

Abstract
A characteristic of highly malignant cells is their increased motility and secretion of proteinases allowing these cells to penetrate surrounding basement membranes and metastasize. Activation of 21 kDa activated kinases (PAKs) is an important mechanism for increasing cell motility. Recently, we reported that binding of receptor-recognized forms of the proteinase inhibitor α2-macroglobulin (α2M*) to GRP78 on the cell surface of 1-LN human prostate cancer cells induces mitogenic signaling and cellular proliferation. In the current study, we have examined the ability of α2M* to activate PAK-1 and PAK-2. Exposure of 1-LN cells to α2M* caused a two-threefold increase in phosphorylated PAK-2 and a similar increase in its kinase activity towards myelin basic protein. By contrast, the phosphorylation of PAK-1 was only negligibly affected. Silencing the expression of the GRP78 gene, using either of two different mRNA sequences, greatly attenuated the appearance of phosphorylated PAK-2 in α2M*-stimulated cells. Treatment of 1-LN cells with α2M* caused translocation of PAK-2 in association with NCK to the cell surface as evidenced by the co-immunoprecipitation of PAK-2 and NCK in the GRP78 immunoprecipitate from plasma membranes. α2M*-induced activation of PAK-2 was inhibited by prior incubation of the cells with specific inhibitors of tyrosine kinases and PI 3-kinase. PAK-2 activation was accompanied by significant increases in the levels of phosphorylated LIMK and phosphorylated cofilin. Silencing the expression of the PAK-2 gene, greatly attenuated the phosphorylation of LIMK. In conclusion, we show for the first time the activation PAK-2 in 1-LN prostate cancer cells by a proteinase inhibitor, α2-macroglobulin. These studies suggest a mechanism by which α2M* enhances the metastatic potential of these cells.
Keywords: PAK-2 autophosphorylation, LIMK, cofilin, NCK, silencing of GRP78 gene expression and PAK-2 translocation, phosphorylation of LIMK, Bad phosphorylation, silencing of PAK-2 gene expression and LIMK phosphorylation
INTRODUCTION
Cancer of the prostate is the most commonly diagnosed malignancy of men (1). In the development of prostate cancer, deregulation of cell growth control often is accompanied by acquisition of androgen independence, a poor prognostic indicator (2,3). Growth factors including EGF, IGF, and FGF play a role in the progression of androgen-independent prostate cancer (2,3). These growth factors induce mitogenic cellular responses by activating their specific receptors. Ligand binding to these receptors induces the autophosphorylation of the receptor on specific tyrosine residues resulting in the assembly of multiprotein complexes which activate the Ras/MAPK and PI 3-kinase signaling pathways (4). In addition to increased activation of signaling pathways that promote cellular proliferation and/or suppression of apoptosis, increased motility is often seen in malignantly transformed cells. This increase in motility, along with increased secretion of proteinases, especially matrix metalloproteinases, enables highly metastatic cancer cells to penetrate surrounding basement membranes and invade blood vessels and lymphatics. One mechanism which promotes increased motility of malignant cells is activation of members of the 21 kDa activated kinase (PAK)1 family.
These proteins are Ser/Thr kinases that mediate Rac and Cdc42 GTPase-dependent signaling (see reviews 5–8 and references therein). The mammalian PAK family consists of six members including PAK-1 and PAK-2. PAK-1 is tissue-specific in its expression, whereas PAK-2 is ubiquitously expressed. The catalytic activity of PAKs is regulated by the binding of active GTPases to the conserved p21 binding motif in the NH2-terminal domain leading to the relief of autoinhibitory interactions with the COOH-terminal catalytic domain (5–8). PAK-2 is also activated by caspase or caspase-like proteinases which generate constitutively active p34 PAK-2, the COOH-terminal catalytic domain (9). Activated full length PAK-2 stimulates cell survival and growth in response to various stress stimulants whereas its proteolytic fragment, p34 protein, stimulates cell death (9,10). Stimulation of cell survival by activated full length PAK-2 is partly mediated by phosphorylation and inhibition of proapoptotic Bad (8–10). The activation of PAK-2 in response to irradiation or cytosine β-D-arabinoside is dependent on protein tyrosine kinase and PI 3-kinase activity (11). PAK-1 mediates signals from the Ras/MAPK and PI 3-kinase signaling pathways to promote cell transformation. PAKs also play important roles in modulating the ability of cancer cells to move and metastasize (5–8). A number of highly metastatic human breast cancer lines exhibit constitutively elevated PAK-1 or PAK-2 activity (12).
α2-macroglobulin (α2M) is a broad specificity proteinase inhibitor which binds to cell surface receptors when activated by proteinases (13). The activated form of α2M (α2M*) is also produced by direct reaction of internal thiol esters present in each of its four identical subunits with small amines or ammonia (13). Binding of α2M* to macrophages (14,15), rheumatoid synovial fibroblasts (16), and 1-LN prostate cancer cells triggers increases in the levels of intracellular inositol (1,3,4-)trisphosphate (IP3) and cytosolic free calcium [Ca2+]i and is followed by activation of components of the Ras/MAPK and PI 3-kinase signaling cascades (17–20). As a consequence of these events, α2M* upregulates DNA synthesis and cellular proliferation (17–20). Based on these and other observations, we hypothesized that α2M* functions like a growth factor and its receptor as a growth factor receptor (13). The low density lipoprotein receptor-related protein (LRP-1) was identified in the 1990’s as an α2M* receptor. Subsequent studies in our laboratory suggested that a receptor distinct from LRP-1 must account for α2M*-dependent signal transduction (14–20). These events require the presence of a small number of sites (~1500/cells) demonstrating very high ligand affinity (Kd 50–100 pM) for cellular binding of α2M* or its receptor binding domain. This second receptor, initially termed the α2M* signaling receptor (α2MSR), was later isolated from murine peritoneal macrophages and 1-LN human prostate cancer cells and identified as cell surface-associated GPR78, a heat shock protein of the HSP70 family (20). This molecular chaperone has been highly characterized for its ability to promote cell survival during endoplasmic reticulum (ER) stress (see reviews 21–26 and references therein). GRP78 is involved in many cellular processes including antigen presentation, translocation of newly synthesized polypeptides across the ER membrane, and their subsequent folding, maturation, transport, or retrotranslocation (21–26). An increased expression of GPR78 is a part of the unfolded protein response (UPR) required to alleviate ER stress, maintain ER function, and protect cells against cell death (21–26). GRP78 is constitutively expressed, but its synthesis can be upregulated by a variety of stressful conditions (21–26,28,29) that perturb protein folding and assembly within the ER including glucose deprivation, acidosis, and hypoxia (21–26). Poorly vascularized solid tumors demonstrate both hypoxia and acidosis and these cells may be viewed as highly stressed.
The presence of GRP78 on the cell surface has only recently been appreciated. Constitutive cell surface expression on normal cells is low, but various treatments upregulate its cell surface expression (18). For example, we have demonstrated in vivo upregulation of GRP78 on the surface of antigen presenting cells when mice are exposed to various stimuli (27). These events appear to have consequences with respect to α2M*-mediated antigen presentation (28). Normal fibroblasts do not show cell surface expression of GRP78 and α2M* treatment does not trigger signaling responses by these cells. Rheumatoid synovial fibroblasts, however, express GRP78 on their cell surface and signal briskly when exposed to α2M* (16). GRP78 also is expressed to a high degree on the surface of a number of cancer cells including the highly metastatic 1-LN human prostate cancer cell line (29). By contrast, GRP78 cell surface localization and α2M*-dependent signal transduction do not occur with PC-3 cells, a cell line of low malignant potential, and the parent line for the 1-LN cell line (see for example 17–19). Recent studies have demonstrated antibodies against GRP78 in the sera of prostate cancer patients and the presence of these antibodies is highly correlated with increased metastatic potential and a poor prognosis (30–31). The appearance of the normally cryptic GRP78 protein on the cell surface in high concentration may be a critical factor in the development of autoantibodies to GRP78.
The circulating concentration of α2M is 1 to 5 μM and α2M* comprises about 200–500 nM of this pool (32). It has been estimated that about 1 g of α2M turns over daily (32). Prostate cancer cells also produce prostate cancer specific antigen (PSA), a proteinase which binds readily to α2M (33,34). Thus highly aggressive prostate cancer may secrete PSA which by binding to α2M generates α2M* further increasing the concentration of α2M* in the tumor microenvironment. Furthermore, tumors may be viewed as existing under ER stress and tumors protect themselves from ER stress by expressing UPR, of which enhanced GRP78 synthesis is a biomarker (13–18). A small pool of newly synthesized GRP78 translocates to cell surface from the ER in association with MTJ-1 (35). Therefore, it could be envisaged that under these conditions, a substantial amount of α2M* would be available to bind to cell surface GRP78 thus triggering the activation of mitogenic signaling-dependent cell proliferation. Since it is known that PAKs can be activated via PI 3-kinase signaling (5–8,11) and membrane localization (5–8, 36), we suggest that activated PAKs may mediate α2M*-induced affects on 1-LN prostate cancer cells. Here we demonstrated that α2M* mediates PAK-2 activation in 1-LN cells, but PAK-1 is only negligibly affected. We then examined the effect of treating 1-LN cells with α2M* on the mechanism of activation of PAK-2, Rac-1, LIMK, and cofilin. We report that exposure of 1-LN cells to α2M* induces autophosphorylation of PAK-2, activation of the kinase activity of PAK-2 towards myelin basic protein in a tyrosine-kinase and PI 3-kinase-dependent manner, and recruitment of PAK-2 to plasma membrane via the adaptor protein NCK. Rac-1 is also activated. We further demonstrate activation of LIMK and cofilin which are essential for regulating cytoskeletal organization.
