

Abstract
Type I interferons (IFNs) play critical roles in the host defense by modulating the expression of various genes via the IFN-dependent activation of STAT (signal transducers and activators of transcription) and NF-κB (nuclear factor kappa B) transcription factors. Previous studies established that IFNα/β activates NF-κB to promote cell survival through a PI-3K/Akt pathway, which involves serine phosphorylation and degradation of IκBα. We now describe a second pathway by which IFNs activate NF-κB that is independent of IκB degradation. This pathway involves NF-κB-inducing kinase (NIK) and the TNF-receptor associated factor-2 (TRAF-2), and results in IFNα/β induced processing of the p100/NF-κB2 precursor into p52. IFNα/β stimulates NF-κB DNA binding and NF-κB-dependent transcription. While expression of NIK and TRAF2 constructs causes NF-κB activation, expression of dominant negative NIK and TRAF2 constructs blocks IFN-promoted NF-κB activation and IFN-stimulated κB-dependent transcription, and IFNα/β induced processing of the p100/NF-κB2 precursor into p52. In contrast, PI-3K does not mediate IFNα/β induced p100 processing although PI3K is involved in the pathway resulting in IκBα degradation. Moreover, while IFN promotes cell survival in lymphoblastoid cells, expression of dominant negative NIK and TRAF2 constructs enhances IFN-induced apoptosis. Our results place for the first time NIK and TRAF2, previously shown to function in TNF signaling, within the IFN signal transduction pathway. Thus, IFN induces NF-κB activation to mediate IFN-dependent cell survival signals through a “canonical” pathway of IκBα proteolysis mediated by PI-3K/Akt and a “noncanonical” pathway of p100 processing mediated by NIK/TRAF.
IFNs are multifunctional cytokines that block viral infection, inhibit cell proliferation, and modulate cell differentiation. In a well-characterized signal transduction pathway, type-1 IFNs (IFN α, β, and ω) bind to their cognate receptors and transduce signals from the cell surface resulting in selective gene induction. This pathway is mediated by the receptor-associated JAK tyrosine kinases and the latent cytosolic STAT transcription factors (1–4). Upon their tyrosine phosphorylation, IFNα/β-activated STATs (STAT1, STAT2, and STAT3) form transcriptionally active dimers and translocate into the nucleus to induce gene transcription. However, other signaling pathways are activated by type I IFNs that are required for IFN action. In a series of recent studies, we defined an important IFN signaling pathway involving NF-κB transcription factors that also leads to altered gene expression (5–7).
NF-κB is a transcription factor that regulates the expression of genes involved in cell survival, as well as immune and inflammatory responses, by binding to cis-acting κB sites in the promoters and enhancers of these genes. NF-κB represents a family of related proteins, which in mammals includes NF-κB1 (p105 processed to p50), NF-κB2 (p100 processed to p52), RelA, RelB and c-Rel. Although p50/RelA and p52/RelB heterodimers are the NF-κB complexes most often observed in cells, other combinations of Rel homodimers and heterodimers also form. The NF-κB1 and NF-κB2 precursor proteins undergo proteolytic processing into the p50 and p52 proteins, respectively. The processing of p105 is constitutive and largely cotranslational, while the processing of p100 is tightly controlled through ligand-inducible phosphorylation and subsequent ubiquitinylation.
Under most circumstances, NF-κB homodimers or heterodimers are bound to IκB inhibitory proteins in the cytoplasm of unstimulated cells. In common with a variety of stimuli, IFNα/β promotes the dissociation of the cytosolic inactive NF-κB/IκB complexes via the serine phosphorylation and degradation of Iκ B, leading to NF-κB translocation to the nucleus and DNA binding (5). IFN-dependent NF-κB activation involves the sequential activation of phosphatidylinositol-3 kinase (PI-3K) and Akt, which mediate IFN-dependent cell survival (6). The pathway leading to proteolysis of IκB is denoted as the canonical NF-κB activation pathway. However, recent studies have identified that the LMP1 protein of Epstein-Barr virus (EBV), B-cell activating factor (BAFF), lymphotoxin-beta (LTβ) and lipopolysaccharide (LPS) induce NF-κB activation through an alternative signaling pathway, which does not involve IκB degradation (8–16). This so-called “noncanonical” pathway involves the linkage of TNF-receptor associated factors (TRAFs) to the activation of the MAP3K related kinase, NF-κB-inducing kinase (NIK), which results in the ubiquitinylation and proteolytic processing of p100/NFκ B2 protein and nuclear translocation of p52:Rel heterodimers to activate transcription.
In the present report, we examined whether IFN induced NF-κ B activation through a signaling pathway independent of IκB degradation. We demonstrate that IFNα/β induced NF-κB activation by a pathway mediated by NIK and TRAF proteins that does not results in IκB degradation. This pathway of NF-κ B activation results from the processing of the p100/NF-κB2 precursor into p52, and is independent of the previously described IFN-induced PI-3K/Akt dependent NF-κB signaling pathway, which results in IκBα degradation. Through this signaling pathway IFN promotes cell survival. Therefore, IFN induces NF-κB through two parallel signaling pathways: the canonical pathway dependent on PI-3K and Akt that results in Iκ B α degradation and the noncanonical pathway dependent on NIK and TRAF that results in NF-κB2 processing into p52.
MATERIALS AND METHODS
Biological reagents and cell culture
Recombinant human IFNα (IFNCon1) was provided by InterMune (Brisbane, CA), and its biological activity expressed in terms of international reference units/ml as described previously (17). Anti-Rel and IκBα antibodies were generously provided by Dr. N. Rice (National Cancer Institute). Human Daudi cells were maintained in static suspension cultures at 2–15 x 105 cells/ml in RPMI-1640 medium supplemented with 10% defined calf serum (DCS) (HyClone Labs, Logan, UT). For experiments, cells were suspended at 0.5–1 x 108 cells per ml in medium prior to the addition of IFN or other agents.
Transfection conditions and constructs
Transient transfection of cells (107) was accomplished by electroporation (capacitance 300 μF, 250 V) with 500 μg of salmon sperm DNA and 20 μg of plasmid DNA for each sample. Dominant negative (DN)-p85 is a p85 mutant in which 35 amino acids from residues 479–513 are deleted and two amino acids (Ser-Arg) are inserted (18). p110* is a constitutively-active mutant of the p110 subunit of PI-3K (19). DN-NIK is a NIK mutant in which Lys-Lys in the ATP binding domain has been replaced by Ala-Ala (20). DN-TRAF2 lacks the NH2-terminal 86 amino acids that comprise the RING finger domain of TRAF2 (21). All constructs were overexpressed in transiently transfected cells (Supplemental Fig. 1). The pUX-CAT 3XHLAκB CAT reporter construct contains three tandemly repeated copies of the NF-κB site from the HLA-B7 gene (22).
Supplemental Figure 1. Expression of PI3K, NIK and TRAF constructs in transiently-transfected Daudi cells.
