

Abstract
Smith-Lemli-Opitz syndrome (SLOS) is an autosomal recessive disorder of cholesterol biosynthesis. It is caused by mutations in the gene encoding the enzyme 7-dehydrocholesterol Δ7-reductase (DHCR7), which catalyzes the final step in cholesterol biosynthesis, usually resulting in cholesterol deficiency. We report a 3.5-year-old girl who has cognition in the low average range and normal behavior, but in whom molecular studies identified two missense mutations in DHCR7: V326L and F284L. She was born at term following an uncomplicated pregnancy and delivery, and presented at 12 days of age with poor feeding, abdominal distention, and jaundice. Colonic biopsy was consistent with Hirschsprung disease. On physical examination she had mild ptosis, a long philtrum, mild micrognathia, a short, upturned nose, and subtle 2,3 syndactyly. Her 7-dehydrocholesterol (7-DHC) level was markedly elevated at 8.7 mg/dl (normal 0.10 ± 0.05), and her cholesterol level was normal at 61 mg/dl (normal for newborn period 50–80 mg/dl). Karyotype analysis was normal, 46,XX. Breast milk feeding was initiated and continued for 18 months. Cholesterol supplementation was implemented at 100 mg/kg/day at 3 months, which resulted in increased cholesterol levels and reduced dehydrocholesterol levels. Neuropsychological testing has shown functioning in the low average range, between the 14th and 18th centiles when compared to peers. This is markedly higher than most children with SLOS. She has no behavioral problems. MRI and MRS testing of the brain revealed no structural abnormalities. This is in contrast to a recently reported case by Prasad et al. [2002: Am J Med Genet 108:64–68] with a mild phenotype, behavioral problems, and abnormal MRI, who is compound heterozygote for both a null and missense mutation. Our case suggests that patients with severe feeding disorders with or without Hirschprung disease and postnatal onset microcephaly may warrant screening for SLOS.
Keywords: Smith-Lemli-Opitz syndrome, 7- and 8-dehydrocholesterol, DHCR7 mutations, Hirschsprung disease
INTRODUCTION
Smith-Lemli-Opitz syndrome (SLOS) is an autosomal recessive disorder of cholesterol biosynthesis. Reduced activity of 7-dehydrocholesterol Δ7-reductase (DHCR7), an enzyme that catalyzes a necessary step in cholesterol biosynthesis, leads to decreased levels of cholesterol and accumulation of the precursors 7-dehydrocholesterol (7-DHC) and 8-dehydrocholesterol (8-DHC) [Tint et al., 1994, 1995; Batta et al., 1995]. Mutations in the gene encoding DHCR7 have since been shown to cause SLOS [Fitzky et al., 1998; Wassif et al., 1998; Waterham et al., 1998; Yu et al., 2000].
The incidence of SLOS is estimated to range from 1 in 20,000 to 50,000 livebirths [Lowry and Yong, 1980; Kelley, 1998; Ryan et al., 1998]. The classic phenotypic manifestations of this malformation syndrome include; anteverted nostrils, ptosis of the eyelids, 2,3 toe syndactyly, ambiguous genitalia, hypospadias, and cryptorchidism in males. A more severe phenotype, which included Hirshsprung disease was described by Curry et al. [1987] and was initially classified as SLOS II, though it is now clear that SLOS I and SLOS II represent two ends of the same disorder. Definitive diagnosis is made by detecting elevated levels of 7-DHC in plasma or in post-mortem tissue samples. The spectrum in biochemically proven cases ranges from severe anomalies such as holoprosencephaly, cleft palate, postaxial polydactyly, pulmonary hypoplasia, complex congenital heart malformations, and renal agenesis, to the previously known mildest case recently reported by Prasad et al. [2002] with abnormal CNS findings of mild hypoplasia of the corpus callosum, prominent cisterna magna, hypoplastic cerebellum, enlargement of the lateral and third ventricles, and thick gyri, in a child with autistic features [Kelley et al., 1996; Cunniff et al., 1997; Nowaczyk et al., 1998; Loffler et al., 2000; Prasad et al., 2002].
