

Abstract
Drug-induced toxicity is an important issue in clinical medicine, which typically results in organ dysfunction and adverse health consequences. The family of NOD-like receptors (NLRs) includes intracellular proteins involved in recognizing pathogens and triggering innate immune responses, including the activation of the NLRP3 inflammasome. The NLRP3 (nucleotide-binding oligomerization domain-like receptor family, pyrin domain-containing 3) inflammasome is a critical component for both innate and adaptive immune responses and has been implicated in various drug-induced toxicities, including hepatic, renal, and cardiovascular diseases. The unusual activation of the NLRP3 inflammasome causes the release of pro-inflammatory cytokines, such as IL-1β and IL-18, which can lead to more damage to tissues. Targeting NLRP3 inflammasome is a potential therapeutic endeavour for suppressing drug-induced toxicity. This review provides insights into the mechanism, drug-induced organ toxicity, therapeutic strategies, and prospective therapeutic approaches of the NLRP3 inflammasome and summarizes the developing therapies that target the inflammasome unit. This review has taken up one of the foremost endeavours in understanding and inhibiting the NLRP3 inflammasome as a means of generating safer pharmacological therapies.
This review aims to shed light on how drugs cause toxicity and summarizes developing therapies and prospective therapeutic approaches that will target the NLRP3 inflammasome unit.
Introduction
Drug-induced toxicity is a serious challenge in clinical medicine and often leads to severe organ damage and limits the therapeutic potential of many drugs.1 Adverse drug reactions can affect many organs, such as the liver, kidney, cardiovascular system, and nervous system, resulting in acute or chronic inflammation, which further aggravates the tissue damage and acts as a driving factor for disease progression.1,2 The NLRP3 (NOD-, LRR-, and pyrin domain-containing protein 3) inflammasome is a fundamental part of the innate immune system, which serves as a cytosolic sensor for danger signals and initiates inflammation (Fig. 1).3 It is crucial for defending the body, but it is also associated with many inflammatory and metabolic conditions. Drug-induced organ toxicity is one of the diseases that arise from inflammation due to abnormal NLRP3 activation.4 To devise effective treatments, it is important to comprehend its structure, how it is activated, and the underlying mechanism of possible therapeutic implications.5 Interactions between proteins within the NLRP3 (nucleotide binding domain and leucine-rich repeat protein-3) molecules are coordinated by three vital domains, among which the NACHT domain facilitates ATP-dependent oligomerization of the pyrin domain (PYD) and is responsible for ASC (apoptosis-associated speck-like protein containing a CARD) recruitment.6 The activation sequence for NLRP3 is accomplished via two essential steps. The first step is priming the signal (signal-1), which is accomplished by toll-like receptors (TLRs) or cytokines that cause the inflammation, such as TNF-α and IL-β, which activate NF-κB signalling to increase pro-IL-1β, pro-IL-18, and NLRP3 expressions.7 Second activation (signal 2) happens when NLRP3 is activated by stressors of the cell, such as ATP, destruction of mitochondria, ROS, and changes in the ion movement (Fig. 2).7 Targeting the NLRP3 inflammasome may offer a revolutionary pharmacological strategy to avert drug-induced toxicity and minimize inflammation-mediated organ damage.8 Several pharmacological inhibitors, including MCC950, glibenclamide, and natural bioactive compounds, have been investigated for their ability to suppress NLRP3 activation.9 Repurposing existing anti-inflammatory drugs, gene therapy, and RNA-based approaches are emerging as novel strategies to regulate inflammasome activity (Fig. 2).10 This review examines the role of the NLRP3 inflammasome in drug-induced toxicity. Some medications have the unintended effect of triggering the NLRP3 inflammasome.11 This can result in organ damage owing to extreme inflammatory reactions. Notable instances include the liver, kidney, heart, and brain damage.12 The NLRP3 inflammasome is one of the grand mediators of inflammatory responses that will definitely play a role in understanding drug-induced organ toxicities.1 Discerning the activation mechanisms facilitates the development of targeted therapies.13 Other strategies including small molecule inhibitors, biologics, and lifestyle interventions can help stop the diseases driven by the inflammasome.13 Future work on precision medicine, novel drug delivery systems, and gene editor technologies will refine the way therapies are delivered while minimizing the chance of toxicity. This review provides insight into the mechanisms underlying drug-induced toxicity, with a particular focus on the role of the inflammasome. It presents an overview of drug structures and mechanisms, highlights organ-specific toxicities, and explores current therapeutic strategies. Furthermore, it outlines future directions for treatment development, emphasizing emerging therapies that target the inflammasome pathway.
Fig. 1. Two-step model of NLRP3 inflammasome activation. The process of NLRP3 inflammasome activation occurs in two steps: (1) priming and (2) activation. In the priming step, pattern recognition receptors such as toll-like receptors (TLRs) and cytokines stimulate the NF-κB signaling pathway, leading to upregulation of inflammasome components. In the activation step, various signals activate the NLRP3 protein, which oligomerizes and recruits ASC (apoptosis-associated speck-like protein containing a CARD) to form the inflammasome complex. This results in cytokine release (e.g., IL-1β and IL-18) and pyroptotic cell death.

Fig. 2. This diagram depicts how the immune system recognises infections and danger signals, resulting in inflammation and cell death to combat pathogens. In step 1 (priming), TLRs, cytokines, and NF-κB signalling activate inflammasome components, preparing the cell. Step 2 (activation): various cellular stress signals cause NLRP3 inflammasome formation, which results in cytokine release and pyroptosis.

NLRP3 inflammasome assembly and activation
NLTRP3 is a key component of the innate immune system that is responsible for detecting pathogenic microorganisms, danger-associated molecular patterns (DAMPs), and environmental irritants.14 An activated NLRP3 triggers an inflammatory response by inducing pro-inflammatory cytokines interleukin 1β (IL-1β) and IL-18 maturation and promoting pyroptotic cell death.15 The NLRP3 inflammasome is a multimeric protein complex that is concerned with the NLRP3 protein (Fig. 2).16 NLRP3 is a pattern recognition receptor (PRR) that senses triggers, while ASC is an adaptor protein that bridges NLRP3 and caspase-1.17 Caspase-1 is a protease that processes pro-IL-1β and pro-IL-18 into their active forms and induces pyroptosis.17
Mechanism of NLRP3 inflammasome in pathogenesis and activation
The NLRP3 inflammasome has emerged as a central player in the pathogenesis of various chronic inflammatory diseases due to its ability to sense a wide array of sterile danger-associated molecular patterns (DAMPs). Its aberrant or prolonged activation promotes tissue injury and exacerbation of numerous non-infectious disorders, including gout, atherosclerosis, and metabolic dysfunction-associated steatohepatitis (MASH). Gout serves as the prototypic NLRP3-driven disease, where monosodium urate (MSU) crystals incite macrophage and neutrophil assembly of the NLRP3 inflammasome complex. This is ultimately responsible for the activation of caspase-1, maturation of IL-1β, and an explosive inflammatory response, culminating in painful joint inflammation. Targeting the NLRP3-IL-1β path for the treatment of acute gout flares has been shown to be beneficial. In the case of atherosclerosis, cholesterol crystals act as endogenous activators of the NLRP3 inflammasome in macrophage-derived foam cells in atherosclerotic plaques. Such activation promotes IL-1β secretion, endothelial dysfunction, and smooth muscle cell proliferation to promote plaque formation and instability. The CANTOS trial provided evidence of the clinical relevance of inflammasome modulation in atherosclerosis when it showed that IL-1β inhibition with canakinumab resulted in fewer recurrent cardiovascular events. In MASH, hepatic lipid accumulation, oxidative stress, and mitochondrial dysfunction can all activate the NLRP3 inflammasome in hepatocytes and Kupffer cells, resulting in hepatic inflammation, cell death, and fibrogenesis. Preclinical studies of pharmacologic inhibition or genetic deletion of NLRP3 alleviate steatohepatitis and fibrosis, suggesting an attractive therapeutic strategy to prevent MASH progression. In summary, these examples highlight the role of NLRP3 inflammasome activation in the pathology of chronic disease and provide a rationale for its targeting in inflammatory disease states that are not infectious or autoimmune.
The steps for NLRP3 assembly and activation are initiated by various menace signals causing cellular stress, leading to NLRP3 assembly.18 Unlike other inflammasomes that directly bind different ligands, NLRP3 is activated indirectly through different cellular disturbances instead of binding to a single ligand.19 Mitochondrial dysfunction and the production of reactive oxygen species are limited and enhanced through cellular stress due to toxins, hypoxia, or infections; potassium efflux due to bacterial toxins, ATP, or various crystalline substances (e.g., silica and uric acid) is one of the biggest triggers that provoke NLRP3 activation (Table 1).20 Ca2+ influx and sodium influx bubbling in calcium and sodium homeostasis propel inflammasome activation.5 Lysosomal damage and cathepsin release occur due to phagocytosis of crystalline substances and liberate cathepsins, which activate NLRP3 inflammasome assembly. Once activated, NLRP3 oligomerizes and recruits ASC, an apoptosis-associated speck-like protein containing a caspase recruitment domain.21 ASC acts as an adaptor protein that brings pro-caspase-1 together, forming the fully developed inflammasome complex (Fig. 2).22 Caspase-1 is then cleaved into its active form, triggering a downstream inflammatory response. Once the inflammasome is fully activated, it drives key inflammatory and cell death processes.23 Caspase-1 also activates gasdermin D (GSDMD), forming membrane pores that lead to pyroptotic cell death, cell lysis, and further inflammation.24 The NLRF3 inflammasome is activated on the basis of signals eliciting cellular stress, transcriptional priming, and assembly of the inflammasome, all of which are tightly regulated processes.25 Its dysregulation is known to cause various inflammatory diseases and toxicity that lead to different drug interactions, establishing it as a powerful target in diseases linked to inflammation.26 The NLRF3 inflammasome is very tightly controlled and regulated. It is activated through cellular stress signals, transcriptional priming, and assembly of the inflammasome (Fig. 2).3 Its dysregulation has been shown to be implicated in many inflammatory diseases and toxicity excreted by drugs, making it a valuable therapeutic target in inflammation-related disorders.27 The key mechanisms of NLRP3 inflammasome activation are shown in Table 1.
Table 1. Key mechanisms of NLRP3 inflammasome activation.
| S. N. | Activation mechanism | Description | Key triggers | References |
|---|---|---|---|---|
| 1 | Canonical pathway | Requires two signals: priming (via NF-κB) and activation (via danger signals) | PAMPs (LPS, bacterial toxins), DAMPs | 28 |
| 2 | Non-canonical pathway | Involves caspase-11 (mouse) or caspase-4/5 (human) activation by intracellular LPS | Intracellular Gram-negative bacteria | 29 |
| 3 | Efflux of potassium ions (K+) | Decrease in intracellular efflux of potassium ions triggers NLRP3 oligomerization | ATP-P2X7 receptor activation | 30 |
| 4 | Calcium (Ca2+) signaling | Increased cytosolic calcium (Ca2+) signaling enhances inflammasome activation | Endoplasmic reticulum stress, ionophores | 31 |
| 5 | Lysosomal rupture | Release of cathepsins and DAMPs from damaged lysosomes | Silica, cholesterol crystals, amyloid-β | 32 |
| 6 | Mitochondrial dysfunction | ROS production and mitochondrial damage promote NLRP3 activation | Oxidative stress, viral infections | 33 |
| 7 | ATP release and P2X7 activation | Extracellular ATP binds to the P2X7 receptor, triggering efflux of potassium ions | Cellular damage, inflammation | 34 |
| 8 | NEK7 interaction | NEK7 binds NLRP3 to promote its oligomerization | ROS, ion flux | 5 |
Structure of NLRP3
NLRP3 is an essential component of the NLRP3 inflammasome, a multiprotein complex that activates inflammatory responses. It consists of three structurally distinct domains: the pyrin domain (also known as the apoptosis-associated speck-like protein), the nucleotide-binding domain (NACHT/NOD domain), which is involved in ATP hydrolysis and is critical for oligomerization and activation, and the leucine-rich repeat (LRR) domain, which is thought to be involved in ligand sensing and autoregulation.17 Cryo-electron microscopy (cryo-EM) analyses revealed that NLRP3 reaches an octameric open structure, exhibiting a high rotation of approximately 90° at the NACHT domain.35 It is supposed that this structural switch is of utmost importance for activation.36 While inactive, NLRP3 exists in a closed conformation. At its activation, it undergoes structural rearmaments that support oligomerization, which favours the assembly of the inflammasome complex.37 This assembly is necessary for the recruitment and activation of downstream effector proteins, contributing to the inflammatory response.17
Several studies have been conducted on the various binding modes of inhibitors to NLRP3 with the goal of finding therapeutic agents for NLRP3-related diseases.38 For example, during studies on MCCC950, the inhibitors were found to bind to the cavity in the NACHT domain, away from the nucleotide-binding site, stabilizing NLRP3 in its closed form, rendering it inactive.39 Several inhibitors have also been identified in structural studies that include NP3-562 binding to the NACHT domain of NLRP3.40 The C-terminal domain is the leucine-rich repeat (LRR) domain, which is postulated to be a regulatory domain.41 The full-length structure of human NLRP3 is still not completely studied; cryo-EM (cryo-electron microscopy) and X-ray crystallography have provided structural models that inform its different conformational states.42 The NACHT domain is targeted by NLRP3 inhibitors, which are responsible for ATP binding and oligomerization.43 The main modes of inhibition are blockade of ATP, stabilization of the inactive confirmation, or targeting of allosteric sites.44 Depending on the inhibitors, many small molecule inhibitors, such as MCC950, block the activation of NLRP3 by binding to the area near the ATP-binding pocket.45 Since MCC950 is a well-studied selective small-molecule inhibitor of the NLRP3-inflammasome, a brief summary of its characteristics and relevance are provided. MCC950 inhibits the NLRP3 inflammasome through its effect on the NACHT domain, which must occur prior to NLRP3 oligomerization and activation. It does not have effects on other inflammasomes such as NLRC4 or AIM2, and is rather selective for NLRP3 activity. It inhibits caspase-1 activation and the cleavage and release of inflammatory cytokines IL-1β and IL-18, and pyroptosis (inflammatory cell death). This has been demonstrated in numerous preclinical models of NLRP3-mediated diseases including gout, atherosclerosis, type 2 diabetes, Alzheimer's disease, multiple sclerosis, Parkinson's disease, non-alcoholic steatohepatitis (NASH/MASH), and inflammatory bowel disease. Other inhibitors make it stable in closed, inactive monomers and stop it from switching into the active oligomeric form. Inhibitors that bind allosterically far away from the ATP-binding pocket are other novel inhibitors that display indirect modulation of NLRP3 activity.46 MCC950 binds to the Walker B motif in the NACHT domain, inhibiting its activation.47 OLT7 is an allosteric target that inhibits ATP hydrolysis. New insights gained into NLRP3 structure and interaction with various inhibitors form a basis for further rational design of small-molecule therapeutics for NLRP3-associated inflammatory disease.43 Increased understanding of the structure of NLRP3, along with important motifs and roles is central to understanding its function in immune response.17 OLT1177 (dapansutrile) is one of the most clinically advanced NLRP3 inhibitors, demonstrating potential as a safe, oral anti-inflammatory agent for a range of acute and chronic inflammatory conditions. Preclinical and early clinical studies suggest that OLT1177 has anti-inflammatory properties that are especially beneficial in diseases that release excessive IL-1β and IL-18. The development of OLT1177 is critical considering the limitations of older molecules like MCC950. INF39 is a synthetic, small-molecule compound characterized as a direct and irreversible inhibitor of the NLRP3 inflammasome. INF39 has been investigated in preclinical studies, particularly in models of inflammatory bowel disease and colitis. Its irreversible mode of action is especially appealing for clinical indications that require sustained inhibition of the inflammasome. INF39 is still in preclinical development and has yet to be tested in human clinical trials. There may be long-term safety concerns with irreversible inhibitors if there are off-target effects discovered. Additional studies are warranted to explore INF39 pharmacokinetics, bioavailability, and organ-specific effects. Sodium-glucose cotransporter 2 (SGLT2) inhibitors are a class of anti-diabetic medications with numerous systemic benefits, including the indirect regulation of NLRP3 inflammasome activation and IL-1β production. Although SGLT2 inhibitors were not originally designed to target inflammation, growing evidence supports their anti-inflammatory and cardio-renal protective properties, partly attributed to the inhibition of NLRP3 activation. GLP-1 receptor agonists can indirectly suppress NLRP3 by improving insulin sensitivity and reducing DAMPs. Statins are likely to modulate cholesterol crystal formation and intracellular ROS, and change the priming events associated with NLRP3 activation. These changes add more clarity for readers and initially prevent possible misinterpretation regarding special targeting of NLRP3 therapies. Through in-depth analysis of structure and function, researchers might uncover therapeutic targets for diseases linked to NLRP3 regulation problems (Table 2).48
Table 2. In-depth breakdown of NLRP3 structure, including key motifs and their roles.
| S. N. | Domain | Location | Function | Key interactions | Structural features | References |
|---|---|---|---|---|---|---|
| 1 | Pyrin domain (PYD) | N-terminal (1–92) | Facilitates protein–protein interactions via homotypic PYD–PYD binding; essential for ASC recruitment | Binds ASC to form an inflammasome complex | Alpha-helical fold: death domain super family | 49 |
| 2 | Walker A motif (P-loop) | NACHT domain | Binds ATP and facilitates hydrolysis for conformational changes | Essential for ATPase activity | Conserved GxxxxGKT/S sequence | 50 |
| 3 | Walker B motif | NACHT domain | Coordinates Mg2+ and hydrolyzes ATP to drive NLRP3 activation | Required for ATPase-dependent activation | Contains hydrophobic residues and aspartate | 51 |
| 4 | Sensor II motif | NACHT domain | Regulates ATPase activity by stabilizing the protein conformation | Modulates ATP hydrolysis | Found near Walker B motif | 52 |
| 5 | Nucleotide-binding and oligomerization domain (NACHT) | Central (120–436) | ATPase activity; responsible for NLRP3 oligomerization and activation | Interacts with NEK7 for oligomerization | Contains Walker A/B motifs for ATP binding | 53 |
| 6 | Helical domain (HD1 & HD2) | Between NACHT and LRR | Acts as a structural bridge between NACHT and LRR, involved in NLRP3 conformational dynamics | Modulates auto inhibition | Alpha-helical bundles | 17 |
| 7 | Leucine-rich repeat (LRR) domain | C-terminal (600–1036) | Regulates NLRP3 activation, possibly through auto inhibition by folding onto NACHT domain | Binds various PAMPs/DAMPs, interacts with NEK7 | Composed of tandem leucine-rich repeats | 54 |
NLRP3 inflammasome pathway inhibitors
In the innate immune arm, the NLRP3 inflammasome consisting of the NOD-LL- and pyrin domain-containing protein-3 aggregates pro-inflammatory responses upon sensing danger signals, culminating in the secretion of pro-inflammatory cytokines such as IL-1β and IL-18.55 Aberrantly activated NLRP3 has been implicated in syndromes and cardiovascular disorders.56 MCC950 or CRID3 are the most selective NLRP3 inhibitors known, and inhibit NLRP3 by blocking ATPase activity.57 OLT1177, or dapansutrile, is an oral small-molecule NLRP3 inhibitor that prevents ATP binding to NLRP3.58 Oridonin is a natural compound that covalently modifies NLRP3 and thereby inhibits its activation.59 CY-09 is a small molecule inhibitor that interacts with NLRP3 and thereby inhibits its activation.60 CY-09 is a small molecule inhibitor that interacts with NLRP3's NACHT domain. Inhibitors located upstream (blocking priming signals) halt the pathway of NF-κB activation, which halts the up-regulation of NLRP3 and pro-IL-1β activation.61 Parthenolide is a sesquiterpene lactone with the ability to inhibit NF-κB activity and prevent subsequent caspase-1 activation.62 Celastrol is a triterpene that inhibits both NF-κB and the NLRP3 inflammasome.63 Downstream inhibitors target IL-1β and caspase-1 to prevent the release or action of IL-1β and IL-18.64 Anakinra is a recombinant IL-1 receptor antagonist that blocks IL-1β signalling. Canakinumab is a monoclonal antibody neutralizing IL-1β.65 VX-765 is a caspase-1 inhibitor preventing IL-1β maturation. There are some indirect modulators that target cellular stress and ion flux and lead to NLRP3 activation.66 DMT (diethyl fumarate) is a modulator of cellular stress responses that results in the inhibition of NLRP activation. Hydroxybutyrate (BHB) is a ketone body that blocks K+ efflux and NLRP3 activation.67 The NLRP3 inflammasome is an important component of the innate immune system, and over activation has been linked to a variety of illnesses.68 Several inhibitors have been discovered and evaluated in NLRP3-dependent disease models.69 The noteworthy NLRP3 inflammasome inhibitors, their targets, and mechanisms are shown in Table 3.
Table 3. Some important NLRP3 inflammasome inhibitors, their targets, and mechanisms.
| S. N. | Inhibitor | Structure | Target(s) | Documented mechanism(s) | References |
|---|---|---|---|---|---|
| 1 | Glyburide |