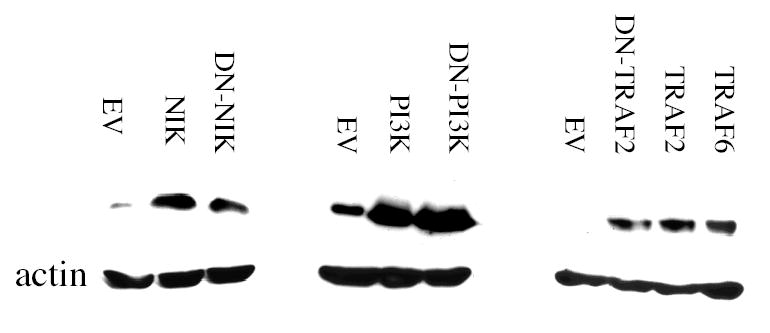
Whole cell extracts were prepared from Daudi cells transiently transfected for 48 hr with empty-vector (EV), PI3K, DN-PI3K, NIK, DNNIK, TRAF2, DN-TRAF2, or TRAF6. The proteins were resolved by SDS-PAGE, blotted onto PVDF membranes and probed with antisera against PI3K (p85), NIK, TRAF2, TRAF6, or actin as a loading control. Blots were visualized by enhanced chemiluminescence.
NF-κB activity measurements
Nuclei were extracted with buffer (20 mM Tris-HCl, pH 7.85, 250 mM sucrose, 0.4 M KCl, 1.1 mM MgCl2, 5 mM β-mercaptoethanol, 1 mM NaF, 1 mM Na3VO4, 1 mM PMSF, 5 μg/ml soybean trypsin inhibitor, 5 μg/ml leupeptin and 1.75 μg/ml benzamidine), and extracts were frozen and stored at −80°C (23). For EMSA, the nuclear extracts were incubated with a [32P]-labeled κ B probe (5′-AGTTGAGGGGACTTTCCCAGG-3′) derived from an NF-κB binding sequence in the immunoglobulin gene promoter (24). To define the presence of specific Rel proteins, nuclear extracts were preincubated with anti-Rel antibodies at 25°C for 20 min and then subjected to EMSA. Gels were quantitated by PhosphorImage autoradiography. For reporter gene assays, Jurkat cells were transiently cotransfected by electroporation with the pUX-CAT 3XHLAκB CAT reporter construct (22) and the appropriate expression vector. After 48 hr the cells were treated with IFNCon1 (1,000 U/ml) for 15 min and assayed for CAT activity. Following thin-layer chromatography, radioactivity was measured by PhosphorImage autoradiography.
IκBα degradation
At various times after IFNα treatment, 2 x 107 cells were lysed directly in Laemmli buffer and equivalent amounts of protein were subjected to SDS-PAGE. Proteins were transferred to PVDF membranes, immunoblotted with specific affinity-purified rabbit anti-Iκ B α and visualized by enhanced chemiluminescence (ECL, Pierce).
GST fusion construct
p65 (RelA) cDNA was subcloned into the HindIII and EcoRI sites of pGEX-KG (25). The construct was confirmed by restriction enzyme digestion. The p65-GST fusion protein was obtained from E. coli transformed with the plasmid construct and affinity-purified on glutathione-Sepharose (Pharmacia) as previously described (26).
Immunoprecipitations and immunoblot analysis
For immunoprecipitation studies, transiently-transfected cells were treated with IFNα (1,000 IU/ml) at 37°C for the indicated periods of time and then washed with ice-cold PBS and lysed for 20 min in lysis buffer (50 mM Tris, pH 7.4, 150 mM NaCl, 1 mM EDTA 0.5 % NP-40; 15% glycerol) containing 1 mM NaF, 1 mM Na3VO4, 1 mM PMSF, 5 μg/ml soybean trypsin inhibitor, 5 μg/ml leupeptin and 1.75 μg/ml benzamidine. Samples were centrifuged (12,000 x g, 15 min) at 4°C and supernates were immunoprecipitated with anti-p65 (Santa Cruz Biotechnology) overnight at 4°C. Immune complexes were collected using Protein A-Sepharose beads (Pharmacia) and eluted in sample buffer. Samples were run on SDS-7.5% PAGE, transferred to PVDF membranes (Millipore) and probed with anti-p52 (Santa Cruz Biotechnology), followed by anti-mouse IgG coupled with horseradish peroxidase (Santa Cruz), or with mouse IgG TrueBlot (eBioscience). Blots were developed using ECL (Pierce).
For GST pull-down assays, lysates from control or IFN-treated Daudi cells were incubated with p65-GST or GST bound to glutathione-agarose beads. The bound proteins were eluted with Laemmli buffer, resolved by SDS-PAGE (7.5%), blotted onto PVDF membranes and probed with anti-p52.
Apoptosis assays
For determining apoptosis, cells were cytospun onto glass slides, fixed with 4% formaldehyde, permeabilized with 0.2% Triton X-100 and processed for terminal deoxynucleotide transferase mediated dUTP nick end labeling (TUNEL) according to the manufacturer’s recommendations (Boehringer-Mannheim). Alternatively, lysates of control and IFN-treated (1000 IU/ml, 24 hr) cells were analyzed for apoptotic DNA by modification of a chemiluminescence-based assay (27). In brief, cells (5 x 106) were lysed in hypotonic buffer, and sequentially digested with RNase and proteinase K. Low molecular weight DNA was extracted and subjected to non-isotopic labeling of 3’ ends with DIG-11-dUTP and Taq DNA polymerase. Labeled DNA was separated by electrophoresis on 1.6% agarose, transferred to nitrocellulose, and fragmented DNA visualized by chemiluminescent detection with alkaline-phosphatase conjugated anti-DIG and CDP-Star substrate (Boehringer-Mannheim).
RESULTS
The Role of NIK in IFN induced NF-κB activity and degradation of IκB
The protein kinase NIK, a member of the MAP3K family, is central to signaling pathways by which TNF and IL-1 activate NF-κB. Previous studies have shown that a kinase-inactive NIK mutant suppresses NF-κB activation mediated by TNF and IL-1 (20). The role of NIK in IFN-promoted NF-κB activation was assessed by expressing NIK or a dominant negative kinase-inactive NIK mutant (DN-NIK) in Daudi cells and examining the effect on IFN-promoted NF-κB activation. As shown in Fig. 1A, NIK expression promoted NF-κB DNA binding activity similar to the level induced by IFN treatment. In contrast, expression of DN-NIK in Daudi cells blocked IFN-promoted NF-κB DNA binding activity (Fig. 1B).
Figure 1. The roles of NIK, TRAF2 and p52 in NF-κB DNA binding activity induced by IFN.