The availability of cholesterol to the developing fetus appears to be one of the determinants of the severity seen in patients with SLOS. Mutations currently recognized in DHCR7 can be characterized as belonging to the following types: missense mutations in (a) transmembrane domains or their border areas (TM), (b) the fourth cytoplasmic loop (4L), (c) the C-terminus (C-T), and null mutations (0). Genotype–phenotype correlations have shown that patients with two TM mutations typically have less severe phenotypes while those homozygous for null mutations are the most severe. Variability has been seen within mutation classes, suggesting that factors in addition to the loss of DHCR7 activity play an important role in the availability of cholesterol and clinical severity [Cunniff et al., 1997; Fitzky et al., 1998; Witsch-Baumgartner et al., 2000; Yu et al., 2000; Prasad et al., 2002]. We report a case of a 3.5-year-old child with minimal physical manifestations of SLOS, who presented with Hirschsprung disease and subtle 2,3 syndactyly of the toes at 2 weeks of age. She has undergone developmental testing as well as behavioral assessments and is currently functioning in the low average range. She appears to have the mildest phenotype identified to date.
CLINICAL REPORT
The proposita was the product of a 38 week gestation to a 32-year-old gravida 4 para 3 white female born by induced vaginal delivery without complications. Amniocentesis was performed antenatally because a multiple marker screen of the mother showed low estriol. Prenatal karyotype was normal, 46,XX. The family history was significant for a previous stillborn delivered at 29 weeks gestation. Autopsy demonstrated intrauterine growth retardation, and no dysmorphic features were identified. Mother and a full sister report chronic constipation, but have no other significant medical history.
Her birth weight was 3,350 g and length was 50.8 cm, both 75th centile for age, and head circumference was 34 cm, 50th–75th centile for age. She presented on day 12 of life with poor feeding, abdominal distention, and jaundice. Colonic biopsy showed aganglionosis consistent with Hirschsprung disease. Her facial features were the most apparent at the initial exam at 2 weeks of age, but were subtle. She had mild ptosis, a short nose, anteverted nares, long philtrum, micrognathia, and 2,3 toe syndactyly which was borderline on the left and 1mm above significance on the right. Syndactyly of the second and third toes has been defined as significant when it extends beyond the proximal crease, or when it is greater than 1/3 the distance from the base of the toes to the tip (Dr. Marilyn Jones, personal communication). She had a normal female karyotype, 46,XX. Her plasma cholesterol level was initially normal at 61 mg/dl, but her 7-DHC level was elevated at 8.7 mg/dl, consistent with SLOS. Parental cholesterol levels were normal. Figures 1 and 2 demonstrate her appearance at 6 months and 3 years 5 months, respectively. The ptosis gradually became significantly less apparent on cholesterol supplementation.
Fig. 1.
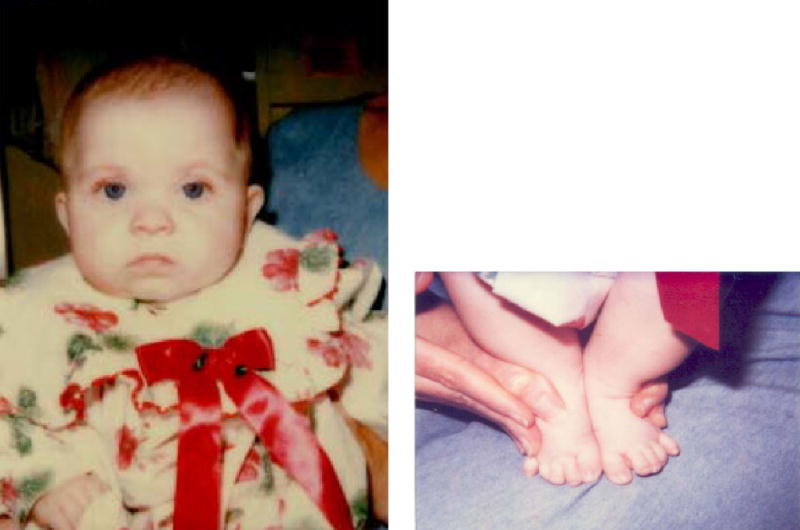
Proband at age 6 months. Left: Mild ptosis, anteverted nares, short nose. Right: 2,3 toe syndactyly.