EXPERIMENTAL PROCEDURES
Materials
Culture media were purchased from Life Technologies, Inc. Antibodies against PAK-1, PAK-2, and Bad, as well as the phosphorylated forms of PAK-1, PAK-2, LIMK, cofilin, and Bad (Ser112 or Ser136), were procured from Cell Signaling Technology, Inc. (Beverly, MA). Myelin basic protein and actin antibodies were from Sigma Chemicals (St. Louis, MO). Anti-GRP78 antibodies were purchased from Stressgen (Victoria, BC, Canada). (γ33P) ATP (specific activity 3000 Ci/mmol) was purchased from Perkin Elmer (Boston, MA). The sources for the inhibitors used have been described previously. α2M* was prepared as described previously. Other reagents used in the study were of AR quality and were procured locally.
The effect of α2M* stimulation on activation of PAK-1 and PAK-2 in 1-LN cells
The highly metastatic human prostate carcinoma cell line 1-LN, derived from less metastatic PC-3 cells was a kind gift from Dr. Philip Walther (Duke University Medical Center, Durham, NC). Confluent 1-LN cells obtained after overnight incubation in 6 well plates (4×106 cells/well) were washed twice with HHBSS, a volume of the HHBSS added to the monolayers. One set of cells was stimulated with different concentrations of α2M* and cells incubated as above for different time periods. The other set of cells was stimulated with different concentrations of α2M* for 10 min. At the end of incubation, medium was aspirated and the cells lysed in lysis buffer containing 20 mM Tris·HCl (pH 8.6), 0.1 M NaCl, 1 mM EDTA, 50 mM NaF, 30 mM Na pyrophosphate, 1 mM Na orthovanadate, 1 mM PMSF, 20 μg/ml leupeptin and 0.5% Nonidet® P40 for 10 min on ice. The DNA strands were broken by passing the lysates through a 27 gauge needle and syringe several times. The lysates were centrifuged at 800 × g for 5 min at 4°C to remove cell debris. The supernatants were transferred to clean tubes and their protein contents determined (37). Equal amounts of lysate proteins were electrophoresed according to Laemmli (38). Proteins from gel (10%) were transferred to Hybond P® membrane and immunoblotted with antibodies against phosphorylated and unphosphorylated PAK-2 and PAK-1 respectively according to the manufacturer’s instructions. Protein bands on the membrane were visualized by ECF (Amersham) and quantified using a Storm 860 Phosphorimager® (Molecular Dynamics, Sunnyvale, CA). The respective membranes were stripped and reprobed for actin according to the manufacturer’s instructions.
α2M*-induced autophosphorylation of PAK-1 and PAK-2 in 1-LN prostate cancer cells
Autophosphorylation of PAK-1 and PAK-2 in α2M* stimulated cells was measured essentially as described (39). 1-LN cells were grown in RPMI 1640 medium containing 10 nM insulin, 2 mM glutamine, 10% fetal bovine serum, 12.5 units of penicillin/ml, and 6.5 μg/ml streptomycin in 6 well plates at 37°C in a humidified CO2 incubator till 80–90% confluent (4×106 cells/well). The medium was aspirated and the monolayers washed with chilled HHBSS buffer (pH 7.4) and a volume of HHBSS added to the monolayer. The cells were stimulated with α2M* (50 pM/10 min) and incubated as above. The reaction was terminated by aspirating the medium. The cells were lysed in a volume of lysis buffer containing 40 mM HEPES (pH 7.4), 1% NP 40, 100 mM NaCl, 1 mM EDTA, 25 mM NaF, 1 mM sodium orthovanadate, 10 μg/ml leupeptin, and 10 μg/ml aprotinin over ice for 15 min. The lysates were pipetted into Eppendorf tubes, DNA strands broken by passing the lysate through 27 gauge needle several times and lysates centrifuged at 1000 rpm for 5 min at 4°C to remove cell debris. The supernatants were transferred to new tubes and their protein contents determined (37). To equal amounts of lysate proteins in respective tubes antibodies against PAK-1 (1:50) and PAK-2 (1:50) were added followed by the addition of Protein A agarose and tubes incubated overnight at 4°C in a rotary shaker. The immunoprecipitates were recovered by centrifugation (2500 rpm/5 min) at 4°C and washed twice with lysis buffer and thrice with kinase buffer containing 50 mM HEPES (pH 7.5) 10 mM MgCl2, 2 mM MnCl2 and 0.2 mM DTT. Autophosphorylation was measured in 50 μl of kinase buffer containing 10 μCi of [γ33P]ATP (specific activity 3000 Ci/mM) for 20 min at 30°C. The reaction was stopped by adding a volume of 4x sample buffer and heating the tubes for 3 min at 90°C. The tubes were centrifuged and protein fractionated on a 10% gel according to Laemmli (38). The protein bands on the gel were transferred to membranes, the membranes dried, and 33P-labeled PAK-1 and PAK 2 detected and quantified by autoradiography in an Storm 860 Phosphorimager®.
PAK-2 kinase activity in PAK-1 and PAK-2 immunoprecipitates from α2M*-stimulated 1-LN cells
The kinase activities of PAK-2 towards myelin basic protein were determined essentially as described (39). Briefly, 1-LN lysates from respective groups were immunoprecipitated with PAK-2 antibodies as above. The immunoprecipitates were washed thrice with lysis buffer and then thrice with 2x kinase buffer as above. To respective immunoprecipitates in 50 μl of kinase buffer were incubated with 5 μg of myelin basic protein (MBP) for 5 min over ice. The kinase reaction of immunoprecipitates was initiated by the addition of 10 μCi of [γ33P]ATP (specific activity 3000Ci/mM) followed by the addition of ATP (20 μM final concentration). The samples were incubated for 10 min at 25°C. The reaction was terminated by the addition of 1 volume of 4x sample buffer. In experiments where the effects of tyrosine kinase and PI 3-kinase inhibitors was examined on PAK kinase activity these were added before stimulation with α2M*. Other details were identical to those described above. The samples were heated for 3 min at 90°C, electrophoresed on 12.5% gel, transferred to membrane and 33P-labeled myelin basic protein visualized and quantitated by autoradiography in an Storm 860 Phosphorimager®.
Modulation of α2M*-induced PAK-2 activation in 1-LN cells by protein kinases
In experiments where the effects of inhibiting tyrosine kinases and of PI 3-kinase was studied on expression of activated PAK-2 by Western blotting, the specific inhibitors of these kinases were added to respective wells (4×106/cells/well) as described above, and cells incubated for specific time period before adding α2M* (50 pM) and incubating cells further for 10 more min. The reaction was stopped by aspirating the medium and lysing the cells in lysis buffer as described above. Equal amounts of lysate proteins were electrophoresed according to Laemmli (38). Proteins from gel (10%) were transferred to Hybond P® membrane and immunoblotted with antibodies against phosphorylated and unphosphorylated PAK-2, respectively according to the manufacturer’s instructions. Protein bands on the membrane were visualized by ECF and quantified using a Storm 860 Phosphorimager®. The respective membranes were stripped and reprobed for actin according to the manufacturer’s instructions.
Assay for Rac·GTP
Active GTP-bound Rac was precipitated employing a PAK-PBD-based assay kit (Upstate Cell Signaling, Lake Placid, NY) (40). Confluent 1-LN cells (4×106 well) in six well plates in RPMI 1640 medium containing 10% FBS, penicillin (12.5 units/ml) streptomycin (6.5 μg/ml), 2 mM glutamine, and 10 nM insulin, were stimulated with α2M* (50 pM/10 min) at 37°C in a humidified CO2 (5%) incubator. The reaction was stopped by aspirating the medium, and washing cells twice with ice-cold HHBSS buffer (pH 7.4). The cells were lysed by adding a buffer containing 50 mM Tris·HCl (pH 7.5), 120 mM NaCl, 25 nM NaF, 1 mM sodium orthovanadate, 1 mM PMSF, 1 mM benzamidine, 10 μg leupeptin/ml, and 1% NP-40 on ice for 10 min. The lysates were transferred into Eppendorf tubes, DNA strands broken by passing the lysate through a 27 gauge needle several times and the lysates centrifuged at 1000 rpm for 5 min at 4°C to remove cell debris. The supernatants were transferred to new tubes and their protein contents determined (37). To equal amounts of lysate proteins in the respective tubes, 40 μl of PAK-PBD agarose was added and the tubes incubated for 1 h at 4°C with gentle rotation. The tubes were centrifuged at 3500 rpm for 10 min at 4°C. The agarose pellet was washed thrice with the above lysate buffer. To the agarose pellets 40 μl of reducing sample buffer was added, the tubes heated at 90°C for 5 min, centrifuged briefly, and the supernatant processed for protein fractionation on a 10% gel according to Laemmli (38). The protein bands on the gel were transferred to Hybond P® membranes and immunoblotted with antibodies against Rac-1 (Santa Cruz Biotechnology, Inc., Santa Cruz, CA). Protein bands on the membranes were visualized by ECF and quantified using a Storm 860 Phosporimager® (Molecular Dynamics, Sunnyvale, CA). An aliquot of lysate was similarly processed for total Rac quantification.