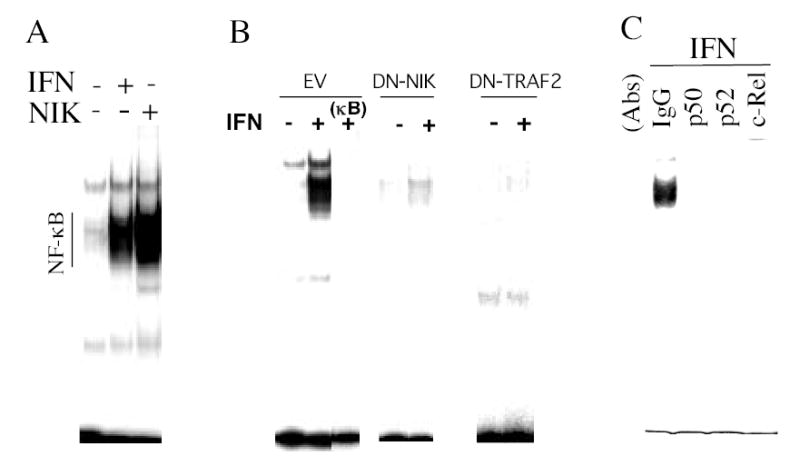
(A) Nuclear extracts were prepared from Daudi cells treated in the absence or presence of IFNCon1 (1,000 IU/ml, 15 min), and were subjected to EMSA. In addition EMSA was performed on nuclear extracts from cells transiently transfected (transfection efficiency of ≈85%) for 48 hr with a NIK construct in the pCMV expression vector. (B) EMSA on nuclear extracts from control and IFN-treated Daudi cells transiently transfected for 48 hr with DN-NIK or DN-TRAF2 expression vectors or with empty vector (EV) in the absence or presence of a 50-fold excess of unlabeled κB oligonucleotide probe (κB). (C) To define the presence of specific Rel proteins in nuclear extracts of IFN-treated Daudi cells (1,000 IU/ml, 15 min), extracts were preincubated with anti-Rel antibodies or isotype-matched control antisera (IgG) prior to EMSA.
Inhibitory IκB proteins bind to NF-κB in the cytoplasm of cells maintaining it in an inactive state. Through a PI-3K dependent pathway, IFNα/β promotes the serine phosphorylation and proteosome-mediated degradation of IκB, thereby inducing NF-κB translocation to the nucleus. Expression of a dominant negative PI-3K mutant blocks IFN-induced NF-κB DNA binding activity and gene reporter activities, as well as the IFN-induced degradation of IκBα (6). The role of NIK and PI-3K in IFN-promoted IκB degradation was assessed by expressing dominant negative NIK or PI-3K in Daudi cells and examining the effect on IFN promoted IκB α degradation by immunoblotting. As shown in Fig. 2, while IFN induced a progressive decrease in cellular levels of IκBα (EV), expression of dominant negative PI-3K blocked IκBα degradation as shown previously (6). In contrast, expression of DN-NIK had no effect on IFN promoted IκBα degradation. This result demonstrates the distinct roles that both NIK and PI-3K play in IFN induced NF-κB activation, and they apparently do so through distinct signaling pathways.
Figure 2. IFN induces IκBα degradation via a NIK-dependent but a PI3K-independent signaling pathway.
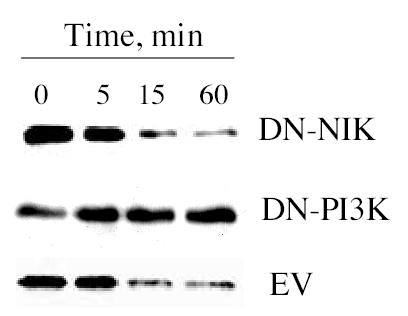
Cell lysates prepared from Daudi cells transiently transfected for 48 hr with DN-NIK, DN-PI3K or empty-vector (EV) treated with IFNCon1 (1,000 IU/ml) were resolved by SDS-PAGE, blotted onto PVDF membranes, probed with anti-IκBα, and visualized by enhanced chemiluminescence.
To assess the functional consequences of NIK on κB-dependent gene transcription, human Jurkat cells were cotransfected with DN-NIK and a CAT reporter construct (pUX-CAT 3XHLAκB CAT) containing three tandemly repeated copies of the NF-κB site from the HLA-B7 gene. Previous studies have shown that this reporter construct is induced by IFN and HLA-B7 is an IFN-induced gene (6). Jurkat cells were used in these assays, because they are IFN-responsive in reporter assays while Daudi cells are relatively nonresponsive (28). As shown in Fig. 3, IFN-treatment stimulated κB-dependent transcription ~6–8-fold. In contrast expression of DN-NIK blocked the stimulation by IFN of κB-dependent transcription by ~85% (Fig. 3). DN-NIK expression had no effect on stimulation by IFN of STAT-dependent transcription (data not shown) demonstrating that the role of NIK is distinct from the well-established ISRE-dependent mechanism in regulating gene expression.
Figure 3. The roles of PI3K, NIK and TRAF in IFN-induced NF-κB-dependent gene transcription induced by IFN.
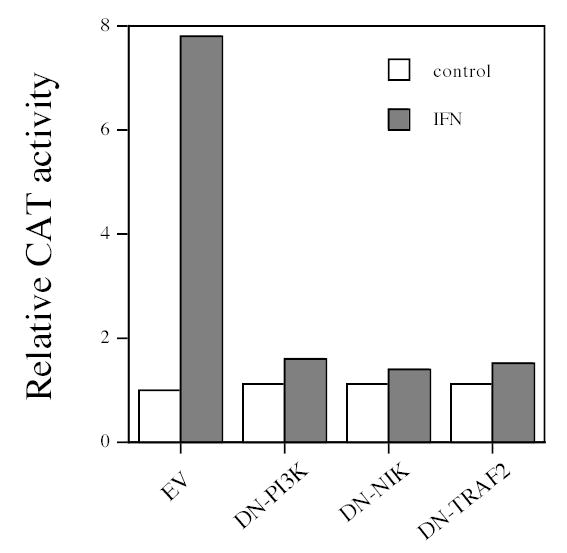
NF-κB-dependent reporter gene activity in control and IFN-treated Jurkat cells transiently cotransfected for 48 hr with DN-PI3K, DN-NIK or DN-TRAF2, and the pUX-CAT 3XHLAκB construct (22). Data shown are from one of three experiments with quantitatively similar results, and expressed relative to CAT activity in cells transfected with empty vector.
The Role of TRAF2 in IFN promoted NF-κB activation
The results described so far demonstrate that NIK plays a role in IFN induced NF-κB activation and cell survival. TRAF2 is a member of the family of TNF-receptor associated factors (TRAFs 1–7) and functions as an adaptor molecule for various members of the TNF superfamily to activate NIK and subsequently mediate NF-κB activation. To assess the role of TRAF2 in IFN-induced NF-κB signal transduction pathway and IFN-mediated cell survival, cells were transfected with an expression plasmid for DN-TRAF2, which lacks the NH2-terminal 86 amino acids of the RING finger domain of TRAF2 and functions as a dominant negative for TRAF2-dependent signaling events (21). The transfected cells were assessed for IFN induced NF-κB activation and NF-κB-dependent gene transcription. As shown in Fig. 1B, expression of DN-TRAF2 in Daudi cells blocked IFN-promoted NF-κB DNA binding activity. Moreover, expression of DN-TRAF2 in Jurkat cells blocked the stimulation by IFN of κB-dependent transcription by ~85%, which is similar to the inhibitory effect of DN-NIK expression (Fig. 3). These results indicate that both TRAF2 and its downstream effector NIK are involved in the IFN-induced NF-κB signal transduction pathway.
IFN induces p100 processing to p52
Our results demonstrate that IFN induces NF-κB through a NIK/TRAF dependent signaling pathway, which is independent of IκBα degradation. Recent studies suggest an alternative pathway for NF-κB activation, in which NIK/TRAF signaling pathway mediates the processing of the p100 NF-κB2 precursor protein into p52. In the “noncanonical pathway” of NF-κB activation p52/Rel heterodimers are formed, which translocate into the nucleus to regulate gene transcription. In a previous study, we determined that IFN treatment of Daudi cells activates NF-κB DNA binding complexes containing cRel but not RelB (5). Preliminary studies of p65 immunoprecipitates indicated that in Daudi cells IFN induced the appearance of a 50–55kda protein that was poorly resolved from heavy chain of IgG in Western blots (data not shown), which we hypothesized might be p52.