Fig. 2.
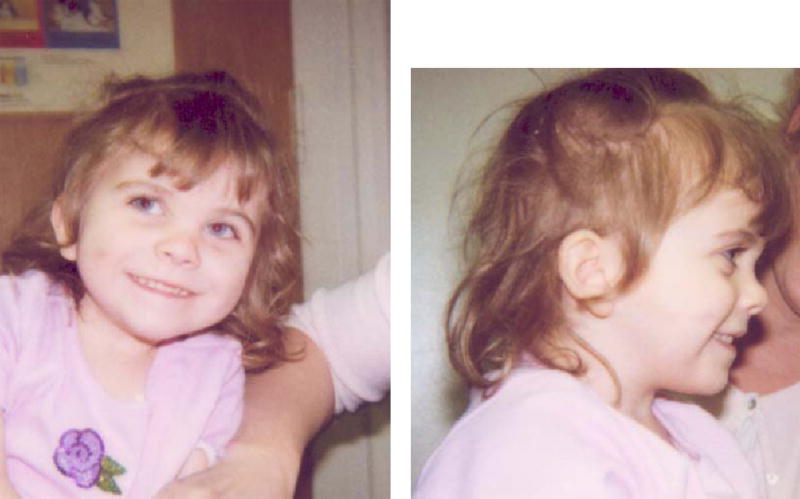
Proband at age 3 years 5 months (front and profile views).
Sigmoid colostomy was performed and breast milk feeding was initiated soon after. At 3 months of age, cholesterol supplementation was initiated at 100 mg/kg/day [Elias et al., 1997; Irons et al., 1997]. She has been maintained at this dose, and her cholesterol levels have remained in the normal ranges for age and each laboratory. Her 7-DHC levels decreased from 8.7 to 0.6 mg/dl (normal 0.037 ± 0.020 mg/dl) and 8-DHC levels from 8.0 to 0.6 mg/dl (normal 0.059 ± 0.038 mg/dl), (Figs. 3,4). Feeding problems, which were present initially, improved gradually over 18 months on cholesterol supplementation and with closure of colostomy. She has continued to do well, except for postnatal onset of microcephaly, which was noted prior to cholesterol supplementation, at 3 months of age (Fig. 5).
Fig. 3.
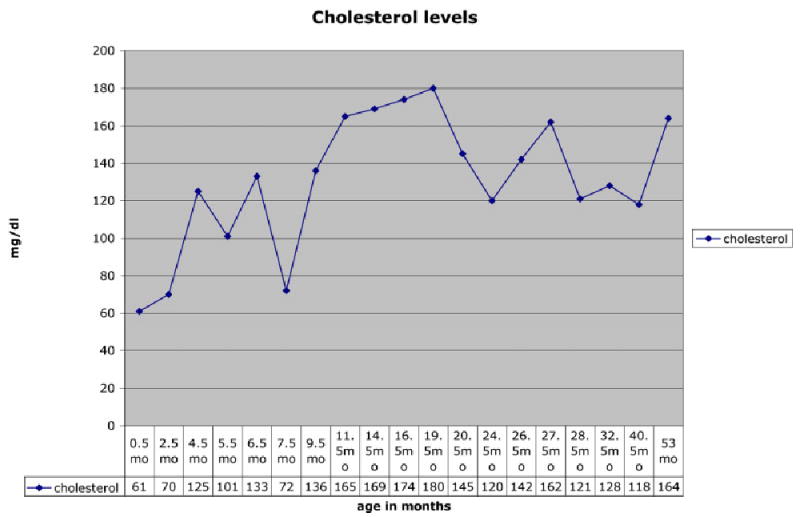
Cholesterol levels.
