

Abstract
The tumor necrosis factor receptor (TNFR) superfamily can induce diverse biological effects, including cell survival, proliferation, differentiation, and apoptosis. The major signal transducers for TNFRs are the family of TNF receptor associated factors (TRAFs). The direct interaction between TRAFs and the intracellular tails of TNFRs is the first step of this signal relay process. Structural studies have revealed a trimeric nature of TRAF2 and a symmetrical mode of receptor binding, suggesting the involvement of trivalent TNFR2-receptor interaction in the signal transduction. In this study, using isothermal titration calorimetry (ITC), we report thermodynamic characterization of the interaction between TRAF2 and monomeric peptide sequences from TNFR members, including TNFR2, CD40, CD30, Ox40, and 4-1BB, and the Epstein–Barr virus (EBV)-transforming protein, latent infection membrane protein-1 (LMP1). The dissociation constants of the interaction were shown to range between 40 μM and 1.9 mM, which are substantially weaker than most protein–peptide interactions. The interaction is entirely driven by exothermic enthalpy, consistent with the abundance of polar contacts. The enthalpy of the interaction has a significant temperature dependence (ΔCp = −245 cal/mol⋅K). The unfavorable entropy in the interaction and the comparison with structural energetics calculations suggest the involvement of conformational rearrangement in the interaction. The low affinity of TRAF2 to monomeric receptor peptides further supports the importance of avidity contribution in TRAF2 recruitment by these receptors upon ligand-induced trimerization or higher order oligomerization.
For a multicellular organism, receptor signal transduction through a membrane barrier is one of the most intriguing aspects of the communication between individual cells. Many elegant studies have revealed that conformational changes and receptor clustering are the two most frequently observed mechanisms of receptor signaling. Neurotransmitters and many peptide ligands bind and induce conformational changes to their transmembrane receptors and activate the G proteins associated with the intracellular domains of these receptors (1). The initiation of signal transduction by many growth factor receptors is by ligand-mediated receptor dimerization, which brings the intracellular tyrosine kinase domains into proximity for autophosphorylation and enzymatic activation (2).
The signal transduction of the tumor necrosis factor receptor (TNFR) superfamily, a group of receptors involved in cell survival and cell death (3), is also thought to be induced by ligand-induced receptor clustering (4). The extracellular domains of TNFRs are composed of differing numbers of cysteine-rich repeats and share extensive sequence homology (5). The ligands for TNFRs belong to the corresponding TNF superfamily and are trimeric in nature (6). The extracellular interaction of a TNF-like ligand with a TNFR indirectly trimerizes the receptor by imposing a 3-fold symmetry to the interaction. In the crystal structure of the complex between TNFR1 and LTα, each chain of TNFR1 binds symmetrically to the groove between pairs of protomers in the LTα trimer (4).
The TNFR-associated factor (TRAF) family of intracellular adapter proteins are recruited to the intracellular tails of many TNFRs in response to receptor trimerization. These TRAF proteins are major signal transducers for TNFRs, leading to activation of kinase cascades and eventually transcription factors in the Rel and AP-1 family for inflammatory and acute phase responses and for protection from apoptosis (7). It has been speculated that the assembly of a multimeric TRAF-signaling complex mediates the activation of downstream kinases through autophosphorylation, as is the case for tyrosine kinase activation of growth factor receptors.
TRAF2 was isolated biochemically from the TNFR2-signaling complex (8) and is the prototype of the six TRAF family members (8–16). Besides TNFR2, a number of other TNFRs such as CD30, CD40, CD27, Ox40, 4-1BB, and ATAR also directly recruit TRAF2 (7). In addition, the transforming effect of the Epstein–Barr virus (EBV) oncoprotein latent infection membrane protein-1 (LMP1) is partly TRAF2 mediated (17, 18). Several linear consensus sequences have been proposed to bind to TRAF2, including the PXQX(T/S/D) (X = any amino acid) motif in LMP1, CD30, CD40, and CD27 (17, 19–25); the ΦSXEE (Φ = large hydrophobe) sequence in TNFR2 and CD30 (8, 22); and the QEE motif in 4-1BB and Ox40 receptors (26).
The primary sequence of TRAFs contains an amino-terminal domain essential for the activation of down-stream effectors and a carboxyl-terminal TRAF domain, which is both necessary and sufficient for self association and receptor interaction (8). The TRAF domain may be subdivided into a TRAF-N domain with propensity for forming coiled-coil structures and a highly conserved TRAF-C domain. We and others have recently determined crystal structures of the TRAF domain of human TRAF2 in complex with various receptor peptides (27–29). The structures revealed a trimeric self association of TRAF2 and a symmetrical mode of receptor binding, suggesting the involvement of trimeric interaction and avidity contribution in ligand-induced TRAF2 recruitment.
Despite the apparent sequence diversity, the receptor peptides bind to a common site on the surface of the TRAF domain of TRAF2 (27–29). Each peptide contacts one protomer of the TRAF domain trimer, unlike the interaction between TNFR1 and LTα. A major portion of the receptor peptides exhibits an extended conformation and forms β-edge main chain hydrogen bonds with the TRAF domain structure. A core of four residues are highly conserved structurally, which are recognized by specific side chain pockets and hydrogen bonds. The structure-based sequence alignment has allowed a unification of existing TRAF2-binding sequences in TNFRs, giving rise to a major consensus motif of (P/S/T/A)X(Q/E)E (28). The EBV-transforming protein LMP1 exhibits a variation in its interaction with TRAF2, which forms an alternative TRAF2-binding motif of PXQXXD (28).
