

Abstract
For a number of reasons, and unlike most other cancers, the mortality rate of PDAC is set to increase and, as such, it is predicted to become the second most common cause of cancer related mortality in the western world by the end of the current decade. One of the main reasons for this is the dire lack of robust therapeutic options. The clinical landscape of PDAC therapeutics is changing at an encouraging pace, exemplified by the breakthroughs in targeting not only KRAS but developing mutant-specific drugs against it. Nevertheless, the clinical community is still faced with a dire lack of effective therapeutics. The targeting of mitotic kinases – here limited to CDKs, Wee1, Chk1, Plk1 and the Aurora kinases – offers one potential avenue for exploitation. Here, we discuss ongoing efforts to target the mitotic kinases and present the advances that have been made for each, whilst also presenting the clinical and therapeutic perspectives for each category.
Here, we look at recent efforts towards the discovery and optimisation of small-molecules targeting the constellation of mitotic kinases for the treatment of pancreatic cancer.
Introduction
At the time of writing, pancreatic ductal adenocarcinoma (PDAC), the most common form of PDAC, despite accounting for only around 3% of cancer diagnoses every year, is the fourth leading cause of cancer death. For a number of reasons, and unlike most other cancers, its mortality rate is set to increase and, as such, it is predicted to become the second most common cause of cancer related mortality in the western world by the end of the current decade.2,3 One of the main reasons for this is the dire lack of robust therapeutic options. In fact, while certain cancers, such as melanoma, have seen a revolution in treatment modalities such as the use of immune checkpoint blockade, PDAC still suffers from a severe lack of options for pharmacological intervention and, for patients, surgical resection is the only curative treatment option for patients.4,5 This problem is, however, compounded by the fact that almost all patients are diagnosed in an advanced stage of the disease, which eliminates the option for surgery. As a result, the prognosis for PDAC patients is notoriously dismal, with a five-year overall survival around 13%, with significant variation according to disease stage and treatment regimens.6 Metastases are the primary cause of death for most PDAC patients, and even for operable patients death from metastatic disease occurs often within 12–24 months of surgical resection.7 On top of this, PDAC is notorious for its resistance to radio- and chemotherapy.8 While adjuvant chemo- and radiotherapy have been used, this has resulted in limited enhancements in patient survival in the past two decades.5 Hence, the highly aggressive, metastatic and heterogeneous character of this disease poses a critical challenge for the improvement of patient outcomes.
PDAC is characterized by the frequent presence of oncogenic driver mutations in four genes: KRAS, TP53, SMAD4 and CDKN2A (cyclin dependent kinase inhibitor 2A, encodes two tumour suppressor proteins). Of these, mutations in KRAS are by far the most prevalent in PDAC.9 Accumulation of driver mutations in these genes form the core of PDAC development and progression, contributing to the extensive genetic heterogeneity observed among PDAC patients falling into a range of different subtypes.9 Two of the major molecular subtypes have been defined – the ‘classical’ and ‘basal-like’ subtypes – which seem to differ in their susceptibility to therapeutics.10 Signal transduction cascades activated by the constitutively ‘on’ oncogenic KRAS protein converge on the cell cycle machinery and promote the cell's transition from G1 (or G0) phase into S phase (see below).11 Since PDAC carry activating mutations in KRAS in more than 90% of tumours, controlling cell cycle machinery – that is, mitotic kinases – might significantly impact PDAC.12
For a long period, the clinical landscape of molecular agents against PDAC has changed very little. Nucleoside analogues such as 5-flurouracil and gemcitabine are a core component of chemotherapeutic regimes against PDAC and have represented the standard of care since their introduction into clinical practice.13,14 Now, however, the clinical landscape might change. For instance, the KRAS gene product was for a long time considered undruggable on account of its flat interaction surfaces and lack of targetable pockets. However, seminal work by Ostrem and co-workers revealed a revolutionary ‘secret’ pocket,15 which, since its discovery, has been targeted by a number of agents and has even led to the development of mutant-specific drugs (e.g., sotorasib against KRASG12C or MRTX1133 against KRASG12D) and, much more recently, multi- or even pan-KRAS inhibitors, which are capable of inhibiting multiple KRAS mutant isoforms. While KRASG12C is rare in PDAC, KRASG12D is prevalent. The rise of contemporary KRAS inhibitors is not, however, without its drawbacks, as development of resistance to these therapies has already been reported.16–18 Researchers and clinicians will still have to explore additional therapies. Hence, the development of targeted therapies – such as the development of single- or multi-kinase inhibitors, particularly against kinases uncovered by recent phosphoproteomics efforts19,20 – offers an avenue to develop potentially more effective (combination) strategies.5 The targeting of mitotic kinases – including but not limited to CDKs, Wee1, Chk1, Plk1 and the Aurora kinases, which are described in this manuscript – offers but one potential approach for exploitation. Collectively, these kinases are key players in an intricate network of mitotic regulators whose pharmacological inhibition not only impairs tumour proliferation but also enhances the efficacy of chemoradiotherapy, underscoring their relevance as components of rational combination regimens in pancreatic cancer treatment (vide infra).
The cell cycle
Cellular divisions are not continuous but rather controlled by external stimuli. For a eukaryotic cell to divide, it must pass through the series of well-orchestrated phases, which together constitute the cell cycle (summarised in Fig. 1). The cell cycle is organised into a series of dependent pathways, whereby the initiation of each event is dependent upon successful completion of previous events.
Fig. 1. Overview of the mitotic kinases featured in this review, where they act on a heavily simplified representation of the cell cycle (mitosis) and where the agents described in the text act upon them. The actions of Chk1, Wee1 and Aurora A are presented in separate panels. Through the phosphorylation of various target proteins, cyclin-dependent kinases are master regulators of the cell cycle. CDKs are only active when complexed with a cyclin whose abundance in the cell oscillates over the course of the cell cycle. With the exception of CDK1, the roles of each CDK–cyclin dyad are shown in the figure. CDK1 has a wide repertoire of regulatory roles in the cell cycle including the control of mitotic chromosome behaviour, kinetochore function and spindle microtubule dynamics. The actions of these CDK–cyclin complexes are regulated through an intricate network of inhibitory kinases (not shown fully), which are, in turn, also regulated. For instance, the addition of inhibitory phosphates to CDK1–cyclin B is mediated, among others, by Wee1 kinase. Plk1 plays a coordinating role in various events between mitotic entry and cytokinesis. Chk1 functions to allow the G1/S transition as well as entry into mitosis (G2 to M). The Aurora kinases are key mediators of the later stages of the cell cycle where they control the actual division of the cell into two daughter cells and the mechanical steps leading up to it. For clarity, G0 is omitted from this figure.

The trigger of cellular division arises from mitogenic (i.e., proliferative) signals that lead to an increase in the levels of cyclin proteins that bind and activate cyclin-dependent kinases (CDKs). The MAPK pathway, for instance, headed by RAS, promotes cell cycle progression by causing increased expression of cyclin D.21 Mutations which cause the constitutive activation of KRAS, thus, lead to dysregulation of the cell cycle.22 CDKs, when bound to their cognate cyclin (although they can have more than one), are able to phosphorylate various intracellular targets in order to modulate their activities in the cell cycle and consequently orchestrate and coordinate all elements of the cell cycle. MAPK does not, however, act alone this regard, and other mitogenic pathways also converge on the regulation of cell cycle progression. CDKs are present in all nucleated eukaryotic cells, and their regulatory and accessory functions in the cell cycle have been evolutionarily conserved. Several members of the CDK family (e.g., CDK1, CDK2, CDK4, CDK6) are central players in coordinating the cell cycle itself while others are involved in regulating other CDKs (CDK7) or transcription (CDK9).23 The roles of the other CDKs – from CDK3 and CDK5 up to CDK12 and CDK13 – are less well understood.
At the same time, the progression of the cell cycle does not proceed unhindered and the activity of CDKs and cyclins is balanced by inhibitory proliferative checkpoints that separate the cell cycle into four distinct phases (Fig. 1): replicating cells traverse these distinct phases of the cell cycle consecutively.
During G1 (growth 1) phase, the cell grows and synthesizes the mRNA and proteins that are required for DNA synthesis and determines whether a cell commits to division or to leaving the cell cycle. The ensuing S (synthesis) phase allows the cellular DNA to be replicated; because of the importance of this, this phase is tightly regulated and highly conserved. A cell uses G2 phase to prepare for mitosis (such as by synthesising the required proteins) and repair DNA damage that arose during or after S phase before cell division occurs. Finally, during M (mitosis) phase, replicated cellular apparatus and chromosomes are separated into two new cells whilst maintaining appropriate ploidy.
The checkpoints between these phases determine the correctness of the preceding steps before continuing into the next proliferative phase and allows the cell to delay progression of the cycle in response to intra- or extracellular stressors. One of these checkpoints, at the G1/S transition, the so-called ‘restriction point’ or ‘DNA damage checkpoint’ (cf.Fig. 1) is a critical checkpoint that is controlled by the CDK4/CDK6–cyclin D pathway.24 The role of the G1/S checkpoint is to allow cells to commit to a cellular division free of damage to their genome and hence enter S phase. Disruption in the regulation at the G1/S checkpoint is consequently a typical event in oncogenesis. Another checkpoint is to be found at the end of G2 phase; it serves to prevent entry into mitosis with damaged DNA, thereby maintaining genomic stability. The proliferating cells are thus forced to stop proliferation they have had the chance to repair the damage. Under normal conditions, cell cycle checkpoints are regulated by the activities of mitotic kinases (as well as other proteins such as RB and p53 that are beyond the scope of this manuscript) and serve to prevent neoplastic or else aberrant cellular division. The actions of these CDK–cyclin complexes are regulated through an elaborate network of activating and inhibitory proteins. For instance, the addition of inhibitory phosphates to CDK1–cyclin B is mediated by Wee1 kinase among other actors. These regulatory kinases are, themselves, often regulated by an additional layer of regulatory kinases. Proper functioning of the DNA damage checkpoint is impaired in many types of neoplastic cells due to mutations in various tumour suppressor genes, including retinoblastoma protein (RB) and p53. This also means that cells lacking a well-functioning G1 checkpoint are thus dependent on the S and G2 checkpoints for DNA repair. Checkpoint kinase-1 (Chk1) is an active transducer kinase at operating at both the S and G2 checkpoints. Chk1 inhibitors are also able to potentiate the efficacy of DNA damaging agents in cancer cells.25 Since approximately 50% of all human cancers are deficient in p53, Chk1 is a particularly attractive therapeutic target for cancer treatment.
At the end of G2, the chromatid pairs condense and are aligned along the centre of the cell, attached to newly formed apparatus called the spindle and separated into the two daughter cells. The Aurora kinases are the foremost coordinators of these processes. Oncogenesis, therefore, arises from an imbalance of these pathways in favour of unhindered cell division, where one or more of the inhibitory checkpoints described above fails to keep proliferation in check.26 As a consequence, it remains the aspiration of medicinal chemists and oncologists to develop inhibitors or else modulators of cell cycle regulatory proteins to shift the balance away from aberrant mitotic divisions,27 or else forcing the cancer cells into mitotic catastrophe i.e., unrepaired DNA damage that results in cell death.
Structure and function of CDKs
For the sake of the discussion that follows, we present a (much simplified) overview of CDK structure. In the absence of their cognate cyclins, CDKs are catalytically inactive (Fig. 2). When a cyclin is not bound, the αC helix of the CDK – in the context of CDKs, sometimes known as the PSTAIRE helix for a conserved motif within it responsible for recognition of the cyclin(s) – is rotated such that the conserved catalytic Glu is held outwards, away from the active site, allowing the additional interaction of important Lys and Asp residues. This arrangement holds the activation loop in an inhibitory conformation. Upon cyclin binding, however, the αC helix is reoriented so that the catalytic Glu residue ‘swings’ into the active site (shown with a circular arrow in Fig. 2).
Fig. 2. (Left) Schematic structure of a free, inactive CDK showing the activation loop in bold, the αC helix and important residues mentioned in the text. The catalytic glutamate residue is labelled Glucat. (Right top) Structural changes seen in the CDK upon cyclin binding; the αC helix is reoriented in such a way that the catalytic glutamate residue ‘swings’ into the active site. This leads to changes in the activation loop which moves “out” and makes space for ATP and the CDK substrate to bind. (Right bottom) Binding of an inhibitor keeps the CDK in an inactive conformation similar to that on the left. This figure was adapted on the basis of an illustration by Huse and Kuriyan.28.