Fig. 4.
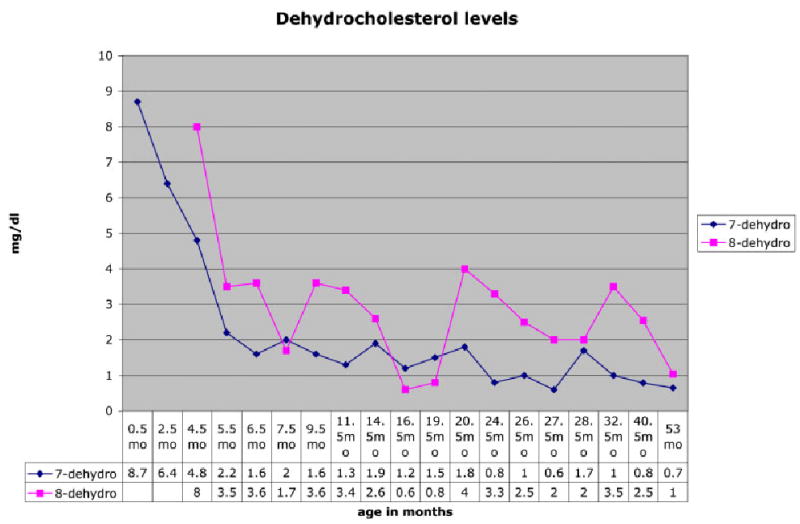
Dehydrocholesterol levels.
Fig. 5.
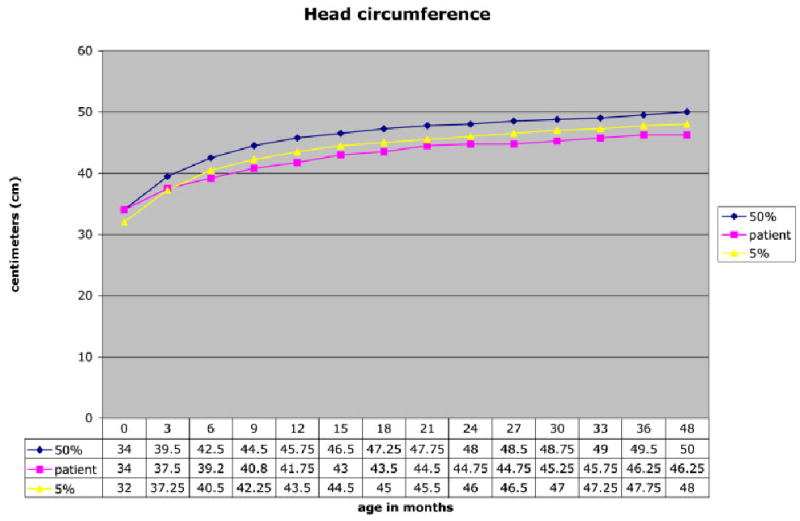
Head circumference.
Molecular analyzes of DHCR7 have subsequently identified that she is a compound heterozygote for two missense mutations [Yu et al., 2000]. These are F284L, just outside the TM domain on exon 8, and the more common V326L in the TM domain on exon 9 (Fig. 6).
Fig. 6.