Translocation of PAK-2 to plasma membrane in 1-LN cells stimulated with α2M*
Confluent monolayers of 1-LN cells grown as above in RPMI medium in 4 well plates (12×104 cells/well) were washed with HHBSS twice and a volume of HHBSS added to the monolayers. The cells were treated with α2M* (50 pM) and incubated for 10 min as above. At the end of incubation, the medium was aspirated and to the cells was added a volume of chilled HHBSS buffer containing 10 mM Tris.HCl pH 7.5, 10 mM NaCl, 1 mM PMSF, 10 μM benzamidine, and 10 μM leupeptin. The cells were scraped into chilled glass homogenizing tube and the membrane fractions highly enriched in plasma membrane was isolated as described previously. Briefly, the cells were homogenized by 30 up and down strokes with a Teflon pestle at 4°C. The homogenate was centrifuged at 600 × g for 5 min at 4°C and the pellet discarded. The supernatant was layered onto a sucrose step gradient of 50% and 30% (3 ml each) and centrifuged at 200,000 × g for 75 min in a Beckman Coulter Ultracentrifuge (Model Optima LE80) at 4°C. The membrane fraction at the interface between the sucrose layers was removed and suspended in a volume of incubation buffer containing 25 mM HEPES, pH 7.4, 10 mM KCl, 3 mM NaCl, 5 mM MgCl2, 2 μM leupeptin, and 1 μM Ca2+. The suspension was centrifuged at 400,000 × g for 90 min as above. The pellet was suspended in a volume of incubation buffer. The enrichment of membrane preparation for plasma membrane was assessed as described previously including by electron microscopy (41). These analyses showed this membrane fraction was highly enriched in plasma membranes (92–95%) and hence we designated this “membrane fraction” as the “plasma membrane fraction”. The membrane pellet was lysed in lysis buffer, the lysate immunoprecipitated with antibodies against GRP78 (1:100) and GRP78 in the membranes quantified as above. The membranes on which these preparations were transferred after electrophoresis, were reprobed and quantified for phosphorylated PAK-2 and NCK according to the manufacturer’s instructions.
Chemical synthesis of dsRNA homologous in sequence to the target GRP78 gene
The chemical synthesis of dsRNA homologous in sequence to the target GRP78 (1) K370IQQLVK376, mRNA sequence 5′AAAATACAGCAATTAGTAAAG3′ and (2) K521NKITIT527 mRNA sequence 5′AAGAATAAAATAACAATAACA3′ peptides (Swiss-Prot. GRP primary sequence accession number P11021) was performed by Ambion (Austin, TX). For making dsRNA of the first mRNA sequence, the sense (5′AAUACAGCAAUUAGUAAAGTT3′) and the antisense (5′CUUUACUAAUUGCUGUAUUTT3′) oligonucleotides and the second (mRNA sequence, the sense (5′GAAUAAAAUAACAAUAACATT-3′) and the antisense (5′UGUUAUUGUUAUUUUAUUCTT) were annealed according to the manufacturer’s instructions. Through the entire period of experimentation, handling of reagents was performed in an RNAse-free environment. Briefly, equal amounts of sense and antisense oligonucleotides were mixed in annealing buffer and heated at 90°C for 1 min then maintained for 1 h at 37°C in an incubator. The dsRNA preparation was stored at −20°C before use.
Transfection of 1-LN cells with dsRNA homologous in sequence to GRP78 gene and effect of α2M* on PAK-2 and NCK
Confluent 1-LN cell monolayers (1.5 106/well in six well plates) incubated as described above were washed twice with HHBSS and 2 ml of DMEM medium containing 10% of FBS and the above mentioned antibiotics added, and cells incubated as above for 16 h. Just before each transfection, 25 μg of both GRP78 dsRNA was diluted to 100 μl of serum- and antibiotic-free DMEM in a tube. In another tube, 10 μl of lipofectamine was diluted into 100 μl of serum- and antibiotic-free medium. The two solutions were combined, mixed gently and incubated for 45 min at room temperature followed by the addition of 800 μl of serum- and antibiotic-free medium to each tube in separate experiments. The monolayers were washed twice with serum-antibiotic-free DMEM medium, layered in each well with 1 ml of lipofectamine-DMEM or lipid dsRNA mixtures containing 25 μg of dsRNA of each GRP78 target mRNA gently mixed and incubated for 5 h at 37°C in a humidified CO2 incubator in separate experiments. At the end of incubation, 1 ml of antibiotic-free DMEM containing 10% FBS was added to each well and cells incubated for 16 h as above. Microscopic observations of the monolayers did not show evidence of toxicity consistent with previous studies. The medium was replaced with DMEM containing antibiotics and 10% FBS 24 h following the start of the transfection. The monolayers were incubated for a further 24 h as above. At the end of incubation, medium was aspirated and monolayers washed with the above medium once and a volume of the same medium added and the cells were used for the experiment outlined below. To demonstrate that the transfection of 1-LN prostate cancer cells with dsRNA homologous in sequence to target GRP78 gene does not produce any nonspecific effects on target gene expression, the 1-LN cells were transfected with equimolar concentrations of scrambled siRNA (Silencer™ negative control, catalog number 4610, Ambion, Austin, TX) under identical conditions as described above for transfection with GRP78 dsRNA. At the end of transfection period (48h), the medium was aspirated and a volume of DMEM medium added and cells either stimulated with buffer or α2M* (50 pM) for 10 min. The reaction was stopped by aspirating the medium and adding a volume of lysis buffer as described above. Equal amounts of lysate proteins (37) were electrophoresed according to Laemmli (38). Proteins from gel (10%) were transferred to Hybond P membrane and immunoblotted with antibodies against GRP78, unphosphorylated PAK-2 and NCK, respectively according to the manufacturer’s instructions. Protein bands on the membrane were visualized by ECF (Amersham, Piscataway, NJ) and quantified using a Storm 860 Phosphorimager®. The respective membranes were stripped and reprobed for actin according to the manufacturer’s instructions.
Chemical synthesis of dsRNA homologous in sequence to the target PAK-2 genes
The chemical synthesis of dsRNA homologous in sequence to the target PAK-2 K382LTDFGF388, mRNA sequence, 5′-AAATTAACAGATTTTGGATTT-3 peptide (Swiss Prot primary accession number Q8CIN4) was performed by Ambion (Austin, TX). For making dsRNA, the sense 5′AUUAACAGAUUUUGGAUUUTT-3 and antisense (5′-AAAUCCAAAAUCUGUUAAUTT--3) oligonucleotides were annealed according to the manufacturer’s instructions as described above for GRP78 above.
Transfection of 1-LN cells with dsRNA homologous in sequence to PAK-2 gene and effect of α2M* on p-LIMK
Confluent 1-LN cells monolayers (1.5 × 106/well in six well plates) incubated as described above were washed twice with HHBSS and 2 ml of DMEM medium containing 10% FBS and the above mentioned antibiotics added and cells incubated as above for 16 h. For each transfection, 25 μg of PAK-2 dsRNA was used and cells transfected as described above for dsGRP78 dsRNA with lipofectamine with or without dsPAK-2. The medium was replaced with DMEM containing antibiotics and 10% FBS 24 h following the start of the transfection. The monolayers were incubated for a further 24 h above. Microscopic observation of the transfected monolayers did not show evidence of toxicity expect that 1-LN cells transfected with dsPAK-2 RNA showed impairments in the uniformity of spreading of monolayers. At the end of incubation, medium was aspirated, monolayers washed with the above medium once, a volume of same medium added, the cells stimulated with α2M*, cells lysed and lysates processed as above for phosphorylated LIMK, PAK-2, and LIMK protein assay by Western blotting as described above. Protein bands on the membrane were visualized by ECF (Amersham, Piscataway, NJ) and quantified using a Storm 860 Phosphorimager®. To demonstrate that the transfection of 1-LN prostate cancer cells with dsRNA homologous in sequence to target PAK-2 gene does not produce any non-specific effects on target gene expression, the 1-LN cells with transfected with equimolar concentrations of scrambled siRNA, under identical conditions as described above. At the end of transfection (48 h), the medium was aspirated and a volume of DMEM medium added and cells stimulated with either buffer or α2M* and processed as above for the quantification of phosphorylated LIMK, PAK-2 and LIMK protein by Western blotting.
RESULTS
Binding of α2M* to cell surface-associated GPR78 enhances phosphorylation of PAK-2 in 1-LN cells
PAK activity is stimulated in response to a variety of extracellular stimuli including chemoattractants acting on G protein-coupled receptors, growth factors interacting with receptor tyrosine kinases, cytokines, and extracellular matrix molecules binding to integrins (5–8). We have previously shown that α2M* promotes cellular growth of 1-LN prostate cancer cells (17,18). To understand the possible involvement of PAK-1 and PAK-2 in α2M*-stimulated cellular growth, we first examined the levels of phosphorylated PAK-1 and PAK-2 in 1-LN cells treated with α2M* for different periods of time and with varying concentrations of α2M* (Figure 1, and Table 1). In the Western blot, phosphorylated PAK-2 is seen as a doublet 58–60 kDa (Figure 1). This appears to represent differential phosphorylation of PAK-2 as previously reported. The maximal increase in phosphorylated PAK-2 occurred at about 10 min of incubation and declined slowly thereafter while the maximal increase in levels of phosphorylated-PAK-2 occurred at about 50–100 pM of α2M*. Surprisingly, incubation of 1-LN cells with α2M* under these conditions only showed a negligible effect on phosphorylation of PAK-1. Similar to the results described above, stimulation of 1-LN cells with α2M* predominantly caused autophosphorylation of PAK-2, but negligible autophosphorylation of PAK-1 (Figure 2A).
Figure 1.
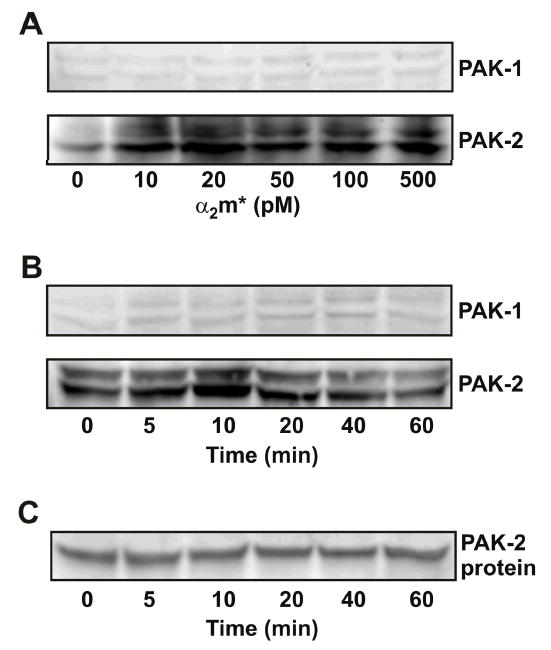
Effect of α2M* on phosphorylation of PAK-2 in 1-LN cells. Western blotting was performed as described under EXPERIMENTAL PROCEDURES. Panel A: Effect of α2M* concentration on the levels of phosphorylated PAK-1 and PAK-2. Panel B: Effect of time of incubation with α2M* on the levels of phosphorylated PAK-1 and phosphorylated PAK-2. Panel C: The protein loading control for PAK-2 is shown. Not shown is the actin loading control. The immunoblots shown here are representative of four to five independent experiments.