A conserved DFG motif is found within the activation loop and this can adopt either an ‘DFG-in’ conformation (Fig. 2, left), which occurs when the correct cyclin binds and so renders the CDK constitutionally active, or is otherwise held in the inactive ‘DFG-out’ conformation (Fig. 2, top right) in which a ‘gatekeeper’ phenylalanine side chain is oriented inwards into the active-site cleft (where inhibitors would otherwise bind).29 The preference for inhibitors that occupy a catalytically competent CDK over those occupying inactivated CDKs derives in part from the bulky character of this gatekeeper residue. In addition, the vast majority of structures that have been determined for complexes of ATP-competitive inhibitors and a CDK feature a ‘DFG-in’ conformation.30
Differences in amino acid sequence found immediately outside the active site can be probed by extending out from the purine binding site and allow more effective discrimination between individual CDKs.30 For instance, the surface of the purine binding site on the C-terminal lobe possesses key differences between CDK pairs: CDK1/2, CDK4/6 and CDK8/9 pairs. As can be expected, various CDK4/6-selective inhibitor series have been designed to exploit the purine binding site, for example offering substituted piperazine moieties capable of favourable polar interactions with the hydroxyl side chain of CDK4 Thr102 or CDK6 Thr107, but being repulsed by the charge on CDK2 Lys89 such as by AMG 925 (not shown),31palbociclib or ribociclib (vide infra).
Inhibitors of cyclin-dependent kinases
The unrestrained proliferation of cells is a hallmark of cancer and the aberrant activity of CDKs has been shown to be frequently increased in cancerous cell.32 Signal transduction cascades activated by KRAS (among other cellular actors beyond the scope of this review) intersect on the cell cycle and promote the transition into S phase.24 Given that more than 90% of pancreatic tumours bear activating mutations to KRAS, pharmacologic modulation of CDK activities is thought to offer significant scope for the treatment of PDAC. This, as well as their pivotal role in harmonising the cell cycle described above makes it hardly surprising that considerable research effort is dedicated to the development of small-molecule inhibitors against various CDKs for the treatment of neoplastic diseases.33 So far, however, inhibitors that are completely selective for a specific CDK remain out of reach of medicinal chemists and clinicians due not only to the extremely high homology within the CDK family and the high homology among kinases in general, but also to the lack of three-dimensional structures of important CDKs.
Inhibitors of CDK4/CDK6
CDK4 and CDK6 are critical mediators of cellular transition from G1 into S phase (where the cell will replicate its entire genome) and they are important for the initiation, growth and survival of many cancer types.26 Inhibitors of CDK4/CDK6 function by hindering the transition from G1 phase to S phase by blocking the phosphorylation of RB by CDK4/CD6; this keeps the transcription factor E2F sequestered to Rb, which induces cell cycle arrest in G1, thereby preventing tumour cell growth.34 Inhibitors of CDK4/CDK6 can limit the proliferation of neoplastic cells and such inhibitors have already revolutionized the treatment of hormone receptor-positive breast cancer and have quickly become the new standard of care.35 In addition, PDAC frequently suffers from the loss of the endogenous CDK4/6 inhibitor p16.36 Salvador-Barbero and colleagues have shown that CDK4/6 inhibitors (see below) are able to prevent the repair of damaged DNA following administration of mitotic poisons (i.e., nab-paclitaxel) and, as such, showed that the maximum benefit from CDK4/6 inhibitors could be obtained when they are administered after and not before rounds of cytotoxic chemotherapy.37
Three different generations of CDKs inhibitors have been developed so far and offer high in vivo activity with only limited toxicity.1 To date, three compounds have entered the clinic: palbociclib, ribociclib and abemaciclib (Fig. 3).
Fig. 3. Structures of palbociclib, ribociclib and abemaciclib.

Palbociclib (Fig. 3) was first described in 2005.38 The pyrido[2,3-d]pyrimidin-7-one scaffold provided an effective platform for the inhibition of a broad cross-section of kinases, including CDKs. It was demonstrated that a series of modifications to this scaffold yielded inhibitors with selectivity for CDK4/CDK6 when tested in vitro.
These modifications included: a) the introduction of a 2-aminopyridine side chain at the C2-position; b) the C6 group which projects into the back of the ATP pocket and which needed to be small and polar; and c) the introduction of a piperazine on the pyridine group; (d) a cyclopentyl group at N8, which provided the best balance of potency and selectivity for CDK4; and (e) introduction of a methyl group at C5 which further increased selectivity for CDK4/CDK6. Following impressive in vitro results, palbociclib was forwarded as a formal candidate for clinical development and, ultimately, received regulatory approval from the FDA in 2015 and the EMA in 2016.
Ribociclib (Fig. 3) bears the same tetrahydropyrido[2,3-d]pyrimidine and cyclopentyl groups as palbociclib but differs in that they are attached to a different core heterocycle – in this case to a pyrrolopyrimidine – and that ribociclib bears an acetamide rather than palbociclib's acetyl group. The utility of the pyrrolopyrimidine scaffold for CDK4/CDK6 inhibition was patented by Novartis in 2007.1Ribociclib binds to the ATP binding site of the inactive conformation of both kinases with similar potency to palbociclib, although ribociclib exhibits five-fold greater in vitro potency against CDK4, as compared to CDK6.39 Crystallographic analyses revealed three hydrogen bonds between the molecule and Val101 and Asp163 of the CDK. The replacement of the aniline moiety with a 2-aminopyridine had a pivotal role to improve selectivity profile, as was previously seen in the development of palbociclib.38
The third-generation CDK4/CDK6 inhibitor, abemaciclib (Fig. 3), displayed high activity in PDAC cell lines and decreased growth through inhibition of phospho-Rb, G1 cell-cycle arrest and ensuing apoptosis. Abemaciclib appears to bind more readily to the ATP cleft and forms a hydrogen bond with a catalytic residue (Lys43) that is conserved among kinases, suggesting that it binds with less selectivity than ribociclib and palbociclib.40 In contrast, ribociclib and palbociclib appear to have a lower lipophilicity and larger side chains than abemaciclib, implying, because of their larger overall size, a smaller number of off-target kinase ATP-binding pockets with which they are able to interact. The ability of abemaciclib to inhibit other members of the CDK family provides efficacy in inhibiting cell growth even in cell lines with Rb deficiency.41 On the other hand, the higher lipophilicity of abemaciclib allows for better tumour penetration in treating various solid tumours and even cerebral tumours.41 Nevertheless, which of these three agents is chosen for a patient remains largely determined by the patient and their toxicity profile, the potential drug interactions that may occur with other agents the patient receives, and, importantly, the physician's own experience with these agents.41
Abemaciclib has been described as therapeutically relevant for the treatment in PDAC.42Abemaciclib monotherapy for PDAC is currently being investigated in phase II trials (NCT03891784), and combination therapy with gemcitabine and samotolisib (an inhibitor of phosphoinositide 3-kinase (PI3K)) is also underway (NCT02981342). Since, in PDAC, monotherapy with an inhibitor of CDK4/CDK6 is ineffective due to RAS-mediated activation of alternative pathways, the combination of ribociclib and everolimus (an mTOR inhibitor) has been investigated in a phase I study in patients with PDAC that is refractory to standard chemotherapy. Ribociclib was well tolerated and associated with decreased CDK4/CDK6-regulated gene expression. This combination was not effective as a third-line therapy but does pharmacologically target CDK4/CDK6 in metastatic PDAC, revealing the potential for benefit in other settings.43
Structurally, these three molecules share a number of common features, described in detail by Poratti and co-workers (see also Fig. 4).1 They bind the inactive conformation of the kinase(s) and establish two key H-bonds; the first between the pyrimidine 3′-position and the backbone amine of His100, the second between the carbonyl of Val101 and the exocyclic NH of the side chain.1Palbociclib and ribociclib go further than this and establish an additional hydrogen-bond between their carbonyl function and the DFG motif. As anticipated by the SAR on palbociclib,38 the steric hindrance caused by the methyl function at position 5 forces the acetyl group into the most adequate orientation for interacting with the enzyme. The steric features of the heterocyclic core constitute a major difference between the three compounds: ribociclib and palbociclib are characterized by larger substituents (dimethylamide and acetyl functions, respectively) than abemaciclib (fluorine atom). As a result, abemaciclib more readily buries itself in the ATP pocket. Nevertheless, the steric features of ribociclib and palbociclib account for their higher selectivity (as described above).1 Superimposition of crystallographic structures shows that, despite the very high structural similarity, the amino side chains occupy rather different positions, whereas the pyrimidine cores overlap well.
Fig. 4. Binding modes of palbociclib (left; PDB: 5L2I), ribociclib (centre; PDB: 5L2T) and abemaciclib (right; PDB: 5L2S). Ligands, gatekeeper residues, DFG domains and hinge residues are depicted as stick while the rest of the protein is represented as a ribbon. Not all molecular interactions are shown. CDK6 is shown in pink, gold and green respectively. Asp163 corresponds to the aspartate of the DFG domain. This figure was inspired by Poratti and co-workers.1.

Despite their clinical utility, these three cornerstone CDK4/CDK6 inhibitors also suffer from problems with toxicity, where there are also differences between the three. Patients receiving palbociclib or ribociclib, for example, present predominantly with bone marrow toxicity, while administration of abemaciclib is more often associated with gastrointestinal symptoms and less-pronounced hematologic toxicity.44 On top of this, ribociclib has been shown to induce QT interval prolongation in up to 3% of patients.45
In addition to problems with the therapeutic profile of these agents, patients also often become acquire insensitivity to these drugs, to which they can acquire drug resistance.46 Xu and co-workers have highlighted the three main mechanisms that can cause acquired resistance. Firstly, through the amplification or overexpression of the components of the main CDK4/CDK6 signalling pathway; secondly, through the activation of alternative signalling pathways; thirdly, and of particular interest in PDAC, through the modulation by these molecules of the tumour-immune microenvironment inhibitors, such as producing changes in PD-L1 expression.46 The immunotherapeutic angle is discussed further in the Perspectives section towards the end of this review.
It is no surprise that considerable research efforts are being undertaken to overcome these issues with CDK4/CDK6 inhibitors, either through the exploration of novel drug combinations, which may extend the therapeutic potential of these molecules or else through the development of novel agents with superior properties that can overcome the limitations currently held by the approved inhibitors. Based on the scaffold of palbociclib, one study designed and synthesized a series of covalent CDK4/CDK6 inhibitors that targeted amino acid Thr107, located on the edge of the binding pocket.47 The distal piperazine nitrogen of palbociclib is not involved in binding (cf.Fig. 4) and is therefore suitable for attachment of additional groups. The distance between it and the Thr107 hydroxyl is 3.4–3.6 Å, allowing the strategic placement of an electrophilic group in order to form a covalent bond with Thr107. The derived compound 1 (Fig. 5) showed an improved inhibitory concentration (with IC50palbociclib 11 nM (CDK4) and 16 nM (CDK6) vs. compound 1 with 6 nM (CDK4) and 14 nM (CDK6)). When tested in vitro, 1 showed enhanced cytotoxic effects compared to palbociclib.
Fig. 5. Structure of palbociclib and its derivation through the addition of an electrophilic group to yield 1 and 2.