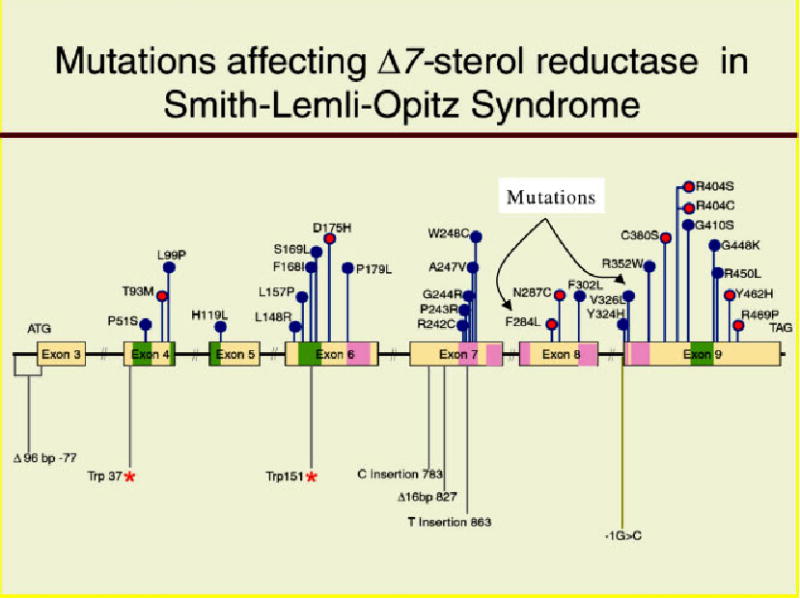
Mutations found in SLOS. Transmembrane domains are dark pink. Note proband’s mutations F284L and V326L.
Developmentally, she demonstrated mild delays in gross motor skills associated with mild hypotonia. She sat alone at 8 months, pulled to stand at 15 months, and walked without support at 20 months. Her first words were noted at 12 months. At 2 years, 9 months she was evaluated using the Bayley Scales of Infant Development (BSID-II) and the Vineland Adaptive Behavior Scales (VABS) (Fig. 7). Her cognitive, language, and motor abilities all were in the low average range for her age, between the 14th and 18th centiles. Her performance in all areas is markedly better than that of other children with SLOS. To date she has not shown the behavioral problems often seen in other children with SLOS, such as temper tantrums, motor stereotypes, sleep disturbance, or hypersensitivity to light and sound. MRI and MRS testing of the brain have revealed no structural abnormalities.
Fig. 7.
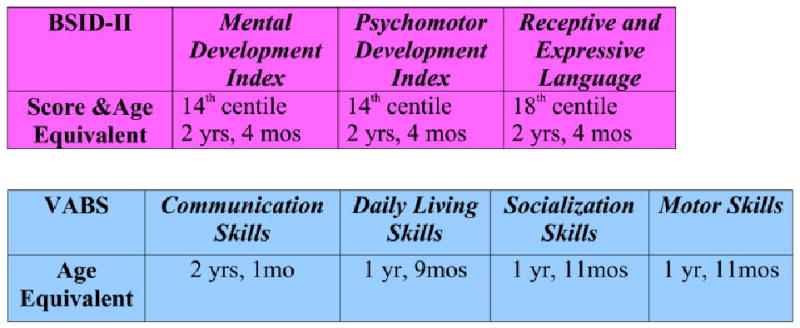
Developmental assessment at age 2 years and 9 months.
DISCUSSION
In a patient with the classic pattern of malformation, SLOS is relatively easy to recognize. However, since the discovery of the underlying biochemical defect and available testing, milder phenotypes have been recognized. Our patient had minimal physical findings, however, the findings of Hirshsprung disease and mild 2,3 syndactyly of the toes suggested the possible diagnosis. Previously though, SLOS patients with Hirschsprung have been reported as being severely affected, often consistent with SLOS II [Patterson et al., 1983; Kim et al., 1986; Lipson et al., 1995].
Our patient had an initial cholesterol level that was in the low normal range, but her 7-DHC level was markedly elevated, confirming the diagnosis of SLOS. Mutational analyzes demonstrating two TM mutations would predict that she would be mildly affected. We hypothesize, based on the normal cholesterol level and an elevated 7-DHC level at presentation, that her combination of mutations has resulted in higher residual enzyme activity than other mutation combinations. On cholesterol supplementation her sterol levels and feeding problems gradually improved. Our patient has cognitive, motor, and language abilities in the low average range for her age, and has no behavioral problems, which is atypical for SLOS patients. The impact of other genetic and environmental factors on the variability of the SLOS phenotype will most likely be better understood as more mildly affected individuals are identified.