To assess whether IFN induces p100 processing to p52, p65 was expressed as a glutathionine S-transferase fusion protein (p65-GST) and GST pull-down assays were performed. Daudi cells were treated with IFN and at various times after IFN addition nuclear and cytoplasmic extracts were prepared, incubated with p65-GST or GST alone, and GST bound material was subjected to immunoblotting with anti-p52. As shown in Fig. 4, in control Daudi cells no detectable p52 was bound to p65-GST. However, within 5 min of addition IFN induced the appearance of p52 in both nuclear and cytoplasmic extracts prepared from Daudi cells, which was bound to p65-GST (Fig. 4C). The maximal binding of p52 in nuclear and cytoplasmic extracts occurred between 15 and 30 min after addition, and p52 was detectable in nuclear extracts as long as 2 hr after IFN addition. To further validate the interaction of p65 with p52 in vitro, anti-p65 immunoprecipitates were prepared from Daudi cell lysates at various times after IFN addition, and western blotted with anti-p52. As shown in Figure 4C, IFN induced the interaction of p65 with p52 after IFN addition as determined by their co-immunoprecipitation from whole cell extracts. Moreover, the presence of p52, as well as p50 and c-Rel, in IFN induced NF-κB complexes was demonstrated by supershift assays with Rel-specific antisera (Fig. 1C).
Figure 4. IFN induces p100 processing to p52 in Daudi cells and human fibrosarcoma cells.
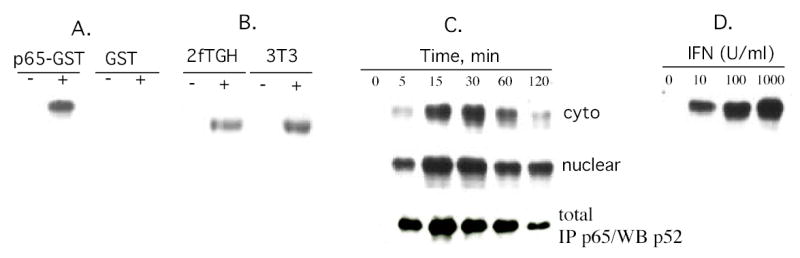
(Panel A) Lysates prepared from control or IFN-treated (1,000 IU/ml; 15 min) Daudi cells were incubated with either GST or p65-GST fusion protein bound to glutathione-agarose beads (pull-down assays). The proteins were resolved by SDS-PAGE, blotted onto PVDF membranes and probed with anti-p52. Blots were visualized by enhanced chemiluminescence. (Panel B) p65-GST pull down assays on nuclear extracts from human 2fTGH fibrosarcoma cells or mouse 3T3 cells treated for 15 min with human IFNCon1 or murine IFNβ (1,000 U/ml), respectively. (Panel C) p65-GST pull down assays on cytoplasmic and nuclear extracts from Daudi cells treated with IFNCon1 (1,000 U/ml) for varying times. Alternatively (lower panel), whole cell extracts were immunoprecipitated with anti-p65 Ab and probed with anti-p52 Ab. (Panel D) p65-GST pull down assays on nuclear extracts from Daudi cells treated with IFN (0, 10, 100 or 1,000 U/ml for 15 min).
The specificity of the interaction between p52 and p65-GST is further demonstrated by the finding that pull-down assays with GST alone did not bring down p52 from IFN-treated Daudi cells (Fig. 4A). Moreover, as shown in Fig. 4B IFN induced p100 processing to p52 in human fibrosarcoma cells and mouse embryo fibroblasts, which are cells previously found to be sensitive to IFN induction of NF-κB activation (5).
IFNs are extremely bioactive substances with effects induced at low concentrations, To further assess the biological significance of IFN induced processing of p100 into p52 we determined whether dose response between IFN and p52 appearance exists. Daudi cells were treated with IFN for 15 min at concentrations, varying from 10 to 1000 U/ml, nuclear extracts were prepared and assayed for p52 binding to p65-GST. As illustrated in Fig. 4D, p52 appearance in nuclear extracts of Daudi cells was detectable at an IFN concentration of 10 U/ml and its appearance increased with increasing IFN concentrations. Moreover, the dose-response relationship for p52 appearance is similar to our previous findings of IFN-induced NF-κB activation in Daudi cells as well as the induction of STAT-dependent DNA binding activity (5).
IFN-induced p100 processing is dependent on NIK and TRAF, but is independent of PI3K
The role of NIK and TRAF in IFN-induced p100 processing was evaluated by expressing a dominant negative NIK or TRAF construct and examining the effect on IFN-induced processing of p100 to p52 by GST pull-down assays. As shown in Fig. 5A, a dominant negative NIK construct completely blocked IFN-induced p52 appearance in nuclear extracts. NIK expression alone induced low levels of p52 processing and enhanced IFN-induced p52 processing. Moreover, expression of TRAF2 or TRAF6 had no effect on IFN-induced appearance of p52 in nuclear extracts (Fig. 5B). In contrast, expression of a dominant negative TRAF2 constructs completely blocked the IFN-induced appearance of p52 in nuclear extracts. These results demonstrate the roles of NIK and TRAF in IFN-induced p100 processing.
Figure 5. IFN-induced p100 processing is dependent on NIK and TRAF.
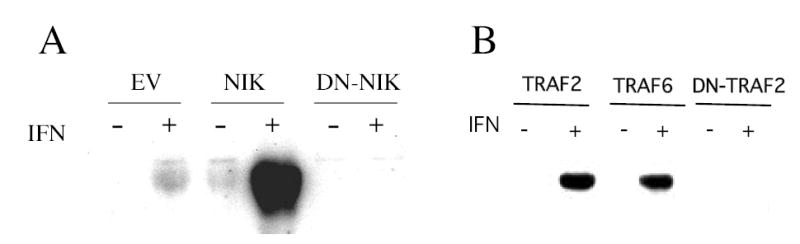
Lysates prepared from control or IFN-treated (1000 U/ml; 15 min) Daudi cells transiently transfected for 48 hr with NIK, DN-NIK or empty vector (Panel A), or TRAF2, TRAF6 or DN-TRAF2 (Panel B) were either incubated with p65-GST fusion protein bound to glutathione-agarose beads. The proteins were resolved by SDS-PAGE, blotted onto PVDF membranes and probed with anti-p52. Blots were visualized by enhanced chemiluminescence.
We previously demonstrated the role of PI-3K on IFN induced NF-κB activation (6). To assess the role of PI3K in IFN-induced p100 processing, PI3K activity in Daudi cells was inhibited either by expression of a dominant negative PI3K construct or with the pharmacological PI3K inhibitor LY294002 and examining the effect on IFN-induced processing of p100 to p52 by GST pull-down assays. As shown in Figure 6A expression of a constitutively active form of PI3K, p110*, alone had no effect on either basal or IFN-promoted appearance of p52 in nuclear extracts. Cells transfected with empty vector served as an internal control. Moreover, expression of DN-PI3K had no effect on IFN-induced p52 nuclear appearance, although its expression inhibited IFN-induced NF-κB activation and N F - κB dependent gene transcription (Figs. 1 and 3). Further evidence that PI3K does not play a role in IFN-induced p100 processing is provided by the finding that the PI3K inhibitor LY294002, which inhibits IFN induced PI3K-dependent signaling events in Daudi cells (6), has no effect on IFN-induced p100 processing (Fig. 6B). Thus, these results demonstrate that IFN-induced p100 processing to p52 is dependent on NIK/TRAF signaling but is independent of PI3K.