In this study, we report thermodynamic characterization of the solution interaction of TRAF2 with various monomeric receptor peptides using isothermal titration calorimetry (ITC). ITC has frequently been used for dissecting protein–peptide and protein–protein interactions, such as peptide recognition by Src homology 2 (SH2) and SH3 domains (30–32), and the T cell antigen receptor (TCR)-MHC interaction (33). It offers the direct determination of a complete thermodynamic profile of an interaction, including the free energy, the enthalpy, the entropy, and the heat capacity change (34). Our results revealed the extreme low affinity associated with the monomeric interaction and the potential conformational changes in the receptor peptides on binding. These results complement the earlier structural studies and provide further support on the importance of avidity in TRAF2 recruitment and signal transduction for the TNFR superfamily.
Materials and Methods
Protein Production and Purification.
The TRAF domain of human TRAF2 (residues 310–501) was expressed and purified similarly as described in the earlier structural studies (27, 28). The cDNA encoding human TRAF2 was purchased from the library of expressed sequence tags through Genome Systems (St. Louis). The DNA sequence for residues D310–L501 of human TRAF2 was amplified by PCR and inserted between the NcoI and the XhoI sites in the pET24D vector (Novagen). The construct contains an additional methionine residue at the amino terminus and residues EHHHHHH at the carboxyl terminus. Freshly transformed BL21 (DE3) cells were grown to OD600 0.5 at 37°C in LB media and induced with 0.5 mM isopropyl β-d-thiogalactoside (IPTG) at 20°C overnight. The cells were harvested and frozen at −80°C.
The cell pellets were resuspended in 50 mM phosphate buffer at pH 8.0 plus 300 mM NaCl, 20 mM imidazole, 10 mM β-mercaptoethanol, and 20 μM PMSF, and subsequently lysed by repeated bursts of sonication. After removing the insoluble materials by high-speed centrifugation, the soluble fraction of the cell lysate was filtered and loaded onto a Ni-nitrilotriacetic acid (NTA) agarose resin (Qiagen, Chatsworth, CA). The resin was washed thoroughly with 50 mM phosphate buffer at pH 6.0 and eluted with 200 mM of imidazole in the same buffer. The eluted fractions were further purified by gel filtration (Superose 12, Amersham Pharmacia) in 50 mM sodium phosphate, pH 7.5.
Peptide Synthesis and Purification.
All peptides used in this study were chemically synthesized by the University of Maryland Biopolymer Core Facility with amino-terminal acetylation and carboxyl-terminal amidation to mimic the intact protein. They were purified by reverse phase HPLC using a C18 column (Vydac, Hesperia, CA) and lyophilized. The molecular mass of each peptide was verified by matrix-assisted laser desorption ionization time-of-flight (MALDI-TOF) mass spectrometry.
Sample Preparation.
Purified TRAF domain of TRAF2 was concentrated by using Centricon 30 (Amicon). Chemically synthesized receptor peptides were dissolved at 10–20 mM concentrations in 50 mM sodium phosphate at pH 7.5. The protein and peptides were dialyzed in the same beaker against 50 mM sodium phosphate at pH 7.5 for at least 2 days at 4°C to ensure buffer equilibration. Accurate concentrations of the protein and peptide samples after dialysis were determined by quantitative amino acid analysis.
Isothermal Titration Calorimetry (ITC) Experiments.
The Micro Calorimetry System (Microcal, Amherst, MA) was used to perform the ITC measurements for the interaction between the TRAF domain of TRAF2 and six receptor peptides. Titration experiments were performed at 20°C for determination of binding enthalpy and affinity, and at 10°C, 20°C, and 30°C for determination of heat capacity (ΔCp). Each peptide ligand was injected into the 1.3-ml sample cell containing either the TRAF2 protein or buffer alone in 2- to 6-μl volumes at 4-min intervals. Approximately 20 to 45 injections were titrated for each measurement. These titration data were analyzed by the origin data analysis software (Microcal Software, Northampton, MA). The heat of dilution obtained from injecting a ligand into buffer was subtracted before the fitting process. Nonconstraint fitting was performed for the interaction of TRAF2 with CD40, CD30, and Ox40 peptides, in which the directly measured heat changes on addition of small volumes of each ligand permit extraction of the enthalpy (ΔH), the binding affinity (Ka), and the stoichiometry (N) of the interaction (34). The accuracy of each ΔH, Ka, and N determination was estimated from the fitting error of nonlinear least squares. The remaining thermodynamic parameters, including the binding free energy (ΔG) and the entropy (ΔS) of the interaction were calculated by using the relationship
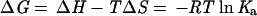 |
where T is the absolute temperature and R is the gas constant. For the weaker interactions of TRAF2 with peptides from TNFR2, 4-1BB and LMP1, an accurate determination of enthalpy and binding stoichiometry were not possible because of the low protein concentrations used in the experiments, in comparison with the binding constants. To obtain a realistic binding affinity from these data, constraint fitting was performed at two extreme stoichiometry values of 0.5 and 1.5. The differences on extracted Ka and the associated errors were both used to obtain the average Ka and the deviation.