With the same threonine residue in mind, Fang and co-workers designed and synthesized a derived series of palbociclib analogues bearing a Michael-acceptor on the piperazine moiety (Fig. 5).48 Among a general preponderance of the authors' library for antiproliferative activity, 2 showed the highest inhibitory activity in both MDA-MB-231 and MCF-7 cells (both breast cancer cell lines) (IC50 0.51 μM and 0.48 μM respectively, cf. 5.62 μM for palbociclib). The Michael acceptor in 2 was the one tested that performed the best in cellular assays. One of the significant observations was that 2 showed strong inhibition against palbociclib-resistant MCF-7 cells, indicating that it functions even when cells have become resistant to palbociclib. The computational experimental analyses showed that the distance between the hydroxyl group of Thr107 and the carbon–carbon double bond of 2 was found to be 1.5–2.0 Å. Subsequent HPLC analyses showed that a covalent bond was formed between 2 and the CDK. In enzymatic assays, 2 produced a significant decrease in the phosphorylation level of both CDK4 and CDK6 as well as displaying selective inhibitory activity against both (ICCDK450 18 nM, ICCDK650 13 nM) and was shown to efficiently cause cell cycle arrest in the G1 phase and induce apoptosis.
While 1 and 2 were not yet tested in models of PDAC, the covalent inhibitor strategy is worth highlighting, since this offers a way to circumvent problems with decreased response and increased drug resistance, especially with palbociclib, that are commonly seen in chemotherapeutic regimens with these agents.48 Given the recent reports of these two agents – 2021 and 2023 respectively – we can expect that a further exploration of the molecular scope of these molecules, as well as a broadening of the cancer types they are assessed in.
A number of reports have been described of PROTAC-based approaches featuring one of the approved inhibitors. These have been reviewed by Adon and colleagues.49 Other approaches currently being investigated also include novel CDK4/6 and PARP (poly (ADP-ribose) polymerase) dual inhibitors that have so far only been tested in breast and ovarian cancers.50 However, given the clinical significance of PARP in PDAC,51 it is likely that such agents will be tested in relevant disease models in the near future.
Inhibitors of CDK7
Unlike CDK4 and CDK6, CDK7 is not directly involved in the regulation of cell cycle progression but is rather implicated as a CDK-activating kinase and, more crucially, as a regulator of transcription, where it phosphorylates the C-terminal domain of RNA polymerase II. It does however function as the kinase that activates CDKs 1, 2, 4 and 6.52 Consequently, CDK7 has received attention as a potential target for cancer.53,54
Many CDK7-selective inhibitors across all cancer types fall into one of two categories: those based on BS-181 and those based on THZ1 (Fig. 6). The docking models of these inhibitors reveal that Asp155 and Met94 may be the key residues of binding.55 Investigation of THZ1 revealed the role of CDK7 in PDAC.56 The molecule itself was first described by Kwiatkowski and colleagues in 2014.52 They had performed cell-based screening and kinase selectivity profiling of a library of known and novel ATP-site-directed kinase inhibitors and identified the phenylaminopyrimidine THZ1 as a low-nanomolar inhibitor of cell proliferation and biochemical CDK7 activity. The molecule bears a cysteine-reactive acrylamide moiety. To investigate the functional relevance of the acrylamide moiety, a saturated analogue was prepared, 3, that cannot react with cysteine (Fig. 6). As expected, compound 3 showed comparatively less CDK7 activity (IC50 146 nM) than THZ1 (3.2 nM) in the LanthaScreen Eu Kinase Binding Assay, as well as a reduced anti-proliferative activity. Profiling with KiNativ™ (a proprietary biochemical kinase profiling assay) established CDK7 as the primary intracellular target (but not of 3). Expanding the profiling to the whole kinome identified additional kinase targets of THZ1. It was however confirmed that CDK7 is the only target showing time-dependent inhibition, suggesting covalent binding of the molecule to its target. Mass spectrometry results pointed to Cys312, which is located outside of the kinase domain, as the site of covalent modification. Sequence alignment of the 20-member CDK family revealed that Cys312 is unique to CDK7.
Fig. 6. Structures of BS-181 (left) and THZ1 or 24 (right).

According to the crystal structure (PDB: 1UA2, not represented here), Asp155 is in proximity to the indole and Met94 is close to the central amide. Other key residues in the ATP-binding site include Phe91, Phe93 and Val100. Altogether, these mechanistic and structural insights will be useful in the design of subsequent generations of CDK7 inhibitors, for which high sequence and shape homology in the ATP pocket has posed a challenge to achieve selectivity with conventional ATP-competitive inhibitors.52
In recent work of 2022, Zeng and co-workers – through kinome-wide CRISPR-Cas9 loss-of-function screening on the KPC mouse model-derived PDAC cell line TB32047 – identified several cell cycle checkpoint kinases and DNA damage-related kinases as targets for overcoming chemoresistance.57 Among them, CDK7 ranked highly in both screenings. It was demonstrated that both gene knockout and THZ1-mediated inhibition of CDK7 result in cell cycle arrest, the induction of apoptosis, and DNA damage through the STAT3-MCL1-Chk1 axis. Furthermore, THZ1 synergized with gemcitabine and paclitaxel (structures not shown) both in vitro and in vivo, resulting in enhanced antitumor effects. Furthermore, Lu and co-workers showed56 that prolonged treatment of PDAC cells with a sublethal concentration of THZ1 concentration resulted in acquired resistance of the cells to the drug through downregulation of the MYC gene, whose protein product regulates cellular proliferation and apoptosis.56
Olson and co-workers set out to design selective inhibitors of CDK7 based off the THZ1 scaffold but, when they were initially unsuccessful, their attention turned to the PAK4 inhibitor PF-3758309, since PAK4 inhibitors were already shown to be competent inhibitors of CDK7.58 The hypothesis was that by creating a hybrid structure based off the covalent warhead present in THZ1 and the pyrrolidinopyrazole scaffold in PF-3758309 – would lead to more selective CDK7 inhibitors. Initial compounds only showed moderate potency and offered only minimal anti-proliferative effects on the cancer cell lines tested. However, the authors' continued medicinal chemistry efforts, which dealt with the acidity of the aminopyrazole core, improving binding interactions with the protein pocket, and optimising the length and trajectory of the covalent warhead that targets the crucial Cys312 residue, which, all together, yielded YKL-5-124 (Fig. 7).
Fig. 7. Structure of PF-3758309 and its derivation through the introduction of a reactive electrophilic warhead to yield YKL-5-124.

The molecule was described as a “CDK7-selective chemical probe” that can elucidate the still unknown pharmacological workings of CDK7. Nevertheless, it induces a strong cell-cycle arrest: YKL-5-124 inhibited CDK7 with an IC50 of 9.7 nM (which compares favourably to CDK2 and CDK9, showing IC50 values of 1300 nM and 3020 nM respectively). When compared to THZ1, both molecules show similar ki values (1.9 nM for YKL-5-124 and 2.1 nM for THZ1) showing that they achieved nearly equivalent inhibition of CDK7. However, YKL-5-124 exhibited a faster kinact of 103 ms−1 nM−1 as compared with kinact of 9 ms−1 nM−1 for of THZ1, implying an 11-fold faster rate of covalent modification of CDK7 for YKL-5-124 than THZ1. Beyond the chemistry of their molecule, the authors were able to draw a number of conclusions about the more complete biophysical role of CDK7 in the cell cycle, but these are beyond the scope of this review.
Following on from this work, Yang and co-workers more recently showed that selective inhibition of CDK7 with YKL-5-124, in combination with gemcitabine, is efficacious both in vitro and in vivo, supporting the notion that YKL-5-124 – but also selective CDK7 inhibition more broadly – is a promising therapeutic strategy for the treatment of PDAC.59 The team showed that YKL-5-124 negatively affects DNA damage repair pathways and evokes genomic instability in PDAC cells.
TCN-1062 (structure not disclosed) is a novel, orally available agent that reversibly and selectively inhibits CDK7 with an IC50 of 33 nM.60In vitro studies demonstrated that PDAC cell lines were sensitive to TGN-1062 with a GI50 (concentration for 50% of maximal inhibition of cell proliferation) of less than 0.4 μM. In addition, phosphorylation of RNA Pol II and expression of c-Myc were suppressed by TGN-1062 in MiaPaca2 PDAC cells. The molecule's preclinical evaluation (kinome profiling, super-enhancer binding protein activities, ADME-Tox and efficacy) is currently underway.
Marineau and co-workers have recently described a selective inhibitor of CDK7.55 Their medicinal chemistry campaign started from compound 4 (Fig. 8), a potent inhibitor of CDK7 but which lacked an adequate selectivity profile. Initial development efforts focused on improving the selectivity through substitution of 7′-position (marked in blue in Fig. 8) but were unsuccessful. The introduction of a methyl sulfone, however, provided an increase in potency (from KD 0.15 to 0.07 nM) and with a slight increase in selectivity. The increased potency likely derives from multiple polar contacts in the sugar binding pocket, where one of the sulfonyl oxygens makes contact with the catalytic Lys33. Installation of a nitrile at the 6-position of the indole ring allowed the molecule to project towards the phosphorylation surface of the active site (the ‘P-loop’). As regards the piperidine group, substitution with gem-dimethyl groups increased the selectivity but resulted in a slight decrease in CDK7 affinity.
Fig. 8. Structure of lead compound 4 and SY-5609 which derives from it. The 7-position and the uncommon dimethyl phosphine oxide (highlighted in yellow), which is an uncommon isostere of a sulfone, are shown in the two structures respectively.

Further development efforts resulted in SY-5609, the most striking aspect of which is the incorporation of a dimethyl phosphine oxide (highlighted in yellow in Fig. 8), an uncommon isostere of a sulfone, at the 7-position of the indole. The strong propensity for hydrogen bonding and the high stability shown by this group have increased its importance in preclinical discovery programs and even in agents that have reached the market (consider the ALK inhibitor brigatinib). This introduction of this group led to an increase in selectivity over CDK2, CDK9, and CDK12. As determined in subsequent computational and crystallographic studies, the key indole substitutions – the nitrile at the 6-position and the rigid dimethylphosphine oxide at the 7-position – likely drive selectivity by exploiting the conformational preferences of the CDK P-loop, thereby allowing CDK7 to accommodate the larger, substituted indole ring of SY-5609 better than the other CDKs could, while still preserving the key polar interactions. Both of these indole substitutions make water-mediated hydrogen bonds; the 7-phosphine makes contact with Lys41 and Asp155 (as does the indole nitrogen) and the 6-nitrile with Glu20. In addition, intermediates bearing the dimethyl phosphine oxide also exhibited more potent antiproliferative activity in HCC70 cells (a breast cancer cell line). After balancing their SAR analyses against the ADME and pharmacokinetic properties of each of their intermediate compounds. Out of these final studies SY-5609 was identified.
In 2023, SY-5609 completed phase I clinical trials for advanced solid tumours and in second and/or third line pancreatic ductal adenocarcinoma (NCT04247126) in combination with gemcitabine and nab-paclitaxel. Encouraging preliminary clinical activity and the emerging exposure–activity relationship was reported and further studies are planned.61
Inhibitors of CDK1
Among other minor roles, CDK1, with its partner cyclin A, is largely responsible for allowing passage of a cell through the G2/M checkpoint (Fig. 1) and activating homologous recombination DNA repair pathways.62 At times, CDK1 has even been described as the “master regulator of the cell cycle”, because its functions cannot be superseded by other CDKs.63 Consequently, the inhibition of CDK1 is now seen as a potentially novel therapeutic strategy against PDAC.64 In the development of PDAC specifically, the role of CDK1 is two-fold. Firstly, since CDK1 activity regulates the G2/M cell cycle checkpoint, overexpression of CDK1 can lead to progression into mitosis even in cells with DNA damage, a clearly tumorigenic process. Carbone and co-workers, using a set of derivatives of the sponge-derived natural products nortopsentin A–C, showed that the compounds inhibited CDK1 and led to cell cycle arrest in their in vitro models.65 Interestingly, the inhibition of CDK1 has also been linked to increased radiosensitivity of PDAC cells.66 Secondly, CDK1 overexpression leads to the stimulation of a range of proteins that induce stem cell properties, which can contribute to the development of cancer stem cells (CSCs). CSCs are a distinct subpopulation of malignant cells with their own defined properties: they exist in a de-differentiated state, they are capable of self-renewal, show an inherent resistance to chemo-/radiotherapy and present a higher tumorigenicity than other cancer cells.67 A full discussion of CSCs is beyond the scope of this manuscript but readers are directed towards a review on this topic by Malumbres and Barbacid.32
Milciclib (Fig. 9) is an inhibitor of CDKs 1, 2, 4 and 5 and was first described in 2009.68 Efforts were started from a 5 (Fig. 9), specifically aimed towards inhibition of CDK2. The molecule binds to the ATP pocket of the kinase in its active conformation. The heterocyclic core binds to the ATP pocket where adenine would normally bind, while the phenyl group points outward towards the solvent accessible region. Incorporation of the dimethyl moiety at the 4-position resulted in a significant improvement of selectivity against Aurora A. For CDK1, the proposed binding mode of this drug was confirmed by X-ray. The pyrazoloquinazoline moiety forms hydrogen bonds with the backbone amine of Leu83, while the adjacent amino group binds to the carbonyl oxygen of Leu83. The different binding mode compared to CDK4/CDK6 inhibitors is partially explained by the {6-6-5} tricyclic structure that features in the molecule, which was previously described as a scaffold for inhibitors of Wee1 kinase (vide infra).69 In a phase I study, on patients with refractory solid tumours, milciclib was tested in combination with gemcitabine and was reportedly able to overcome gemcitabine-resistance in PDAC patients.70 The combination treatment was well-tolerated with manageable toxicities. Currently no clinical trials for PDAC are scheduled or in progress. However, after these results, the combination of CDK inhibitors – including milciclib with CDK4/6 inhibitors – has been patented for ‘tumour suppression’ in a range of solid tumours such as PDAC (WO2021205363A1).
Fig. 9. Structure of lead compound 5 and milciclib, which derives from it, with structural changes highlighted.

Despite being designed against CDK2, dinaciclib (Fig. 10) is a potent and selective inhibitor of CDK1 (but also CDKs 2, 5 and 9) with an IC50 value of 3 nM for CDK1.71
Fig. 10. (Left) Structure of dinaciclib. (Right) Crystal structure of dinaciclib (grey) bound to CDK1 (aqua) with key residues labelled (PDB: 4KD1). Note the interaction of the pyridyl N-oxide with Lys89.