Figure 6. IFN-induced p100 processing is independent of PI3K.
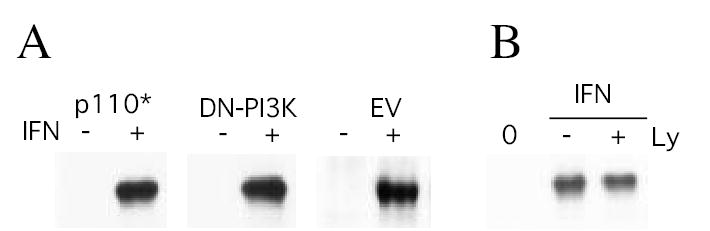
Lysates prepared from control or IFN-treated (1000 U/ml; 15 min) Daudi cells transiently transfected for 48 hr with constitutively-active PI3K (p110*), DN-PI3K or empty vector (Panel A), or control or IFN-treated (1000 U/ml; 15 min) Daudi cells pretreated with 5 μM LY294002 (Panel B) were either incubated with p65-GST fusion protein bound to glutathione-agarose beads. The proteins were resolved by SDS-PAGE, blotted onto PVDF membranes and probed with anti-p52. Blots were visualized by enhanced chemiluminescence.
The Role of NIK and TRAF2 in IFN mediated cell survival
NF-κB plays a critical role in regulating cell survival. While IFN has demonstrated pro-apoptotic activity on some tumor cells (29,30), IFN also protects cells from pro-apoptotic stimuli (5,31,32). The strategy of expressing in Daudi cells dominant negative constructs to block IFN-promoted NF-κB activation and to sensitize cells to pro-apoptotic signals generated by IFN itself has been used previously to define the role of IκBα, PI3K and Akt in the IFN promoted cell survival signals (5,6). To assess the role of NIK in IFN mediated cell survival, Daudi cells were transfected with expression plasmids for DN-NIK, and assessed for apoptosis by terminal deoxynucleotide transferase mediated dUTP nick end labeling (TUNEL) assays. In empty vector- and mock-transfected Daudi cells, IFN induced a negligible increase in apoptosis, as determined by TUNEL assays. However, expression of DN-NIK resulted in a marked sensitization to IFN-induced cell death (Fig. 7A), demonstrating that NIK plays a role in the pathway that protects cells against IFN’s own pro-apoptotic action. A prominent feature of apoptosis is the formation of DNA ladders, which reflects DNA cleavage into discrete multimers of ~200 bp. When cell lysates of IFN-treated Daudi cells expressing DN-NIK were examined by a highly sensitive chemiluminiscent-based DNA fragmentation assay, the formation of the telltale DNA ladder was clearly evident when compared to lysates of IFN-treated Daudi cells transfected with an empty vector (Fig. 7B). In addition, expression of DN-TRAF2 sensitized Daudi cells to IFN-induced cell death as determined by TUNEL or DNA fragmentation assays, while expression of the plasmid alone had no effect. These results indicate that the NIK/TRAF dependent pathway leading to NF-κB activation protects cells against IFN’s pro-apoptotic action. Thus, IFN also generates a strong cell survival signal through the noncanonical NF-κB pathway dependent on NIK/TRAF, in addition to the canonical NF-κB pathway dependent on Akt/PI3K (6).
Figure 7. The roles of PI3K, NIK and TRAF in the promotion of cell survival by IFN.
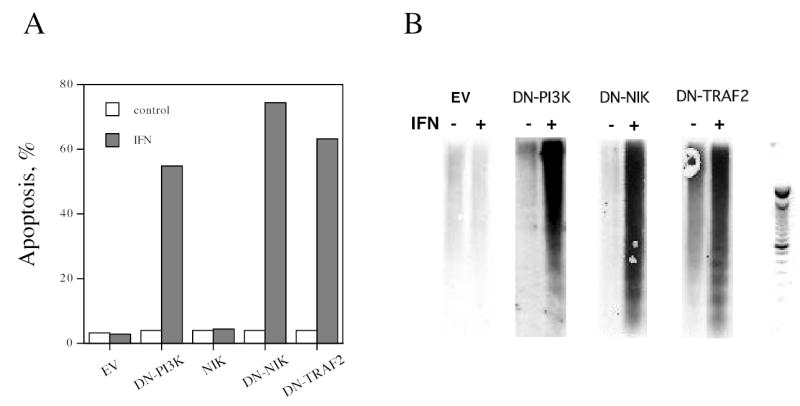
Daudi cells transiently transfected for 48 hr with DN-PI3K, DN-NIK or DN-TRAF2 were pretreated overnight with IFN (1,000 U/ml) and analyzed for apoptosis by TUNEL assays (Panel A), or for apoptotic DNA by a chemiluminescent assay with a DNA ladder provided for reference (Panel B). The data shown in Panel A are the average of two experiments performed in duplicate.
DISCUSSION
IFNs by definition are antiviral cytokines, but they also regulate cell survival and the immune/inflammatory system. IFNα elicits biological activity by inducing gene expression through the JAK tyrosine-kinase dependent activation of STAT transcription factors (1–3,17,23,24,33–35). Upon IFN binding the JAKs, TYK2 and JAK1, become activated and phosphorylate specific tyrosine residues in STAT proteins (23,36). Phosphorylated STAT1 and STAT2 complex with the IRF-9 DNA-binding protein, translocate into the nucleus and bind to a highly conserved IFN-stimulus response element (ISRE) in ISG promoters to directly activate these genes (1,37). The activation of STAT1 and STAT2 proteins has been assumed to account for nearly all of IFN’s manifold biologic actions. However, IFN has been shown to regulate gene expression in both STAT1 and STAT2 knockout mouse cells (38,39). Thus, in addition to the classic JAK-STAT pathway IFN must activate other signaling pathways to regulate gene expression and induce its biological effects. We have previously established that IFNα activates the NF-κB pathway that promotes cell survival in a diverse variety of human cells (5–7).
The family of NF-κB transcription factors regulates the expression of a wide spectrum of genes involved in immunity, inflammation, and cell growth by binding to cis-acting κB sites (7,40–43). However, the analysis of NF-κB deficient mice and cells have led to the identification of a novel function for this versatile transcription factor, the inhibition of apoptosis. In most cells, NF-κB lies dormant in the cytoplasm through the binding of IκB inhibitory proteins. Stimulating agents such as IFN promote the serine phosphorylation and subsequent degradation of IκB thereby unmasking of the nuclear localization sequence of NF-κB. The liberated Rel dimers translocate into the nucleus, bind DNA and regulate κB dependent gene transcription. This NF-κB signaling pathway is described as the canonical pathway and is activated by viruses, cytokines, and lipopolysaccharides. IFN induces the canonical N F - κB pathway involving PI3K-dependent IκBα degradation and liberation of p50: Rel protein complexes (6). We describe in the present study an additional IFN signaling pathway, which leads to NF-κB activation that is independent of IκBα degradation.