Structural Energetics Calculations.
Structure-based calculations were based on previously published formulations using changes in polar and apolar surface area on binding (35, 36).
Results and Discussion
We used ITC to characterize the interaction between TRAF2 and TRAF2-binding peptides from representative members of the TNFRs, including TNFR2, CD40, CD30, Ox40, and 4-1BB, as well as the EBV oncoprotein LMP1 (Fig. 1). In most cases, the exact peptides have also been used successfully to obtain crystals with the TRAF domain of TRAF2. The CD30 peptide used in the ITC measurement has three additional residues at the amino terminus, compared with that used in crystallization; these three residues do not contact TRAF2 (28). Because the thermodynamic property of an interaction often reflects its structural characteristic, the availability of these crystal structures provides a unique opportunity for us to correlate the structures with the thermodynamic measurements and to infer further understanding of the interaction.
Figure 1.

Peptide sequences used in the ITC analysis. (A) Structure-based sequence alignment of receptor peptides and the two consensus motifs. Three residues at the N terminus of CD30 and eight residues at the C terminus of CD40 were not observed in the crystal structures. (B) A schematic representation of the interactions between major side chains of each motif and TRAF2.
Low-Affinity Interaction.
The heat release during the titration of receptor peptides into a TRAF2 solution exhibited excellent agreement with ideal binding (Fig. 2), indicating the presence of a single type of binding sites and the lack of cooperativity in the interaction. All titration experiments were performed under high ratios of peptide-to-protein concentrations. This increased the sensitivity of the experiments while reducing the demand on protein concentrations, which are limited by the yield, solubility, and stability of the TRAF2 protein. The heat of peptide dilution was measured by titrating each peptide into buffer alone. This was generally small and subtracted from each binding titration curve. The measured dissociation constants (Kd) were in the range of 40–60 μM for the CD30, the Ox40, and the CD40 peptides, and in the range of 0.5–1.9 mM for the TNFR2, the mouse 4-1BB, and the LMP1 peptides (Table 1). The Kd values for CD30, CD40, and Ox40 are within the ideal range for study by ITC. However, the measurements on the weak interactions of TRAF2 with TNFR2, m4-1BB, and LMP1 peptides were only made possible by significantly increasing both protein and peptide concentrations (Table 1).
Figure 2.

Representative titration data analysis. ITC data for titrating the CD40 peptide (A) and the LMP1 peptide (B) into the solutions containing the TRAF domain of TRAF2 are shown. A total of 20 and 45 injections were performed respectively for the CD40–TRAF2 and LMP1–TRAF2 interactions.
Table 1.
Thermodynamic parameters in the interaction between TRAF2 and receptor peptides as measured by ITC
| Receptor peptide | Sequence | T, °C | Ka, 103⋅M−1 | Kd, mM | ΔG, kcal/mol | ΔH, kcal/mol | −TΔS, kcal/mol | N | Ligand, mM | Protein, mM |
|---|---|---|---|---|---|---|---|---|---|---|
| hCD30 (573–583) | SDVMLSVEEEG | 20 | 28.5 ± 3.5 | 0.04 | −5.97 | −14.0 ± 0.8 | 8.03 | 0.86 ± 0.05 | 6.7 | 0.056 |
| hCD40 (250–266) | PVQETLHGCQPVTQEDG | 20 | 15.9 ± 1.5 | 0.06 | −5.63 | −9.5 ± 1.0 | 3.87 | 1.1 ± 0.1 | 5.9 | 0.056 |
| hOX40 (262–266) | PIQEE | 20 | 20.8 ± 2.5 | 0.05 | −5.78 | −13.0 ± 0.9 | 7.22 | 0.86 ± 0.07 | 12.9 | 0.056 |
| hTNFR2 (420–428) | QVPFSKEEC | 20 | 1.76 ± 0.04– | 0.5* | −4.35 | ND | ND | ND | 9.7 | 0.16 |
| 2.0 ± 0.05* | ||||||||||
| m4-1BB (231–236) | GAAQEE | 20 | 0.91 ± 0.014– | 1.0* | −3.96 | ND | ND | ND | 13.4 | 0.16 |
| 1.11 ± 0.011* | ||||||||||
| hLMP1 (204–210) | PQQATDD | 20 | 0.50 ± 0.017– | 1.9* | −3.62 | ND | ND | ND | 17.2 | 0.16 |
| 0.55 ± 0.019* | ||||||||||
| hCD30 (573–583) | SDVMLSVEEEG | 10 | 36.0 ± 5.0 | 0.03 | −5.90 | −11.5 ± 0.5 | 5.60 | 0.63 ± 0.02 | 6.7 | 0.28 |
| hCD30 (573–583) | SDVMLSVEEEG | 20 | 23.7 ± 2.0 | 0.04 | −5.86 | −14.9 ± 0.7 | 9.04 | 0.63 ± 0.02 | 6.7 | 0.28 |
| hCD30 (573–583) | SDVMLSVEEEG | 30 | 14.4 ± 0.4 | 0.07 | −5.76 | −16.4 ± 0.2 | 10.64 | 0.62 ± 0.006 | 6.7 | 0.28 |
Ka, association constant; Kd, dissociation constant; h, human; ND, not determined.