|
NLRP3 (indirect action) | Abrogation of ASC agglomeration acting downstream of P2X7 and inhibition of ATP-sensitive potassium (K(ATP)) channels | 70 |
| MCC950 |

|
NLRP3 | Specifically targets and inhibits the ATP-hydrolysis motif in both canonical and non-canonical NLRP3 inflammasomes | 71 | |
| CY-09 |
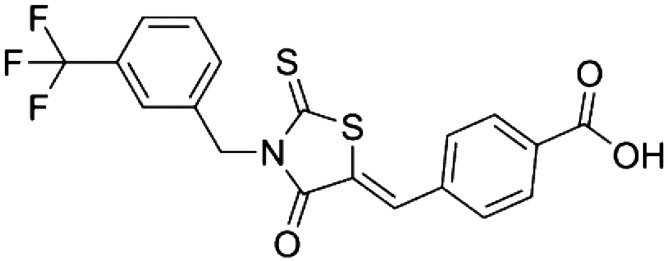
|
NLRP3 | Effective and direct suppressor of the NLRP3 inflammasome; inhibits NLRP3 ATPase activities | 72 | |
| Edaravone |

|
NLRP3 | Reduces reactive oxygen species (ROS) and reduces NLRP3-induced IL-1β, caspase-1, and NF-κB-mediated inflammation signaling | 73 | |
| Arsenic trioxide |

|
NLRP3 | Reduces NLRP3 inflammasome activation and subsequent IL-1β and IL-18 release | 74 | |
| Colchicine |

|
NLRP3 | Reduces IL-1β, IL-6, and IL-18 levels by inhibiting the activation of the NLRP3 inflammasome | 75 | |
| Metformin |
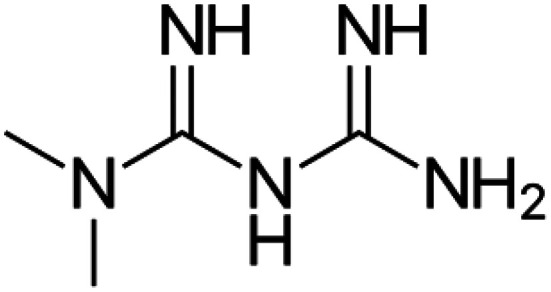
|
NLRP3 | Reduces NLRP3 expression and activates the NLRP3 inflammasome signalling pathway through AMPK regulation | ||
| Liraglutide |

|
NLRP3 (hepatic) | Repression of the liver's NLRP3 inflammasome | 76 | |
| Atorvastatin |

|
NLRP3 | Reduces NLRP3, caspase-1, and IL-1β levels while suppressing NF-κB signalling and lowering inflammatory cytokines | 77 | |
| Dapagliflozin |
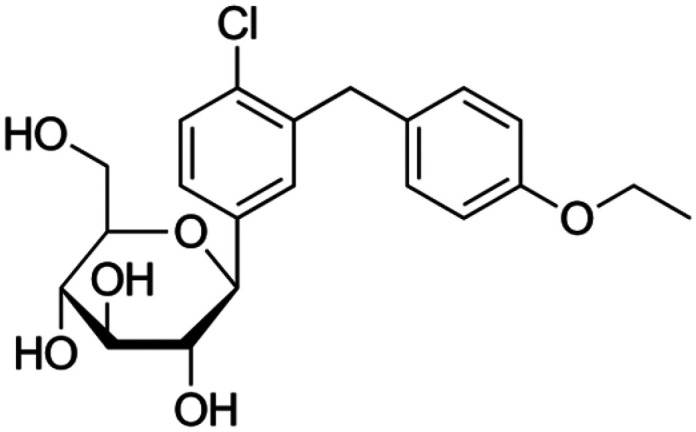
|
NLRP3 | Reduces inflammation-induced kidney damage by inhibiting NLRP3 inflammasome activation; requires AMPK activation | 78 | |
| Empagliflozin |
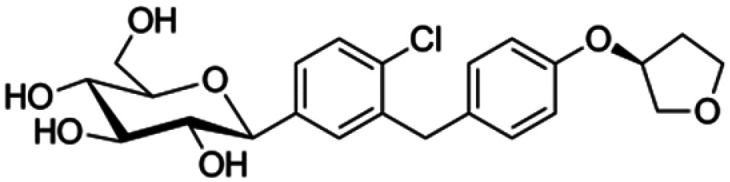
|
NLRP3 | Inhibits the activation of the NLRP3 inflammasome and decreases downstream inflammatory signalling in diabetic kidneys | 79 | |
| Ticagrelor |

|
NLRP3 | Represses NLRP3 inflammasome activation; requires AMPK activity | 80 | |
| Allopurinol |
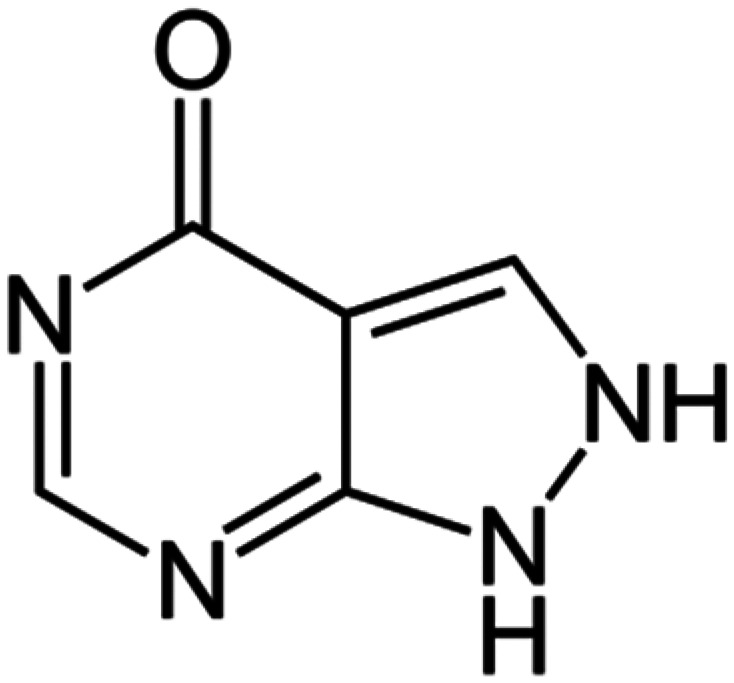
|
NLRP3, XOD | Suppresses xanthine oxidase activity, lowering uric acid and ROS formation, which are known to activate the NLRP3 pathway | 81 | |
| BAY11-7082 |

|
NLRP3, IKK, E2/3 enzymes, PTPs | Leads to cysteine alkylation of the NLRP3 inflammasome ATPase domains; inhibits NLRP3 ATPase activities | 82 | |
| MNS |

|
NLRP3 | Leads to cysteine alteration of NLRP3 inflammasome ATPase domains; represses NLRP3 inflammasome actions | 83 | |
| INF39 |

|
NLRP3 | Abrogates NLRP3 inflammasome ATPase activities and suppresses priming | 84 | |
| EMD638683 |

|
NLRP3 | Suppression of NLRP3 and IL-1β expression | 85 | |
| FC11A-2 |

|
NLRP3 (indirect effect) | Reduces pro-caspase-1 autocleavage and inhibits IL-1β/18 release | 86 | |
| Dapansutrile (OLT1177) |
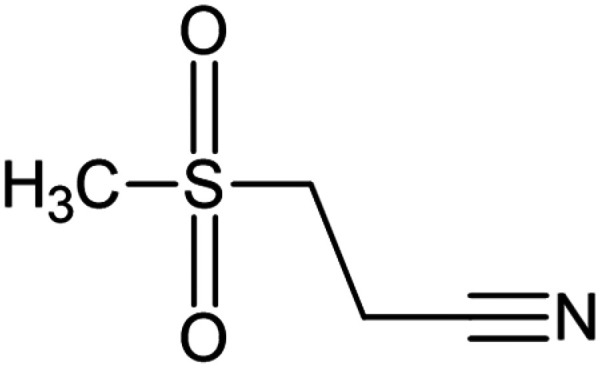
|
NLRP3 | Inhibits NLRP3 inflammasome ATPase activity and reduces NLRP3 inflammasome activation | 87 | |
| BOT-4-one |