Dinaciclib binds to the ATP site through the pyrazolopyrimidine moiety that makes a hydrogen bond with Leu83 of the hinge region. The 3-ethyl group of the pyrazolopyrimidine moiety establishes hydrophobic interactions with the gatekeeper residues Phe80. The pyridine oxide ring is exposed to the solvent region and interacts with the ε-amino group of Lys89.72 This pyridine N-oxide has since been featured in analogues of ribociclib where the nitrogen atom of the pyridine was replaced with an N-oxide moiety73 and in novel inhibitors of CDK2 for the treatment of leukaemia.74
Since dinaciclib inhibits multiple CDKs, the in vitro results – swaying between the effects of CDK1 and/or CDK2 inhibition – suggested that the effect of the drug is dependent on the mutations present in the PDAC cell type used.64 In preclinical studies, dinaciclib was able to inhibit the growth and progression of PDAC murine xenograft models.75 When tested in humans, however, dinaciclib, while well tolerated by patients, failed to show clinical benefit in combination with the Akt inhibitor MK-2206.76
More recently, however, in tumour-bearing mice, dinaciclib (here acting as an inhibitor of CDK5) was shown to significantly inhibit the growth and motility of PDAC cells growth in vivo, inducing apoptosis and cell cycle arrest in the G2/M phase.77 Li and co-workers noted a strong correlation between CDK5 expression and tumour size and staging. High CDK5 expression can therefore be a prognosticator of poor survival in PDAC patients. It was also noted that dinaciclib led to a down-regulation of the mRNA of yes-associated protein (YAP) and its subsequent protein expression through a concomitant reduction of β-catenin expression.
CDK1 remains an important target in PDAC and its substrates have been shown to be important for the proliferation of PDAC cells.78,79 It can be expected that work will continue on the design of CDK1 inhibitors.
Inhibitors of CDK9 or other CDKs
Immunohistochemical comparisons of CDK9 expression in normal tissue and pancreatic tumours has revealed an overexpression of CDK9 in the latter.80 This is associated with significantly shortened survival, especially in well-differentiated tumours. Kretz and co-workers went on to show that pharmacological inhibition of CDK9 reduced the cell viability of PDAC cells, suppresses their long-term survival and enhances the therapeutic efficacy of other chemotherapeutic agents.
Around the same time as, Lücking and co-workers described the first CDK9-selective inhibitor atuveciclib (Fig. 11).81 With regards to its development, efforts started from the lead compound BAY-958, which had displayed potent inhibitory activity of PTEFb – a transcription regulatory complex comprising CDK9 and other proteins – and high kinase selectivity in vitro. The team's lead optimization efforts eventually led to the identification of the orally available clinical candidate, which is structurally characterized by an unusual benzylsulfoximine group (highlighted in Fig. 11).
Fig. 11. Development of atuveciclib from BAY-958, with the benzylsulfoximine group highlighted.

While BAY-958 had shown good in vitro activity against CDK9 and very high kinase selectivity it suffered from limited aqueous solubility, only moderate permeability and high efflux resulting in low oral bioavailability. Optimization efforts finally led to the incorporation of the benzylsulfoximine, which shows comparable in vitro potency and selectivity as BAY-958, but with much-improved properties that furnished good bioavailability in animal models. Moreover, the decision to switch from the benzyl-sulfonamide in BAY-958 to the benzylsulfoximine in atuveciclib removed a potential metabolic liability through CYP1A2 induction.82 The sulfur stereochemistry of the sulfoximine group was determined by X-ray crystallography, revealing the (R)-sulfoximine as the superior enantiomer in terms of potency (ICCDK950 13 nM vs. 16 nM). Atuveciclib has been shown to sensitise PDAC cells to TRAIL-induced cell death (tumour necrosis factor-related apoptosis-inducing ligand) through the concomitant suppression of cFlip and Mcl-1 (both anti-apoptotic proteins).83 A gemcitabine-resistant PDAC cell-line and patient-derived xenograft cell lines were also suppressed by this combinatorial approach.
Atuveciclib was tested in two phase I trials, where in each study the molecule led to high incidences of neutropenia across different dose schedules. This led to dose-limiting toxicity at sub-therapeutic doses that could not be managed with granulocyte colony-stimulating factor and, consequently, the drug was discontinued. However, further optimization of atuveciclib led to the development of a highly potent and more selective CDK9 inhibitor, enitociclib (structure not shown).84 This molecule was only discovered in 2023 but given the improved properties it shows over atuveciclib, it may well come to be tested in PDAC models and patients. For the time being, however, atuveciclib continues to be used as an agent in pre-clinical studies.
Fadraciclib (Fig. 12) is a second-generation clinical candidate reported by Frame and co-workers in 2020 based on the aminopurine scaffold of the CDK inhibitor seliciclib.85 The precursor seliciclib is an ATP-competitive inhibitor of CDK2, CDK7 and CDK9, but which is rapidly converted to a less potent metabolite through first-pass oxidative metabolism.
Fig. 12. Structures of seliciclib and fadraciclib, which derives from it, with key structural changes highlighted.

As part of medicinal chemistry efforts to increase target CDK inhibitory potency and enhance the metabolic stability of seliciclib, a range of 6-pyridylmethyl-aminopurines was prepared with improved metabolic stability, significantly improved target inhibition and superior anti-proliferative activity. The clinical development candidate selected from this series was fadraciclib, which exhibited a more than 33-fold enhancement in anti-proliferative activity over seliciclib. It was determined that fadraciclib inhibited CDK2 and CDK9 with IC50 values are 4.5 and 26 nM respectively. Fadraciclib is currently being tested in two phase 1/2 trials including one for the treatment of advanced solid tumours, including PDAC (NCT04983810).
Inhibition of Wee1 kinase
As described above, in normal cells DNA damage can be repaired at checkpoints at the end of G1 and G2. The G1/S checkpoint does not function correctly in tumour cells with a lack or deficiency of p53 or RB and, as such, the cells become highly dependent on the G2/M checkpoint to maintain genomic stability. Wee1 belongs to a family of protein kinases that activate the G2/M checkpoint of the cell cycle in response to double-stranded DNA breaks (DDBs).86 Wee1 kinase is a critical regulator of the G2/M checkpoint, functioning through phosphorylation of CDK1, which, as we have seen, inhibits the activity of the CDK1/cyclin B dyad. This inhibition of Wee1 leads to G2 checkpoint escape, where tumour cells proceed into mitosis with incompletely replicated or damaged DNA resulting in cell death, ultimately undergoing apoptosis via mitotic catastrophe after successive rounds of cell division.87 As a result, Wee1 kinase is seen as an attractive target that can enhance the effects of chemotherapeutic agents that generate DNA damage (e.g., platinum drugs).88 Given recent progress in development of KRAS-mutant inhibitors, the importance of understanding the compensatory activities that can drive resistance to KRAS suppression has become all the more important. A recent study revealed that several kinases – including Wee1 – that are upregulated in PDAC represent compensatory mechanisms for KRAS suppression.89
Wee1, which when inhibited with adavosertib (Fig. 13), was capable of inhibiting the proliferation of six pancreatic cancer cell lines evaluated. Previous studies reported that the highly selective, small molecule Wee1 inhibitor adavosertib is able to force cells past the G2/M checkpoint and through into mitosis and in doing so is able to sensitize them to the effects of immune checkpoint blockade.90Adavosertib can potentially synergize with DNA-damaging therapies commonly used in PDAC treatment such as 5-fluorouracil and irinotecan, both components of the FOLFIRINOX regime.91Adavosertib was discovered from high-throughput screening of a small chemical compound library. It shows considerable overlap with the structure of milciclib (vide supra). The initial hit compounds were optimized through SAR-based approaches, giving adavosertib as a first-in-class and ATP-competitive specific small molecule inhibitor of Wee1 (KD 3.2 nM).92 The crystal structure of Wee1 kinase bound to the inhibitor adavosertib (PDB: 5V5Y) defined two key residues for the hydrogen bonding interactions with Wee1: Cys379 and Asn376. While the authors of the original discovery do not delineate their actual design process, Zhu and co-workers assessed the structural determinants of Wee kinase functionality with adavosertib;93 they obtained high-resolution X-ray co-crystal structures of the kinase domains of all three Wee kinases (Wee1, Wee2 and Wee3) in complex with adavosertib.
Fig. 13. (Left) Structure of adavosertib. (Right) 2D representation of adavosertib bound to Wee1. The N-allyl group is marked in a pink box and its interaction with Lys328 is shown by a dotted black line.

They were able to show that the molecule interacts primarily with the main chain atoms of a conserved cysteine residue (Cys379) of the hinge region. An additional hydrogen bond exists between the pyrazolopyrimidinone oxygen of adavosertib and the side chain of the gatekeeper residue, which, in Wee1, is Asn376 (Fig. 13, right). Adavosertib is stabilized by additional hydrophobic interactions in the ATP site, where the residues are almost entirely conserved between Wee1 and Wee2; Asp386Wee1-to-Ala is the exceptional substitution in the solvent exposed front specificity pocket. Here, the tertiary alcohol on adavosertib is important for making the interaction with Asp386.
As regards the N2-allyl group, the original publication downplayed its role in binding to Wee1, describing it as providing “slight hydrophobic interactions with Val313, Ala326, and Lys328”. In addition, it was considered to constitute a metabolic liability, with which it would undergo CYP450-mediated conversion to the primary alcohol or diol. Matheson and co-workers at the time hypothesized that through the removal of this allyl group, the molecule could benefit from deeper access into the ATP-binding domain and consequently allow tighter binding.94
Since then, however, the significance of this N2-allyl group has been shown and subsequent studies by other groups have demonstrated its importance (see below). Guler and co-workers, in particular, have shown that its modification can improve the selectivity of Wee1 kinase inhibitors over Plk1 inhibitors (vide infra). This is especially pertinent as adavosertib itself has previously been reported to inhibit Plk1 (see below).96 A number of Wee1 kinase inhibitors were synthesized, replacing this N-allyl group with various other functions. The structure-based drug design efforts began by using the X-ray crystal structure of adavosertib bound to Wee1 kinase (PDB: 5V5Y) – where adavosertib binds into the canonical ATP pocket, as described above – and Plk1 (PDB: 8BJT). In each structure, adavosertib binds in the canonical kinase ATP pocket in a similar orientation and making the same key interactions. The authors then reasoned that selectivity for Wee1 over Plk1 could be achieved by targeting residues that are not conserved in these three pockets, namely substitution of the N-allyl group; since the gatekeeper residue of Plk1 is the bulky Leu130 gatekeeper residue, larger groups will not be tolerated in the selectivity pocket, particularly aliphatic substitutions that have more sp3 character due to the shape of this pocket.
As part of their SAR studies, the same authors noted that the R2 pyridine nitrogen group (shown in blue in Fig. 14) was not necessary for Wee1 inhibition, but the geminal dimethyl groups were important for orienting the hydroxyl group toward the catalytic/activation loop region. Although this region is conserved in Plk1, the hydroxyl group in Plk1 points toward the G-loop instead and makes a hydrogen-bond with backbone carbonyl of Lys61; this interaction substantially changes the conformation of the kinase G-loop. They also explored the R3 region (shown in a pink box in Fig. 14) but deferred to other authors who have undertaken more extensive work in this area. The most potent of the compounds described by Guler and co-workers was compound 6 (Fig. 14), which demonstrated a Plk1/Wee1 selectivity ratio of more than 5800 (ICPlk150 > 25 000 vs. ICWee150 I2.3 nM), which is vastly more selective than adavosertib (IC50s 124 vs. 3.0 nM respectively). Through crystallographic analyses, it was shown that compound 6 binds very similarly to adavosertib. At the same time, the Asn376 gatekeeper residue of Wee1 kinase makes contact the carbonyl group of the heterocyclic core forming a hydrogen bond with the amide NH2 side chain of the Asn376. Crucially, the allyl group also binds making a π–π interaction with the amide plane of the Asn376 gatekeeper side chain located in the pocket – alternatively termed the selectivity pocket – framed by structural elements of the kinase (including the αC helix region (cf.Fig. 2)).
Fig. 14. Structure of compound 6 after medicinal chemistry development with the three R groups discussed in the text highlighted.

Sticking with the same N2 group, SC0191 (Fig. 15, left) is a Wee1 inhibitor that bears a unique cyclisation between the N2 group and the distal tertiary alcohol. Little has been described about its design and conception, but Alli and co-workers have posited that the cyclization approach may also disrupt the π–π stackings of SC0191 with Phe433.97 As part of their preclinical evaluation of the molecule, Yang and co-workers found potent kinase inhibiting activity for Wee1 with an IC50 of 22.3 nM. The cellular anti-proliferative activity of SC0191 in TP53-mutant BxPC-3 PDAC cell lines was also determined and showed significant inhibition with IC50 value of 224 nM.98SC0191 is scheduled to start phase Ib/II clinical trials to determine its the safety, pharmacokinetic characteristics, but limited to ovarian cancer (NCT06055348).
Fig. 15. (Left) Structure of SC0191 with the macrocyclic link that replaces the N-allyl group of adavosertib highlighted in bold. (Right) Structure of azenosertib, with the bulky bicyclic motif highlighted in purple. This group is important for orienting the hydroxyl group in the appropriate conformation for interaction with the kinase.