Our patient extends the spectrum of mild phenotypes in SLOS, and suggests that cases far milder than those previously recognized exist. This patient could have remained undiagnosed if a genetics consultation had not been requested. Due to the frequent occurrence of more than one affected individual in extended pedigrees in nonconsanguineous couples, SLOS is thought to have a high carrier frequency. This fact also suggests that there are many mild patients in the community who have not been diagnosed.
Minimal diagnostic criteria to suggest laboratory testing for SLOS have not been established. Previous recommendations include testing of 7-DHC levels in children with developmental delays, especially in association with mild dysmorphic features suggestive of SLOS. We suggest that the diagnostic criteria for testing of 7-DHC include infants presenting with severe feeding abnormalities, with or without Hirschsprung disease, as well as the presence of significant 2,3 syndactyly of the toes. Postnatal onset of microcephaly along with any of these features may also warrant testing for SLOS. Early diagnosis, medical therapy, and developmental intervention in mild SLOS patients could lead to improved outcomes.
References
- Batta AK, Tint GS, Shefer S, Abuelo D, Salen G. Identification of 8-dehydrocholesterol (cholesta-5, 8-dien-3 β-ol) in patients with Smith-Lemli-Opitz syndrome. J Lipid Res. 1995;36:705–713. [PubMed] [Google Scholar]
- Cunniff C, Kratz L, Moser A, Natowicz M, Kelley R. Clinical and biochemical spectrum of patients with RSH/SLOS and abnormal cholesterol metabolism. Am J Med Genet. 1997;68:263–269. [PubMed] [Google Scholar]
- Curry C, Carey J, Holland J, Chopra D, Fineman R, Golabi M, Sherman S, Pagon R, Allanson J, Shulman S, Barr M, McGarvey V, Dabiri C, Schimke N, Ives E, Hall B. SLOS type II: Multiple congenital anomalies with male pseudohermaphroditism and frequent early lethality. Am J Med Genet. 1987;26:45–57. doi: 10.1002/ajmg.1320260110. [DOI] [PubMed] [Google Scholar]
- Elias E, Irons M, Hurley A, Tint GS, Salen G. Clinical effects of cholesterol supplementation in six patients with SLOS. Am J Med Genet. 1997;68:305–310. doi: 10.1002/(sici)1096-8628(19970131)68:3<305::aid-ajmg11>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- Fitzky B, Witsch-Baumgartner M, Erdel M, Lee J, Paik Y, Glossmann H, Utermann G, Moebius F. Mutations in the human sterol Δ7-sterol reductase gene in patients with the Smith-Lemli-Opitz syndrome. Proc Natl Acad Sci. 1998;95:8181–8186. doi: 10.1073/pnas.95.14.8181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irons M, Elias E, Abuelo D, Bull M, Greene C, Johnson V, Keppen L, Schanen C, Tint GS, Salen G. Treatment of SLOS: Results of a multicenter trial. Am J Med Genet. 1997;68:311–314. [PubMed] [Google Scholar]
- Kelley R. RSH/Smith-Lemli-Opitz syndrome: Mutations and metabolic morphogenesis. Am J Hum Genet. 1998;63:322–326. doi: 10.1086/301987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley R, Herman G. Inborn errors of sterol biosynthesis. Annu Rev Genomics Hum Genet. 2001;2:299–341. doi: 10.1146/annurev.genom.2.1.299. [DOI] [PubMed] [Google Scholar]
- Kim EH, Boutwell WC. Smith-Lemli-Opitz syndrome associated with Hirschsprung disease, 46XY female karyotype, and total anomalous pulmonary venous drainage. J Pediatr. 1985;106:861. doi: 10.1016/s0022-3476(85)80382-6. [DOI] [PubMed] [Google Scholar]