The ranges of Ka for these three receptor peptides were determined at N = 0.5 and N = 1.5. The corresponding Kd is calculated from the median value of the Ka range.
Three of the receptor peptides (those from CD30, Ox40, and CD40) exhibit the highest affinity to TRAF2 with dissociation constants of 40, 50, and 60 μM, respectively (Table 1). The ability of the short five-residue peptide of Ox40 to confer an affinity to TRAF2 as high as the longer peptides is consistent with the structural observation that a core of five residues appears to dominant the interaction with TRAF2 (28). The CD40 and Ox40 peptides contain the most prevalent TRAF2-binding sequence PXQE (positions P−2 to P1), which is a subset of the major TRAF2-binding consensus sequence (P/S/T/A)X(Q/E)E. The CD30 peptide, on the other hand, contains the SXEE (positions P−2 to P1) subset of the major TRAF2-binding motif (28). Although Pro is by far the most frequently observed residue at the P−2 position, the substitution to Ser in CD30 does not decrease the affinity of the peptide to TRAF2. Similarly, the difference between Gln and Glu at the P0 position does not result in differential affinities, even though structurally the Glu side chain can make only one hydrogen bond to TRAF2 whereas the Gln side chain may possibly make three hydrogen bonds with three Ser residues of TRAF2. The solvent exposure of these potential hydrogen bonds may decrease their contribution to the overall binding energy because the alternative interactions with solvent may also be favorable.
The receptor peptides from TNFR2 and mouse 4-1BB exhibit affinities to TRAF2 that are approximately an order of magnitude lower than the receptor sequences from CD30, CD40, and Ox40. The mouse 4-1BB peptide has an Ala at the P−2 position instead of the more favorable Pro or Ser (Fig. 1). The Ala side chain appears to be too small for the P−2 pocket on TRAF2 for optimal interaction, as suggested by the crystal structure of the complex. The corresponding human sequence has a Thr at this position and may confer higher affinity to human TRAF2 than the mouse homologue. It is not clear from structural observations why the TNFR2 peptide possesses much lower affinity than the homologous CD30 peptide, both of which belong to the SXEE subset of the major TRAF2-binding motif. One possibility would be that the P−1 residue in CD30 (Val) is more preferred than the corresponding residue in TNFR2 (Lys), even though no strict consensus is observed at this position. Consistent with this hypothesis, the TRAF2 surface that contacts the P−1 side chain is rather hydrophobic.
The human EBV LMP1 peptide, bearing the minor consensus sequence PXQXXD, exhibits the lowest affinity measured in our ITC experiments. It is possible that this alternative motif confers weaker affinity to TRAF2. The major structural difference between the two TRAF2-binding motifs resides on the last acidic residue in each motif. Whereas both residues (E or D) form hydrogen-bond and salt-bridge interactions with R393 and Y395 of TRAF2, the different positions of these residues in each motif lead to different interaction geometries and potential differences in the effectiveness of the interaction. LMP1 constitutively engages TRAFs through aggregation of its transmembrane domain, leading to persistent NF-κB activation and growth-transformation effects (19). Interestingly, most of the simian EBV LMP1 sequences contain the major consensus sequence motif rather than the minor consensus motif in human LMP1. It is likely that the constitutive clustering of LMP1 through membrane patching is sufficient to engage TRAFs regardless of its weak monovalent affinity.
In addition to LMP1, the TRAF-signaling modulator I-TRAF/TANK (37, 38) also contains the minor consensus sequence for TRAF2-binding (28). Sequence analysis suggests that this protein may have an oligomeric structure and therefore may be able to interact with TRAFs in the latent state. On ligand treatment, the trimerized receptors bearing the major consensus sequences will likely compete off the binding of I-TRAF/TANK, releasing its inhibitory effect on TRAFs.
A recent characterization by surface plasmon resonance of the interaction between TRAF2 and monomeric full-length cytoplasmic domain of CD40 gave rise to a dissociation constant of 30 μM (39), similar to the 60 μM dissociation constant derived from our ITC measurement on a CD40 peptide (Table 1). The agreement further confirms that TRAF2-receptor interactions are mediated by short linear sequences. The slight difference between the two methods may reflect potential avidity effects on the binding of trimeric TRAF2 to a CD40-coupled chip in the surface plasmon resonance experiment. ITC, on the other hand, involves only solution phase and is in theory most suitable for measuring monomeric interactions between binding partners.
In general, these affinities between TRAF2 and monomeric TNFR peptides are weaker than most of the known protein–peptide interactions involved in signal transduction (40). The ranges of affinity between SH2 and phosphotyrosine peptides are mostly at least 10- to 100-fold higher than those observed here for TRAF2. Some of the weak SH2-peptide interactions have similar affinity to the TRAF2–peptide interactions; these peptides, however, are not recognized by SH2 in vivo (31). The low affinity of TRAF2 to monomeric receptor peptides supports the importance of avidity in TRAF2 recruitment and signal transduction.
Thermodynamics of the Interaction.
The ITC measurements showed a variable range of enthalpy components (between −9.5 and −14.0 kcal/mol) and entropy components (−TΔS between 3.9 and 8.0 kcal/mol) in the interaction of TRAF2 with the series of receptor peptides (Table 1). The invariably observed favorable enthalpies and unfavorable entropies indicate that the interactions are energetically driven by exothermic enthalpy. This enthalpically driven interaction is in contrast to most protein–protein interactions, which possess favorable entropy because of the similarity of many protein interfaces to the interiors of proteins (41).