Table 1.
Effect of time of incubation of 1-LN prostate cancer cells with α2M* on activation of PAK-2
| Time (min) | Phosphorylated PAK-2 (A.U. × 103) | Phosphorylated PAK-1 (A.U. × 103) |
|---|---|---|
| 0 | 1769 ±17 | 534 ± 66 |
| 5 | 2243 ± 213 | 532 ± 73 |
| 10 | 4009 ± 100 | 540 ± 19 |
| 20 | 3169 ± 686 | 597 ± 43 |
| 40 | 2127 ± 60 | 628 ± 39 |
| 60 | 1358 ± 111 | 656 ± 35 |
The values shown are the mean ± S.E. and have been obtained by quantifying 3–4 different immunoblots by Phosphorimager. A representative blot is shown in Figure 1A, Figure 3A, and B.
Figure 2.
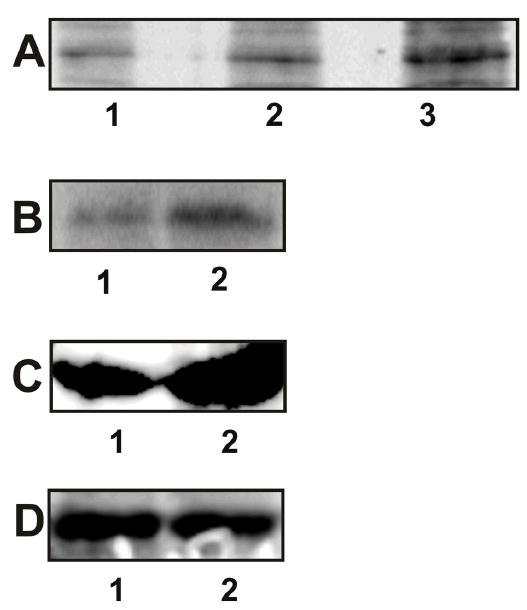
Autophosphorylation and kinase activity of PAKs in 1-LN cells stimulated with α2M*. See EXPERIMENTAL PROCEDURES section for details. Panel A: Autoradiograph showing autophosphorylation of PAK-1 and PAK-2 in the respective immunoprecipitates. The lanes are: (1) buffer control; (2) PAK-1 immunoprecipitate; and lane (3) PAK-2 immunoprecipitate. Panel B: Autoradiograph showing PAK-2 kinase activity towards MBP. The lanes are: (1) buffer control and (2) PAK-2 immunoprecipitate. Panel C: Rac-1·GTP levels in 1-LN-cells treated with (1) buffer and (2) α2M* (50 pM)/10 min); Panel D: Levels of total Rac-1 protein in 1-LN cells treated with (1) buffer and (2) α2M* (50 pM/10 min). Autoradiographs shown are representative of three independent experiments. The immunoblots shown are representative of two individual experiments performed in duplicate.
α2M* stimulates the kinase activity of PAK-2 in 1-LN prostate cancer cells
In the next series of experiments we studied the activation of only PAK-2 by measuring its kinase activity towards MBP (Figure 2B). Treatment of 1-LN prostate cancer cells with α2M* increased PAK-2-dependent phosphorylation of MBP by two-threefold (Figure 2B).
α2M* activates Rac-1 in 1-LN cells
p21-activated protein kinases are phosphorylated by small G proteins such as Rac-1 and CdC42 in the presence of GTP, which binds to the G protein binding site in the NH2-terminal regulatory domain. In the preceding section we showed that stimulation of 1-LN cells with α2M* induces phosphorylation and the kinase activity of PAK-2 (Figure 2A&B). We next determined the activation of Rac-1, by quantifying the levels of Rac-1·GTP by Western blotting (Figure 2C). Indeed α2M* treatment of 1-LN cells elevated the levels of Rac-1·GTP by about twofold compared to unstimulated cells (Figure 2C). The results show the importance of small G proteins in α2M*-induced activation of PAK-2 in 1-LN cells.
Recruitment of PAK-2 to plasma membranes via interaction with the adaptor protein NCK in α2M*-stimulated 1-LN prostate cancer cells
In response to external stimuli, PAKs which contain variable SH3 binding motifs/sites, can interact with the SH3 containing adaptor protein NCK (5–8). This protein is involved in the recruitment of PAKs to activated tyrosine kinase receptors in plasma membranes. NCK is either constitutively associated with PAK-2 or its association is induced (5–8, 36, 42). Interaction of the adaptor protein NCK and PAKs has been implicated in its translocation and stimulation of PAK activity by growth factors (see 5–8, 36, 42). We have evaluated the involvement of the adaptor protein NCK in the translocation of PAK-2 to plasma membrane GRP78 in 1-LN cells upon stimulation of cells with α2M* (Figure 3A,B, and C). GRP78 immunoprecipitate of plasma membrane lysates showed that GRP78, NCK and PAK-2 are co-immunoprecipitated in 1-LN cells stimulated with α2M* (Figure 4A,B, and C). These results demonstrate that in plasma membranes isolated from 1-LN cells, stimulated with α2M*, PAK-2 exists in complex with the adaptor protein NCK and the receptor GRP78.
Figure 3.
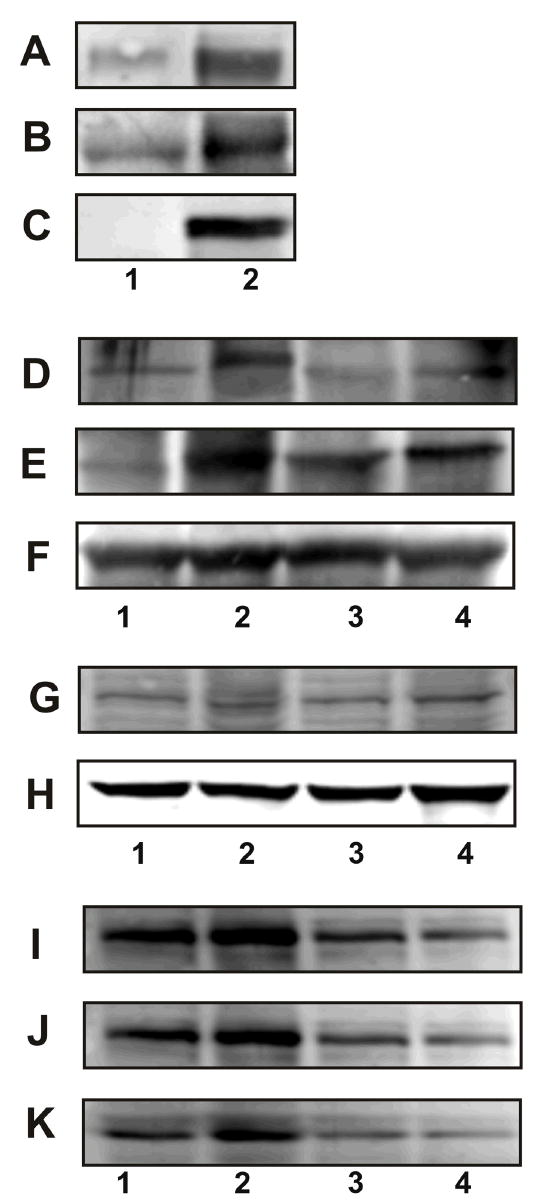
Plasma membrane association of GRP78, NCK, and PAK-2 in 1-LN cells stimulated with α2M*. See EXPERIMENTAL PROCEDURES section for details. Panel A: GRP78 in the GRP78 immunoprecipitate from plasma membrane. Panel B: PAK-2 in the GRP78 immunoprecipitate from plasma membrane. Panel C: NCK in GRP78 immunoprecipitate from plasma membrane. The lanes are: (1) buffer and (2) α2M* (50 pM/10 min). Panel D amd J: Effect of silencing the expression of the GRP78 gene on association of PAK-2. Panel E and K: Effect of silencing the expression of the GRP78 gene on the association of NCK. Panel F: Protein loading control, actin. Panel G and I: Effect of silencing the expression of the GRP78 gene on GRP78 protein levels, and Panel H protein loading control GADPH. The lanes in Panel D, E, F, and G are: (1) buffer; (2) α2M*; (3) dsRNA GRP78 then α2M*; and (4) scrambled dsRNA then α2M*. The lanes in Panel I, J, and K are: (1) buffer; (2) α2M*; (3) dsRNA GRP78 and (4) dsRNA GRP78 then α2M*. The immunoblots shown are representative of three to four independent experiments.
Figure 4.