|
NLRP3 | Inhibits NLRP3 ATPase activity; prevents priming | 88 | |
| Tranilast |
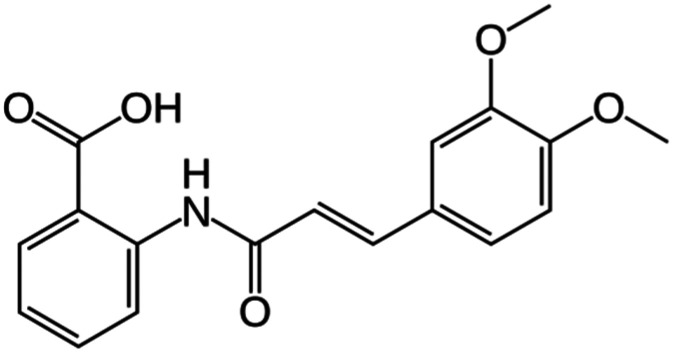
|
NLRP3 | Interacts with the NACHT region of NLRP3 to prevent NLRP3–NLRP3 and NLRP3–ASC interactions | 89 | |
| BHB |

|
NLRP3 (indirectly) | Eliminates outward migration of ATP-sensitive potassium (K(ATP)), reduces ASC aggregation and IL-1β/18 release | 90 | |
| Parthenolide |

|
NLRP1 and 3, caspase-1, NF-κB, IKKβ kinase activity | Alkyl modification of cysteine moieties in ATPase regions of NLRP3 and caspase-1 inhibits NLRP3 ATPase activity | 62 | |
| Oridonin |

|
NLRP3 | Selectively suppresses NLRP3 inflammasome stimulation; interacts with NLRP3's cysteine 279 residue and inhibits NLRP3-NEK7 interaction | 59 | |
| Pralnacasan (VX-740) |

|
Caspase-1 | Covalent modification of the catalytic cysteine moiety in the caspase-1 active site eliminates caspase-1 actions and splits pro-IL-1β/18 | 91 | |
| Emricasan (VX-765) |