Azenosertib (Fig. 15, right) was described by Huang and co-workers in 2021, whose work focused on the R2 position (as corresponds to Fig. 14 and the work of Guler and co-workers).99 The 3D-structures of adavosertib in complex with a number of kinases inhibited by the pyrazolopyrimidinone chemical series – including Wee1, Plk, DDR2, and others – were superimposed and compared. It was noted that Tyr378 from the hinge binding region and Phe310 from the G-loop stood out for their potential for leveraging superior protein–ligand interaction for Wee1 selectivity improvement. It should be pointed out that these are different residues than targeted by Guler and co-workers. However, the bulky bicyclic motif orients the important hydroxyl group in the appropriate conformation, just as the gem-dimethyl groups had done in compound 6 (Fig. 14).
Since their initial strategy, which was to introduce bulkier bicyclic tail groups into the pyrazolopyrimidine scaffold, did not help to improve the overall kinase selectivity as anticipated, their attention turned instead to the introduction of a bicyclic head group to improve the lipophilic interactions with a G-loop through Phe310. It was quickly found that the highlighted group in Fig. 15 afforded the best balance of potency (IC50 3.8 nM), polarity, and clearance. The ethyl group, in particular, was superior to smaller methyl or bulkier cyclopropyl and could offer good cellular potency (IC50 103 nM), acceptable polarity (log D 2.4), as well as reasonable pharmacokinetic parameters (clearance, AUC and rodent oral exposures). The co-crystal structure (PDB: 7N3U) showed, among other interactions, that the chiral ethyl motif interacted with Phe310 from the G-loop very efficiently. The same structural analyses showed, as expected, that the allyl group of azenosertib perfectly fills a lipophilic pocket formed by Ala326, Lys328, Ile374, and Asn376. In contrast to 6 and SC0191, a simple allyl group provides the optimal interaction efficiency and its modification results in a significant decrease of potency.
As of November 2023, azenosertib is currently being tested in a phase II study in patients with PDAC to determine the safety and effectiveness of combination therapy with gemcitabine (NCT06015659).
While Guler and colleagues deferred substitution of the tail region of adavosertib to other groups, Matheson and co-workers, have specifically looked at modifications of the solvent-exposed tail region.100 They found that substitution on the piperazine ring, as in 7 (Fig. 16), reduces cellular toxicity of the molecule with a retention of its inhibitory activity. In view of the aim of their work, which centred on paediatric medulloblastoma, the researchers conceded there is little chance that 7 would be able to cross the blood–brain barrier (BBB). They propose, however, that there may be scope to improve BBB permeability of these molecules through further examination of rationally placed N-substitutions of the piperazinyl side chain.
Fig. 16. Structure of 7, derived from adavosertib by Matheson and co-workers.
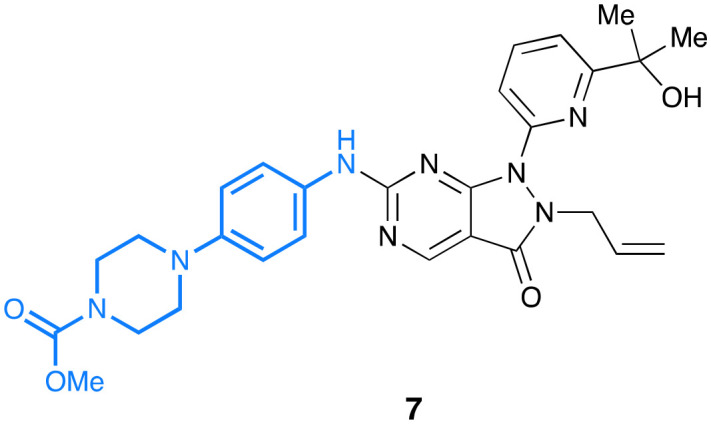
The various positions around the foundational adavosertib scaffold have each been examined in detail, as in azenosertib, 6 and 7, but to the best of our knowledge there have been no reports of molecules that fully combine these into an agent that benefits from the advantages of each modification.
Inhibition of Plk1
Because of its role in regulating a cell's entry into mitosis, chromosomal condensation, spindle assembly, and other cytokinetic functions, polo-like kinase 1 (Plk1) is considered a master mitotic regulator.101 Hence overexpression of this kinase causes the overriding of mitotic checkpoints, which can lead to immature cell division. In a more recent study by Li and co-workers, the combination of Plk1 inhibition and gemcitabine led to a significant increase in cellular apoptosis and, in an orthotopic PDAC xenograft model, was able to reduce tumour growth.102 In addition, it has been shown that the viability of cancer cells carrying a mutant KRAS is dependent on Plk1 and that silencing Plk1 leads to the death of cells containing mutant KRAS;103 as described for CDK4/6 inhibitors above, compounds that inhibit Plk1 would be useful in treating cancers that arise from KRAS mutations. Because of its many roles, Plk1 is tightly linked to the actions of other mitotic kinases including Wee1, CDK1 (see above), Chk1 and Aurora A (see below). High expression levels of Plk1 are closely associated with reduced survival in PDAC patients.104
A number of groups have sought to develop Plk1-specific inhibitors, despite the initial clinical promise, the first trials in patients with Plk1 inhibitors have been disappointing.105 In fact, adavosertib, described above, was shown to also bind to Plk1. So much so that, in 2017, Wright and co-workers demonstrated that adavosertib is a potent dual Wee1 and Plk1 inhibitor, which limits its use as a specific molecular probe for Wee1.106 They noted, however, that Plk1 inhibition nevertheless makes important contributions to the single-agent mechanism of action of adavosertib and enhances its anticancer effects. Later, however, Serpico and colleagues revealed that, in the nanomolar concentration range, the pharmacological effects of adavosertib in human cells derive solely from inhibition of Wee1 rather than of concomitant inhibition of Plk1.95 The team showed that phosphorylation of Plk1 substrates is not inhibited by adavosertib and that cells enter mitosis through an overriding of the DNA-repair checkpoint.
Perhaps the best described Plk1 inhibitor is onvansertib (Fig. 17), which was first described by Beria and co-workers in 2011.107 Out of a HTS campaign followed by medicinal chemistry optimisation, the 4,5-dihydro-1H-pyrazolo[4,3-h]quinazoline template had been identified as a good scaffold to obtain potent and selective Plk1 inhibitors. The 5′-position of the aniline was investigated but found that while their test compounds offered an increase in solubility, there was a drastic decrease in Plk1 and cytotoxic activities. When alternative substitution of the pyrazole 1′-position was tested, the researchers obtained increased solubility and an improved selectivity for Plk1 not only over Plk2 and Plk3 but also over a larger panel of kinases, where at least 300-fold selectivity was found.
Fig. 17. Development pathway of onvansertib starting from the pyrazoloquinazoline hit 8 that arose from a high-throughput screening campaign. Specific molecular features of onvansertib are highlighted and discussed in the text.

Onvansertib inhibits Plk1 with IC50 of 2 nM and is more than 1000-fold more selective for Plk1 than its sister isoforms Plk2 and Plk3. The addition of the 2-hydroxyethyl group (red in Fig. 17) also afforded superior pharmacokinetic properties (higher AUC and Cmax, lower clearance, and acceptable oral bioavailability (F = 24%)). A crystal structure of their compound in complex with Plk1 was obtained about which a number of observations could be made. As expected, onvansertib bound in the ATP-pocket where the pyrazolo-quinazoline core is sandwiched between Cys67 and Phe183 and the attached ethyl hydroxyl extends into the ribose pocket. The 2′-trifluoromethoxy group binds in a pocket formed by Arg57 and the hinge segment Leu132-Cys133-Arg134 and multipolar interactions are present between fluorine atoms and the guanidinium group of Arg57 and the backbone carbonyl of Arg134. It was reasoned that the ‘fit’ of the 2′-trifluoromethoxy group in this pocket is the origin of the Plk selectivity over kinases, which typically feature a much bulkier residue at the corresponding position. In addition, the 5′-methylpiperazine moiety contributes to the Plk1 selectivity with respect to Plk2–Plk3 since it establishes a polar interaction with the side chain of Glu140, which is present as a histidine in the other isoforms. The 2′-trifluoromethoxy (shown in blue) makes contact with Leu132 and is essential for providing Plk-family selectivity, since other members have bulkier residues in the position corresponding to Leu132.
The efficacy of onvansertib, in combination nanoliposomal irinotecan, leucovorin, and fluorouracil (standard chemo-therapeutic agents in PDAC), is currently being assessed in a phase II study (NCT04752696) comprising participants with histologically confirmed metastatic PDAC.
Lu and co-workers sought to improve the properties of onvansertib.108 On the basis of the existing structure–activity relationships and crystallographic information obtained from Plk1–onvansertib co-crystal (PDB 2YAC), a set of strategies was proposed to improve Plk1 activity: aromatisation of the pyrazolo[4,3-h]quinazoline core and the replacement of the 2-hydroxyethyl with a more hydrophobic group (blue and yellow respectively in Fig. 18). These changes, which provided a marked improvement in the activity and permeability of the molecule, furnished 9. They also attempted to improve the Plk1 selectivity through modifications made to the 5′-methylpiperazine, since it is here where selectivity for Plk1 over Plk2/Plk3 can be won, but, despite thorough assessment of this group, ultimately no changes were made with respect to onvansertib. As a consequence of their other modifications, however, it was determined that 9 had an IC50 of 0.66 nM (cf. IC50onvansertib 1.06 nM). Further in vitro testing showed additional improvements over onvansertib: an improved antiproliferative activity against HCT116 (human colorectal carcinoma) cells (IC50 = 5 nM versus 45 nM), improved cellular permeability and efflux ratio and more favourable antitumor activity with 116.2% tumour growth inhibition (cf. 43.0% for onvansertib).
Fig. 18. Structure of the Plk1 inhibitor 9, developed by Lu and co-workers, whose aim was to improve the properties of onvansertib.

Structural studies that followed showed that the 1H-pyrazolo[4,3-h]quinazoline core formed a π–π interaction with Phe183. Particularly interesting is the fact that the fluoroethyl group of 9 inserts into the glycine-rich pocket and makes hydrophobic interaction with Leu59, which was not possible with onvansertib, thereby increasing the binding activity. It was also shown that the hydrogen bond formed between the aniline NH and Cys133 is crucial for maintaining activity at Plk1.
In tissue distribution studies, a more significant accumulation of 9 in the colorectum, pancreas, and lung was observed than would otherwise be seen with onvansertib, which raised the possibility of using 9 against tumours of these types.
BI2536 (Fig. 19, box) is an ATP-competitive Plk1 inhibitor with an IC50 value of 0.83 nM for treating acute myeloid, leukaemia and various solid tumours. Unfortunately, multiple phase II clinical studies indicated that BI2536 lacked advanced solid tumour activity, which might be attributed to its low intratumoral drug levels and short half-life in patients.109 Nevertheless, Li and co-workers envisioned that BI2536 represented a good starting point for further optimization to yield new Plk1 inhibitors with improved potency and metabolic stability. Their derived molecule, volasertib (Fig. 19, box) originates from a lead optimisation program centred on BI2536 to improve its potency, selectivity, activity and pharmacokinetic characteristics (Patent Application WO 2004/076454).110Volasertib is highly potent (enzyme IC50 = 0.87 nM, EC50 = 11–37 nM across a panel of cancer cell lines). Volasertib was then tested in a phase I trial in solid tumours and showed positive results: favourable preliminary antitumour activity and a PK profile of volasertib, with manageable toxicities. The most common adverse events were haematological (thrombo-cytopenia and neutropenia) as expected. Further clinical development of volasertib is in progress and phase II trials as combination and monotherapy are scheduled.
Fig. 19. (Left) Structures of BI2536 and volasertib related to their parent structure. (Right) Structure of 10.