In this pathway upon IFN addition the p100 NF-κB2 precursor protein is processed into p52, which dimerizes with p65 (RelA) and translocates to the nucleus to regulate κB-dependent gene transcription. We show that IFN induces the rapid appearance of p52:p65 complexes in the cytoplasm, which rapidly translocate into the nucleus, in lymphoblastoid and fibrosarcoma cells. IFNα/β induced processing of the p100/NF-κB2 precursor into p52 is dependent on NIK, the NF-κB inducing kinase, but is independent of PI3K. We show that DN-PI3K expression or LY294002 (a pharmacological inhibitor of PI3K) treatment has no affect on the IFN-induced processing of p100 precursor. In contrast, DN-NIK expression blocks IFN-induced p100 processing and IFN-induced NF-κB -dependent gene transcription. Thus while the canonical NF-κB pathway is PI3K-dependent, this alternate NF-κB pathway is independent of PI3K. A number of cytokines activate NF-κB through a PI-3K/Akt pathway (6,44,45), which our data would indicate is through the canonical NF-κB pathway of IκBα degradation and liberation of p50:Rel protein complexes.
Recent studies have identified that members of the TNF superfamily such as CD40, LT-β, and BAFF activate an alternate NF-κB pathway. This noncanonical pathway is mediated by activation of NIK, which binds to the C-terminal portion of p100 precursor protein resulting in its proteolytic processing to p52 and the preferential release of p52: RelB dimers (13,46). Mutant mice with defects in this pathway such as mice deficient in NIK, lymphotoxin or its receptor, B-cell activating factor or its receptor, and NF-κB2 exhibit similar immune defects, suggesting that proper activation of this pathway plays a critical role in the development, organization, and function of the immune system (47–53). Moreover, uncontrolled processing of p100 may contribute to the development of human malignancies (54,55). In contrast to the formation of p52: RelB dimers, we find that IFN induces the translocation of p52:p65 (RelA) into the nucleus through the noncanonical pathway. The involvement of RelA dimers, not RelB dimers, in the noncanonical NF-κB pathway is not without precedent. For example, CD28 stimulation leads to preferential nuclear translocation of RelA and p52 (56). Moreover, p52 and RelA are recruited to the promoters of CD28 target genes, while RelB and p50 are not recruited. Transcriptionally active p52: RelA heterodimers have been observed after CD40 and LPS stimulation of B cells (13,14).
In addition, we show that the IFN-induced noncanonical NF-κB signaling pathway also involves the TRAF protein, TRAF2. While TRAF2 constructs induce NF-κB activation, expression of DN-TRAF2 blocks IFN-promoted NF-κB activation, IFN-induced processing of p100 into p52, and IFN-stimulated κB-dependent transcription. The TRAF proteins are critical mediators of NF-κB pathways activated by members of the TNF superfamily that have TRAF binding domains (57). TRAF proteins (six members of the TRAF family have been found to date) form heterodimers with one another and activate NIK leading to the ubiquitinylation and proteasomal processing of p100 into p52. We focused on the potential role of TRAF2 in IFN-induced NF-κB activation because a conserved TRAF binding site (58), a (P/S/A/T)X(Q/E)E motif, is present within the cytoplasmic tail of the IFNAR1 subunit of the IFNα/β receptor. Moreover, in preliminary studies TRAF2 was found in anti-IFNAR1 precipitates, and a DN-TRAF2 construct blocked IFN-induced NF-κB activity. TRAF2 plays a direct role in p100 processing by TNF receptor family members CD40 and CD120 (59). Since TRAF2 forms heterodimers with other TRAF proteins, we anticipate the involvement of other TRAF proteins in the activation of the NF-κB pathway by IFN.
NF-κB plays a critical role in cell survival through regulation of anti-apoptotic and pro-apoptotic signaling pathways. IFNα/β promotes the survival of activated T cells (32), protects CD4+ cells from HIV-induced cell death (31), and protects lymphoblastoid cells against several pro-apoptotic stimuli (5). However, IFN can also efficiently induce the apoptosis of certain tumor cells (29,30). Thus, the clinical efficacy of IFNα in the treatment of human cancer may be limited by its inability to induce marked cell death because of activation of the NF-κB cell survival pathway. Therefore, understanding the molecular mechanisms underlying the NF-κB pathway activated by IFN that regulate apoptosis is an important goal. We have previously shown that expression of super-repressor IκB α, DN-PI3K, and DN-Akt constructs or the use of pharmacological inhibitors of PI3K (LY294002 and wortmannin) to inhibit the canonical NF-κB pathway potentiates the ability of IFN to induce cell death (5,6). In the present study we show that inhibition of the IFN-induced noncanonical NF-κB pathway by expression of DN-NIK or DN-TRAF2 also potentiates IFN-induced cell death. These results suggest that the canonical and noncanonical NF-κB pathways are functionally redundant for IFN-induced cell death.
The results reported here demonstrate that IFN activates two distinct NF-κB signaling pathways to promote cell survival. One unanswered question is why does IFN specifically, and other cytokines in general, activate multiple NF-κB pathways. These different NF-κB pathways result in the activation of different combination of Rel dimers. For example, in Daudi cells IFN induces the canonical N F - κB pathway resulting in the activation c-Rel/p50 heterodimers while the non-canonical pathway results in RelA/p52 dimers. The generation of knockout mice has defined the overlapping role of Rel proteins to processes such as proliferation and cell survival, nonetheless they have identified the distinct roles of individual Rel proteins (52,60–63). Therefore, the differential activation of canonical and noncanonical NF-kB pathways has potential important ramifications for receptor-mediated gene induction. A number of IFN-regulated genes have κB elements (64), but the role of NF-κB in IFN-regulated gene expression has been largely unexplored (7). Recent studies with fibroblasts in which the p65 and p50 NF-κB proteins have been knocked out demonstrate that NF-κB plays a complex role in IFN-regulated gene expression (7). Specifically, NF-κB enhances the expression of some IFN-regulated genes, while diminishing the expression of other IFN-regulated genes. In future studies it will be important to define the roles of the “canonical” p50: Rel dimers and the “noncanonical” p52: Rel dimers in IFN-regulated gene expression.
Thus, in summary, our studies reveal that IFN’s antiapoptotic activity requires the dual activation of the canonical and noncanonical pathways. While the canonical pathway requires the activation of PI3K, the noncanonical pathway is PI3K-independent. In contrast, the noncanonical pathway induced by IFN is NIK dependent, while the canonical pathway is NIK-independent. Thus, these parallel pathways require distinct signaling components. It is of particular interest that blockage (using either dominant-negative constructs or by pharmacological inhibitors) of either the “canonical” PI3K pathway or the “non-canonical” NIK/TRAF pathway results in nearly complete inhibition of both NF-κB DNA binding and NF-κB dependent gene activities. These findings are consistent with the hypothesis that these IFN-induced signaling pathways function synergistically to regulate NF-κB activity. Moreover, our results place for the first time NIK and TRAF2, previously shown to function in signaling by members of the TNF cytokine superfamily, within the IFN signal transduction pathway. Continued investigation is required to understand how these signaling circuits cooperate in IFN-signal transduction and the diversity of IFN’s biological actions. Our results are a reminder that the mechanisms by which cells execute diverse functions are dependent on different combinations of distinct as well as shared signals to elicit diverse biological responses.