|
Caspase-1 | Covalent modification of the catalytic cysteine moiety in the caspase-1 active site eliminates caspase-1 actions and splits pro-IL-1β/18 | 92 |
Role of NLRP3 in drug-induced organ toxicity
Some drugs promote NLRP3 inflammasome activation, resulting in inflammatory-mediated organ damage.1 Drugs such as paracetamol (acetaminophen), anti-tuberculosis drugs, and others, lead to oxidative injury to the liver, which is coupled with mitochondrial dysfunction leading to enhanced NLRP3 activation.93 These changes contribute to increased IL-1β release and ultimately death by hepatocyte apoptosis or cytokine dysregulation, finally culminating in liver failure.94 Nephrotoxic medications like cisplatin, aminoglycosides, and non-steroidal anti-inflammatory drugs overcome oxidative stress and cause mitochondrial damage to renal cells, thereby inducing inflammasome activation.95 This leads to pyrolysis of cells followed by fibrosis and heart failure. The role of NLRP3 in drug-induced organ toxicity with the affected organs, drug triggers, and their mechanisms is summarized in Table 4.96 The inflammation also aggravates injury to the kidney and opens up pathways to chronic kidney disease (CKD) progression.97 NLRP3 inflammasome inhibitors are shown in Table 5, including their IC50 values in cells and disease models. NLRP3 activation within cardiac cells has been reported in some chemotherapeutic drugs (such as doxorubicin) and a few cardiovascular drugs.98 Neuroinflammation from drugs such as methamphetamine and certain anaesthetics is associated with NLRP3 activation, contributing to neurodegeneration and cognitive impairments. Nephrotoxic drugs like cisplatin, aminoglycosides, and NSAIDs cause oxidative stress and mitochondrial damage in renal cells, which is important in initiating inflammasome activation.99 It leads to inflammation, further aggravating kidney injury and promoting CKD progression.100 Certain chemotherapy drugs, such as doxorubicin, and some cardiovascular drugs have been implicated in cardiac cell NLRP3 activation, leading towards pyroptotic cell death, fibrosis, and heart failure.101 Neuroinflammation from drugs such as methamphetamine and certain anaesthetics is related to NLRP3 activation, furthering neurodegeneration and cognitive impairments.102
Table 4. Role of NLRP3 in drug-induced organ toxicity, summarizing the affected organs, drug triggers, and mechanisms.
| Organ system | Drug trigger(s) | Mechanism involving NLRP3 | References |
|---|---|---|---|
| Liver (hepatotoxicity) | Acetaminophen (APAP), isoniazid, methotrexate | ROS-mediated activation of NLRP3, hepatocyte pyroptosis, IL-1β release | 103 |
| Kidney (nephrotoxicity) | Cisplatin, cyclosporine A, vancomycin | NLRP3-driven renal tubular injury, inflammation, K+ efflux | 104 |
| Heart (cardiotoxicity) | Doxorubicin, trastuzumab | Mitochondrial ROS activates NLRP3, cardiomyocyte apoptosis, IL-18 release | 105 |
| Lung (pulmonary toxicity) | Bleomycin, amiodarone, methotrexate | NLRP3 activation leads to pulmonary fibrosis, IL-1β-mediated inflammation | 106 |
| Brain (neurotoxicity) | Methamphetamine, MPTP (Parkinson's model) | NLRP3 activation in microglia, neuroinflammation, neuronal apoptosis | 106 |
Table 5. NLRP3 inflammasome inhibitors, with their IC50 values in cells and disease models.
| Inhibitor name | IC50 value in cells | Disease model | References |
|---|---|---|---|
| MCC950 | ∼7.5 nM (BMDMs) | Sepsis, gout, MS, T2D, atherosclerosis | 45 |
| Oridonin | ∼2.1 μM (THP-1) | Rheumatoid arthritis, sepsis | 107 |
| CY-09 | ∼6 μM (THP-1) | Gout, T2D, atherosclerosis | 61 |
| INF39 | ∼15 μM (THP-1) | Sepsis, peritonitis | 43 |
| OLT1177 | ∼0.9 μM (THP-1) | Osteoarthritis, gout | 108 |
| BAY 11-7082 | ∼10 μM (THP-1) | ALI, sepsis | 109 |
| Ac-YVAD-CMK | ∼1.3 μM (BMDMs) | Gout, peritonitis | 110 |
| Glyburide | ∼20 μM (THP-1) | Type 2 diabetes, atherosclerosis | 111 |
| VX-765 | ∼0.5 μM (THP-1) | Epilepsy, inflammatory diseases | 112 |
Drug-induced hepatotoxicity by the NLRP3 inflammasome
Drug-induced hepatotoxicity (DIH), usually arising from excessive inflammation and immune activation, classifies as a leading cause of liver injury and failure.113 The NLRP3 inflammasome has a vital involvement in mediating liver inflammation during hepatic cellular damage caused by different hepatotoxic drugs.94 It aggravates liver injury via enhanced release of pro-inflammatory cytokines and induction of pyroptotic cell death.94 Hepatotoxic drugs directly induce NLRP3 inflammasome activation via different routes originating from cellular stress. Many hepatotoxic drugs, such as acetaminophen, isoniazid, and methotrexate, induce oxidative stress and mitochondrial injury, leading to ROS and mtDNA release, which serve as important NLRP3 activators.113 ROS drive cell death and inflammation by disrupting the homeostasis of hepatocytes.114 Drug-induced necrotic death of the hepatocyte leads to the release of ATP into the extracellular space, stimulating the P2X7 receptor, and potassium efflux ensues.115 The hepatocyte necrosis releases ATP outside the cells, activating the P2X receptor and leading to K+ efflux. Certain drugs, particularly certain chemotherapeutics and antibiotics, will lead to the instability of lysosomes.116 Drugs that induce such processes include tunicamycin, diclofenac, and statins, causing ER stress that activates the unfolded protein response (UPR).117 Prolonged ER stress activates the inflammasome, which aggravates liver injury.118 Therefore, the activation of caspase-1 leads to the maturation and release of IL-1β and 18, further amplifying hepatic inflammation.118 The released cytokines will recruit immune cells (neutrophils and macrophages), prompting further damage to the liver.119 Activation of caspase-1 by the NLRP3 inflammasome will also cleave gasdermin D, producing a membrane pore that initiates pyroptosis.120 Pyroptosis leads to cell swelling and lysis of hepatocytes with the release of further DAMPs, which will further escalate inflammation.121 However, chronic NLRP3 activation that occurs upon removal of stimuli still contributes to hepatic fibrosis, a precursor to cirrhosis.122 Continued activation of IL-1β signalling promotes the activation of satellite cells and collagen deposition, resulting in liver scarring and dysfunction. Several commonly used medications have been associated with hepatotoxicity via NLRP3 activation (Table 6).123
Table 6. Hepatotoxic drugs involving NLRP3 activation.
| S. N. | Drug class | Examples | Mechanism of NLRP3 activation | References |
|---|---|---|---|---|
| 1 | Analgesics | Acetaminophen (APAP) | ROS, mitochondrial dysfunction, ATP release | 124 |
| 2 | Antibiotics | Isoniazid, rifampin | ER stress, oxidative stress | 125 |
| 3 | NSAIDs | Diclofenac, ibuprofen | Mitochondrial damage, K+ efflux | 126 |
| 4 | Chemotherapeutics | Methotrexate, doxorubicin | ROS, lysosomal rupture | 1 |
| 5 | Antidepressants | Fluoxetine, sertraline | Mitochondrial dysfunction, inflammasome priming | 127 |
| 6 | Statins | Atorvastatin, simvastatin | ER stress, inflammasome activation | 128 |
Drug-induced nephrotoxicity by the NLRP3 inflammasome
Drug-induced nephrotoxicity (DIN) is an important cause of acute kidney injury (AKI) and CKD and often results in irreversible renal damage.129 The role of the NLRP3 inflammasome in mediating kidney inflammation and injury is pertinent since its activation, upon nephrotoxin exposure, aggravates drug-induced nephrotoxicity through enhancement of cytokine release, immune-inflammatory cell recruitment, and cell death by pyroptosis.130 The activation of the NLRP3 inflammasome occurs in renal cells under the stress of nephrotoxic drugs like cisplatin, aminoglycosides (e.g., gentamicin), and NSAIDs, which induce excessive generation of ROS in renal tubular cells.131 Mitochondrial damage and release of mitochondrial DNA are potent activators for NLRP3 activation. Calcium (Ca2+) and sodium (Na+) perturbations trigger the assembly of the inflammasome and renal inflammation.5 Adverse renal effects are caused by aminoglycosides and radiocontrast medicines whenever they are used, and lysosomes are the most notorious; the first step of the process involves the cathepsins being set free.131 In tandem with these processes, the NLRP3 inflammasome is also simultaneously set into action. Cyclosporine and tacrolimus are calcineurin inhibitors that use endoplasmic reticulum dysfunction, which signals UPPR and directly activates the inflammasome, unleashing inflammatory stimuli responsible for kidney injuries and obliterating it.132 Activation of the local and systematic inflammatory response is facilitated by a member of the interleukin family, IL-1β, which, along with another pro-inflammatory cytokine, IL-18, is cleaved and secreted from the activated caspase-1.133 All of these receptors bind to immune system components, such as neutrophils and macrophages, causing additional damage to the kidneys.134 Gasdermin D (GSDMD) is cleaved by activated caspase-1, leading to the generation of membrane pores, where pyroptotic cell death occurs as a result of pyroptosis, leading to worsened acute kidney injury (AKI) and subsequent renal fibrosis.135 By stimulating renal fibroblasts and causing the aforementioned changes, chronic NLRP3 paves the way for renal interstitial fibrosis.136 The combination of these factors leads to an enhanced TGF-β response that causes chronic kidney disease, or CKD. Several regulatory used medicines have been associated with nephrotoxicity via NLRP3 activation (Table 7).100
Table 7. Nephrotoxic drugs involving NLRP3 activation.
| S. N. | Drug class | Examples | Mechanism of NLRP3 activation | References |
|---|---|---|---|---|
| 1 | Chemotherapeutics | Cisplatin, methotrexate | ROS, mitochondrial dysfunction, K+ efflux | 137 |
| 2 | Aminoglycosides | Gentamicin, amikacin | Lysosomal rupture, ROS, cathepsin release | 138 |
| 3 | NSAIDs | Ibuprofen, diclofenac | Mitochondrial stress, K+ efflux, inflammation | 139 |
| 4 | Radiocontrast agents | Iohexol, iodixanol | Endothelial dysfunction, ROS, tubular injury | 140 |
| 5 | Immunosuppressants | Cyclosporine, tacrolimus | ER stress, inflammasome activation | 141 |
| 6 | Diuretics | Furosemide | Tubular stress, K+ efflux, inflammation | 142 |
Drug-induced cardiotoxicity by the NLRP3 inflammasome
Drug-induced cardiotoxicity is a serious side effect related to many pharmaceutical classes, including chemotherapeutics, non-steroidal anti-inflammatory drugs (NSAIDs), antibiotics, and other biologics.143 The NLRP3 inflammasome mediates cardiac inflammation and injury, which can lead to myocardial dysfunction, fibrosis, and heart failure.144 It is activated in cardiomyocytes, endothelial cells, and macrophages in response to drug-induced cellular stress and inflammation, resulting in myocardial damage.132 Several cardiotoxic agents, including doxorubicin, trastuzumab, and proteasome inhibitors, lead to mitochondrial abnormalities and excess production of ROS, which increase mitochondrial permeability transition pore (mPTP) opening, leading to the release of mtDNA, a potent NLRP3 activator.145 Mitochondrial dysfunction follows the depletion of ATP, resulting in compromised cardiac function. Among the cardiotoxic agents are anthracyclines and tyrosine kinase inhibitors (TKIs), which effectuate ion disarray.146 Activation of the P2X7 receptors by ATP will foster the efflux of potassium, an important trigger for NLRP3.115 Aberrant calcium influx plays a contributing role in the contractile dysfunction, represented by cardiomyocytes afflicted by inflammasome activation.147 Trastuzumab (an HER2 inhibitor) and proteasome inhibitors induce ER stress, stimulating the activation of the unfolded protein response (UPR). Sensors of ER stress, such as PERK, IRE1, and ABTF6, activate the inflammasome, further aggravating cardiomyocyte apoptosis.148 Drugs that alter lipid metabolism, such as antipsychotic agents, palmitate, and ceramides, act as damage-associated molecular patterns (DAMPs), which are prototypical triggers for the assembly and activation of the inflammasome.149
Within instigated and active NLRP3, the NLRP3 inflammasome promotes cardiotoxicity via multiple inflammatory pathways.150 Activation of caspase-1 cleaves pro-IL-1β and pro-IL-18 to their active form, enhancing myocardial inflammation.16 The recruited neutrophils and macrophages prefer to inflict further tissue damage within the myocardium.151 Caspase-1 activates gasdermin D (GSDMD), leading to pore formation and causing pyroptotic cell death in cardiomyocytes.1 Pyroptosis leads to contractile dysfunction and cardiac remodelling, thereby enhancing the body burden for subsequent heart failure.150 Chronic NLLRP3 activation leads to fibroblast proliferation and deposition of extracellular matrix, leading to cardiac fibrosis. IL-1β upregulates TGF-β, a key mediator of fibrosis, contributing toward diastolic dysfunction and heart failure.152 Several frequently used medicines linked to NLRP3-mediated cardiotoxicity are shown in Table 8.
Table 8. Cardiotoxic drugs involving NLRP3 activation.
| S. N. | Drug class | Examples | Mechanism of NLRP3 activation | References |
|---|---|---|---|---|
| 1 | Anthracyclines | Doxorubicin, daunorubicin | Mitochondrial ROS, K+ efflux, ER stress | 153 |
| 2 | Tyrosine kinase inhibitors (TKIs) | Trastuzumab, imatinib | ER stress, mitochondrial dysfunction | 154 |
| 3 | Proteasome inhibitors | Bortezomib, carfilzomib | ER stress, ROS generation | 155 |
| 4 | NSAIDs | Ibuprofen, celecoxib | Mitochondrial dysfunction, oxidative stress | 156 |
| 5 | Antipsychotics | Clozapine, olanzapine | Lipotoxicity, metabolic stress | 157 |
| 6 | Antiretrovirals | Zidovudine, lopinavir | Mitochondrial dysfunction, inflammasome priming | 158 |
| 7 | Statins (high dose) | Atorvastatin, simvastatin | Lipotoxicity, ER stress | 159 |
Drug-induced neurotoxicity by the NLRP3 inflammasome