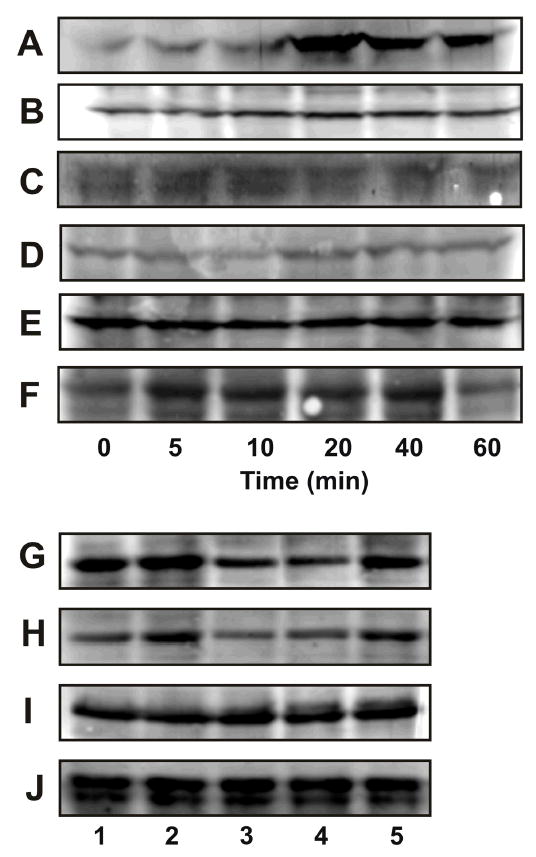
α2M* and the phosphorylation of LIMK and cofilin in 1-LN cells. Panel A: Effect of time of incubation on phosphorylation of LIMK. Panel B: Effect of time of incubation on LIMK protein. Panel C: Protein loading control actin; Panel D: Effect of time of incubation on phosphorylation of cofilin; Panel E: Effect of time of incubation on cofilin protein levels; Panel F: Protein loading control, actin; Panel G: Effect of silencing the expression of the PAK-2 gene on PAK-2 protein levels; Panel H: Effect of silencing the expression of the PAK-2 gene on phosphorylated LIMK; Panel I: Effect of silencing the expression of the PAK-2 gene on LIMK protein levels and Panel J: protein loading control GADPH. Immunoblots shown are representative of three to four independent experiments.
Silencing of GRP78 gene expression with RNAi attenuates the association of PAK-2 and NCK in α2M*-stimulated cells
In the preceding section, we demonstrated the presence of PAK-2 in plasma membranes in association with the adaptor protein NCK and GPR78 after α2M* stimulation (Figure 3A,B, and C). Ligand-induced tyrosine phosphorylation of the receptor causes the recruitment of SH3 and SH2 containing adaptor proteins and form a multiprotein complex responsible for generating and propagating intracellular signaling responsible for cellular responses. To assess the role of ligand-activated GRP78 in the recruitment of NCK-PAK-2 complex to the plasma membrane, we silenced the expression of GRP78 gene with dsRNA homologous in sequence to the target gene mRNA sequence 5′AAAATACAGCAATTAGTAAAG3′ and assayed the association of PAK-2 and NCK in cell lysates in α2M*-stimulated 1-LN cells (Figure 3D and E and Table 2). Silencing GRP78 gene expression with this target sequence, greatly attenuated the association of PAK-2 and NCK in these cells. To further ascertain, that these effects are specific to GRP78 gene silencing, we next employed dsRNA of a second target gene sequence 5′AAGAATAAAATAACAATAACA3′ to silence the expression of GRP78 gene. This approach also profoundly reduced the levels of GRP78 protein (Figure 3G) compared to cells treated with α2M* alone (Figure 3G). If GRP78 is in complex with NCK and PAK-2, then limitations imposed on the availability of GRP78, would also limit the levels of associated NCK and PAK-2. Indeed this was found in studies where silencing of GRP78 gene expression with dsRNA greatly attenuated the levels of GRP78 (Figure 3I), PAK-2 (Figure 3I), and NCK (Figure 3K) employing the immunoprecipitation protocol described above. The specificity of silencing GPR78 gene expression as a cause of these effects is further supported by the use of a scramble dsRNA in control experiments (Figure 3) under identical conditions. Scrambled dsRNA showed none or negligible effects on GRP78, PAK-2, or NCK (Figure 4D, E,G,I,J,K and Table 2). The results presented confirm that ligand-induced activation of GRP78 is required for recruitment of PAK-2 in association with NCK to the plasma membrane. The mechanism by which PAK-2 is activated at the membrane is not clearly understood; however, it has been reported that mere membrane localization of PAK-2 is sufficient for its activation.
Table 2.
Effect of silencing the expression of GRP78 gene on PAK-2 and NCK association in 1-LN cells stimulated with α2M*
| GRP78 (A.U. × 103) | PAK-2 (A.U. × 103) | NCK (A.U. × 103) | ||||
|---|---|---|---|---|---|---|
| Additions | A | B | A | B | A | B |
| Lipofectamine | 276 ± 47 | 448 ± 55 | 612 ± 60 | 2227 ± 73 | 1617 ± 24 | 1780 ± 160 |
| Lipofectamine + α2M* (50 pM/10 min) | 459 ± 28 | 834 ± 18 | 1163 ± 60 | 3795 ± 138 | 4219 ± 534 | 3310 ± 100 |
| dsRNAGRP78 (25 μg) | 192 ± 16 | 482 ± 43 | n.d. | 2253 ± 108 | n.d. | 1020 ± 150 |
| dsRNAGRP78 (25μg) + α2M 50 pm/10 min | 188 ± 23 | 500 ± 33 | 682 ± 183 | 1112 ± 72 | 2196 ± 190 | 490 ± 60 |
| Scrambled dsRNA (25 μg) + α2M | n.d. | 902 ± 87 | 1175 ± 218 | n.d. | 3013 ± 198 | n.d. |
A = First GRP78 dsRNA; B = Second GRP78 dsRNA. Values shown are the mean ± S.E. and have been obtained by quantifying 2–3 different immunoblots by Phosphorimager. A representative blot is shown in Figure 3. n.d. = not done.
α2M* upregulates levels of phosphorylated LIMK and cofilin in 1-LN prostate cancer cells
Reorganization of the cytoskeleton is an essential feature of motility, detachment, and invasion by cancer cells. Formation and stabilizing of actin filaments provide the protrusive force for cellular extension of the leading edge of migratory cells (43–45). The Rho family of GTPases, Rho, Rac, and Cdc42 regulate cell morphology, cytokinesis and cell motility through reorganization of actin filaments. The interplay between these GTPases is critically involved in the regulation of cell morphology and motility. Activation of Rac enhances cell spreading and migration by stimulation of actin polymerization at the plasma membrane and promoting lamellipodia formation. Rho stimulates contractibility and adhesion by inducing the formation of actin stress fibers and focal adhesions. Rho kinases ROCK-I and ROCK-II are downstream effectors of Rho to form stress fibers and focal adhesions. Rho kinases also activate LIM kinase which phosphorylates cofilin and thereby stabilizes actin stress fibers. LIM kinases are dual specificity (Ser/Thr and Tyr) kinases that contain two NH2-terminal LIM domains which are commonly associated with the actin cytoskeleton (46,47). LIMK mediates specifically Rac-induced actin cytoskeleton reorganization and focal adhesion complexes. Rac-induced activation of LIMK is mediated by PAK-1 which phosphorylates LIMK at its Thr508 residue. Activated LIMK specifically phosphorylates cofilin, an actin binding protein that promotes the disassembly of actin filaments (5–8, 42–48). Phosphorylation of cofilin inhibits its actin depolymerizing action and hence provides a mechanism by which LIMK could regulate the assembly of actin (49). LIMK mRNA is upregulate in prostate adenocarcinomas and is correlated with the aggressiveness of these cells (48). Active PAKs greatly enhanced phosphorylation of both LIMK and cofilin in vitro (20–23, 48,49). In the proceeding sections we demonstrated that treatment of 1-LN prostate cancer cells with α2M* activates PAK-2. 1-LN cells are highly malignant, motile, and invasive; therefore, to understand the involvement of LIMK in cytoskeleton reorganization in 1-LN cells upon treatment of α2M*, we have determined the levels of phosphorylated LIMK (Figure 4A and Table 3) and phosphorylated cofilin (Figure 4D and Table 3) by Western blotting. A two-threefold increase in the levels of phosphorylated LIMK and phosphorylated cofilin was observed in 1-LN cells treated with α2M*. The results indicate the involvement of phosphorylated-PAK-2 in cytoskeleton reorganization and possible protrusive behavior of 1-LN prostate cancer cells treated with α2M* via phosphorylation of LIMK and cofilin. The results indicate that upon treatment of 1-LN cells with α2M*, the phosphorylation of LIMK and cofilin is markedly increased and suggests that these effects are possibly mediated by LIMK under our experimental conditions. Since LIMK is also activated by Rho kinases, to further understand the role of PAK-2 in LIMK activation, we silenced the expression of the PAK-2 gene by RNA interference (Figure 4). Silencing of PAK-2 gene expression (Figure 4G) attenuated the levels of phosphorylated LIMK (Figure 4H) without affecting the levels of LIMK protein (Figure 4I) compared to cells treated with α2M* alone (Figure 4G, H and I) or cells transfected with scramble dsRNA and treated with α2M* (Figure G,H, and I). The reductions in the protein levels of PAK-2 and phosphorylated LIMK were nearly comparable which suggest that in 1-LN cells prostate cancer cells, α2M*-induced PAK-2 is involved in reorganization of the cytoskeleton via LIMK and cofilin. However, the role of Rho kinase in phosphorylation of LIMK in 1-LN cells is not ruled out.
Table 3.