Given that the aniline nitrogen in BI2536 is a potential metabolic liability, it was hypothesised that modification of this site may afford optimised potency, solubility, and metabolic stability. A 1,3,4-oxadiazole was introduced to combat the potential metabolic problems of amide but found that the 1,2,4-regioisomer allows appropriate conformational restriction to allow the molecule to bind to Plk1 correctly. The piperazinyl group attached to the oxadiazole was also investigated and found that the piperazine without N-methylation, as in 10 (Fig. 19, right), was the superior candidate from their cohort of molecules. In a kinase profiling assay, 10 exhibited excellent kinase selectivity: of the 217 kinases profiled, only EGFR and highly homologous Plk3 were inhibited by >90%. The optimised compound 10 showed excellent inhibitory activity against Plk1 (IC50 = 0.45 nM) and antiproliferative activity was demonstrated in various breast cancer and leukaemia cell lines. Subsequent cell cycle arrest and cell apoptosis assays indicated that 10 produced cell cycle arrest in the G2 phase and subsequently induced apoptosis.
The development of another Plk1 inhibitor GSK461364 (Fig. 20) was first described in 2009 by Kuntz and co-workers.111 Their high-throughput screen for Plk1 identified several potential starting points. However, a very selective chemotype was discovered from the authors' kinase cross-screening efforts: novel thiophene benzimidazole 11 was the scaffold identified as a promising lead with good enzyme potency and selectivity (Plk1 IC50 61 nM). Hence, their efforts were placed solely on optimising this screening hit.
Fig. 20. Development of GSK461364 from the initial hit compound 11.

SAR analysis of the benzyl ether portion revealed that larger groups at the ortho-position were approximately ten-fold less potent than small lipophilic groups – especially with electron – withdrawing groups – and that substitution at either the meta- or para-positions also resulted in a drop in potency irrespective of the substituent's size. The benzimidazole portion of the molecule was also examined, which offered space at the solvent front for further modification. The desire to develop their Plk1-inhibitor for acceptable IV formulation led to the incorporation of solubility-enhancing substituents on the benzimidazole; substitution at the 6-position of the benzimidazole improved both the solubility and the cellular potency of the molecule, as well as providing selectivity over Plk3. Of the assessed groups, the methylene piperazine provided the best balance of potency and pharmacokinetics. One of the breakthroughs of this medicinal chemistry campaign was the addition of a methyl group to the benzyl carbon of the benzyl ether (marked in Fig. 20). Substitution to give the (R)-enantiomer was significantly more potent than the (S)-enantiomer (IC50 0.63 vs. 25 nM respectively). While small substitutions on the chiral methyl group were tolerated, the simple, unsubstituted methyl proved most potent (Plk1 IC50 1.0 nM). All of these SAR analyses taken together, with development issues accounted for, yielded GSK461364 (Plk1 IC50 1.6 nM). In another study, when combined with the standard-of-care drug gemcitabine, GSK461364 significantly increased cellular apoptosis in cells and reduced tumour growth in an orthotopic PDAC xenograft tumour model.112
Importantly, and more recently, Plk1 inhibition with GSK461364 has been shown to sensitise pancreatic tumours to immune checkpoint therapy.113 It was show that in an inducible transgenic mouse line with specific expression of Plk1 in the pancreas, Plk1 inhibition caused the upregulation of PD-L1 by activating the NFκB pathway. This led to the sensitisation of the tumour to immune checkpoint blockade therapy and suppressed PDAC progression, thereby highlighting the role of Plk1 in potentiating the efficacy of immunotherapy in PDAC.
Inhibitors of Chk1
Checkpoint kinase 1 (Chk1) is a coordinator of the DNA damage response as well as the transition from G1 to S phase and mitotic entry.7 Activation of Chk1 initiates cell cycle checkpoints and leads to cell cycle arrest, DNA repair and cell death in order to impede the progression of damaged cells into mitotic commitment. Inhibition of Chk1 as a therapeutic strategy aims to selectivity potentiate the cytotoxicity of DNA-damaging chemotherapeutics in cell-cycle checkpoint-defective tumour cells while minimizing toxicity to normal cells that are checkpoint-competent.114 A number of these research efforts have resulted in Chk1 inhibitors being evaluated in early phase clinical trials. Unfortunately, however, most of these were terminated for unfavourable toxicity profile or side effects115 that outweigh the modest therapeutic efficacy and, as such, no agent within this class of kinase inhibitors has reached phase III evaluation or FDA approval.116 Nevertheless, targeting Chk1 remains a very promising therapeutic avenue holding considerable potential. Klomp and colleagues have shown that Chk1 protects KRAS-expressing cells from DNA damage and defined this protein as a target for PDAC treatment.117 The inhibition of Chk1 causes compensatory activation of ERK and autophagy pathways.
To the best of our knowledge, no description of the design or discovery process around the Chk1–Wee1 inhibitor LY2880070 other than its structure (Fig. 21) has been made publicly available. It is, however, currently being assessed in a multicentre phase I/II study (Canada and USA) in different solid tumours including advanced and metastatic PDAC (NCT02632448). The aim of this study is to assess the efficacy of LY2880070 both alone and in combination with gemcitabine, since previous phase Ib studies have shown that the combination of these two agents was synergistic and allowed the dosing to be reduced.118 A recent phase I expansion study performed by Huffman and co-workers involved a cohort of patients with metastatic PDAC treated with a combination of low-dose gemcitabine and LY2880070.119 Here, however, despite the in vitro efficacy shown by this combination, the researchers unfortunately found no evidence of clinical activity for this combination.
Fig. 21. Structures of LY2880070 and rabusertib.

Rabusertib (Fig. 21) is another Chk1 inhibitor that has been tested in combination with gemcitabine.120 65 PDAC patients received the combination of rabusertib–gemcitabine while another 34 received gemcitabine alone. However, while the severity of adverse events with the rabusertib–gemcitabine combination was comparable to gemcitabine alone, the combination group did not benefit from any significant improvements in progression-free survival, objective response rate or duration of response. The authors concluded that the combination of rabusertib–gemcitabine was not superior to gemcitabine for the treatment of patients with PDAC. In other cancers, however, rabusertib has shown improved properties and, for example, increases the in vivo response to irinotecan in colon cancer cells.121
Wiechmann and co-workers have shown that combination therapy of irradiation with rabusertib or the FAK inhibitor defactinib offers a substantial sensitisation of radioresistant PDAC cells to radiation, seen in the lowered fraction of cells treated with these agents that survive irradiation.122 Given the selectivity of rabusertib, its radiosensitisation was ascribed to the inhibition of Chk1.
Taken together, though, in comparison to the other targets described here, there are only a limited number of agents that have so far been tested for use against PDAC. That being said, a much larger number are currently in pre-clinical development, such as the picolinonitrile compound described by Jin and co-workers,123 which, in its case, is currently being assessed for use in leukaemia. It is, however, foreseeable that this and other agents would eventually be tested in PDAC models and patients.
Guo and co-workers have, however, shown that repeated treatments of Capan-1 PDAC cells with PARP1 and Chk1 inhibitors leads to drug resistance, migration and invasion.124 The efficacious combination of Chk1 and PARP inhibitors has previously been described, but it was described here that the G2/M arrest and apoptosis induced by the Chk1 inhibitors decreased significantly in the resistant variants of Capan-1 cells when also exposed to olaparib; the combination conferred enhanced migratory and invasive capabilities on the variants. As they point out, this observation will undoubtedly have an impact on the possibility of dosing PARP1 and Chk1 inhibitors and their combinations.
Inhibition of Aurora kinase A
The family of Aurora kinases – of which there are three, A, B and C, in metazoans – have come to be seen as the key orchestrators of mitotic events from centrosome maturation to chromosomal congression and cytokinesis.125 Aurora A is heavily involved in the early events of mitosis such as controlling the assembly and maturation of the centrosome and spindle (Fig. 1), being a critical regulator of the spindle assembly checkpoint. Inhibitors of Aurora kinase, as a consequence, lead to a stalling of cell division. Aurora A is overexpressed in a variety of tumour types including PDAC.126 Since the initial observation of their expression in human cancer tissue in 1998, the Aurora kinases have been the subject of successful research efforts among the academic and industrial oncology communities,127 which have led to more than ten Aurora inhibitors entering early clinical assessment. Two of these have been assessed in PDAC.
Using KRAS-positive lung cancer (H358 and A549) cell lines, Dos Santos and co-workers have shown that targeting Aurora kinase leads to loss of cell viability by promoting cell cycle arrest at G2 and through the subsequent activation of apoptosis in an oncogenic KRAS-dependent manner.128 This backs up their hypothesis that inhibition of Aurora kinases specifically targets KRAS-transformed cells. The same team later showed that Aurora kinase A may act as a biomarker in PDAC that is even indicative of poorer prognoses.129 The authors went on to propose, based on their results, that targeting Aurora kinase A in PDAC cells may reduce their transformed phenotype.
Alisertib (Fig. 22) is a potent inhibitor of Aurora kinase A that was discovered in 2015. Sells and co-workers discovered alisertib starting from the pyrimidobenzazepine BBL22, which was reported to induce G2/M cell cycle arrest and apoptosis in human tumour cell lines.130
Fig. 22. Development of both MLN8054 and alisertib from BBL22.

The authors showed that the introduction of an additional substituent on the 7-phenyl ring in the ortho position (shown in Fig. 22) led to improvements in cellular potency: upon the introduction of fluorine, MLN8054 (Fig. 22) was obtained, which demonstrated an increased potency in cells and a 150-fold selectivity for Aurora A over Aurora B in HCT116 cells (a human colorectal carcinoma cell line) but which exhibited reversible somnolence as dose-limiting toxicity, which was attributed to GABAA binding. The authors then engaged in efforts to widen the therapeutic window of MLN8054 by decreasing the affinity of the molecule for GABAA and to reduce the degree of brain partitioning in Sprague-Dawley rats. Installation of a methoxy group at R1 led to a 3-fold brain AUC reduction but when an additional methoxy group was introduced at R2 (yielding alisertib) an additional reduction in mouse brain partitioning was observed. Alisertib is significantly more potent than MLN8054 (IC50 = 1.2 nM) and is competitive with ATP (Ki 0.3 nM, apparent Koff 2 × 10−4 s−1). Alisertib has a generally tolerable safety profile in humans and has advanced into multiple clinical studies including an open-label phase III trial for patients with relapsed and refractory peripheral T-cell lymphoma (NCT). It is now being investigated in a phase Ib study in patients with advanced solid tumours including pancreatic tumours (NCT02719691).
Alisertib has since been tested in combination with methyltransferase inhibitors where it was found to be effective in reducing the viability of PDAC cells both in vitro and in vivo through the induction of mitotic catastrophe (cell death occurring as a result of improper progression of or entrance to the cell cycle).131 A phase I dose escalation study of alisertib in combination with gemcitabine revealed that the combination shows potential for disease control in heavily pre-treated tumours, despite apparent gastrointestinal and hematologic toxicity.132 The study also established a recommended phase 2 dose of 50 mg alisertib.
Fancelli and co-workers identified danusertib (Fig. 23) – another inhibitor of Aurora kinase A – from optimization efforts on a previously discovered Aurora kinase inhibitor PHA-680632, which had demonstrated high in vitro antiproliferative activity on a wide range of cancer cell lines and significant tumour growth inhibition in different animal tumour models.133 It was hypothesized that the introduction of a pro-R hydrogen-bond acceptor on the methylene of the phenylacetyl would improve binding through favourable interactions with Lys162. On the other hand, substitutions at the pro-S position were thought to be less favourable due to the proximity of Val147. Crystallographic studies confirmed the binding mode of danusertib, in which case the hydrogen-bond acceptor was the R-methoxy group, proximal to Lys162. Danusertib was described as “ATP competitive” and an apparent Ki of 2.5 nM. It was tested in a number of human cancer cell lines and showed potent antiproliferative activity. In 2014, danusertib completed phase II trials for metastatic hormone refractory prostate cancer, but unfortunately showed only minimal efficacy, where only 14% of treated patients had stable disease for ≥6 months.
Fig. 23. Development of danusertib from PHA-680632.