Neurotoxicity is a major concern in pharmacotherapy because it may cause cognitive impairment, neuroinflammation, and neurodegeneration.160 The NLRP3 inflammasome is the principal mechanism for regulating neuroinflammation during neuronal damage and death.161 Chemotherapeutics, anaesthetics, antidepressants, and psychostimulants are some of the examples of neurotoxic drugs that activate NLRP3 through mitochondrial stress, oxidative damage, and ER stress.162 The activation of the NLRP3 inflammasome in microglia, astrocytes, and neurons triggers neuroinflammation leading to neuronal death.163 Neurotoxic drugs like methamphetamine, doxorubicin, and anaesthetics generate oxidative stress on mitochondria, leading to tremendous production of ROS.164 ROS opens the mPTP, leading to the release of mtDNA, potent NLRP3 activators. Mitochondrial dysfunction leads to ATP depletion, impairing neuronal function and synaptic plasticity.165 Psychostimulants (like methamphetamine and cocaine) cause dopamine release, resulting in ion channel dysfunction with K+ efflux.166 This ionic imbalance primes NLRP3 for activation and eventually leads to the inflammatory cascade.167 Anaesthetics, antipsychotic drugs and proteasome inhibitors trigger ER stress and activate the UPR.168 ER stress primes the inflammasome through the PERK, ATF6, and IRE1 signalling pathways.118 Cisplatin, doxorubicin, and methamphetamine disrupt the integrity of the BBB, permitting the entry of peripheral immune cells into the brain.169 Thereafter, increases in microglial activation and NLRP3 activation of inflammation occur. Drugs like ketamine, cocaine, and some antidepressants invoke glutamate excitotoxicity-induced Ca2+ overload that leads to NLRP3 activation.170 The excess release of glutamate is known to enhance ROS production, thus aggravating neurotoxicity.170 This leads to the activation of pro-caspase-1, which cleaves pro-IL-1β and pro-IL-18, culminating in neuroinflammation. Sustained IL-1β signalling contributes to synaptic dysfunction, cognitive impairment, and neurodegeneration.171 Activated caspase-1 processes GSDMD into pore-forming proteins, leading to pyroptotic neuronal death.24 Pyroptotic death recapitulates axonal degeneration and loss of neural circuits. Chronic activation of NLRP3 may compromise the integrity of the BBB, allowing infiltrating toxic substances and immune cells into the brain.172 Such a condition sets off a vicious cycle of neuroinflammation and neurodegeneration. Prolonged inflammasome activation catalyzes the deposition of amyloid beta (Aβ), an Alzheimer's disease (AD) trait.173 IL-1β also alters dopaminergic neurons, adding to PD pathology.174 Several frequently used medicines linked to NLRP3-mediated neurotoxicity are shown in Table 9.
Table 9. Neurotoxic drugs involving NLRP3 activation.
| S. N. | Drug class | Examples | Mechanism of NLRP3 activation | References |
|---|---|---|---|---|
| 1 | Chemotherapeutics | Doxorubicin, cisplatin | Mitochondrial ROS, BBB disruption | 175 |
| 2 | Psychostimulants | Methamphetamine, cocaine | Dopamine imbalance, oxidative stress | 176 |
| 3 | Anaesthetics | Ketamine, isoflurane | Glutamate excitotoxicity, ER stress | 177 |
| 4 | Antidepressants | Fluoxetine, amitriptyline | Mitochondrial dysfunction, ROS generation | 178 |
| 5 | Antipsychotics | Clozapine, haloperidol | Lipotoxicity, ER stress | 179 |
| 6 | Proteasome inhibitors | Bortezomib, carfilzomib | Protein misfolding, ER stress | 180 |
Therapeutic targeting of NLRP3 in drug-induced toxicity
Targeting the NLRP3 inflammasome may be a promising therapeutic strategy in light of its role in drug-induced toxicity.1 Small molecules such as MCC950 and OLT1177 block NLRP3 activation and have demonstrated protective effects in various preclinical models.45 Monoclonal antibodies such as anakinra (IL-1 receptor antagonist) and canakinumab (IL-1β inhibitor) are instrumental in reducing inflammasome-driven inflammation.65 Other compounds such as N-acetylcysteine (NAC), resveratrol, and melatonin can alleviate oxidative stress and suppress NLRP3 activation.181 Statins, colchicine, and certain anti-diabetic drugs (e.g., glyburide) exhibit NLRP3 inhibitory effects and may offer protective benefits against drug-induced toxicity.157 Small molecule inhibitors targeting NLRP3 in drug-induced toxicity and natural compounds targeting NLRP3 in drug-induced toxicity are shown in Tables 10 and 11.182
Table 10. Small molecule inhibitors targeting NLRP3 in drug-induced toxicity.
| Inhibitor | Target/mechanism | Affected organ/system | Drug-induced toxicity model | References |
|---|---|---|---|---|
| MCC950 | Selective NLRP3 inhibitor, blocks ATPase activity | Liver, kidney, heart, brain | APAP, cisplatin, doxorubicin, MPTP | 183 |
| CY-09 | Directly binds NLRP3 ATP-binding domain | Liver, kidney, brain | Acetaminophen, cyclosporine A, MPTP | 60 |
| Oridonin | Covalently modifies NLRP3 at Cys279 | Liver, kidney, heart | Methotrexate, cisplatin, doxorubicin | 59 |
| VX-765 | Caspase-1 inhibitor, blocks IL-1β and IL-18 release | Liver, brain, pancreas | APAP, methamphetamine, alcohol | 66 |
| Glyburide | Blocks ATP-sensitive K+ channels, prevents NLRP3 activation | Kidney, liver | Cisplatin, APAP | 70 |
| Dapansutrile (OLT1177) | Prevents inflammasome oligomerization | Heart, lung | Doxorubicin, bleomycin | 87 |
| BAY 11-7082 | NF-κB inhibitor, prevents NLRP3 priming | Liver, gut | NSAID-induced liver/GI toxicity | 184 |
Table 11. Natural compounds targeting NLRP3 in drug-induced toxicity.
| Compound | Mechanism | Affected organ | Drug-induced toxicity model | References |
|---|---|---|---|---|
| Resveratrol | Suppresses ROS production, inhibits NLRP3 activation | Liver, brain | APAP, methamphetamine | 185 |
| Curcumin | Inhibits NF-κB/NLRP3 signaling, reduces IL-1β | Liver, kidney | Methotrexate, cisplatin | 80 |
| Quercetin | Blocks mitochondrial ROS, suppresses IL-18 | Liver, heart | APAP, doxorubicin | 186 |
| Emodin | Inhibits TLR4/NF-κB/NLRP3 signaling | Lung, liver | Bleomycin, APAP | 187 |
| Melatonin | Reduces oxidative stress, suppresses NLRP3 priming | Liver, kidney, brain | APAP, cisplatin, MPTP | 188 |
| Berberine | Inhibits caspase-1/NLRP3 pathway | Gut, liver | NSAID-induced GI toxicity | 189 |
Therapeutic strategies targeting NLRP3 in drug-induced hepatotoxicity
NLRP3 inflammasome activation is key in determining drug-induced hepatotoxicity with concurrent inflammatory cell death and fibrosis in the liver.94 The insults leading to drug-induced hepatotoxicity are numerous, including acetaminophen, antibiotics, and chemotherapeutics, which impact oxidative stress, mitochondrial damage, and ion flux disturbances, resulting in the activation of NLRP3.190 The use of inhibitors, antioxidants, and anti-inflammatory agents to target NLRP3 holds great therapeutic potential against drug-induced liver injury and improves patient outcomes.1 However, there is still a need for further research to make NLRP3 therapeutic applications safe and effective in the prevention and treatment of hepatotoxicity.191 Considering the involvement of NLRP3 in hepatotoxicity, several therapeutic strategies are explored.1 MCC950 is a selective NLRP3 inhibitor that inhibits NLRP3 inflammasome assembly and IL-1β secretion.192 OLT1177 is a novel non-competitive small-molecule inhibitor that has been shown to inhibit NLRP3 activation. Anakinra (IL-1 receptor antagonist) and canakinumab (anti-IL-1β monoclonal antibody) serve to reduce inflammation and liver injury.193N-Acetylcysteine (NAC) is used for the treatment of acetaminophen overdose; it replenishes glutathione and reduces ROS levels.194 Resveratrol and curcumin are used in therapeutic strategies as natural compounds with antioxidant and anti-inflammatory properties.195 Rapamycin increases autophagosome formation for autophagic removal of damaged mitochondria and inhibition of excess NLRP3 activation in the liver.196 Colchicine (an anti-gout drug) acts on the fibroblast and inhibits the assembly of different proteins involved in the inflammatory response and liver fibrosis. Colchicine disrupts microtubule polymerization by binding to tubulin, which interferes with the microtubule network necessary for the spatial arrangement and assembly of the NLRP3 inflammasome complex. This disruption prevents the proper localization and interaction of NLRP3 with the adaptor protein ASC and pro-caspase-1, thereby inhibiting inflammasome activation. Additionally, colchicine reduces the generation of reactive oxygen species (ROS) and suppresses mitochondrial damage, both of which are upstream signals necessary for NLRP3 activation. These mechanisms collectively contribute to the anti-inflammatory effects of colchicine in NLRP3-mediated pathologies. Statins (low-dose) may counteract NLRP3-induced hepatotoxicity.197
Therapeutic strategies targeting NLRP3 in drug-induced nephrotoxicity
On the one hand, the activation of the NLRP3 inflammasome is a key mediator of drug-induced nephrotoxicity, leading to kidney inflammation, pyroptotic cell death, and fibrosis.198 Nephrotoxic drugs like cisplatin, aminoglycosides, NSAIDs, and radiocontrast media activate NLRP3 by various means, including oxidative stress, disturbances in ion flux, and ER stress.199 Monoclonal antibodies against NLRP3 or its component proteins will mitigate renal injury and avoid CKD progression when delivered in conjunction with antioxidants and anti-inflammatory agents.100 There is ongoing research on the use of anakinra (an IL-1 receptor antagonist) and canakinumab (an anti-IL-1β monoclonal antibody) to inhibit inflammation and preserve renal function.65 Oxidative stress in contrast-induced nephropathy is reduced by N-acetylcysteine (NAC).200 Natural products with anti-inflammatory and mitochondrial-protective properties are represented best by resveratrol and curcumin.201 Rapamycin and metformin stimulate autophagy to clear the damaged mitochondria and prevent excessive inflammasome activation.202 Statin use exhibited anti-inflammatory effects and is effective in reducing NLRP3-mediated kidney damage.203 Colchicine inhibits inflammasome activation and restrains fibrosis in models of nephrotoxicity.204
Therapeutic strategies targeting NLRP3 in drug-induced cardiotoxicity
The NLRP3 inflammasome's facilitatory action reflects drug-induced cardiotoxicity, thereby influencing myocardial inflammation, pyroptotic death of cardiomyocytes, and development into fibrosis.98 The common toxic pharmaceuticals comprise anthracyclines, tyrosine kinase inhibitors, NSAIDs, and antipsychotics, activating NLRP3 owing to oxidative stress, mitochondrial dysfunction, and ER stress.205 NLRP3-specific inhibitors, antioxidants, and metabolic modulators targeting the NLRP3 inflammasome hold much promise in protecting against drug-induced damage to the heart.206 Future development holds promise for therapies that will have wide clinical applications in the prevention and treatment of cardiotoxicity, targeting the NLRP3 inflammasome.207 Anakinra (an IL-1 receptor antagonist) is being tested for heart failure and myocardial inflammation.208 Canakinumab (an IL-β monoclonal antibody) can reduce inflammatory responses and myocardial fibrosis.209N-Acetylcysteine (NAC) scavenges ROS and improves mitochondrial function. Coenzyme Q10 (Co-Q10) enhances mitochondrial ATP production and reduces oxidative stress.210 Resveratrol and curcumin suppress NLRP3 activation via antioxidant and anti-inflammatory properties.211 Rapamycin and metformin enhance autophagy to clear damaged mitochondria, preventing inflammasome activation.212 PPARγ agonists (pioglitazone) reduce lipotoxicity and inflammasome priming. Omega-3 fatty acids suppress NENP3 activation and improve cardiac metabolism.213
Therapeutic strategies targeting NLRP3 in drug-induced neurotoxicity
The NLRP3 inflammasome predominates in the mediation of drug neurotoxicity by driving neuroinflammation, pyroptosis, and cognitive impairment.206 Cancer chemotherapies, psychostimulants, anesthetics, and antidepressants can activate NLRP3 through mechanisms such as oxidative stress, mitochondrial dysfunction, and ER stress. These pathways represent potential targets for neuroprotective strategies using NLRP3 inhibitors, mitochondrial antioxidants, or autophagy enhancers.214 Advancing NLRP3-targeting therapies could reduce drug-induced brain damage, thereby improving neurologic outcomes.215 A promising neuroprotective strategy targets the NLRP3. Anakinra downregulates neuroinflammation in neurodegenerative disease. Canakinumab is an anti-IL-1β monoclonal antibody that attenuates NLRP3-driven neurotoxicity.216N-Acetylcysteine reduces oxidative stress and mitochondrial function. Resveratrol and curcumin are naturally derived inhibitors of NLRP3 that have neuroprotective effects.217 Coenzyme Q10 boosts ATP production and reduces ROS.218 Rapamycin and metformin activate autophagy, allowing for the clearing of damaged mitochondria and reducing inflammasome activation.196 Omega-3 fatty acids downregulate neuroinflammation and boost synaptic plasticity. Melatonin reduces ROS and modulates NLRP3 activation.219
Future perspectives of therapeutic strategies via inhibiting the NLRP3 inflammasome for drug-induced toxicity
The targeted inhibition of NLRP3 may have significant potential for ameliorating drug-induced toxicity to multiple organ systems.1 Future work should aim to develop safer, more selective, and organ-specific inhibitors; optimize combination therapies; and establish AI and biomarker-based approaches to further develop the field.220 Overcoming these challenges will ensure that therapies targeting NLRP3 could bring about a major revolution in drug safety and improve patient outcomes in multiple pharmacotherapy fields.1
NLRP3 inhibition is promising for treating drug-induced toxicities for various organs. Nevertheless, several challenges and opportunities remain for NLRP3-targeted therapies to be used clinically, safely, and effectively in the future.221 MCC950 and OLT1177 are small-molecule inhibitors endowed with meaningful pharmacokinetics, bioavailability, and safety characteristics.222 Research should continue with an emphasis on developing allosteric NLRP3 inhibitors, preventing inflammasome assembly without compromising the physiological functions of immunity.223 Drug delivery systems in nanotechnology (e.g., liposomes and nanoparticles) can improve target inhibition of NLRP3 for specific organs while minimizing off-target effects. Novel NLRP3 inhibitors can also be designed using biomaterial carriers that selectively release drugs at the target inflamed sites, reducing system toxicity.224 The combination of NLRP3 with antioxidants, autophagy inducers, or mitochondrial protectors may strengthen the protective effect.225 Personalized approaches utilizing genetic and biomarker-based screening could identify patients who would benefit most from combination therapies. There is a need for greater clinical consideration of the long-term safety and efficacy of NLRP3-targeted therapies in humans.226 Large trials should also study the interactions between agents, optimal doses, and treatment length to maximize patient benefit and minimize toxicity.227 Drug development aided by AI technology is speeding up the identification and optimization of new NLRP3 inhibitors with a better profile of selectivity and minimal side effects.223 Systems biology approaches facilitate a better understanding of the complex immune–metabolic interactions that are involved in NLRP3-mediated toxicity. The identification of biomarkers (e.g., circulating IL-β) can be indicative of early diagnosis, lead to patient stratification, and facilitate therapy response monitoring.228 NLRP3 polymorphism studies may reveal the genetic basis for individual susceptibility to drug-induced toxicity and enable personalized treatment strategies.229
Conclusion