Effect of time of incubation of 1-LN prostate cancer cells with α2M* on phosphorylation of LIMK, cofilin, and Bad proteins
| Time (min) | Phosphorylated LIMK (A.U. × 103) | Phosphorylated cofilin (A.U. × 103) | Bad at Ser112 (A.U. × 102) | Bad at Ser136 (A.U. × 102) |
|---|---|---|---|---|
| 0 | 897 ± 69 | 130 ± 9 | 63 ± 15 | 167 ± 7 |
| 5 | 1275 ± 75 | 157 ± 20 | 98 ± 4 | 240 ± 14 |
| 10 | 1772 ± 28 | 127 ± 10 | 83 ± 8 | 224 ± 11 |
| 20 | 3481 ± 223 | 288 ± 14 | 120 ± 10 | 231 ± 2 |
| 40 | 2023 ± 48 | 295 ± 13 | 121 ± 10 | 414 ± 19 |
| 60 | 2545 ± 48 | 248 ± 18 | 106 ± 7 | 299 ± 14 |
α2M* treatment of 1-LN prostate cancer cells upregulate the increased expression of phosphorylated Bad (Ser112 and Ser136)
Different PAK family members regulate the balance between prosurvival and proapoptotic proteins of the Bcl-2 family (8). As noted abvoe, caspase-induced cleavage of PAK-2 releases the constitutively active 34 kDa catalytic COOH terminal fragment in response to multiple stimuli that induce apoptosis; however, full length activated PAK-2 is antiapoptotic (5–8, 11). Members of the Bcl-2 family are intracellular proteins that can either promote survival (Bcl-2, BclXL) or augment cell death (Bad and Bax) (50–53). Bad binds to Bcl-2 and neutralizes the antiapoptotic effects of Bcl-2 and promotes cell death. Phosphorylation of Bad causes its interaction with 14-3-3 protein and prevents its binding to Bcl-2, which then interacts with Bax to inhibit apoptosis. Bad is phosphorylated on Ser112, Ser136, and Ser155 by protein kinases including MAPKs and Akt (54,55). The latter which is the effector of PI 3-kinase, and PAK-1 phosphorylates Bad at Ser136 either directly or indirectly through PAK-2. Treatment of 1-LN prostate cancer cells, with α2M* profoundly elevated the levels of Bad phosphorylated at Ser112 and Ser136 (Figure 5A and B and Table 3) with kinetics similar to that PAK-2 activation (Figure 1). These studies suggest that PAK-2 is involved in the cellular proliferative effects of α2M* observed in 1-LN prostate cancer cells by upregulating the levels of antiapoptotic proteins.
Figure 5.

Phosphorylation of Bad in α2M*-stimulated 1-LN cells. Panel A: Effect of time of incubation of 1-LN cells with α2M* (50 pM) on phosphorylation of BAD at Ser112 and Panel B: Effect of time of incubation of 1-LN cells with α2M* (50 pM) on phosphorylation of BAD at Ser136. Panel C: Bad protein. The immunoblots are representative of three to four independent experiments.
α2M*-induced increased activation of PAK-2 in 1-LN cells is inhibited by tyrosine kinase and PI 3-kinase inhibitors
External stimuli activate members of PAK family by multiple mechanisms including Ras/MAPK and PI 3-kinase-dependent signal transduction (5–8). In our earlier studies, we demonstrated that binding of α2M* to cells also causes activation of these pathways (16–20). Activation of Ras, and PI 3-kinase in macrophages stimulated with α2M* induced increase expression of mitogenic signaling culminating in an increased DNA synthesis and cellular proliferation (17,19). In view of the functional dependence of PAK-2 activation on receptor tyrosine phosphorylation, and PI 3-kinase activation, we examined the effect of inhibiting tyrosine phosphorylation of GPR78 and PI 3-kinase activation consequent to α2M* binding to 1-LN cells on the activation of PAK-2 by measuring its kinase activity towards MBP (Figure 6A and Table 4) and then expression of phosphorylated PAK-2 (Figure 7B and Table 4). Pretreatment of cells with genestin, a specific inhibitor of tyrosine kinases greatly attenuated the α2M*-induced MBP phosphorylating activity of PAK-2 (Figure 6A and Table 4) as well as the levels of activated PAK-2 by Western blotting. Likewise, inhibition of PI 3-kinase with its specific inhibitor LY294004 profoundly inhibited the MBP phosphorylating activity of PAK-2 (Figure 6A and Table 4) as well as the elevated levels of phosphorylated PAK-2 (Figure 6B and Table 4) in 1-LN cells. The data presented show that α2M*-induced increased activation of PAK-2 in 1-LN cells requires tyrosine phosphorylation of the receptor and activation of downstream PI 3-kinase signaling. Prior treatment of cells with genestin drastically attenuated kinase activity of PAK-2. Likewise, LY294002 also inhibited kinase activity of PAK-2 but the magnitude of inhibition was smaller than that of tyrosine kinase inhibition, which suggest that PI 3-kinase-independent mechanism may be involved in PAK-2 activation.
Figure 6.
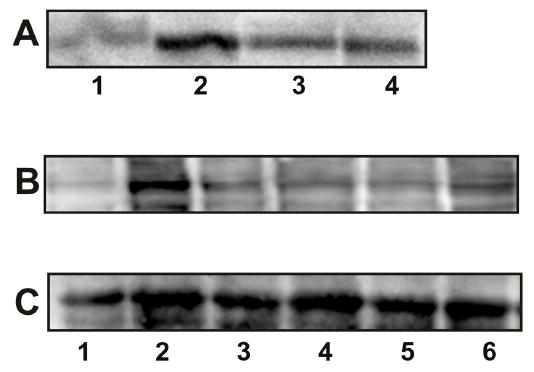
Modulation of PAK-2 activity by tyrosine kinase and PI 3-kinase in 1-LN cells stimulated with α2M*. Panel A: Autoradiograph showing MBP phosphorylation by PAK-2. The lanes are: (1) buffer; (2) PAK-2 immunoprecipitate; (3) PAK-2 immunoprecipitate from 1-LN cells treated with genestin (20 μM/16h) before α2M* (50 pM/10 min) stimulation; and (4) PAK-2 immunoprecipitate from 1-LN cells treated with LY294002 (20 μM/20 min) before α2M* stimulation. Panel B: Immunoblot showing the effect of tyrosine kinase and PI 3-kinase inhibitors on levels of phosphorylated PAK-2. The lanes are: (1) buffer; (2) α2M (50 pM/10 min); (3) genestin (20μM/16 h), then α2M* (50 pM/10 min); (4) LY294002 (20 μM/20 min) then α2M*; (5) wortmannin (30 nM/30 min) then α2M*; and (6) LY353511 (20 μM/20 min) then α2M*. The protein loading controls immunoblots of actin are shown below the immunoblot which is representative of three independent experiments.
Table 4.
Effect of silencing the expression of PAK-2 gene on phosphorylation of LIMK in 1-LN prostate cancer cells stimulated with α2M*
| Additions | PAK-2 | Phosphorylated LIMK | LIMK |
|---|---|---|---|
| Lipofectamine | 745 ± 70 | 530 ± 59 | 435 ± 20 |
| Lipofectamine+ α2M* (50 pm/10 min) | 1501 ± 42 | 1730 ± 200 | 399 ± 52 |
| PAK-2-dsRNA (25 μg) | 776 ± 83 | 520 ± 120 | 410 ± 9 |
| PAK-2-dsRNA + α2M* (50 pm/10 min) | 947 ± 25 | 770 ± 80 | 420 ± 40 |
| Scrambled dsRNA (25 μg) + α2M* | 1350 ± 169 | 2320 ± 210 | 435 ± 17 |
The values shown are the mean ± S.E. and have been obtained by quantifying 4 different immunoblots from 2 individual experiments by Phosphorimager. A representative immunoblot is shown in Figure 4.
Figure 7.
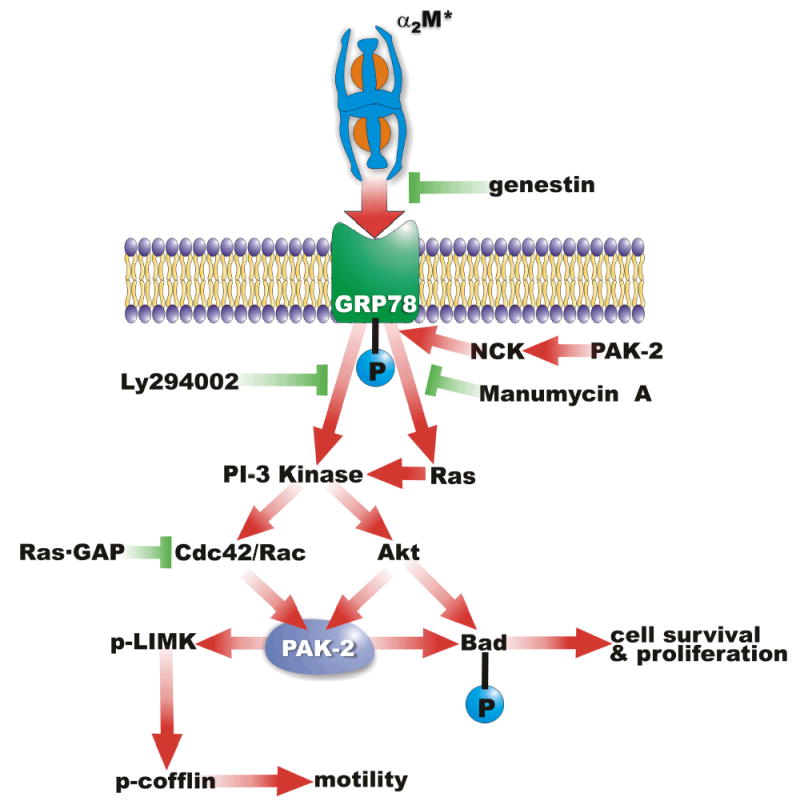
A schematic representation of the mechanisms of PAK-2 activation in 1-LN prostate cancer stimulated with α2M*.