Acknowledgments
Supported by NIH grant CA73753 (L.M.P.) and by funds from the Muirhead Chair Endowment at the University of Tennessee Health Science Center. We thank D. Donner (Walther Oncology Center), M. Kasuga (Kobe University), J. Vilcek (New York University), R. Roth (Stanford University) and L.T. Williams (University of California, San Diego) for providing expression vectors.
References
- 1.Friedman RL, Stark GR. Nature. 1985;314:637–639. doi: 10.1038/314637a0. [DOI] [PubMed] [Google Scholar]
- 2.Larner AC, Chaudhuri A, Darnell JEJ. JBiolChem. 1986;261:453–459. [PubMed] [Google Scholar]
- 3.Darnell JEJ, Kerr IM, Stark GR. Science. 1994;264:1415–1421. doi: 10.1126/science.8197455. [DOI] [PubMed] [Google Scholar]
- 4.Schindler C, Fu XY, Improta T, Aebersold R, Darnell JEJ. ProcNatlAcadSciUSA. 1992;89:7836–7839. doi: 10.1073/pnas.89.16.7836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang CH, Murti A, Basu L, Kim JG, Pfeffer LM. Proc Natl Acad Sci USA. 2000;97:13631–13636. doi: 10.1073/pnas.250477397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang CH, Murti A, Pfeffer SR, Kim JG, Donner DB, Pfeffer LM. J Biol Chem. 2001;276:13756–13761. doi: 10.1074/jbc.M011006200. [DOI] [PubMed] [Google Scholar]
- 7.Pfeffer LM, Kim JG, Pfeffer SR, Carrigan DJ, Baker DP, Wei L, Homayouni R. J Biol Chem. 2004;279:31304–31311. doi: 10.1074/jbc.M308975200. [DOI] [PubMed] [Google Scholar]
- 8.Pomerantz JL, Baltimore D. Mol Cell. 2002;10:693–695. doi: 10.1016/s1097-2765(02)00697-4. [DOI] [PubMed] [Google Scholar]
- 9.Luftig M, Yasui T, Soni V, Kang MS, Jacobson N, Cahir-McFarland E, Seed B, Kieff E. Proc Natl Acad Sci U S A. 2004;101:141–146. doi: 10.1073/pnas.2237183100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xiao G, Harhaj EW, Sun SC. Mol Cell. 2001;7:401–409. doi: 10.1016/s1097-2765(01)00187-3. [DOI] [PubMed] [Google Scholar]
- 11.Xiao G, Fong A, Sun SC. J Biol Chem. 2004;279:30099–30105. doi: 10.1074/jbc.M401428200. [DOI] [PubMed] [Google Scholar]
- 12.Muller JR, Siebenlist U. J Biol Chem. 2003;278:12006–12012. doi: 10.1074/jbc.M210768200. [DOI] [PubMed] [Google Scholar]
- 13.Dejardin E, Droin NM, Delhase M, Haas E, Cao Y, Makris C, Li ZW, Karin M, Ware CF, Green DR. Immunity. 2002;17:525–535. doi: 10.1016/s1074-7613(02)00423-5. [DOI] [PubMed] [Google Scholar]
- 14.Coope HJ, Atkinson PG, Huhse B, Belich M, Janzen J, Holman MJ, Klaus GG, Johnston LH, Ley SC. Embo J. 2002;21:5375–5385. doi: 10.1093/emboj/cdf542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Claudio E, Brown K, Park S, Wang H, Siebenlist U. Nat Immunol. 2002;3:958–965. doi: 10.1038/ni842. [DOI] [PubMed] [Google Scholar]
- 16.Senftleben U, Cao Y, Xiao G, Greten FR, Krahn G, Bonizzi G, Chen Y, Hu Y, Fong A, Sun SC, Karin M. Science. 2001;293:1495–1499. doi: 10.1126/science.1062677. [DOI] [PubMed] [Google Scholar]
- 17.Pfeffer LM, Mullersman JE, Pfeffer SR, Murti A, Shi W, Yang CH. Science. 1997;276:1418–1420. doi: 10.1126/science.276.5317.1418. [DOI] [PubMed] [Google Scholar]
- 18.Hara K, Yonezawa K, Sakaue H, Ano A, Kotani K, Kitamura T, Kitamura Y, Ueda H, Stephens L, Jackson TR, Hawkins PT, Dhand R, Clark AE, Holman GD, Waterfield MD, Kasuga M. ProcNatlAcadSciUSA. 1994;91:7415–7419. doi: 10.1073/pnas.91.16.7415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hu Q, Klippel A, Muslin AJ, Fantl W, Williams LT. Science. 1995;268:100–102. doi: 10.1126/science.7701328. [DOI] [PubMed] [Google Scholar]
- 20.Malinin NL, Boldin MP, Kovalenko AV, Wallach D. Nature. 1997;385:540–544. doi: 10.1038/385540a0. [DOI] [PubMed] [Google Scholar]
- 21.Rothe M, Sarma V, Dixit VM, Goeddel DV. Science. 1995;269:1424–1427. doi: 10.1126/science.7544915. [DOI] [PubMed] [Google Scholar]
- 22.Oliviera IC, Mukaida N, Matsushiam K, Vilcek J. MolCellBiol. 1994;14:5300–5308. doi: 10.1128/mcb.14.8.5300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang CH, Shi W, Basu L, Murti A, Constantinescu SN, Blatt L, Croze E, Mullersman JE, Pfeffer LM. JBiolChem. 1996;271:8057–8061. doi: 10.1074/jbc.271.14.8057. [DOI] [PubMed] [Google Scholar]
- 24.Yang CH, Murti A, Pfeffer LM. Proc Natl Acad Sci USA. 1998;95:5568–5572. doi: 10.1073/pnas.95.10.5568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guan K, Dixon JE. AnalBiochem. 1991;192:262–267. doi: 10.1016/0003-2697(91)90534-z. [DOI] [PubMed] [Google Scholar]
- 26.Smith DB, Johnson KS. Gene. 1988;67:31–40. doi: 10.1016/0378-1119(88)90005-4. [DOI] [PubMed] [Google Scholar]
- 27.Blanco FL, Gonzalez-Reyes J, Fanjul LF, Ruiz de Galarreta CM, Aguiar JQ. Biotechniques. 1998;24:354–358. doi: 10.2144/98243bm04. [DOI] [PubMed] [Google Scholar]
- 28.Yang CH, Murti A, Baker SJ, Frangou-Lazaridis M, Vartapetian AB, Murti KG, Pfeffer LM. Exp Cell Res. 2004;298:197–206. doi: 10.1016/j.yexcr.2004.04.008. [DOI] [PubMed] [Google Scholar]
- 29.Einhorn S, Grander D. JInterferon Cytokine Res. 1996;16:275–281. doi: 10.1089/jir.1996.16.275. [DOI] [PubMed] [Google Scholar]
- 30.Chawla-Sarkar M, Leaman DW, Jacobs BS, Borden EC. J Immunol. 2002;169:847–855. doi: 10.4049/jimmunol.169.2.847. [DOI] [PubMed] [Google Scholar]
- 31.Cremer I, Viellard V, De Maeyer E. Virology. 1999;253:241–249. doi: 10.1006/viro.1998.9470. [DOI] [PubMed] [Google Scholar]