The enthalpy of the interaction showed a linear dependence with temperature, as measured for the TRAF2–CD30 interaction at 10°C, 20°C, and 30°C (Table 1 and Fig. 3). The heat capacity change, ΔCp, determined from the slope of the fitted line, is −245 cal/mol⋅K. This relatively large negative heat capacity change may be indicative of a specific interaction, even though the affinities of these monomeric interactions between TRAF2 and receptors are rather low. As suggested from thermodynamic studies of protein–DNA interactions, a nonspecific weak complex held together by electrostatic forces often exhibits little temperature dependence of enthalpy (42).
Figure 3.

Heat capacity change ΔCp for the interaction between TRAF2 and CD30. The correlation coefficient of the fitting is 0.95, and the slope of the line, ΔCp, is −245cal/mol⋅K.
The favorable enthalpy may arise from the significant number of polar interactions, including main chain hydrogen bonds, side chain hydrogen bonds, and salt bridges in the complex (27, 28). The relatively large negative ΔCp, on the other hand, also suggests the presence of significant hydrophobic component in the interaction. The observed unfavorable entropy appears to contradict the presumably favorable solvation entropy from the burial of hydrophobic surfaces. We suggest that the unfavorable entropy may be largely conformational. This conformational component of unfavorable entropy has been observed for the interaction between TCR and MHC peptides (33). Secondary structure predictions of cytoplasmic tails of most TRAF-interacting TNFRs suggest that these receptor tails do not have a preformed well-ordered three-dimensional structure. Rather, linear peptides from localized regions of the receptors are responsible for TRAF2 interaction. These observations suggest that the peptides, or full-length receptors, are flexible before docking onto the protein and penalized by conformational entropy.
The availability of the crystal structural information permits a correlation of structural energetics with the experimental information (43). The interaction between TRAF2 and the CD30 peptide buries a total of 1177 Å2 surface area. Most of the surface area is apolar (765 Å2) rather than polar (412 Å2), even though there are no observable large hydrophobic surface patches. The current structural energetics formulations assume that the thermodynamic parameters, including ΔH, ΔS, and ΔCp, can be directly related to the surface area burial of polar and apolar groups (35, 36). Unexpectedly, the calculation for the TRAF2–CD30 interaction showed a poor agreement with experimental data (Tables 1 and 2), even though these formulations have been successfully used in predicting energetics of ligand binding in many cases (44, 45). The calculation suggests that the entropic change is the major contributor of the interaction. This predicted entropic contribution is in complete contrast with the actual interaction, which is enthalpy-driven. The ΔCp of the interaction is the only parameter that appears to be correctly predicted, perhaps out of coincidence.
Table 2.
Calculated structural energetics for the interaction between TRAF2 and the CD30 peptide at 20°C
| Parameter | Calculated value |
|---|---|
| ΔASA (Å2) | |
| Polar | −412 |
| Apolar | −765 |
| Total | −1,177 |
| ΔCp, cal/mol⋅K | −236.6 |
| ΔH, kcal/mol | 3.0 |
| −ΔS⋅T, kcal/mol | |
| Solvation | −18.9 |
| Conformation | 5.9 |
| Mix | 2.3 |
| Total | −10.7 |
| ΔG, kcal/mol | −7.7 |
| Kd, μM | 1.8 |
We suggest that the gross deviation between the experimental observation and the calculation reflects the incorrect assumption that goes into the calculation for the TRAF2–CD30 interaction. Our structural studies have established that the TRAF2 structure does not undergo large conformational rearrangement on binding. However, the unbound state for the receptor peptide is entirely unknown and unlikely to be similar to the bound state because of its intrinsic flexibility. Therefore, the actual surface area changes on binding and the estimation of solvation and conformational entropy are likely to deviate significantly from the values obtained by assuming a rigid-body association. This deviation again suggests the involvement of conformational changes and induced fit in the interaction between TRAF2 and receptor peptides.
TRAF-Mediated Signal Transduction.
The dependence of TRAF recruitment on ligand-induced receptor oligomerization has been shown for several TNFRs including TNFR2, CD40, and LTβR (46–48) and is considered a common feature of the TNF receptor superfamily. The thermodynamic characterization of the interaction between TRAF2 and various receptor peptides has revealed the mechanism by which TRAFs respond to receptor oligomerization. The low-affinity nature of TRAF2 to monomeric receptors ensures that TRAFs do not interact with inactive receptors. Ligand binding brings receptors into proximity and provides an opportunity for trimeric TRAFs to engage in multivalent interactions. Both affinity and specificity of the interaction will be greatly amplified by the avidity contribution from this oligomeric association, transforming a low-affinity and somewhat promiscuous interaction into a tight and highly specific one.