More recently, however, Kirbiyik and co-workers investigated the cytotoxicity, apoptotic potential and its impact on the cell cycle of danusertib in human CFPAC-1 ductal adenocarcinoma cells.134 An IC50 value of approximately 400 nM was noted, at which concentration-dependent apoptosis was induced in CFPAC-1 cells. They also noted significant populations of cells arrested in the S (32%) and G2 (11%) phases. This led them to the conclusion that danusertib induced a significant effect of cytotoxic, apoptotic and cell cycle arrest in CFPAC-1 cells.
Perspectives
One of the biggest turning points in modern cancer therapeutics has been the advent of immune checkpoint blockade (i.e., an immunotherapy). Checkpoint blockade has now become standard-of-care therapy for melanoma, increasing the five-year overall survival rate across all stages to around 50% of all patients (compared to only 10% in the past).135 It is also applied to the treatment of various other cancers with varying degrees of efficacy.
Despite these successes, however, a significant proportion of patients fail to achieve any long-term benefit. A number of cancers, including PDAC, do not respond to immunotherapy at all.35 The exact mechanisms surrounding this failure are still not fully understood.
While the primary consequence of CDK4/CDK6 inhibition is the establishment of cell cycle arrest, various reports have shown that CDK4/6 inhibitors are also able to promote various immunomodulatory effects through their effects not only on malignant cells but also on immune cell populations of the tumour microenvironment (Fig. 24).136 Numerous mouse models have shown that the inhibition of CDK4/6 leads to a significant enhancement of the anti-tumour efficacy of immune checkpoint (PD-1/PD-L1) inhibition.136,137 The authors of these studies have attributed the desirable outcome of this combination to the intrinsic immunomodulatory activity of CDK4/6 inhibitors,138 such as through altered expression levels of MHC I and PD-L1 as well as increased production of T cell chemoattractants by the tumour cells. In certain other oncological settings, CDK4 itself has been shown to regulate PD-L1 protein stability and/or promote PD-L1 degradation (effect).139 Other studies have demonstrated a suppression of tumour growth by CDK4/6 inhibitors by additional mechanisms including through improved anti-tumour immune responses.140Dinaciclib, for example, has been shown to enhance anti-PD1 mediated tumour suppression.141
Fig. 24. Immunomodulatory effects of CDK4/6 inhibitors. Numerous mouse models have shown that the inhibition of CDK4/6 leads to a significant enhancement of the anti-tumour efficacy of immune checkpoint inhibition by increasing antigen presentation (1), reducing the proliferation of regulatory T cells (2, here shown by the transcription of proliferative genes) or the increased infiltration of T cells into the tumour through the production and release of T cell chemoattractants (3). In addition, by way of example, Wee1 kinase has been shown to sensitise cancer cells to immunotherapy by reversing the activation of G2/M cell cycle checkpoint (4).

Immunomodulatory activity has also been observed in other mitotic kinases. For instance, the inhibition of Plk1 has been shown to upregulate the expression of PD-L1 in PDAC, which stimulates antitumour immunity and increases the sensitivity of these tumours to immunotherapy.142 Wee1 is also implicated in immunotherapeutic options: Sun and co-workers have shown that inhibition of Wee1 inhibition sensitises cancer cells to immunotherapy by reversing the activation of G2/M cell cycle checkpoint.90Adavosertib in combination with the anti-PD-L1 antibody durvalumab has also shown evidence of antitumor activity in a phase I trial to assess safety and tolerability in solid tumours (NCT02617277).143 In melanoma cells, Aurora kinase inhibition has been shown to cause sensitisation to T-cell-mediated cytotoxicity.144 While the precise mechanisms surrounding these immunomodulatory phenomena remain to be fully elucidated, the encouraging pre-clinical results hold promise for the design of newer and more robust therapeutic regimens that combine these treatments.
Signalling through the Ras axis, headed by one of several cell-surface receptors, leads, among other cellular outcomes, to the transcriptional and functional regulation of CDK4/6 (and cyclin D).145 Mounting preclinical evidence points to a possible role of the Ras proteins – such as KRAS – in the induction of resistance to CDK inhibitors.145 Approximately 10% of patients with a resistance to CDK inhibitors bore activating mutation of Ras, which were absent in tumours that were sensitive to CDK inhibition.146 At the head of the Ras signalling axis is, of course, KRAS. It is worth reiterating that >90% of pancreatic tumours bear activating mutations to KRAS.12 Emerging data highlights the action of mutant KRAS on CDK5 (which was not discussed in detail in this manuscript) and its activators to advance the malignant progression, migration, and invasion of PDAC cells.24
As we have described above, KRAS signalling intersects on the actions of a constellation of mitotic kinases including CDKs, Wee1, Plk1 and Aurora A (vide supra). Combination therapies that target KRAS as well as one of these targets may offer further benefit to patients. The combination therapy with CDK4/6 inhibitors to treat KRAS-mutant PDAC has already shown promising preclinical efficacy.147 While currently limited in scope to lung cancer, Yamamoto and co-workers have recently shown that adavosertib is capable of enhancing the anti-tumour activity of sotorasib in both in vitro and in vivo preclinical models.148
Conclusions
The constellation of mitotic kinases are central players in the proper functioning of the cell cycle and without them, or without their proper functioning, cells can begin to proliferate indiscriminately, which leads to tumour formation. The central role of these kinases has been recognised in breast cancer, where palbociclib, ribociclib and abemaciclib are approved and are used in combination with hormone therapy, but concerted efforts are being made not only to extend the scope of these agents to other cancers but also to introduce new agents that can circumvent the problems associated with them (i.e., toxicity and selectivity issues). In addition, and especially given the heterogeneous nature of pancreatic tumours, numerous studies have pointed to the benefit of combination therapies. Beneficial, additive effects of inhibiting CDKs – or other mitotic kinases – alongside other cellular protein targets have been observed in studies described herein and continue to offer promise through the exploration of this approach as an alternative to conventional treatment strategies, particularly against targets that may have more relevance for one cancer type over another e.g., KRASG12D in PDAC. KRAS, in particular, because of the large extent to which mitotic kinases intersect on its signalling pathways, may offer the largest potential for successful combination therapy in the future.
It was shown that immunotherapy, despite being notoriously ineffective in PDAC, stands poised to have its effects boosted by combination therapies with mitotic kinases; inhibition of CDK4/6, Wee1 or Plk1 has shown encouraging pre-clinical results that have sensitised pancreatic tumours to anti-PD-L1 therapy. If the appropriate molecular partner for combination can be leveraged, we can remain hopeful that a certain proportion of PDAC patients may become responsive to this type of therapy. It should be pointed out that there remains a large potential for the exploration of other mitotic kinases (e.g., Bub1, Nek1, or less well described CDKs such as CDK12 and CDK13); these targets are less well described in PDAC, but as their roles become clearer so too may their potential to offer more benefit to pancreatic patients.
Finally, the current therapeutic landscape should be compared with that of ten years ago, which was very different. 2024 marks ten years since the landmark approval of the anti-PD-1 antibody nivolumab and, since then, the clinical landscape around several cancers, melanoma first among them, has been transformed by this and other checkpoint inhibitors. While there have not been any new approvals against PDAC in this period, we can remain optimistic for a similar similarly transformative breakthrough in the treatment of PDAC over the years to come and to imagine how different the pre-clinical environment will be in another ten years.
Author contributions
Thomas M. A. Barlow: conceptualisation, writing – original draft, review & editing; Ilse Rooman: supervision, writing – reviewing and editing; Steven Ballet: supervision, writing – reviewing and editing.
Conflicts of interest
There are no conflicts to declare.
Acknowledgments
TMAB is funded by the Flemish Government's Industrial Research Fund (IOF). SB is supported by the Strategic Research Program (SRP95) of the Research Council of the Vrije Universiteit Brussel. IR is funded by Research Foundation Flanders Senior Research Project G0A7322N, by the Senior Research Fellowship OZRSRF44 of the Research Council Vrije Universiteit Brussel, and by a Strategic Research Program (SRP65) of the Research Council of the Vrije Universiteit Brussel. IR and SB are funded by TRANSCAN-3 ERA-NET JTC 2021 (FWO & ERA-NET)(G0L0722N), by the Interdisciplinary Research Project IRP9 of the Research Council of the Vrije Universiteit Brussel, by the Stichting Tegen Kanker project grant (InterCePPT) and by the Scientific Fund Willy Gepts (WFWG SPARTAN). Fig. 1, 2 and 24 were created using BioRender (https://www.biorender.com). The authors thank Dr. Kevin Van holsbeeck for the creation of modelling images in Fig. 4, 10 and 13.
Biographies
Biography
Thomas M. A. Barlow.

Thomas M. A. Barlow completed his doctoral research in medicinal chemistry in the Research Group of Organic Chemistry (ORGC) at the Vrije Universiteit Brussel under Prof. Steven Ballet, which investigated the use of tandem multicomponent-‘click’ reactions to generate privileged scaffolds and other drug-like macrocycles. He now supports medicinal chemistry and other fundamental research within the group.
Biography
Ilse Rooman.

Ilse Rooman, PhD, is a Full Professor at Vrije Universiteit Brussel and Director of the Visual and Spatial Tissue Analysis core facility. She has focused her career on pancreatic biology, transitioning from cell therapy for diabetes to pancreatic cancer research. After leading a team at Australia's Garvan Institute, she returned to Belgium with an Odysseus fellowship to establish her current lab. Her work explores exocrine cell plasticity, tumour initiation, and novel pancreatic cell types, using integrative – omics approaches. Her team also investigates axon guidance genes in cancer, contributing to both fundamental and translational oncology research.
Biography
Steven Ballet.