The NLRP3 inflammasome is key in drug-induced toxicity in several organ systems, including the liver, kidney, heart, and brain. Its activation produces excessive inflammation, pyroptotic cell death, and tissue damage in the induction of severe adverse drug reactions. Various therapeutics have been shown to be capable of triggering the activation of NLRP3, including mitochondrial dysfunction, oxidative stress, ER stress, and ionic imbalances. Targeting the NLRP3 inflammasome represents a very promising strategy in reducing drug-induced toxicity. Innovative, yet modern approaches of interventions include direct NLRP3 inhibitors (MCC950 and OLT1177), IL-1β blockers (anakinra and canakinumab), antioxidants (NAC, resveratrol, and CoQ10), and autophagy inducers (rapamycin and metformin), all of which have performed well in preclinical and clinical studies in reducing inflammation, preventing pyroptosis, and restoring cellular homeostasis, which provides an upper hand in protecting organs from the toxic effects of drugs.
Data availability
No primary research results, software or code have been included, and no new data were generated or analyzed as part of this review.
Author contributions
DDS: conceptualization, validation, methodology, formal analysis, writing – original draft, and approval of the manuscript.
Conflicts of interest
The author declares that there is no conflict of interest.
Acknowledgments
The author acknowledges the Department of Science & Technology; Government of India grant for DST-FIST 2019 (SR/FIST/LS-1/2019/502) and DST-PURSE (SR/PURSE/2021/77) at Amity University Rajasthan for providing necessary facilities. D. D. S. is thankful to DST-PURSE and DST-FIST-AIMT at Amity University Rajasthan, Jaipur, and India and thankful to https://Biorender.com for their graphical support.
References
- Wei S. Ma W. Zhang B. Li W. Front. Cell Dev. Biol. 2021;9:634607. doi: 10.3389/fcell.2021.634607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran R. Manan A. Kim J. Choi S. Exp. Mol. Med. 2024;56:1488–1500. doi: 10.1038/s12276-024-01261-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley N. Jeltema D. Duan Y. He Y. Int. J. Mol. Sci. 2019;20:3328. doi: 10.3390/ijms20133328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leemans J. C. Cassel S. L. Sutterwala F. S. Immunol. Rev. 2011;243:152–162. doi: 10.1111/j.1600-065X.2011.01043.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson K. V. Deng M. Ting J. P.-Y. Nat. Rev. Immunol. 2019;19:477–489. doi: 10.1038/s41577-019-0165-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirota J. A. Im H. Rahman M. M. Rumzhum N. N. Manetsch M. Pascoe C. D. Bunge K. Alkhouri H. Oliver B. G. Ammit A. J. Am. J. Respir. Cell Mol. Biol. 2013;49:517–524. doi: 10.1165/rcmb.2013-0047OC. [DOI] [PubMed] [Google Scholar]
- Hamilton C. Olona A. Leishman S. MacDonald-Ramsahai K. Cockcroft S. Larrouy-Maumus G. Anand P. K. ImmunoHorizons. 2022;6:642–659. doi: 10.4049/immunohorizons.2200058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao X. Han Y. Shen C. Liu J. Wang Y. Phytother. Res. 2023;37:5622–5638. doi: 10.1002/ptr.8009. [DOI] [PubMed] [Google Scholar]
- Zhan X. Li Q. Xu G. Xiao X. Bai Z. Front. Immunol. 2023;13:1109938. doi: 10.3389/fimmu.2022.1109938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaffey L. E. Roberti A. Bowman A. O'Brien C. Jo. Som L. Purvis G. Sd. Greaves D. R. Eur. J. Pharmacol. 2024;969:176437. doi: 10.1016/j.ejphar.2024.176437. [DOI] [PubMed] [Google Scholar]
- Sharma B. Satija G. Madan A. Garg M. Alam M. M. Shaquiquzzaman M. Khanna S. Tiwari P. Parvez S. Iqubal A. Haque S. E. Khan M. A. Inflammation. 2023;46:56–87. doi: 10.1007/s10753-022-01730-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L. Deng H. Cui H. Fang J. Zuo Z. Deng J. Li Y. Wang X. Zhao L. Oncotarget. 2018;9:7204–7218. doi: 10.18632/oncotarget.23208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L. Hauenstein A. V. Mol. Aspects Med. 2020;76:100889. doi: 10.1016/j.mam.2020.100889. [DOI] [PubMed] [Google Scholar]
- Roh J. S. Sohn D. H. Immune Netw. 2018;18:e27. doi: 10.4110/in.2018.18.e27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y. Xu W. Zhou R. Cell. Mol. Immunol. 2021;18:2114–2127. doi: 10.1038/s41423-021-00740-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Que X. Zheng S. Song Q. Pei H. Zhang P. Genes Dis. 2024;11:819–829. doi: 10.1016/j.gendis.2023.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu J. Wu H. Annu. Rev. Immunol. 2023;41:301–316. doi: 10.1146/annurev-immunol-081022-021207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott E. I. Sutterwala F. S. Immunol. Rev. 2015;265:35–52. doi: 10.1111/imr.12286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casali E. Serapian S. A. Gianquinto E. Castelli M. Bertinaria M. Spyrakis F. Colombo G. Int. J. Biol. Macromol. 2023;246:125609. doi: 10.1016/j.ijbiomac.2023.125609. [DOI] [PubMed] [Google Scholar]
- Jomova K. Raptova R. Alomar S. Y. Alwasel S. H. Nepovimova E. Kuca K. Valko M. Arch. Toxicol. 2023;97:2499–2574. doi: 10.1007/s00204-023-03562-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Q. Lim C. S. Annu. Rev. Pharmacol. Toxicol. 2024;64:417–433. doi: 10.1146/annurev-pharmtox-031023-125300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu A. Magupalli V. G. Ruan J. Yin Q. Atianand M. K. Vos M. R. Schröder G. F. Fitzgerald K. A. Wu H. Egelman E. H. Cell. 2014;156:1193–1206. doi: 10.1016/j.cell.2014.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng D. Liwinski T. Elinav E. Cell Discovery. 2020;6:36. doi: 10.1038/s41421-020-0167-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo H. M. Rathkey J. Boyd-Tressler A. Katsnelson M. A. Abbott D. W. Dubyak G. R. J. Immunol. 2016;197:1353–1367. doi: 10.4049/jimmunol.1600699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y. Kong Y. Chen H. Xia J. Zhao J. Zhou Y. Int. Immunopharmacol. 2025;146:113821. doi: 10.1016/j.intimp.2024.113821. [DOI] [PubMed] [Google Scholar]
- Abdallah Y. E. H. Chahal S. Jamali F. Mahmoud S. H. J. Pharm. Pharm. Sci. 2023;26:11137. doi: 10.3389/jpps.2023.11137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R. Yin C. Fang J. Liu B. J. Neuroinflammation. 2021;18:84. doi: 10.1186/s12974-021-02131-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H. Lin L. Zhang Z. Zhang H. Hu H. Signal Transduction Targeted Ther. 2020;5:209. doi: 10.1038/s41392-020-00312-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi Y. Immunology. 2017;152:207–217. doi: 10.1111/imm.12787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muñoz-Planillo R. Kuffa P. Martínez-Colón G. Smith B. L. Rajendiran T. M. Núñez G. Immunity. 2013;38:1142–1153. doi: 10.1016/j.immuni.2013.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feliziani C. Fernandez M. Quassollo G. Holstein D. Bairo S. M. Paton J. C. Paton A. W. De Batista J. Lechleiter J. D. Bollo M. Cell Calcium. 2022;106:102622. doi: 10.1016/j.ceca.2022.102622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou Y. He H. Ma M. Zhou R. Front. Immunol. 2023;14:1128700. doi: 10.3389/fimmu.2023.1128700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salminen A. Ojala J. Kaarniranta K. Kauppinen A. Cell. Mol. Life Sci. 2012;69:2999–3013. doi: 10.1007/s00018-012-0962-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rissiek B. Haag F. Boyer O. Koch-Nolte F. Adriouch S. Front. Immunol. 2015;6:204. doi: 10.3389/fimmu.2015.00204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X. Matico R. E. Miller R. Chauhan D. Van Schoubroeck B. Grauwen K. Suarez J. Pietrak B. Haloi N. Yin Y. Tresadern G. J. Perez-Benito L. Lindahl E. Bottelbergs A. Oehlrich D. Van Opdenbosch N. Sharma S. Nat. Commun. 2024;15:1164. doi: 10.1038/s41467-024-45396-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuah J. J. Y. Rexroad M. S. Smith D. M. Commun. Biol. 2023;6:733. doi: 10.1038/s42003-023-05123-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y. Lee S. Park Y. H. Int. J. Mol. Sci. 2024;25:9018. [Google Scholar]
- Zulfat M. Hakami M. A. Hazazi A. Mahmood A. Khalid A. Alqurashi R. S. Abdalla A. N. Hu J. Wadood A. Huang X. Heliyon. 2024;10:e34410. doi: 10.1016/j.heliyon.2024.e34410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohto U. Kamitsukasa Y. Ishida H. Zhang Z. Murakami K. Hirama C. Maekawa S. Shimizu T. Proc. Natl. Acad. Sci. U. S. A. 2022;119:e2121353119. doi: 10.1073/pnas.2121353119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Sayed S. McMahon E. Musleh S. Freeman S. Brough D. Kasher P. R. Bryce R. A. Bioorg. Chem. 2024;153:107909. doi: 10.1016/j.bioorg.2024.107909. [DOI] [PubMed] [Google Scholar]
- Thomas T. P. Grisanti L. A. Front. Physiol. 2020;11:529075. doi: 10.3389/fphys.2020.529075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreeva L. David L. Rawson S. Shen C. Pasricha T. Pelegrin P. Wu H. Cell. 2021;184:6299–6312. doi: 10.1016/j.cell.2021.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]; , e22
- Ma Q. Pharmacol. Rev. 2023;75:487–520. doi: 10.1124/pharmrev.122.000629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao J. Sterling K. Wang Z. Zhang Y. Song W. Signal Transduction Targeted Ther. 2024;9:10. doi: 10.1038/s41392-023-01687-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coll R. C. Robertson A. A. B. Chae J. J. Higgins S. C. Muñoz-Planillo R. Inserra M. C. Vetter I. Dungan L. S. Monks B. G. Stutz A. Croker D. E. Butler M. S. Haneklaus M. Sutton C. E. Núñez G. Latz E. Kastner D. L. Mills K. H. G. Masters S. L. Schroder K. Cooper M. A. O'Neill L. A. J. Nat. Med. 2015;21:248–255. doi: 10.1038/nm.3806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moasses Ghafary S. Soriano-Teruel P. M. Lotfollahzadeh S. Sancho M. Serrano-Candelas E. Karami F. Barigye S. J. Fernández-Pérez I. Gozalbes R. Nikkhah M. Orzáez M. Hosseinkhani S. Int. J. Mol. Sci. 2022;23:1651. doi: 10.3390/ijms23031651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X. Yang J. Wu J. Yang X. Biomed. Pharmacother. 2024;172:116261. doi: 10.1016/j.biopha.2024.116261. [DOI] [PubMed] [Google Scholar]
- Dai Y. Zhou J. Shi C. MedComm. 2023;4:e391. doi: 10.1002/mco2.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vajjhala P. R. Kaiser S. Smith S. J. Ong Q.-R. Soh S. L. Stacey K. J. Hill J. M. J. Biol. Chem. 2014;289:23504–23519. doi: 10.1074/jbc.M114.553305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkschulte R. Fußhöller D. M. Hoss F. Rodríguez-Alcázar J. F. Lauterbach M. A. Kolbe C.-C. Rauen M. Ince S. Herrmann C. Latz E. Geyer M. Commun. Biol. 2022;5:1176. doi: 10.1038/s42003-022-04120-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandall C. F. Ziehr B. K. MacDonald J. A. Molecules. 2020;25:4572. doi: 10.3390/molecules25194572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekker C. Mattes H. Wright M. Boettcher A. Hinniger A. Hughes N. Kapps-Fouthier S. Eder J. Erbel P. Stiefl N. Mackay A. Farady C. J. J. Mol. Biol. 2021;433:167309. doi: 10.1016/j.jmb.2021.167309. [DOI] [PubMed] [Google Scholar]
- Sharma M. De Alba E. Int. J. Mol. Sci. 2021;22:872. doi: 10.3390/ijms22020872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan Y. Wang J. Cai J. Kelley N. He Y. J. Biol. Chem. 2022;298:102717. doi: 10.1016/j.jbc.2022.102717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colunga Biancatelli R. M. L. Solopov P. A. Catravas J. D. Am. J. Pathol. 2022;192:837–846. doi: 10.1016/j.ajpath.2022.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leu S.-Y. Tsang Y.-L. Ho L.-C. Yang C.-C. Shao A.-N. Chang C.-Y. Lin H.-K. Tsai P.-J. Sung J.-M. Tsai Y.-S. J. Endocrinol. 2023;257:e220184. doi: 10.1530/JOE-22-0184. [DOI] [PubMed] [Google Scholar]
- Hull C. Dekeryte R. Buchanan H. Kamli-Salino S. Robertson A. Delibegovic M. Platt B. Neuropharmacology. 2020;180:108305. doi: 10.1016/j.neuropharm.2020.108305. [DOI] [PubMed] [Google Scholar]
- Toldo S. Mauro A. G. Cutter Z. Van Tassell B. W. Mezzaroma E. Del Buono M. G. Prestamburgo A. Potere N. Abbate A. J. Cardiovasc. Pharmacol. 2019;73:215–222. doi: 10.1097/FJC.0000000000000658. [DOI] [PubMed] [Google Scholar]
- He H. Jiang H. Chen Y. Ye J. Wang A. Wang C. Liu Q. Liang G. Deng X. Jiang W. Zhou R. Nat. Commun. 2018;9:2550. doi: 10.1038/s41467-018-04947-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han S. He Z. Hu X. Li X. Zheng K. Huang Y. Xiao P. Xie Q. Ni J. Liu Q. Antioxidants. 2023;12:722. doi: 10.3390/antiox12030722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H. He H. Chen Y. Huang W. Cheng J. Ye J. Wang A. Tao J. Wang C. Liu Q. Jin T. Jiang W. Deng X. Zhou R. J. Exp. Med. 2017;214:3219–3238. doi: 10.1084/jem.20171419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L. Feng L. Gao J. Hu J. Li A. Zhu Y. Zhang C. Qiu B. Shen Z. Int. Immunopharmacol. 2023;119:110229. doi: 10.1016/j.intimp.2023.110229. [DOI] [PubMed] [Google Scholar]
- Jing M. Yang J. Zhang L. Liu J. Xu S. Wang M. Zhang L. Sun Y. Yan W. Hou G. Wang C. Xin W. Int. Immunopharmacol. 2021;98:107879. doi: 10.1016/j.intimp.2021.107879. [DOI] [PubMed] [Google Scholar]
- Exconde P. M. Hernandez-Chavez C. Bourne C. M. Richards R. M. Bray M. B. Lopez J. L. Srivastava T. Egan M. S. Zhang J. Yoo W. Shin S. Discher B. M. Taabazuing C. Y. Cell Rep. 2023;42:113581. doi: 10.1016/j.celrep.2023.113581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinarello C. A. Simon A. Van Der Meer J. W. M. Nat. Rev. Drug Discovery. 2012;11:633–652. doi: 10.1038/nrd3800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amand M. Adams P. Schober R. Iserentant G. Servais J.-Y. Moutschen M. Seguin-Devaux C. eLife. 2023;12:e83207. doi: 10.7554/eLife.83207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tastan B. Arioz B. I. Tufekci K. U. Tarakcioglu E. Gonul C. P. Genc K. Genc S. Front. Immunol. 2021;12:737065. doi: 10.3389/fimmu.2021.737065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y. Ye X. Escames G. Lei W. Zhang X. Li M. Jing T. Yao Y. Qiu Z. Wang Z. Acuña-Castroviejo D. Yang Y. Cell. Mol. Biol. Lett. 2023;28:51. doi: 10.1186/s11658-023-00462-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q.-L. Yin H.-R. He Q.-Y. Wang Y. Biomed. Pharmacother. 2021;138:111442. doi: 10.1016/j.biopha.2021.111442. [DOI] [PubMed] [Google Scholar]
- Lamkanfi M. Mueller J. L. Vitari A. C. Misaghi S. Fedorova A. Deshayes K. Lee W. P. Hoffman H. M. Dixit V. M. J. Cell Biol. 2009;187:61–70. doi: 10.1083/jcb.200903124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coll R. C. Hill J. R. Day C. J. Zamoshnikova A. Boucher D. Massey N. L. Chitty J. L. Fraser J. A. Jennings M. P. Robertson A. A. B. Schroder K. Nat. Chem. Biol. 2019;15:556–559. doi: 10.1038/s41589-019-0277-7. [DOI] [PubMed] [Google Scholar]
- Zhang Y. Lin Z. Chen D. He Y. Biochem. Biophys. Res. Commun. 2021;553:119–125. doi: 10.1016/j.bbrc.2021.03.055. [DOI] [PubMed] [Google Scholar]