Many of the TNF-like cytokine ligands are membrane bound and therefore may be capable of inducing membrane patching and higher order of receptor aggregation. Even though the minimal aggregation state of TRAF2 appears to be trimeric, higher orders of aggregation may be possible as well in response to the higher order of receptor aggregation. This higher order of aggregation would increase the avidity in the TRAF2–receptor interaction and the strength of the signal transduction. In keeping with this hypothesis, soluble trimeric CD40 ligand can be fairly inefficient in inducing CD40 signaling under certain circumstances, compared with cell-bound or crosslinked hexameric CD40 ligand (39). In addition, TNFR2 is mostly activated by membrane-bound form of TNFα (49). In some instances, multiple TRAF2-binding sequences in a single protein may also allow additional avidity from the interaction of TRAF2 with neighboring TRAF2-binding sites. Therefore, TRAF-mediated signal transduction may be modulated at several levels including affinity and avidity.
Even though that the intracellular domains of the TNFRs may contain up to several hundred residues, our structural studies suggest that the TRAF2-binding sites of these receptors are within a short stretch of linear sequences. Therefore, the affinities measured here between TRAF2 and short-receptor peptides should be representative of the interaction between TRAF2 and full-length receptors. For example, full-length CD40 (39) and the 17-residue CD40 peptide used in our study possess similar binding affinity to TRAF2. This 17-residue CD40 peptide has also been shown to bind TRAFs and activate NF-κB when linked immediately after the transmembrane domain of the CD40 receptor (37). In addition, it has been proposed that longer constructs, and possibly full-length LMP1, may possess comparable or lower affinity to TRAF proteins than the short peptide we used in the study (17).
It has been suggested that TRAF down-stream signaling may couple to a mitogen-activated protein kinase cascade involving kinases such as NF-κB-inducing kinase (NIK) (50), MEK kinase-1 (MEKK1) (51), and apoptosis signal-regulating kinase-1 (ASK1) (52). The assembly of a multivalent TRAF signaling complex may result in activation of these kinases through an induced-proximity mechanism. This mode of intracellular signaling has frequently been used for enzymatic activation of receptor tyrosine kinases (2) and proposed for receptor-mediated caspase activation (53, 54).
Implications in Designing TRAF Inhibitors.
TRAF2 may serve as a potential therapeutic target because of its role in inflammation and tumorigenesis. Several aspects of the interaction between TRAF2 and receptor peptides may assist the design of high-affinity TRAF2-binding ligands. First of all, the low-affinity interaction indicates a nonideal steric or chemical complementarity between TRAF2 and these receptor peptides, increasing the possibility for affinity improvement. Secondly, the ability of short peptides such as the five-residue Ox40 peptide to bind as well as longer peptides or even full-length receptors may indicate the likelihood of isolating small-molecule TRAF2 inhibitors. In addition, because reduction of conformational entropy may contribute negatively to the interaction, an increase in affinity may be achieved by rigidifying potential TRAF2-binding moieties.
Because signal transduction processes are usually transient, it is possible that potent inhibitors would only need to possess higher affinity to TRAF2 than monomeric receptors. These inhibitors may have the opportunity to presaturate the receptor-binding sites on TRAF proteins and slow down the trivalent association of TRAFs with trimerized receptors, leading to repression of TRAF recruitment and activation.
Acknowledgments
We greatly appreciate the generosity of the structural biology groups at the Memorial Sloan-Kettering Cancer Center for access to the Micro Calorimetry System instrument. We thank Dr. Liang Tong for comments and discussions. This work was supported by the National Institutes of Health (AI47831, to H.W.) and the departmental startup fund. H.W. is a Pew Scholar of Biomedical Sciences.
Abbreviations
- TNF
tumor necrosis factor
- TRAF
TNF receptor-associated factor
- ITC
isothermal titration calorimetry
- EBV
Epstein–Barr virus
- SH2
Src homology 2
- LMP1
latent infection membrane protein-1