- Lipson A, Hayes A. Smith-Lemli-Opitz syndrome and Hirschsprung disease. J Pediatr. 1983;105:177. doi: 10.1016/s0022-3476(84)80406-0. [DOI] [PubMed] [Google Scholar]
- Loffler J, Trojovsky A, Casati B, Kroisel P, Utermann G. Homozygosity for the W151X mutation in the Δ7-sterol reductase gene (DHCR7) causing a lethal form of Smith-Lemli-Opitz syndrome: Retrospective molecular diagnosis. Am J Med Genet. 2000;95:174–177. doi: 10.1002/1096-8628(20001113)95:2<174::aid-ajmg16>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- Lowry R, Yong S. Borderline normal intelligence in the Smith-Lemli-Opitz syndrome. Am J Med Genet. 1980;5:137–143. doi: 10.1002/ajmg.1320050205. [DOI] [PubMed] [Google Scholar]
- Nowaczyk M, Whelan D, Hill R. Smith-Lemli-Opitz syndrome: Phenotypic extreme with minimal clinical findings. Am J Med Genet. 1998;78:419–423. doi: 10.1002/(sici)1096-8628(19980806)78:5<419::aid-ajmg5>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- Patterson K, Toomey K, Chandra R. Hirschsprung’s disease in a 46,XY female phenotypic infant girl with Smith-Lemli-Opitz syndrome. J Pediatr. 1983;103:425. doi: 10.1016/s0022-3476(83)80422-3. [DOI] [PubMed] [Google Scholar]
- Prasad C, Marles S, Prasad A, Nikkel S, Longstaffe S, Peabody D, Eng B, Wright S, Waye J, Nowaczyk M. SLOS: New mutations with a mild phenotype. Am J Med Genet. 2002;108:64–68. doi: 10.1002/ajmg.10211. [DOI] [PubMed] [Google Scholar]
- Ryan A, Bartlett K, Clayton P, Eaton S, Mills L, Donnai D, Winter R, Burn J. Smith-Lemli-Opitz sydrome: A variable clinical and biochemical phenotype. J Med Genet. 1998;35:558–565. doi: 10.1136/jmg.35.7.558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tint GS, Irons M, Roy E, Batta S, Frieden R, Chen T, Salen G. Defective cholesterol biosynthesis associated with the SLOS. NEJM. 1994;330:107–113. doi: 10.1056/NEJM199401133300205. [DOI] [PubMed] [Google Scholar]
- Tint GS, Seller M, Hughes-Benzie R, Batta AK, Shefer S, Genest D, Irons M, Elias E, Salen G. Markedly increased tissue concentrations of 7-dehydrocholesterol combined with low levels of cholesterol are characteristic of the Smith-Lemli-Opitz syndrome. J Lipid Res. 1995;36:705–713. [PubMed] [Google Scholar]
- Wassif C, Maslen C, Kachilele-Linjewile S, Lin D, Linck L, Connor W, Steiner R, Porter F. Mutations in the human. Am J Hum Genet. 1998;63:55–62. doi: 10.1086/301936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterham H, Wijburg F, Hennekam R, Vreken P, Poll-The B, Dorland L, Duran M, Jira P, Smeitink J, Wevers R, Wanders R. Smith-Lemli-Opitz syndrome is caused by mutations in the 7-dehydrocholesterol reductase gene. Am J Hum Genet. 1998;63:329–338. doi: 10.1086/301982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witsch-Baumgartner M, Fitzky B, Ogorelkova M, Kraft H, Moebius F, Glossman H, Seedorf U, Gillessen-Kaesbach G, Hoffmann G, Clayton P, Kelley R, Utermann G. Mutational spectrum in the Δ7-sterol reductase gene and genotype–phenotype correlation in 84 patients with Smith-Lemli-Opitz syndrome. Am J Hum Genet. 2000;66:402–412. doi: 10.1086/302760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H, Lee M, Starck L, Elias E, Irons M, Salen G, Patel S, Tint GS. Spectrum of Δ7-dehydrocholesterol reductase mutations in patients with SLOS. Hum Mol Genet. 2000;9:1385–1391. doi: 10.1093/hmg/9.9.1385. [DOI] [PubMed] [Google Scholar]