- Zeng Y. Zhu G. Zhu M. Song J. Cai H. Song Y. Wang J. Jin M. Oxid. Med. Cell. Longevity. 2022;2022:1–11. doi: 10.1155/2022/6908884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier N. K. Crown D. Liu J. Leppla S. H. Moayeri M. J. Immunol. 2014;192:763–770. doi: 10.4049/jimmunol.1301434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amaral N. B. Rodrigues T. S. Giannini M. C. Lopes M. I. Bonjorno L. P. Menezes P. I. S. O. Dib S. M. Gigante S. L. G. Benatti M. N. Rezek U. C. Emrich-Filho L. L. Sousa B. A. Almeida S. C. L. Luppino-Assad R. Veras F. P. Schneider A. H. Leiria L. O. S. Cunha L. D. Alves-Filho J. C. Cunha T. M. Arruda E. Miranda C. H. Pazin-Filho A. Auxiliadora-Martins M. Borges M. C. Fonseca B. A. L. Bollela V. R. Del-Ben C. M. Cunha F. Q. Santana R. C. Vilar F. C. Zamboni D. S. Louzada-Junior P. Oliveira R. D. R. Inflammation Res. 2023;72:895–899. doi: 10.1007/s00011-023-01718-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang F. Qin Y. Wang Y. Meng S. Xian H. Che H. Lv J. Li Y. Yu Y. Bai Y. Wang L. Int. J. Biol. Sci. 2019;15:1010–1019. doi: 10.7150/ijbs.29680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D. Sui L. Chen C. Liu S. Sun X. Guan J. Aging. 2022;14:462–476. doi: 10.18632/aging.203824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Y. Bajaj M. Yang H.-C. Perez-Polo J. R. Birnbaum Y. Cardiovasc. Drugs Ther. 2017;31:119–132. doi: 10.1007/s10557-017-6725-2. [DOI] [PubMed] [Google Scholar]
- Byrne N. J. Matsumura N. Maayah Z. H. Ferdaoussi M. Takahara S. Darwesh A. M. Levasseur J. L. Jahng J. W. S. Vos D. Parajuli N. El-Kadi A. O. S. Braam B. Young M. E. Verma S. Light P. E. Sweeney G. Seubert J. M. Dyck J. R. B. Circ.: Heart Failure. 2020;13:e006277. doi: 10.1161/CIRCHEARTFAILURE.119.006277. [DOI] [PubMed] [Google Scholar]
- Huang B. Qian Y. Xie S. Ye X. Chen H. Chen Z. Zhang L. Xu J. Hu H. Ma S. Héroux P. Wang D. Shen H.-M. Wu Y. Xia D. Cell. Mol. Immunol. 2021;18:1278–1289. doi: 10.1038/s41423-020-0444-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Q.-H. Zhang X. Pan Y. Li Y.-C. Kong L.-D. Biochem. Pharmacol. 2012;84:113–125. doi: 10.1016/j.bcp.2012.03.005. [DOI] [PubMed] [Google Scholar]
- Juliana C. Fernandes-Alnemri T. Wu J. Datta P. Solorzano L. Yu J.-W. Meng R. Quong A. A. Latz E. Scott C. P. Alnemri E. S. J. Biol. Chem. 2010;285:9792–9802. doi: 10.1074/jbc.M109.082305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y. Varadarajan S. Muñoz-Planillo R. Burberry A. Nakamura Y. Núñez G. J. Biol. Chem. 2014;289:1142–1150. doi: 10.1074/jbc.M113.515080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y. Lv Q. Zheng M. Sun H. Shi F. Int. Immunopharmacol. 2021;92:107358. doi: 10.1016/j.intimp.2020.107358. [DOI] [PubMed] [Google Scholar]
- Gan W. Ren J. Li T. Lv S. Li C. Liu Z. Yang M. Biochim. Biophys. Acta, Mol. Basis Dis. 2018;1864:1–10. doi: 10.1016/j.bbadis.2017.10.001. [DOI] [PubMed] [Google Scholar]
- Liu W. Guo W. Wu J. Luo Q. Tao F. Gu Y. Shen Y. Li J. Tan R. Xu Q. Sun Y. Biochem. Pharmacol. 2013;85:1504–1512. doi: 10.1016/j.bcp.2013.03.008. [DOI] [PubMed] [Google Scholar]
- Sánchez-Fernández A. Skouras D. B. Dinarello C. A. López-Vales R. Front. Immunol. 2019;10:2578. doi: 10.3389/fimmu.2019.02578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shim D.-W. Shin W.-Y. Yu S.-H. Kim B.-H. Ye S.-K. Koppula S. Won H.-S. Kang T.-B. Lee K.-H. Sci. Rep. 2017;7:15020. doi: 10.1038/s41598-017-15314-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y. Jiang H. Chen Y. Wang X. Yang Y. Tao J. Deng X. Liang G. Zhang H. Jiang W. Zhou R. EMBO Mol. Med. 2018;10:e8689. doi: 10.15252/emmm.201708689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youm Y.-H. Nguyen K. Y. Grant R. W. Goldberg E. L. Bodogai M. Kim D. D'Agostino D. Planavsky N. Lupfer C. Kanneganti T. D. Kang S. Horvath T. L. Fahmy T. M. Crawford P. A. Biragyn A. Alnemri E. Dixit V. D. Nat. Med. 2015;21:263–269. doi: 10.1038/nm.3804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanase D. M. Valasciuc E. Gosav E. M. Ouatu A. Buliga-Finis O. N. Floria M. Maranduca M. A. Serban I. L. Int. J. Mol. Sci. 2023;24:8162. doi: 10.3390/ijms24098162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Y. Liu Y. Xu L. Xu J. Xiong Y. Peng Y. Ding K. Zheng S. Yang N. Zhang Z. Li L. Tan L. Song H. Fu J. Cell Death Dis. 2022;13:512. doi: 10.1038/s41419-022-04966-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen X.-L. Guo Y.-N. Lu M.-H. Ding K.-N. Liang S.-S. Mou R.-W. Yuan S. He Y.-M. Tang L.-P. Ecotoxicol. Environ. Saf. 2023;252:114590. doi: 10.1016/j.ecoenv.2023.114590. [DOI] [PubMed] [Google Scholar]
- Wree A. Eguchi A. McGeough M. D. Pena C. A. Johnson C. D. Canbay A. Hoffman H. M. Feldstein A. E. Hepatology. 2014;59:898–910. doi: 10.1002/hep.26592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos N. A. G. Catão C. S. Martins N. M. Curti C. Bianchi M. L. P. Santos A. C. Arch. Toxicol. 2007;81:495–504. doi: 10.1007/s00204-006-0173-2. [DOI] [PubMed] [Google Scholar]
- Yu C. Chen P. Miao L. Di G. Int. J. Mol. Sci. 2023;24:3067. doi: 10.3390/ijms24043067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapa S. F. Di Iorio B. R. Campiglia P. Heidland A. Marzocco S. Int. J. Mol. Sci. 2019;21:263. doi: 10.3390/ijms21010263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toldo S. Mezzaroma E. Buckley L. F. Potere N. Di Nisio M. Biondi-Zoccai G. Van Tassell B. W. Abbate A. Pharmacol. Ther. 2022;236:108053. doi: 10.1016/j.pharmthera.2021.108053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson F. L. Biggs K. E. Rankin B. E. Havrda M. C. Transl. Res. 2023;252:21–33. doi: 10.1016/j.trsl.2022.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang G. Zhang Y. Zhang Y. Ma Y. Biochem. Biophys. Rep. 2023;33:101417. doi: 10.1016/j.bbrep.2022.101417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L. Fan C. Jiao H.-C. Zhang Q. Jiang Y.-H. Cui J. Liu Y. Jiang Y.-H. Zhang J. Yang M.-Q. Li Y. Xue Y.-T. Oxid. Med. Cell. Longevity. 2022;2022:1–15. doi: 10.1155/2022/1733834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu E. Liu J. Liu H. Wang X. Xiong H. J. Neuroimmune Pharmacol. 2018;13:237–253. doi: 10.1007/s11481-018-9780-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han X. Sun S. Sun Y. Song Q. Zhu J. Song N. Chen M. Sun T. Xia M. Ding J. Lu M. Yao H. Hu G. Autophagy. 2019;15:1860–1881. doi: 10.1080/15548627.2019.1596481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McSweeney K. R. Gadanec L. K. Qaradakhi T. Ali B. A. Zulli A. Apostolopoulos V. Cancers. 2021;13:1572. doi: 10.3390/cancers13071572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei S. Ma W. Yang Y. Sun T. Jiang C. Liu J. Zhang B. Li W. Biochem. Pharmacol. 2023;214:115662. doi: 10.1016/j.bcp.2023.115662. [DOI] [PubMed] [Google Scholar]
- Biswas R. Bunderson-Schelvan M. Holian A. Pulm. Med. 2011;2011:1–8. doi: 10.1155/2011/105707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He S. Huang C. Tan N. Zhang J. DNA Cell Biol. 2023;42:711–719. doi: 10.1089/dna.2023.0244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchetti C. Swartzwelter B. Koenders M. I. Azam T. Tengesdal I. W. Powers N. De Graaf D. M. Dinarello C. A. Joosten L. A. B. Arthritis Res. Ther. 2018;20:169. doi: 10.1186/s13075-018-1664-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J. Rhee M. H. Kim E. Cho J. Y. Mediators Inflammation. 2012;2012:1–11. doi: 10.1155/2012/416036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldrighi M. Doreth C. Li Y. Zhao X. Warner E. Chenoweth H. Kishore K. Umrania Y. Minde D.-P. Thome S. Yu X. Lu Y. Knapton A. Harrison J. Clarke M. Latz E. De Cárcer G. Malumbres M. Ryffel B. Bryant C. Liu J. Lilley K. S. Mallat Z. Li X. J. Clin. Invest. 2023;133:e162129. doi: 10.1172/JCI162129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuletic S. Dong W. Wolfbauer G. Tang C. Albers J. J. Biochim. Biophys. Acta, Mol. Cell Res. 2011;1813:1917–1924. doi: 10.1016/j.bbamcr.2011.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao M. Wang J. Liu W. Zhao X. Qin Y. Zhang C. Yin H. Zhao C. Ann. Hepatol. 2023;28:101082. doi: 10.1016/j.aohep.2023.101082. [DOI] [PubMed] [Google Scholar]
- Liao J. Lu Q. Li Z. Li J. Zhao Q. Li J. Front. Pharmacol. 2023;14:1122632. doi: 10.3389/fphar.2023.1122632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittal M. Siddiqui M. R. Tran K. Reddy S. P. Malik A. B. Antioxid. Redox Signaling. 2014;20:1126–1167. doi: 10.1089/ars.2012.5149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoque R. Sohail M. A. Salhanick S. Malik A. F. Ghani A. Robson S. C. Mehal W. Z. Am. J. Physiol. 2012;302:G1171–G1179. doi: 10.1152/ajpgi.00352.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W. Purinergic Signalling. 2021;17:151–162. doi: 10.1007/s11302-020-09761-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokołowska P. Siatkowska M. Jóźwiak-Bębenista M. Komorowski P. Koptas M. Kowalczyk E. Wiktorowska-Owczarek A. Molecules. 2022;27:3449. doi: 10.3390/molecules27113449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X. Guo X. Ge Q. Zhao Y. Mu H. Zhang J. Oxid. Med. Cell. Longevity. 2019;2019:1–18. doi: 10.1155/2019/3462530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arango Duque G. Descoteaux A. Front. Immunol. 2014;5:491. doi: 10.3389/fimmu.2014.00491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C. Yang T. Xiao J. Xu C. Alippe Y. Sun K. Kanneganti T.-D. Monahan J. B. Abu-Amer Y. Lieberman J. Mbalaviele G. Sci. Immunol. 2021;6:eabj3859. doi: 10.1126/sciimmunol.abj3859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y. Pan R. Ouyang Y. Gu W. Xiao T. Yang H. Tang L. Wang H. Xiang B. Chen P. Signal Transduction Targeted Ther. 2024;9:245. doi: 10.1038/s41392-024-01958-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brahadeeswaran S. Dasgupta T. Manickam V. Saraswathi V. Tamizhselvi R. Front. Immunol. 2023;14:1215333. doi: 10.3389/fimmu.2023.1215333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inzaugarat M. E. Johnson C. D. Holtmann T. M. McGeough M. D. Trautwein C. Papouchado B. G. Schwabe R. Hoffman H. M. Wree A. Feldstein A. E. Hepatology. 2019;69:845–859. doi: 10.1002/hep.30252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran A. and Jaeschke H., in Advances in Pharmacology, Elsevier, 2019, vol. 85, pp. 195–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Q. Kuang W. Hao W. Liang J. Wu L. Tang C. Wang Y. Liu T. Mediators Inflammation. 2021;2021:1–13. doi: 10.1155/2021/8086253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels M. J. D. Rivers-Auty J. Schilling T. Spencer N. G. Watremez W. Fasolino V. Booth S. J. White C. S. Baldwin A. G. Freeman S. Wong R. Latta C. Yu S. Jackson J. Fischer N. Koziel V. Pillot T. Bagnall J. Allan S. M. Paszek P. Galea J. Harte M. K. Eder C. Lawrence C. B. Brough D. Nat. Commun. 2016;7:12504. doi: 10.1038/ncomms12504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu W. Xu G. Wei Z. Wang Z. Qin Q. Lin L. Ren L. Liu T. Fang Z. Yang Y. Zhao J. Wang J. Zhan X. Xiao X. Bai Z. Cell Death Discovery. 2022;8:313. doi: 10.1038/s41420-022-01109-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koushki K. Shahbaz S. K. Mashayekhi K. Sadeghi M. Zayeri Z. D. Taba M. Y. Banach M. Al-Rasadi K. Johnston T. P. Sahebkar A. Clin. Rev. Allergy Immunol. 2021;60:175–199. doi: 10.1007/s12016-020-08791-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagi T. Inoue A. Noguchi T. Suzuki W. Takano S. Otani K. Naganuma R. Sekiguchi Y. Hirata Y. Shindo S. Hwang G.-W. Matsuzawa A. J. Immunol. 2024;212:1807–1818. doi: 10.4049/jimmunol.2300193. [DOI] [PubMed] [Google Scholar]
- Vilaysane A. Chun J. Seamone M. E. Wang W. Chin R. Hirota S. Li Y. Clark S. A. Tschopp J. Trpkov K. Hemmelgarn B. R. Beck P. L. Muruve D. A. J. Am. Soc. Nephrol. 2010;21:1732–1744. doi: 10.1681/ASN.2010020143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett W. M. Luft F. Porter G. A. Am. J. Med. 1980;69:767–774. doi: 10.1016/0002-9343(80)90450-7. [DOI] [PubMed] [Google Scholar]
- Bronner D. N. Abuaita B. H. Chen X. Fitzgerald K. A. Nuñez G. He Y. Yin X.-M. O'Riordan M. X. D. Immunity. 2015;43:451–462. doi: 10.1016/j.immuni.2015.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ihim S. A. Abubakar S. D. Zian Z. Sasaki T. Saffarioun M. Maleknia S. Azizi G. Front. Immunol. 2022;13:919973. doi: 10.3389/fimmu.2022.919973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imig J. D. and Ryan M. J., in Comprehensive Physiology, ed. Y. S. Prakash, Wiley, 1st edn, 2013, pp. 957–976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y., Shi J. and Shao F., in Innate Immune Activation, ed. D. De Nardo and C. M. De Nardo, Springer New York, New York, NY, 2018, vol. 1714, pp. 131–148 [Google Scholar]
- Farris A. B. Colvin R. B. Curr. Opin. Nephrol. Hypertens. 2012;21:289–300. doi: 10.1097/MNH.0b013e3283521cfa. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marullo R. Werner E. Degtyareva N. Moore B. Altavilla G. Ramalingam S. S. Doetsch P. W. PLoS One. 2013;8:e81162. doi: 10.1371/journal.pone.0081162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karasawa T. Steyger P. S. Integr. Biol. 2011;3:879. doi: 10.1039/c1ib00034a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thai P. N. Ren L. Xu W. Overton J. Timofeyev V. Nader C. E. Haddad M. Yang J. Gomes A. V. Hammock B. D. Chiamvimonvat N. Sirish P. Cardiovasc. Drugs Ther. 2023;37:25–37. doi: 10.1007/s10557-021-07253-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreucci M. Faga T. Pisani A. Sabbatini M. Michael A. BioMed Res. Int. 2014;2014:1–21. doi: 10.1155/2014/362725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yilmaz D. E. Kirschner K. Demirci H. Himmerkus N. Bachmann S. Mutig K. J. Biol. Chem. 2022;298:101589. doi: 10.1016/j.jbc.2022.101589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon B. A. Chawla L. S. Renal Failure. 2021;43:830–839. doi: 10.1080/0886022X.2021.1906701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh B. K. Haque S. E. Pillai K. K. Expert Opin. Drug Metab. Toxicol. 2014;10:143–156. doi: 10.1517/17425255.2014.856881. [DOI] [PubMed] [Google Scholar]
- Zhang X. Qu H. Yang T. Kong X. Zhou H. Biomed. Pharmacother. 2021;143:112219. doi: 10.1016/j.biopha.2021.112219. [DOI] [PubMed] [Google Scholar]
- Yin Y. Shen H. Front. Cardiovasc. Med. 2021;8:739095. doi: 10.3389/fcvm.2021.739095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorini S. De Angelis A. Berrino L. Malara N. Rosano G. Ferraro E. Oxid. Med. Cell. Longevity. 2018;2018:7582730. doi: 10.1155/2018/7582730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami T. Ockinger J. Yu J. Byles V. McColl A. Hofer A. M. Horng T. Proc. Natl. Acad. Sci. U. S. A. 2012;109:11282–11287. doi: 10.1073/pnas.1117765109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumandan S. Mahadevan N. R. Chiu K. DeLaney A. Zanetti M. Cancer Lett. 2013;329:236–242. doi: 10.1016/j.canlet.2012.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding P. Song Y. Yang Y. Zeng C. Front. Pharmacol. 2024;15:1368835. doi: 10.3389/fphar.2024.1368835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y. Xu L. Dong N. Li F. Front. Cardiovasc. Med. 2022;9:927061. doi: 10.3389/fcvm.2022.927061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee D. Tian R. Cai S. Curr. Cardiol. Rep. 2023;25:631–640. doi: 10.1007/s11886-023-01897-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humeres C. Frangogiannis N. G. JACC Basic Transl. Sci. 2019;4:449–467. doi: 10.1016/j.jacbts.2019.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J. Wu R. Chen L. Yang Z. Yan D. Li M. Front. Pharmacol. 2022;13:811406. doi: 10.3389/fphar.2022.811406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez-Hernández M. A. De La Cruz-Ojeda P. López-Grueso M. J. Navarro-Villarán E. Requejo-Aguilar R. Castejón-Vega B. Negrete M. Gallego P. Vega-Ochoa Á. Victor V. M. Cordero M. D. Del Campo J. A. Bárcena J. A. Padilla C. A. Muntané J. Redox Biol. 2020;36:101510. doi: 10.1016/j.redox.2020.101510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X. Chen Y. Ou Y. Min W. Liang S. Hua L. Zhou Y. Zhang C. Chen P. Yang Z. Hu W. Sun P. Biochem. Pharmacol. 2022;206:115326. doi: 10.1016/j.bcp.2022.115326. [DOI] [PubMed] [Google Scholar]