DISCUSSION
The development, progression, and metastasis of prostate cancer is a multistage phenomenon where the role of cellular metabolism, environment, and intracellular signaling play crucial roles. In this study, we have examined PAK activation in 1 LN cells treated with α2M* and various PAK-induced downstream signaling events. The salient observations of this study are that α2M* binding to 1-LN cells: (1) upregulates activation of PAK-2; (2) induces autophosphorylation of PAK-2, and the tyrosine kinase- and PI 3-kinase-dependent kinase activity of PAK-2; (3) induces activation of Rac-1; (4) increases phosphorylation of LIMK and cofilin which are greatly reduced upon silencing of PAK-2 gene expression; (5) promotes NCK-mediated recruitment of PAK-2 to plasma membrane-localized GPR78; (6) regulates phosphorylation of Bad at Ser112 and Ser136. Finally, silencing expression of the GRP78 gene nearly abolishes the association of PAK-2 and NCK in the plasma membranes of α2M*-stimulated 1-LN cells. The data presented suggest that PAK-2 is involved in α2M*-induced cellular growth, migration, and the survival of 1-LN prostate cancer cells. The mechanism by which α2M* activates PAK-2 appears to be dependent on receptor tyrosine phosphorylation, and PI 3-kinase activation since pretreatment of cells with genestein, LY294002, and wortmannin greatly attenuated the activation of PAK-2. Since silencing of the expression of the GRP78 gene also greatly attenuated the activation of PAK-2 in α2M* treated 1-LN cells, ligand-induced activation of GRP78 is a prerequisite for PAK-2 activation in these cells. That the activation of PAK-2 in 1-LN cells under these conditions was not completely abolished also suggests the involvement of other mechanisms in PAK-2 activation.
Highly malignant and invasive androgen-independent human 1-LN prostate cancer cells were derived from less malignant and noninvasive PC-3 cells at this University a number of years ago. Unlike PC-3 prostate cancer cells, 1-LN cells in nude mice migrate and metastasize. 1-LN prostate cancer cells also differ from PC-3, LnCap, and DU 145 cancer cells in one very important respect; namely when 1-LN, cells but not the other cell lines, are exposed to picomolar concentrations of α2M* cell proliferation increases substantially (16,18,19). The expression of GRP78 on the surface of PC3, DU145, and LnCap cells was either absent or minimal (17,18). By contrast, the highest levels that we have detected are on the surface of 1-LN cells which were employed to purify α2MSR and identify it as GRP78 (20). Preincubation of 1-LN cells with antibodies against GRP78, silencing of the expression of the GRP78 gene and silencing of the expression of MTJ-1, with which GRP78 associates, greatly attenuated α2M*-induced mitogenic signaling and cellular responses (16,17,19,35). These observations suggest a functionally close relationship between cell surface-associated GRP78 receptor activation and growth, metastasizing, and the invasive potential of 1-LN prostate cancer cells. Ligation of this cell surface-associated GRP78 on 1-LN cells with α2M* triggers the activation of mitogenesis and cell proliferative secondary to IP3/Ca2+ signaling, Ras/MAPK signaling, and PI 3-kinase signaling (17,18). In our previous reports we have shown that cellular responses elicited post α2M* binding to cell surface-associated GRP78 are analogous to various growth factors and its receptor behaves like a growth factor receptor. In androgen-independent 1-LN prostate cancer cells, α2M* could function as a growth factor (16–20). That this is so, is evidenced by increased DNA synthesis and cellular proliferation observed in 1-LN cells treated with α2M* (16). Both these events are dependent upon tyrosine phosphorylation of GRP78, and receptor downstream Ras/MAPK and PI 3-kinase signaling. Based on positive correlation between circulating GRP78 antibodies and prostate cancer, GRP78 has been suggested as a diagnostic biomarker of prostate cancer (30,31). Prostate cancer cells also produce PSA and matrix metalloproteinases (33,34), which bind readily to α2M whose daily turnover has been estimated to be about 1 g (32). Thus highly aggressive prostate cancer may produce PSA converting α2M to α2M* which would then be available to bind to cell surface-associated GRP78 on prostate cancer cells promoting their growth, metastasizing and invasive potential mediated by mitogenic signaling elicited consequent to α2M* binding to GRP78. Taken together with the clinical data cited above, it could be argued that upregulation of α2M* binding receptor α2MSR (GRP78), is part of the aggressive phenotype in prostate cancer.
PAK-2 is ubiquitously expressed whereas the expression of PAK-1 is tissue specific (5–8). In dividing cells PAK-2 is inactive, but is transiently activated when cells are subjected to moderate stress conditions such as hyperosmolarity, ionizing radiations, DNA damaging drugs (5–8). Under these conditions, PAK-2 activation requires upstream tyrosine kinase and PI 3-kinase activities. GRP78 is a ER resident protein which is involved in many cellular processes including antigen presentation, translocation of newly synthesized polypeptides across the ER membrane, and their subsequent folding, maturation, transport, or retrotranslocation (21–26). An increased expression of GRP78 protein is a part of UPR required to alleviate ER stress, maintain ER function and protect cells against cell death. GRP78 is constitutively expressed, but its synthesis can be induced by a variety of stressful conditions that perturb protein folding and assembly within the ER including glucose deprivation, acidosis, hypoxia, conditions which are generally present in poorly vascularized solid tumors (21–26). The kinase activity of PAKs has been implicated in proliferative signaling by growth factor receptor kinases which in turn regulate cell survival, programmed cell death and malignant transformation (5–8). The dominant role of PI 3-kinase signaling in cell survival and proliferation is well documented (53). Akt a downstream effector of PI 3-kinase attenuates the apoptotic events by phosphorylating apoptotic proteases, apoptotic protein Bad and transcription factor FOX01/FOX02/FOX03 and thus promotes cell survival (53). In 1-LN cells α2M* promotes cellular proliferation by activating Ras/MAPK and PI 3-kinase signaling and protects cells from cell death by phosphorylating Bad at Ser112 and Ser136. Reorganizing of cytoskeleton is an essential feature of motility, detachment and invasion of cancer cells. Formation and stabilization of actin filaments provide the protrusive force for cellular extension of the leading edge of migratory cells (43–45). PAKs effect cytoskeletal reorganization via phosphorylating LIMK which phosphorylates cofilin. These events are essential in the dynamics of actin assembly and disassembly (5–8). It can be inferred from these studies that α2M*-mediated activation of PAK-2 confers migratory properties to these cells thus promoting their invasive and metastatic properties. PI 3-kinases and their products have also been implicated in the regulation of the cytoskeleton (5–8).
In conclusion we show here for the first time that binding of α2M* to cell surface associated GRP78 activates PAK-2 in highly metastatic and invasive prostate cancer cells. α2M*-induced activation of PAK-2 requires tyrosine phosphorylation of GRP78, activation of Ras/MAPK and PI 3-kinase downstream signaling, and recruitment of PAK-2 via the adaptor protein NCK to the plasma membrane. The increased expression of phosphorylated LIMK and cofilin confers motility, necessary for metastasis, in 1-LN cells. α2M* treatment of 1-LN cells also protects these cell from cell death by inhibiting the proapoptotic protein Bad.
Table 5.
Modulation of PAK-2 activation of 1-LN cells stimulated with α2M*
| Additions | By Kinase Activity (A.U. × 103) | By Western Blotting (A.U. × 103) |
|---|---|---|
| None | 105 ± 2 | 1080 ± 101 |
| α2M* (50 pM/10 min) | 302 ± 7 | 4217 ± 782 |
| Genestin (20 μM/16 min) then α2M | 154 ± 4 | 2459 ± 278 |
| LY294002 (20 μM/20 min) then α2M | 154 ± 7 | 2659 ± 356 |
| Wortmannin (30 μM/30 min) then α2M | n.d. | 2212 ± 380 |
| LY393511 (20 μM/20 min) then α2M* | n.d. | 3423 ± 103 |
n.d. - not done.
Values shown are the mean + S.E. and have been obtained by quantifying 2–3 autoradiography/immunoblots by Phosphorimager. Representative blots are shown in Figure A and B.
Footnotes
This work was supported by grant HL-24066 from the National Heart, Lung and Blood Institute.
The abbreviations are: α2M, α2-macroglobulin; α2M*, “activated” α2-macroglobuin which binds to cell surface receptors; α2MSR, α2M* signaling receptor; PAK-1, p21 activated protein kinase-1 (α); p21 activated protein kinase-2 (γ); GRP78, glucose regulated proteins 78; MAPKs, mitogen activated protein kinases; PI 3-kinase, phosphatidylinositol dependent protein kinase; LRP-1, low density lipoprotein receptor related protein-1; HHBSS, Hanks’ balanced salt solution; ECF; enhanced chemifluorescence; MBP, myelin basic protein; and A.U., arbitrary units.
