, Pendas AM, Fueyo A, Lopez-Otin C. J Biol Chem. 1998;273:16816–16823. doi: 10.1074/jbc.273.27.16816. [DOI] [PubMed] [Google Scholar]
- 124.Santamaria I, Velasco G, Pendas AM, Paz A, Lopez-Otin C. J Biol Chem. 1999;274:13800–13809. doi: 10.1074/jbc.274.20.13800. [DOI] [PubMed] [Google Scholar]