Footnotes
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.160241997.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.160241997
References
- 1.Iismaa T P, Shine J. Curr Opin Cell Biol. 1992;4:195–202. doi: 10.1016/0955-0674(92)90033-9. [DOI] [PubMed] [Google Scholar]
- 2.Ullrich A, Schlessinger J. Cell. 1990;61:203–212. doi: 10.1016/0092-8674(90)90801-k. [DOI] [PubMed] [Google Scholar]
- 3.Gravestein L A, Borst J. Semin Immunol. 1998;10:423–434. doi: 10.1006/smim.1998.0144. [DOI] [PubMed] [Google Scholar]
- 4.Banner D W, D'Arcy A, Janes W, Gentz R, Schoenfeld H J, Broger C, Loetscher H, Lesslauer W. Cell. 1993;73:431–445. doi: 10.1016/0092-8674(93)90132-a. [DOI] [PubMed] [Google Scholar]
- 5.Naismith J H, Sprang S R. Trends Biochem Sci. 1998;23:74–79. doi: 10.1016/s0968-0004(97)01164-x. [DOI] [PubMed] [Google Scholar]
- 6.Eck M J, Sprang S R. J Biol Chem. 1989;264:17595–17605. doi: 10.2210/pdb1tnf/pdb. [DOI] [PubMed] [Google Scholar]
- 7.Arch R H, Gedrich R W, Thompson C B. Genes Dev. 1998;12:2821–2830. doi: 10.1101/gad.12.18.2821. [DOI] [PubMed] [Google Scholar]
- 8.Rothe M, Wong S C, Henzel W J, Goeddel D V. Cell. 1994;78:681–692. doi: 10.1016/0092-8674(94)90532-0. [DOI] [PubMed] [Google Scholar]
- 9.Hu H M, O'Rourke K, Boguski M S, Dixit V M. J Biol Chem. 1994;269:30069–30072. [PubMed] [Google Scholar]
- 10.Cheng G, Cleary A M, Ye Z, Hong D I, Lederman S, Baltimore D. Science. 1995;267:1494–1498. doi: 10.1126/science.7533327. [DOI] [PubMed] [Google Scholar]
- 11.Mosialos G, Birkenbach M, Yalamanchili R, VanArsdale T, Ware C, Kieff E. Cell. 1995;80:389–399. doi: 10.1016/0092-8674(95)90489-1. [DOI] [PubMed] [Google Scholar]
- 12.Regnier C H, Tomasetto C, Moog-Lutz C, Chenard M-P, Wendling C, Basset P, Rio M-C. J Biol Chem. 1995;270:25715–25721. doi: 10.1074/jbc.270.43.25715. [DOI] [PubMed] [Google Scholar]
- 13.Ishida T K, Tojo T, Aoki T, Kobayashi N, Ohishi T, Watanabe T, Yamamoto T, Inoue J. Proc Natl Acad Sci USA. 1996;93:9437–9442. doi: 10.1073/pnas.93.18.9437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nakano H, Oshima H, Chung W, Williams-Abbott L, Ware C F, Yagita H, Okumura K. J Biol Chem. 1996;271:14661–14664. doi: 10.1074/jbc.271.25.14661. [DOI] [PubMed] [Google Scholar]
- 15.Cao Z, Xiong J, Takeuchi M, Kurama T, Goeddel D V. Nature (London) 1996;383:443–446. doi: 10.1038/383443a0. [DOI] [PubMed] [Google Scholar]
- 16.Ishida T, Mizushima S, Azuma S, Kobayashi N, Tojo T, Suzuki K, Aizawa S, Watanabe T, Mosialos G, Kieff E, Yamamoto T, Inoue J. J Biol Chem. 1996;271:28745–28748. doi: 10.1074/jbc.271.46.28745. [DOI] [PubMed] [Google Scholar]
- 17.Devergne O, Hatzivassiliou E, Izumi K M, Kaye K M, Kleijnen M F, Kieff E, Mosialos G. Mol Cell Biol. 1996;16:7098–7108. doi: 10.1128/mcb.16.12.7098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kaye K M, Devergne O, Harada J N, Izumi K M, Yalamanchili R, Kieff E, Mosialos G. Proc Natl Acad Sci USA. 1996;93:11085–11090. doi: 10.1073/pnas.93.20.11085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Franken M, Devergne O, Rosenzweig M, Annis B, Kieff E, Wang F. J Virol. 1996;70:7819–7826. doi: 10.1128/jvi.70.11.7819-7826.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gedrich R W, Gilfillan M C, Duckett C S, Van Dongen J L, Thompson C B. J Biol Chem. 1996;271:12852–12858. doi: 10.1074/jbc.271.22.12852. [DOI] [PubMed] [Google Scholar]
- 21.Aizawa S, Nakano H, Ishida T, Horie R, Nagai M, Ito K, Yagita H, Okumura K, Inoue J, Watanabe T. J Biol Chem. 1997;272:2042–2045. doi: 10.1074/jbc.272.4.2042. [DOI] [PubMed] [Google Scholar]
- 22.Boucher L, Marengere L E M, Lu Y, Thukral S, Mak T W. Biochem Biophys Res Comm. 1997;233:592–600. doi: 10.1006/bbrc.1997.6509. [DOI] [PubMed] [Google Scholar]
- 23.Brodeur S R, Cheng G, Baltimore D, Thorley-Lawson D A. J Biol Chem. 1997;272:19777–19784. doi: 10.1074/jbc.272.32.19777. [DOI] [PubMed] [Google Scholar]
- 24.Sandberg M, Hammerschmidt W, Sugden B. J Virol. 1997;71:4649–4656. doi: 10.1128/jvi.71.6.4649-4656.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Akiba H, Nakano H, Nishinaka S, Shindo M, Kobata T, Atsuta M, Morimoto C, Ware C F, Malinin N L, Wallach D, Yagita H, Okumura K. J Biol Chem. 1998;273:13353–13358. doi: 10.1074/jbc.273.21.13353. [DOI] [PubMed] [Google Scholar]