- 32.Marrack P, Kappler J, Mitchell T. JExpMed. 1999;189:521–530. doi: 10.1084/jem.189.3.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Muller M, Briscoe J, Laxton C, Guschin D, Ziemiecki A, Silvennoinen O, Harpur AG, Barbieri G, Witthuhn BA, Schindler C, Pellegrini S, Wilks AF, Ihle JN, Stark GR, Kerr IM. Nature. 1993;366:129–135. doi: 10.1038/366129a0. [DOI] [PubMed] [Google Scholar]
- 34.Velazquez L, Fellous M, Stark GR, Pellegrini S. Cell. 1992;70:313–322. doi: 10.1016/0092-8674(92)90105-l. [DOI] [PubMed] [Google Scholar]
- 35.Schindler C, Shuai K, Prezioso VR, Darnell JEJ. Science. 1992;257:809–813. doi: 10.1126/science.1496401. [DOI] [PubMed] [Google Scholar]
- 36.Greenlund AC, Farrar MA, Viviano BL, Schreiber RD. EMBO J. 1994;13:1591–1600. doi: 10.1002/j.1460-2075.1994.tb06422.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reich NC, Evans B, Levy DE, Fahey D, Knight E, Darnell JEJ. ProcNatlAcadSciUSA. 1987;84:6394–6398. doi: 10.1073/pnas.84.18.6394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gil MP, Bohn EOGAK, Ramana CV, Levine B, Stark GR, Virgin HW, Schreiber RD. Proc Natl Acad Sci USA. 2001;98:6680–6685. doi: 10.1073/pnas.111163898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Park C, Li S, Cha E, Schindler C. Immunity. 2000;13:795–804. doi: 10.1016/s1074-7613(00)00077-7. [DOI] [PubMed] [Google Scholar]
- 40.Beg AA, Sha WC, Bronson RT, Ghosh S, Baltimore D. Nature. 1995;376:167–170. doi: 10.1038/376167a0. [DOI] [PubMed] [Google Scholar]
- 41.Beg AA, Baltimore D. Science. 1996;274:782–784. doi: 10.1126/science.274.5288.782. [DOI] [PubMed] [Google Scholar]
- 42.Van Antwerp DJ, Martin SJ, Kafri T, Green DR, Verma IM. Science. 1996;274:787–789. doi: 10.1126/science.274.5288.787. [DOI] [PubMed] [Google Scholar]
- 43.Wang CY, Mayo MW, Baldwin AS., Jr Science. 1996;274:784–787. doi: 10.1126/science.274.5288.784. [DOI] [PubMed] [Google Scholar]
- 44.Ozes ON, Mayo LD, Gustin JA, Pfeffer SR, Pfeffer LM, Donner DB. Nature. 1999 Sep 2;401:82–85. doi: 10.1038/43466. [DOI] [PubMed] [Google Scholar]
- 45.Romashkova JA, Makarov SS. Nature. 1999;401:86–90. doi: 10.1038/43474. [DOI] [PubMed] [Google Scholar]
- 46.Matsushima A, Kaisho T, Rennert PD, Nakano H, Kurosawa K, Uchida D, Takeda K, Akira S, Matsumoto M. J Exp Med. 2001;193:631–636. doi: 10.1084/jem.193.5.631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Miyawaki S, Nakamura Y, Suzuka H, Koba M, Yasumizu R, Ikehara S, Shibata Y. Eur J Immunol. 1994;24:429–434. doi: 10.1002/eji.1830240224. [DOI] [PubMed] [Google Scholar]
- 48.De Togni P, Goellner J, Ruddle NH, Streeter PR, Fick A, Mariathasan S, Smith SC, Carlson R, Shornick LP, Strauss-Schoenberger J, et al. Science. 1994;264:703–707. doi: 10.1126/science.8171322. [DOI] [PubMed] [Google Scholar]
- 49.Futterer A, Mink K, Luz A, Kosco-Vilbois MH, Pfeffer K. Immunity. 1998;9:59–70. doi: 10.1016/s1074-7613(00)80588-9. [DOI] [PubMed] [Google Scholar]
- 50.Thompson JS, Bixler SA, Qian F, Vora K, Scott ML, Cachero TG, Hession C, Schneider P, Sizing ID, Mullen C, Strauch K, Zafari M, Benjamin CD, Tschopp J, Browning JL, Ambrose C. Science. 2001;293:2108–2111. doi: 10.1126/science.1061965. [DOI] [PubMed] [Google Scholar]
- 51.Schiemann B, Gommerman JL, Vora K, Cachero TG, Shulga-Morskaya S, Dobles M, Frew E, Scott ML. Science. 2001;293:2111–2114. doi: 10.1126/science.1061964. [DOI] [PubMed] [Google Scholar]
- 52.Caamano JH, Rizzo CA, Durham SK, Barton DS, Raventos-Suarez C, Snapper CM, Bravo R. J Exp Med. 1998;187:185–196. doi: 10.1084/jem.187.2.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Franzoso G, Carlson L, Poljak L, Shores EW, Epstein S, Leonardi A, Grinberg A, Tran T, Scharton-Kersten T, Anver M, Love P, Brown K, Siebenlist U. J Exp Med. 1998;187:147–159. doi: 10.1084/jem.187.2.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rayet B, Gelinas C. Oncogene. 1999;18:6938–6947. doi: 10.1038/sj.onc.1203221. [DOI] [PubMed] [Google Scholar]
- 55.Karin M, Cao Y, Greten FR, Li ZW. Nat Rev Cancer. 2002;2:301–310. doi: 10.1038/nrc780. [DOI] [PubMed] [Google Scholar]
- 56.Marinari B, Costanzo A, Marzano V, Piccolella E, Tuosto L. Proc Natl Acad Sci U S A. 2004;101:6098–6103. doi: 10.1073/pnas.0308688101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Takeda K, Akira S. Semin Immunol. 2004;16:3–9. doi: 10.1016/j.smim.2003.10.003. [DOI] [PubMed] [Google Scholar]
- 58.Ye H, Wu H. Proc Natl Acad SciUSA. 2000;97:8961–8966. doi: 10.1073/pnas.160241997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Munroe ME, Bishop GA. J Biol Chem. 2004;279:53222–53231. doi: 10.1074/jbc.M410539200. [DOI] [PubMed] [Google Scholar]
- 60.Sha WC, Liou HC, Tuomanen EI, Baltimore D. Cell. 1995;80:321–330. doi: 10.1016/0092-8674(95)90415-8. [DOI] [PubMed] [Google Scholar]
- 61.Snapper CM, Rosas FR, Zelazowski P, Moorman MA, Kehry MR, Bravo R, Weih F. J Exp Med. 1996;184:1537–1541. doi: 10.1084/jem.184.4.1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Grumont RJ, Rourke IJ, O’Reilly LA, Strasser A, Miyake K, Sha W, Gerondakis S. J Exp Med. 1998;187:663–674. doi: 10.1084/jem.187.5.663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yilmaz ZB, Weih DS, Sivakumar V, Weih F. Embo J. 2003;22:121–130. doi: 10.1093/emboj/cdg004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Baeuerle PA, Baltimore D. Cell. 1996;87:13–20. doi: 10.1016/s0092-8674(00)81318-5. [DOI] [PubMed] [Google Scholar]