- Bindu S. Mazumder S. Bandyopadhyay U. Biochem. Pharmacol. 2020;180:114147. doi: 10.1016/j.bcp.2020.114147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillinger T. McCutcheon R. A. Vano L. Mizuno Y. Arumuham A. Hindley G. Beck K. Natesan S. Efthimiou O. Cipriani A. Howes O. D. Lancet Psychiatry. 2020;7:64–77. doi: 10.1016/S2215-0366(19)30416-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullis C. Swartz T. H. Front. Cardiovasc. Med. 2020;7:95. doi: 10.3389/fcvm.2020.00095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Shalchi R. F. Mohammad F. K. Cureus. 2024;16:e54071. doi: 10.7759/cureus.51433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoury Z. S. Sohail F. Wang J. Mendoza M. Raake M. Silat M. T. Bathinapatta M. R. Sadeghzadegan A. Meghana P. Paul J. Cureus. 2024;16:e53112. doi: 10.7759/cureus.62310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irrera N. Russo M. Pallio G. Bitto A. Mannino F. Minutoli L. Altavilla D. Squadrito F. Int. J. Mol. Sci. 2020;21:6204. doi: 10.3390/ijms21176204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Was H. Borkowska A. Bagues A. Tu L. Liu J. Y. H. Lu Z. Rudd J. A. Nurgali K. Abalo R. Front. Pharmacol. 2022;13:750507. doi: 10.3389/fphar.2022.750507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song L. Pei L. Yao S. Wu Y. Shang Y. Front. Cell. Neurosci. 2017;11:63. doi: 10.3389/fncel.2017.00063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayanthi S. Daiwile A. P. Cadet J. L. Exp. Neurol. 2021;344:113795. doi: 10.1016/j.expneurol.2021.113795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litwiniuk A. Baranowska-Bik A. Domańska A. Kalisz M. Bik W. Pharmaceuticals. 2021;14:1221. doi: 10.3390/ph14121221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedges D. M. Obray J. D. Yorgason J. T. Jang E. Y. Weerasekara V. K. Uys J. D. Bellinger F. P. Steffensen S. C. Neuropsychopharmacol. 2018;43:1405–1414. doi: 10.1038/npp.2017.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chivero E. T. Thangaraj A. Tripathi A. Periyasamy P. Guo M.-L. Buch S. Mol. Neurobiol. 2021;58:2215–2230. doi: 10.1007/s12035-020-02184-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ri M. Int. J. Hematol. 2016;104:273–280. doi: 10.1007/s12185-016-2016-0. [DOI] [PubMed] [Google Scholar]
- Ramirez S. H. Potula R. Fan S. Eidem T. Papugani A. Reichenbach N. Dykstra H. Weksler B. B. Romero I. A. Couraud P. O. Persidsky Y. J. Cereb. Blood Flow Metab. 2009;29:1933–1945. doi: 10.1038/jcbfm.2009.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdallah C. G. Sanacora G. Duman R. S. Krystal J. H. Pharmacol. Ther. 2018;190:148–158. doi: 10.1016/j.pharmthera.2018.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajesh Y. Kanneganti T.-D. Cells. 2022;11:1885. doi: 10.3390/cells11121885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao C. Jiang J. Tan Y. Chen S. Signal Transduction Targeted Ther. 2023;8:359. doi: 10.1038/s41392-023-01588-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampel H. Hardy J. Blennow K. Chen C. Perry G. Kim S. H. Villemagne V. L. Aisen P. Vendruscolo M. Iwatsubo T. Masters C. L. Cho M. Lannfelt L. Cummings J. L. Vergallo A. Mol. Psychiatry. 2021;26:5481–5503. doi: 10.1038/s41380-021-01249-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stojakovic A. Paz-Filho G. Arcos-Burgos M. Licinio J. Wong M.-L. Mastronardi C. A. Mol. Neurobiol. 2017;54:4486–4495. doi: 10.1007/s12035-016-9988-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomeli N. Di K. Czerniawski J. Guzowski J. F. Bota D. A. Free Radical Biol. Med. 2017;102:274–286. doi: 10.1016/j.freeradbiomed.2016.11.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundar V. Ramasamy T. Doke M. Samikkannu T. Redox Rep. 2022;27:53–59. doi: 10.1080/13510002.2022.2043224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson S. C. Pan A. Li L. Sedensky M. Morgan P. Neurotoxicol. Teratol. 2019;71:22–31. doi: 10.1016/j.ntt.2018.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bielecka-Wajdman A. M. Ludyga T. Machnik G. Gołyszny M. Obuchowicz E. Cancer Control. 2018;25:1073274818798594. doi: 10.1177/1073274818798594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauressergues E. Bert E. Duriez P. Hum D. Majd Z. Staels B. Cussac D. Neuropharmacology. 2012;62:784–796. doi: 10.1016/j.neuropharm.2011.08.048. [DOI] [PubMed] [Google Scholar]
- Nunes A. T. Annunziata C. M. Semin. Oncol. 2017;44:377–380. doi: 10.1053/j.seminoncol.2018.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milara J. Martínez-Expósito F. Montero P. Roger I. Bayarri M. A. Ribera P. Oishi-Konari M. N. Alba-García J. R. Zapater E. Cortijo J. Int. J. Mol. Sci. 2022;23:14518. doi: 10.3390/ijms232314518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X. Xu A. Lv J. Zhang Q. Ran Y. Wei C. Wu J. Eur. J. Med. Chem. 2020;185:111822. doi: 10.1016/j.ejmech.2019.111822. [DOI] [PubMed] [Google Scholar]
- Qu J. Yuan Z. Wang G. Wang X. Li K. Int. Immunopharmacol. 2019;70:147–155. doi: 10.1016/j.intimp.2019.02.016. [DOI] [PubMed] [Google Scholar]
- Irrera N. Vaccaro M. Bitto A. Pallio G. Pizzino G. Lentini M. Arcoraci V. Minutoli L. Scuruchi M. Cutroneo G. Anastasi G. P. Ettari R. Squadrito F. Altavilla D. Clin. Sci. 2017;131:487–498. doi: 10.1042/CS20160645. [DOI] [PubMed] [Google Scholar]
- Chang Y.-P. Ka S.-M. Hsu W.-H. Chen A. Chao L. K. Lin C.-C. Hsieh C.-C. Chen M.-C. Chiu H.-W. Ho C.-L. Chiu Y.-C. Liu M.-L. Hua K.-F. J. Cell. Physiol. 2015;230:1567–1579. doi: 10.1002/jcp.24903. [DOI] [PubMed] [Google Scholar]
- Dong Q. Chen L. Lu Q. Sharma S. Li L. Morimoto S. Wang G. Br. J. Pharmacol. 2014;171:4440–4454. doi: 10.1111/bph.12795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q. Gao J. Pang X. Chen A. Wang Y. Front. Pharmacol. 2020;11:559607. doi: 10.3389/fphar.2020.559607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farré-Alins V. Narros-Fernández P. Palomino-Antolín A. Decouty-Pérez C. Lopez-Rodriguez A. B. Parada E. Muñoz-Montero A. Gómez-Rangel V. López-Muñoz F. Ramos E. González-Rodríguez Á. Gandía L. Romero A. Egea J. Antioxidants. 2020;9:1299. doi: 10.3390/antiox9121299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J. Zeng Q. Wu Z. Huang L. Sun T. Ling C. Zhang B. Chen C. Wang H. Neurotherapeutics. 2024;21:e00347. doi: 10.1016/j.neurot.2024.e00347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye H. Nelson L. J. Moral M. G. D. Martínez-Naves E. Cubero F. J. World J. Gastroenterol. 2018;24:1373–1385. doi: 10.3748/wjg.v24.i13.1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayaf K. Battistella S. Russo F. P. Int. J. Mol. Sci. 2024;25:4537. doi: 10.3390/ijms25084537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao J. Zhao G. Wang Y. Ren P. Wu M. Front. Mol. Biosci. 2020;7:37. doi: 10.3389/fmolb.2020.00037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchetti C. Swartzwelter B. Gamboni F. Neff C. P. Richter K. Azam T. Carta S. Tengesdal I. Nemkov T. D'Alessandro A. Henry C. Jones G. S. Goodrich S. A. Laurent J. P. St. Jones T. M. Scribner C. L. Barrow R. B. Altman R. D. Skouras D. B. Gattorno M. Grau V. Janciauskiene S. Rubartelli A. Joosten L. A. B. Dinarello C. A. Proc. Natl. Acad. Sci. U. S. A. 2018;115:E1530–E1539. doi: 10.1073/pnas.1716095115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkuri K. R. Mantovani J. J. Herzenberg L. A. Herzenberg L. A. Curr. Opin. Pharmacol. 2007;7:355–359. doi: 10.1016/j.coph.2007.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shindikar A. Singh A. Nobre M. Kirolikar S. J. Oncol. 2016;2016:1–13. doi: 10.1155/2016/9750785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko J. H. Yoon S.-O. Lee H. J. Oh J. Y. Oncotarget. 2017;8:40817–40831. doi: 10.18632/oncotarget.17256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung W. Roxanis I. Sheldon H. Buffa F. M. Li J.-L. Harris A. L. Kong A. Oncotarget. 2015;6:5678–5694. doi: 10.18632/oncotarget.3296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi K. Geng X. Liu C. Cai G. Hong Q. Mediators Inflammation. 2020;2020:1–9. doi: 10.1155/2020/8032797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S. Lin Q. Shao X. Mou S. Gu L. Wang L. Zhang Z. Shen J. Zhou Y. Qi C. Jin H. Pang H. Ni Z. Exp. Cell Res. 2019;383:111488. doi: 10.1016/j.yexcr.2019.07.001. [DOI] [PubMed] [Google Scholar]
- Jo S.-H. Korean Circ. J. 2011;41:695. doi: 10.4070/kcj.2011.41.12.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J. Liang Y. Zhou L. Front. Pharmacol. 2023;14:1142001. doi: 10.3389/fphar.2023.1142001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolff C. A. Reid J. J. Musci R. V. Bruns D. R. Linden M. A. Konopka A. R. Peelor F. F. Miller B. F. Hamilton K. L. J. Gerontol., Ser. A. 2020;75:32–39. doi: 10.1093/gerona/glz058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji Y. Hua H. Jia Z. Zhang A. Ding G. Kidney Dis. 2024;10:369–383. doi: 10.1159/000539496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landau D. Shukri N. Arazi E. Tobar A. Segev Y. Nephron. 2023;147:693–700. doi: 10.1159/000531313. [DOI] [PubMed] [Google Scholar]
- Foufelle F. Fromenty B. Pharmacol. Res. Perspect. 2016;4:e00211. doi: 10.1002/prp2.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan W.-L. Wang X.-J. Ma Y.-P. Sheng Z.-M. Dong H. Zhang L.-Y. Zhang B.-G. He M.-T. Mol. Med. Rep. 2024;29:46. doi: 10.3892/mmr.2024.13170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y. Gao J. Hou Y. Yang H. Wang D. Zhang G. Qin Z. Du P. Wang Z. Wang Y. Chen Q. Sun Z. Li P. Zhang J. Tang J. J. Immunother. Cancer. 2025;13:e010127. doi: 10.1136/jitc-2024-010127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorelik M. Lee Y. Abe M. Andrews T. Davis L. Patterson J. Chen S. Crother T. R. Aune G. J. Noval Rivas M. Arditi M. Clin. Exp. Immunol. 2019;198:101–110. doi: 10.1111/cei.13314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothman A. M. MacFadyen J. Thuren T. Webb A. Harrison D. G. Guzik T. J. Libby P. Glynn R. J. Ridker P. M. Hypertension. 2020;75:477–482. doi: 10.1161/HYPERTENSIONAHA.119.13642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirilli I. Damiani E. Dludla P. V. Hargreaves I. Marcheggiani F. Millichap L. E. Orlando P. Silvestri S. Tiano L. Antioxidants. 2021;10:1325. doi: 10.3390/antiox10081325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du X. Amin N. Xu L. Botchway B. O. A. Zhang B. Fang M. Front. Pharmacol. 2023;14:1249644. doi: 10.3389/fphar.2023.1249644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bharath L. P. Agrawal M. McCambridge G. Nicholas D. A. Hasturk H. Liu J. Jiang K. Liu R. Guo Z. Deeney J. Apovian C. M. Snyder-Cappione J. Hawk G. S. Fleeman R. M. Pihl R. M. F. Thompson K. Belkina A. C. Cui L. Proctor E. A. Kern P. A. Nikolajczyk B. S. Cell Metab. 2020;32:44–55. doi: 10.1016/j.cmet.2020.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]; , e6
- Eraky S. M. Abdel-Rahman N. Eissa L. A. Prostaglandins, Leukotrienes Essent. Fatty Acids. 2018;136:123–129. doi: 10.1016/j.plefa.2017.06.009. [DOI] [PubMed] [Google Scholar]
- Jiang H. Zuo J. Li B. Chen R. Luo K. Xiang X. Lu S. Huang C. Liu L. Tang J. Gao F. Redox Biol. 2023;63:102754. doi: 10.1016/j.redox.2023.102754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Q. Zhao T. Liu M. Cao D. Li J. Li Y. Xia M. Wang X. Zheng T. Liu C. Mu X. Sun P. Front. Pharmacol. 2021;12:707696. doi: 10.3389/fphar.2021.707696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J. Kim H. Kim J. Yook T. Kim K. Lee J. Yang G. Int. J. Mol. Sci. 2021;22:1324. [Google Scholar]
- Yang N. Guan Q.-W. Chen F.-H. Xia Q.-X. Yin X.-X. Zhou H.-H. Mao X.-Y. Oxid. Med. Cell. Longevity. 2020;2020:1–14. doi: 10.1155/2020/6687185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez-Mariscal F. M. Arenas-de Larriva A. P. Limia-Perez L. Romero-Cabrera J. L. Yubero-Serrano E. M. López-Miranda J. Int. J. Mol. Sci. 2020;21:7870. doi: 10.3390/ijms21217870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C. Chao H. Li Z. Xu X. Liu Y. Bao Z. Hou L. Liu Y. Wang X. You Y. Liu N. Ji J. Exp. Neurol. 2017;290:115–122. doi: 10.1016/j.expneurol.2017.01.005. [DOI] [PubMed] [Google Scholar]
- Vora L. K. Gholap A. D. Jetha K. Thakur R. R. S. Solanki H. K. Chavda V. P. Pharmaceutics. 2023;15:1916. doi: 10.3390/pharmaceutics15071916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Özenver N. Efferth T. Pharmacol. Res. 2021;170:105710. doi: 10.1016/j.phrs.2021.105710. [DOI] [PubMed] [Google Scholar]
- Xu Y. Yang Y. Chen X. Jiang D. Zhang F. Guo Y. Hu B. Xu G. Peng S. Wu L. Hu J. Transl. Neurodegener. 2023;12:49. doi: 10.1186/s40035-023-00381-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y. Yang W. Li W. Zhao Y. Front. Immunol. 2021;12:732933. doi: 10.3389/fimmu.2021.732933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patra J. K. Das G. Fraceto L. F. Campos E. V. R. Rodriguez-Torres M. D. P. Acosta-Torres L. S. Diaz-Torres L. A. Grillo R. Swamy M. K. Sharma S. Habtemariam S. Shin H.-S. J. Nanobiotechnol. 2018;16:71. doi: 10.1186/s12951-018-0392-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S. Chen F. Yin Q. Wang D. Han W. Zhang Y. Front. Physiol. 2020;11:571810. doi: 10.3389/fphys.2020.571810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tengesdal I. W. Banks M. Dinarello C. A. Marchetti C. Front. Immunol. 2024;15:1422249. doi: 10.3389/fimmu.2024.1422249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klück V. Jansen T. L. T. A. Janssen M. Comarniceanu A. Efdé M. Tengesdal I. W. Schraa K. Cleophas M. C. P. Scribner C. L. Skouras D. B. Marchetti C. Dinarello C. A. Joosten L. A. B. Lancet Rheumatol. 2020;2:e270–e280. doi: 10.1016/s2665-9913(20)30065-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortellaro A. Front. Immunol. 2023;14:1171138. doi: 10.3389/fimmu.2023.1171138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hitomi Y. Ebisawa M. Tomikawa M. Imai T. Komata T. Hirota T. Harada M. Sakashita M. Suzuki Y. Shimojo N. Kohno Y. Fujita K. Miyatake A. Doi S. Enomoto T. Taniguchi M. Higashi N. Nakamura Y. Tamari M. J. Allergy Clin. Immunol. 2009;124:779–785. doi: 10.1016/j.jaci.2009.07.044. [DOI] [PubMed] [Google Scholar]; , e6
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No primary research results, software or code have been included, and no new data were generated or analyzed as part of this review.