References
- 1.Jemal A, Thomas A, Murray T, Thun M. CA Cancer J Clin. 2002;52:23–47. doi: 10.3322/canjclin.52.1.23. [DOI] [PubMed] [Google Scholar]
- 2.Heinlein CA, Chang C. Endocr Rev. 2004;25:276–308. doi: 10.1210/er.2002-0032. [DOI] [PubMed] [Google Scholar]
- 3.Montano X, Djamgoz M. FEBS Lett. 2004;571:1–8. doi: 10.1016/j.febslet.2004.06.088. [DOI] [PubMed] [Google Scholar]
- 4.Schlessinger J. Cell. 2000;103:193–200. [Google Scholar]
- 5.Sells MA, Chernoff J. Trends Cell Biol. 1997;7:162–167. doi: 10.1016/S0962-8924(97)01003-9. [DOI] [PubMed] [Google Scholar]
- 6.Daniels RH, Bokoch GM. Trends Biochem Sci. 1999;24:350–355. doi: 10.1016/s0968-0004(99)01442-5. [DOI] [PubMed] [Google Scholar]
- 7.Vadlamudi RK, Kumar R. Cancer Metastasis Reviews. 2003;22:385–393. doi: 10.1023/a:1023729130497. [DOI] [PubMed] [Google Scholar]
- 8.Bokoch GM. Annu Rev Biochem. 2003;72:743–81. doi: 10.1146/annurev.biochem.72.121801.161742. [DOI] [PubMed] [Google Scholar]
- 9.Walter BN, Huang Z, Jakobi R, Tuazon PT, Alnemri PT, Traugh JA. J Biol Chem. 1998;273:28733–28739. doi: 10.1074/jbc.273.44.28733. [DOI] [PubMed] [Google Scholar]
- 10.Jakobi R, McCarthy CC, Koeppel MA, Stringer DK. J Biol Chem. 2003;278:38675–38685. doi: 10.1074/jbc.M306494200. [DOI] [PubMed] [Google Scholar]
- 11.Roig J, Thazon PT, Zipfel PA, Pendergast AM, Traugh JA. Proc Natl Acad Sci USA. 2000;97:14346–14351. doi: 10.1073/pnas.97.26.14346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Salh B, Marrota A, Wagey R, Sayed M, Pelech S. Int J Cancer. 2002;98:148–154. doi: 10.1002/ijc.10147. [DOI] [PubMed] [Google Scholar]
- 13.Krieger M, Herz J. Annu Rev Biochem. 1994;63:601–637. doi: 10.1146/annurev.bi.63.070194.003125. [DOI] [PubMed] [Google Scholar]
- 14.Misra UK, Chu CT, Rubenstein DS, Gawdi G, Pizzo SV. Biochem J. 1993;290:885–291. doi: 10.1042/bj2900885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Misra UK, Chu CT, Gawdi G, Pizzo SV. J Biol Chem. 1994;269:18303–18306. [PubMed] [Google Scholar]
- 16.Misra UK, Gonzalez-Gronow M, Gawdi G, Pizzo SV. J Biol Chem. 1997;272:497–502. doi: 10.1074/jbc.272.1.497. [DOI] [PubMed] [Google Scholar]
- 17.Misra UK, Pizzo SV. Cell Signal. 2004;16:487–496. doi: 10.1016/j.cellsig.2003.09.010. [DOI] [PubMed] [Google Scholar]
- 18.Asplin IR, Misra UK, Gawdi G, Gonzalez-Gronow M, Pizzo SV. Arch Biochem Biophys. 2000;383:135–141. doi: 10.1006/abbi.2000.2052. [DOI] [PubMed] [Google Scholar]
- 19.Misra UK, Gonzalez-Gronow M, Gawdi G, Wang F, Pizzo SV. Cell Signal. 2004;16:929–938. doi: 10.1016/j.cellsig.2004.01.003. [DOI] [PubMed] [Google Scholar]
- 20.Misra UK, Gonzalez-Gronow M, Gawdi G, Hart JP, Johnson CE, Pizzo SV. Biol Chem. 2002;277:42082–42087. doi: 10.1074/jbc.M206174200. [DOI] [PubMed] [Google Scholar]
- 21.Kaufman RJ. Genes Dev. 1999;13:1211–1233. doi: 10.1101/gad.13.10.1211. [DOI] [PubMed] [Google Scholar]
- 22.Lee AS. Trends Biochem Sci. 2001;26:504–510. doi: 10.1016/s0968-0004(01)01908-9. [DOI] [PubMed] [Google Scholar]
- 23.Harding HP, Calfon M, Urano F, Novoa I, Ron D. Ann Rev Cell Dev Biol. 2002;18:575–579. doi: 10.1146/annurev.cellbio.18.011402.160624. [DOI] [PubMed] [Google Scholar]
- 24.Zhang K, Kaufman RJ. J Biol Chem. 2004;279:25935–25938. doi: 10.1074/jbc.R400008200. [DOI] [PubMed] [Google Scholar]
- 25.Rutkowski DT, Kaufman RJ. Trends Cell Biol. 2004;14:20–28. doi: 10.1016/j.tcb.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 26.Hendershot LM. Mount Sinai, J Med. 2004;71:289–297. [PubMed] [Google Scholar]
- 27.Bhattarcharjee G, Misra UK, Gawdi G, Cianciolo G, Pizzo SV. J Cell Biochem. 2001;82:260–270. doi: 10.1002/jcb.1152. [DOI] [PubMed] [Google Scholar]
- 28.Adam L, Vadlamudi R, Kondapaka SB, Chernoff J, Mendelsohn J, Kumar R. J Biol Chem. 1998;273:28238–28246. doi: 10.1074/jbc.273.43.28238. [DOI] [PubMed] [Google Scholar]
- 29.Hart JP, Gunn MD, Pizzo SV. J Immunol. 2004;172:70–78. doi: 10.4049/jimmunol.172.1.70. [DOI] [PubMed] [Google Scholar]
- 30.Mintz PJ, Kim J, Do KA, Wang X, Zinner RG, Cristofanilli M, Arap AM, Hong WK, Troncoso P, Logothetis CJ, Pasqualini R, Arap W. Nature Biotechnology. 2003;21:57–63. doi: 10.1038/nbt774. [DOI] [PubMed] [Google Scholar]
- 31.Arap MA, Lahdenranta J, Mintz PJ, Hajitou A, Sarkis AS, Arap W, Pasqualini R. Cancer Cell. 2004;6:275–284. doi: 10.1016/j.ccr.2004.08.018. [DOI] [PubMed] [Google Scholar]
- 32.Pizzo, S.V. and Wu, S.M.: Proteinase Inhibitors: α-Macroglobulins and Kunins In: Hemostasis and Thromboses. Basic Principles and Clinical Practice (Colman RW, Hirsh J, Marder VJ, and Salzman EW, eds.) Fourth Edition, pp. 367–386, J.B. Lippincott, Williams & Wilkins, 2000.
- 33.Otto A, Bar J, Bienmeier G. J Urol. 1998;159:297–303. doi: 10.1016/s0022-5347(01)64085-0. [DOI] [PubMed] [Google Scholar]
- 34.Zhang WM, Finne P, Leinonen J, Vesalainen S, Nordling S, Rannikko S, Stenman UH. Clin Chem. 1998;44:2471–2479. [PubMed] [Google Scholar]
- 35.Misra, U.K., Gonzalez-Gronow, M., Gawdi, G., and Pizzo, S.V. (2004) J. Immunol. (in press).
- 36.Lu W, Mayer BJ. Oncogene. 1999;18:797–806. doi: 10.1038/sj.onc.1202361. [DOI] [PubMed] [Google Scholar]
- 37.Bradford MM. Anal Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- 38.Laemmli UK. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 39.Knaus UG, Morris S, Dong H, Chernoff J, Bokoch GM. Science. 1995;269:221–223. doi: 10.1126/science.7618083. [DOI] [PubMed] [Google Scholar]
- 40.van Triest M, Rooif JD, Bos JL. Methods Enzymol. 2001;333:343–348. doi: 10.1016/s0076-6879(01)33068-9. [DOI] [PubMed] [Google Scholar]
- 41.Misra UK, Pizzo SV. J Biol Chem. 2002;277:4069–4078. doi: 10.1074/jbc.M109764200. [DOI] [PubMed] [Google Scholar]
- 42.Bokoch GM, Reilly AM, Daniels RH, King CC, Olivera A, Spiegel S, Knaus UG. J Biol Chem. 1998;273:8137–8144. doi: 10.1074/jbc.273.14.8137. [DOI] [PubMed] [Google Scholar]
- 43.Nobes CD, Hall A. Cell. 1995:53–62. doi: 10.1016/0092-8674(95)90370-4. [DOI] [PubMed] [Google Scholar]
- 44.Chambers AF, Groom AC, MacDonald IC. Nat Rev Cancer. 2002;2:563–572. doi: 10.1038/nrc865. [DOI] [PubMed] [Google Scholar]
- 45.Small JV, Rottner K, Kaverina I. Curr Opin Cell Biol. 1999;11:54–60. doi: 10.1016/s0955-0674(99)80007-6. [DOI] [PubMed] [Google Scholar]
- 46.Okano I, Hiraoka J, Otera H, Nunone K, Ohashi K, Iwashita S, Hairai M, Mizuno K. J Biol Chem. 1995;270:31321–31330. doi: 10.1074/jbc.270.52.31321. [DOI] [PubMed] [Google Scholar]
- 47.Yoshioka K, Foletta V, Bernard O, Itoh KC. Proc Natl Acad Sci USA. 2003;100:7247–7252. doi: 10.1073/pnas.1232344100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Davila M, Frost AR, Grizzle WF, Chakrabarti R. J Biol Chem. 2003;278:36868–36875. doi: 10.1074/jbc.M306196200. [DOI] [PubMed] [Google Scholar]
- 49.Edwards DC, Sanders LC, Bokoch GM, Gill GN. Nat Cell Biol. 1999;1:253–259. doi: 10.1038/12963. [DOI] [PubMed] [Google Scholar]
- 50.Strasser A, O’Connor L, Dixit VM. Ann Rev Biochem. 2000;69:217–245. doi: 10.1146/annurev.biochem.69.1.217. [DOI] [PubMed] [Google Scholar]
- 51.Reed JC. Oncogene. 1998;17:3225–3236. doi: 10.1038/sj.onc.1202591. [DOI] [PubMed] [Google Scholar]
- 52.Green DR, Reed JC. Science. 1998;281:1309–1312. doi: 10.1126/science.281.5381.1309. [DOI] [PubMed] [Google Scholar]
- 53.Datta SR, Brunet A, Greenberg ME. Genes Develop. 1999;13:2905–2927. doi: 10.1101/gad.13.22.2905. [DOI] [PubMed] [Google Scholar]
- 54.Lizcano JM, Morrice N, Cohen P. Biochem J. 2000;349:547–557. doi: 10.1042/0264-6021:3490547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schurmann A, Mooney AF, Sanders LC, Sells MA, Wang HG, Reed JC, Bokoch GM. Mol Cell Biol. 2000;20:453–461. doi: 10.1128/mcb.20.2.453-461.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]