- 26.Arch R H, Thompson C B. Mol Cell Biol. 1998;18:558–565. doi: 10.1128/mcb.18.1.558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Park Y C, Burkitt V, Villa A R, Tong L, Wu H. Nature (London) 1999;398:533–538. doi: 10.1038/19110. [DOI] [PubMed] [Google Scholar]
- 28.Ye H, Park Y C, Kreishman M, Kieff E, Wu H. Mol Cell. 1999;4:321–330. doi: 10.1016/s1097-2765(00)80334-2. [DOI] [PubMed] [Google Scholar]
- 29.McWhirter S M, Pullen S S, Holton J M, Crute J J, Kehry M R, Alber T. Proc Natl Acad Sci USA. 1999;96:8408–8413. doi: 10.1073/pnas.96.15.8408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bradshaw J M, Grucza R A, Ladbury J E, Waksman G. Biochemistry. 1998;37:9083–9090. doi: 10.1021/bi973147k. [DOI] [PubMed] [Google Scholar]
- 31.Lemmon M A, Ladbury J E. Biochemistry. 1994;33:5070–5076. doi: 10.1021/bi00183a010. [DOI] [PubMed] [Google Scholar]
- 32.Lemmon M A, Ladbury J E, Mandiyan V, Zhou M, Schlessinger J. J Biol Chem. 1994;269:31653–31658. [PubMed] [Google Scholar]
- 33.Willcox B E, Gao G F, Wyer J R, Ladbury J E, Bell J I, Jakobsen B K, van der Merwe P A. Immunity. 1999;10:357–365. doi: 10.1016/s1074-7613(00)80035-7. [DOI] [PubMed] [Google Scholar]
- 34.Bundel D R, Sigurskjold B W. Methods Enzymol. 1994;247:288–305. doi: 10.1016/s0076-6879(94)47022-7. [DOI] [PubMed] [Google Scholar]
- 35.Murphy K P, Freire E. Adv Protein Chem. 1992;43:313–361. doi: 10.1016/s0065-3233(08)60556-2. [DOI] [PubMed] [Google Scholar]
- 36.Baker B M, Murphy K P. J Mol Biol. 1997;268:557–569. doi: 10.1006/jmbi.1997.0977. [DOI] [PubMed] [Google Scholar]
- 37.Cheng G, Baltimore D. Genes Dev. 1996;10:963–973. doi: 10.1101/gad.10.8.963. [DOI] [PubMed] [Google Scholar]
- 38.Rothe M, Xiong J, Shu H B, Williamson K, Goddard A, Goeddel D V. Proc Natl Acad Sci USA. 1996;93:8241–8246. doi: 10.1073/pnas.93.16.8241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pullen S S, Labadia M E, Ingraham R H, McWhirter S M, Everdeen D S, Alber T, Crute J J, Kehry M R. Biochemistry. 1999;38:10168–10177. doi: 10.1021/bi9909905. [DOI] [PubMed] [Google Scholar]
- 40.Kuriyan J, Cowburn D. Annu Rev Biophys Biomol Struct. 1997;26:259–288. doi: 10.1146/annurev.biophys.26.1.259. [DOI] [PubMed] [Google Scholar]
- 41.Janin J, Miller S, Chothia C. J Mol Biol. 1988;204:155–164. doi: 10.1016/0022-2836(88)90606-7. [DOI] [PubMed] [Google Scholar]
- 42.Ladbury J E. Structure. 1995;3:635–639. doi: 10.1016/s0969-2126(01)00197-6. [DOI] [PubMed] [Google Scholar]
- 43.Edgcomb S P, Murphy K P. Curr Opin Biotechnol. 2000;11:62–66. doi: 10.1016/s0958-1669(99)00055-5. [DOI] [PubMed] [Google Scholar]
- 44.Gomez J, Freire E. J Mol Biol. 1995;252:337–350. doi: 10.1006/jmbi.1995.0501. [DOI] [PubMed] [Google Scholar]
- 45.Luque I, Gomez J, Semo N, Freire E. Proteins. 1998;30:74–85. [PubMed] [Google Scholar]
- 46.Kuhne M R, Robbins M, Hambor J E, Mackey M F, Kosaka Y, Nishimura T, Gigley J P, Noelle R J, Calderhead D M. J Exp Med. 1997;186:337–342. doi: 10.1084/jem.186.2.337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shu H B, Takeuchi M, Goeddel D V. Proc Natl Acad Sci USA. 1996;93:13973–13978. doi: 10.1073/pnas.93.24.13973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.VanArsdale T L, VanArsdale S L, Force W R, Walter B N, Mosialos G, Kieff E, Reed J C, Ware C F. Proc Natl Acad Sci USA. 1997;94:2460–2465. doi: 10.1073/pnas.94.6.2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Grell M, Douni E, Wajant H, Lohden M, Clauss M, Maxeiner B, Georgopoulos S, Lesslauer W, Kollias G, Pfizenmaier K, et al. Cell. 1995;83:793–802. doi: 10.1016/0092-8674(95)90192-2. [DOI] [PubMed] [Google Scholar]
- 50.Malinin N L, Boldin M P, Kovalenko A V, Wallach D. Nature (London) 1997;385:540–544. doi: 10.1038/385540a0. [DOI] [PubMed] [Google Scholar]
- 51.Baud V, Liu Z, Bennett B, Suzuki N, Xia Y, Karin M. Genes Dev. 1999;13:1297–1308. doi: 10.1101/gad.13.10.1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nishitoh H, Saitoh M, Mochida Y, Takeda K, Nakano H, Rothe M, Miyazono K, Ichijo H. Mol Cell. 1998;2:389–395. doi: 10.1016/s1097-2765(00)80283-x. [DOI] [PubMed] [Google Scholar]
- 53.Muzio M, Chinnaiyan A M, Kischkel F C, O'Rourke K, Shevchenko A, Ni J, Scaffidi C, Bretz J D, Zhang M, Gentz R, Mann M, Krammer P H, Peter M E, Dixit V M. Cell. 1996;85:817–827. doi: 10.1016/s0092-8674(00)81266-0. [DOI] [PubMed] [Google Scholar]
- 54.Muzio M, Stockwell B R, Stennicke H R, Salvesen G S, Dixit V M. J Biol Chem. 1998;273:2926–2930. doi: 10.1074/jbc.273.5.2926. [DOI] [PubMed] [Google Scholar]