Prof. Steven Ballet obtained his PhD from Vrije Universiteit Brussel in 2007, followed by postdoctoral research in Australia (peptide stapling via metathesis) and Canada (opioid peptide design). Since 2010, he has been a faculty member at VUB, heading the Research Group of Organic Chemistry. His research interests centre on peptide and peptidomimetic research including protein mimicry, peptide hydrogels, peptide secondary structure mimetics, and transition metal-catalyzed peptide modifications. He has authored more than 150 papers, six patents, and several book chapters.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.
References
- Poratti M. Marzaro G. Eur. J. Med. Chem. 2019;172:143–153. doi: 10.1016/j.ejmech.2019.03.064. [DOI] [PubMed] [Google Scholar]
- Rahib L. Smith B. D. Aizenberg R. Rosenzweig A. B. Fleshman J. M. Matrisian L. M. Cancer Res. 2014;74:2913–2921. doi: 10.1158/0008-5472.CAN-14-0155. [DOI] [PubMed] [Google Scholar]
- Hall B. R. Cannon A. Atri P. Wichman C. S. Smith L. M. Ganti A. K. Are C. Sasson A. R. Kumar S. Batra S. K. Oncotarget. 2018;9:19396–19405. doi: 10.18632/oncotarget.25036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. B. Yang Z. H. Guo Q. Q. Cell Commun. Signaling. 2021;19:117. doi: 10.1186/s12964-021-00789-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J. Russell C. C. Scarlett C. J. McCluskey A. RSC Med. Chem. 2020;11:164–183. doi: 10.1039/c9md00447e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al Mutar S. Barrows N. Abunafeesa H. Getchell G. Watson E. Pirzadeh-Miller S. Fentor R. Hill M. Stricker C. T. Herman J. M. Polanco P. M. Aguilera T. A. J. Clin. Oncol. 2025;43:23243. [Google Scholar]
- Dreyer S. B. Pinese M. Jamieson N. B. Scarlett C. J. Colvin E. K. Pajic M. Johns A. L. Humphris J. L. Wu J. Cowley M. J. Chou A. Nagrial A. M. Chantrill L. Chin V. T. Jones M. D. Moran-Jones K. Carter C. R. Dickson E. J. Samra J. S. Merrett N. D. Gill A. J. Kench J. G. Duthie F. Miller D. K. Cooke S. Aust D. Knösel T. Rümmele P. Grützmann R. Pilarsky C. Nguyen N. Q. Musgrove E. A. Bailey P. J. McKay C. J. Biankin A. V. Chang D. K. Ann. Surg. 2020;272:366–376. doi: 10.1097/SLA.0000000000003143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X. Tang W. Marquez R. T. Li K. Highfill C. A. He F. Lian J. Lin J. Fuchs J. R. Ji M. Li L. Xu L. Oncotarget. 2016;7:11708–11723. doi: 10.18632/oncotarget.7336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey P. Chang D. K. Nones K. Johns A. L. Patch A.-M. Gingras M.-C. Miller D. K. Christ A. N. Bruxner T. J. C. Quinn M. C. Nourse C. Murtaugh L. C. Harliwong I. Idrisoglu S. Manning S. Nourbakhsh E. Wani S. Fink L. Holmes O. Chin V. Anderson M. J. Kazakoff S. Leonard C. Newell F. Waddell N. Wood S. Xu Q. Wilson P. J. Cloonan N. Kassahn K. S. Taylor D. Quek K. Robertson A. Pantano L. Mincarelli L. Sanchez L. N. Evers L. Wu J. Pinese M. Cowley M. J. Jones M. D. Colvin E. K. Nagrial A. M. Humphrey E. S. Chantrill L. A. Mawson A. Humphris J. Chou A. Pajic M. Scarlett C. J. Pinho A. V. Giry-Laterriere M. Rooman I. Samra J. S. Kench J. G. Lovell J. A. Merrett N. D. Toon C. W. Epari K. Nguyen N. Q. Barbour A. Zeps N. Moran-Jones K. Jamieson N. B. Graham J. S. Duthie F. Oien K. Hair J. Grützmann R. Maitra A. Iacobuzio-Donahue C. A. Wolfgang C. L. Morgan R. A. Lawlor R. T. Corbo V. Bassi C. Rusev B. Capelli P. Salvia R. Tortora G. Mukhopadhyay D. Petersen G. M. Munzy D. M. Fisher W. E. Karim S. A. Eshleman J. R. Hruban R. H. Pilarsky C. Morton J. P. Sansom O. J. Scarpa A. Musgrove E. A. Bailey U.-M. H. Hofmann O. Sutherland R. L. Wheeler D. A. Gill A. J. Gibbs R. A. Pearson J. V. Waddell N. Biankin A. V. Grimmond S. M. I. Australian Pancreatic Cancer Genome Nature. 2016;531:47–52. doi: 10.1038/nature16965. [DOI] [PubMed] [Google Scholar]
- Martens S. Lefesvre P. Nicolle R. Biankin A. V. Puleo F. Van Laethem J. L. Rooman I. Ann. Oncol. 2019;30:1428–1436. doi: 10.1093/annonc/mdz181. [DOI] [PubMed] [Google Scholar]
- Ju H.-Q. Ying H. Tian T. Ling J. Fu J. Lu Y. Wu M. Yang L. Achreja A. Chen G. Nat. Commun. 2017;8:14437. doi: 10.1038/ncomms14437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant K. L. Mancias J. D. Kimmelman A. C. Der C. J. Trends Biochem. Sci. 2014;39:91–100. doi: 10.1016/j.tibs.2013.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teague A. Lim K. H. Wang-Gillam A. Ther. Adv. Med. Oncol. 2015;7:68–84. doi: 10.1177/1758834014564775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burris, 3rd H. A. Moore M. J. Andersen J. Green M. R. Rothenberg M. L. Modiano M. R. Cripps M. C. Portenoy R. K. Storniolo A. M. Tarassoff P. Nelson R. Dorr F. A. Stephens C. D. Von Hoff D. D. J. Clin. Oncol. 1997;15:2403–2413. doi: 10.1200/JCO.1997.15.6.2403. [DOI] [PubMed] [Google Scholar]
- Ostrem J. M. Peters U. Sos M. L. Wells J. A. Shokat K. M. Nature. 2013;503:548–551. doi: 10.1038/nature12796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singhal A. Styers H. C. Rub J. Li Z. Torborg S. R. Kim J. Y. Grbovic-Huezo O. Feng H. Tarcan Z. C. Sahin Ozkan H. Hallin J. Basturk O. Yaeger R. Christensen J. G. Betel D. Yan Y. Chio I. I. C. de Stanchina E. Tammela T. Cancer Discovery. 2024;14:2122–2134. doi: 10.1158/2159-8290.CD-24-0740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dilly J. Hoffman M. T. Abbassi L. Li Z. Paradiso F. Parent B. D. Hennessey C. J. Jordan A. C. Morgado M. Dasgupta S. Uribe G. A. Yang A. Kapner K. S. Hambitzer F. P. Qiang L. Feng H. Geisberg J. Wang J. Evans K. E. Lyu H. Schalck A. Feng N. Lopez A. M. Bristow C. A. Kim M. P. Rajapakshe K. I. Bahrambeigi V. Roth J. A. Garg K. Guerrero P. A. Stanger B. Z. Cristea S. Lowe S. W. Baslan T. Van Allen E. M. Mancias J. D. Chan E. Anderson A. Katlinskaya Y. V. Shalek A. K. Hong D. S. Pant S. Hallin J. Anderes K. Olson P. Heffernan T. P. Chugh S. Christensen J. G. Maitra A. Wolpin B. M. Raghavan S. Nowak J. A. Winter P. S. Dougan S. K. Aguirre A. J. Cancer Discovery. 2024;14:2135–2161. doi: 10.1158/2159-8290.CD-24-0177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasko U. N. Jiang J. Dalton T. C. Curiel-Garcia A. Edwards A. C. Wang Y. Lee B. Orlen M. Tian S. Stalnecker C. A. Drizyte-Miller K. Menard M. Dilly J. Sastra S. A. Palermo C. F. Hasselluhn M. C. Decker-Farrell A. R. Chang S. Jiang L. Wei X. Yang Y. C. Helland C. Courtney H. Gindin Y. Muonio K. Zhao R. Kemp S. B. Clendenin C. Sor R. Vostrejs W. P. Hibshman P. S. Amparo A. M. Hennessey C. Rees M. G. Ronan M. M. Roth J. A. Brodbeck J. Tomassoni L. Bakir B. Socci N. D. Herring L. E. Barker N. K. Wang J. Cleary J. M. Wolpin B. M. Chabot J. A. Kluger M. D. Manji G. A. Tsai K. Y. Sekulic M. Lagana S. M. Califano A. Quintana E. Wang Z. Smith J. A. M. Holderfield M. Wildes D. Lowe S. W. Badgley M. A. Aguirre A. J. Vonderheide R. H. Stanger B. Z. Baslan T. Der C. J. Singh M. Olive K. P. Nature. 2024;629:927–936. doi: 10.1038/s41586-024-07379-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pecoraro C. Carbone D. Cascioferro S. M. Parrino B. Diana P. Curr. Med. Chem. 2023;30:776–782. doi: 10.2174/0929867329666220729152741. [DOI] [PubMed] [Google Scholar]
- Vallés-Martí A. Mantini G. Manoukian P. Waasdorp C. Sarasqueta A. F. de Goeij-de Haas R. R. Henneman A. A. Piersma S. R. Pham T. V. Knol J. C. Giovannetti E. Bijlsma M. F. Jiménez C. R. Cell Rep. 2023;42:112581. doi: 10.1016/j.celrep.2023.112581. [DOI] [PubMed] [Google Scholar]
- Chambard J. C. Lefloch R. Pouysségur J. Lenormand P. Biochim. Biophys. Acta. 2007;1773:1299–1310. doi: 10.1016/j.bbamcr.2006.11.010. [DOI] [PubMed] [Google Scholar]
- Wang S. Zheng Y. Yang F. Zhu L. Zhu X.-Q. Wang Z.-F. Wu X.-L. Zhou C.-H. Yan J.-Y. Hu B.-Y. Kong B. Fu D.-L. Bruns C. Zhao Y. Qin L.-X. Dong Q.-Z. Signal Transduction Targeted Ther. 2021;6:249. doi: 10.1038/s41392-021-00659-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorda R. Hendrychová D. Voller J. i. Řezníčková E. Gucký T. s. Kryštof V. r. J. Med. Chem. 2018;61:9105–9120. doi: 10.1021/acs.jmedchem.8b00049. [DOI] [PubMed] [Google Scholar]
- García-Reyes B. Kretz A.-L. Ruff J.-P. Von Karstedt S. Hillenbrand A. Knippschild U. Henne-Bruns D. Lemke J. Int. J. Mol. Sci. 2018;19:3219. doi: 10.3390/ijms19103219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao Z. F. Lin N. H. Anti-Cancer Agents Med. Chem. 2006;6:377–388. doi: 10.2174/187152006777698132. [DOI] [PubMed] [Google Scholar]
- Matthews H. K. Bertoli C. de Bruin R. A. M. Nat. Rev. Mol. Cell Biol. 2022;23:74–88. doi: 10.1038/s41580-021-00404-3. [DOI] [PubMed] [Google Scholar]
- Goel S. Bergholz J. S. Zhao J. J. Nat. Rev. Cancer. 2022;22:356–372. doi: 10.1038/s41568-022-00456-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huse M. Kuriyan J. Cell. 2002;109:275–282. doi: 10.1016/s0092-8674(02)00741-9. [DOI] [PubMed] [Google Scholar]
- Kornev A. P. Taylor S. S. Trends Biochem. Sci. 2015;40:628–647. doi: 10.1016/j.tibs.2015.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M. P. Endicott J. A. Noble M. E. M. Essays Biochem. 2017;61:439–452. doi: 10.1042/EBC20170040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z. Wang X. Eksterowicz J. Gribble, Jr. M. W. Alba G. Q. Ayres M. Carlson T. J. Chen A. Chen X. Cho R. Connors R. V. DeGraffenreid M. Deignan J. T. Duquette J. Fan P. Fisher B. Fu J. Huard J. N. Kaizerman J. Keegan K. S. Li C. Li K. Li Y. Liang L. Liu W. Lively S. E. Lo M.-C. Ma J. McMinn D. L. Mihalic J. T. Modi K. Ngo R. Pattabiraman K. Piper D. E. Queva C. Ragains M. L. Suchomel J. Thibault S. Walker N. Wang X. Wang Z. Wanska M. Wehn P. M. Weidner M. F. Zhang A. J. Zhao X. Kamb A. Wickramasinghe D. Dai K. McGee L. R. Medina J. C. J. Med. Chem. 2014;57:3430–3449. doi: 10.1021/jm500118j. [DOI] [PubMed] [Google Scholar]
- Malumbres M. Barbacid M. Nat. Rev. Cancer. 2009;9:153–166. doi: 10.1038/nrc2602. [DOI] [PubMed] [Google Scholar]
- Asghar U. Witkiewicz A. K. Turner N. C. Knudsen E. S. Nat. Rev. Drug Discovery. 2015;14:130–146. doi: 10.1038/nrd4504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Q. Guo X. Wang M. Li Y. Sun X. Li Q. J. Hematol. Oncol. 2020;13:41. doi: 10.1186/s13045-020-00880-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullman N. A. Burchard P. R. Dunne R. F. Linehan D. C. J. Clin. Oncol. 2022;40:2789–2805. doi: 10.1200/JCO.21.02616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiggens E. Mortoglou M. Grant G. H. Uysal-Onganer P. Healthcare. 2021;9:1478. doi: 10.3390/healthcare9111478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvador-Barbero B. Álvarez-Fernández M. Zapatero-Solana E. El Bakkali A. Menéndez M. D. C. López-Casas P. P. Di Domenico T. Xie T. VanArsdale T. Shields D. J. Hidalgo M. Malumbres M. Cancer Cell. 2020;37:340–353. doi: 10.1016/j.ccell.2020.01.007. [DOI] [PubMed] [Google Scholar]
- Toogood P. L. Harvey P. J. Repine J. T. Sheehan D. J. VanderWel S. N. Zhou H. Keller P. R. McNamara D. J. Sherry D. Zhu T. Brodfuehrer J. Choi C. Barvian M. R. Fry D. W. J. Med. Chem. 2005;48:2388–2406. doi: 10.1021/jm049354h. [DOI] [PubMed] [Google Scholar]
- Shah A. Bloomquist E. Tang S. Fu W. Bi Y. Liu Q. Yu J. Zhao P. Palmby T. R. Goldberg K. B. Clin. Cancer Res. 2018;24:2999–3004. doi: 10.1158/1078-0432.CCR-17-2369. [DOI] [PubMed] [Google Scholar]
- Chen P. Lee N. V. Hu W. Xu M. Ferre R. A. Lam H. Bergqvist S. Solowiej J. Diehl W. He Y. A. Yu X. Nagata A. VanArsdale T. Murray B. W. Mol. Cancer Ther. 2016;15:2273–2281. doi: 10.1158/1535-7163.MCT-16-0300. [DOI] [PubMed] [Google Scholar]
- Cejuela M. Gil-Torralvo A. Castilla M. Domínguez-Cejudo M. Falcón A. Benavent M. Molina-Pinelo S. Ruiz-Borrego M. Salvador Bofill J. Int. J. Mol. Sci. 2023;24:8488. doi: 10.3390/ijms24108488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhir T. Schultz C. W. Jain A. Brown S. Z. Haber A. Goetz A. Xi C. Su G. H. Xu L. Posey III J. Mol. Cancer Res. 2019;17:2029–2041. doi: 10.1158/1541-7786.MCR-19-0589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg B. A. Wang H. Witkiewicz A. K. Marshall J. L. He A. R. Vail P. Knudsen E. S. Pishvaian M. J. J. Pancreat. Cancer. 2020;6:45–54. doi: 10.1089/pancan.2020.0005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marra A. Curigliano G. npj Breast Cancer. 2019;5:27. doi: 10.1038/s41523-019-0121-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlovic D. Niciforovic D. Papic D. Milojevic K. Markovic M. Ther. Adv. Med. Oncol. 2023;15:17588359231205848. doi: 10.1177/17588359231205848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X. Q. Pan X. H. Wang T. T. Wang J. Yang B. He Q. J. Ding L. Acta Pharmacol. Sin. 2021;42:171–178. doi: 10.1038/s41401-020-0416-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan H. Ma X. Yan G. Luo M. Zhong X. Lan S. Yang J. Liu Y. Pu C. Tong Y. Li R. Eur. J. Med. Chem. 2021;219:113432. doi: 10.1016/j.ejmech.2021.113432. [DOI] [PubMed] [Google Scholar]
- Fang L. Chu M. Yan C. Liu Y. Zhao Z. Bioorg. Med. Chem. 2023;84:117263. doi: 10.1016/j.bmc.2023.117263. [DOI] [PubMed] [Google Scholar]
- Adon T. Shanmugarajan D. Kumar H. Y. RSC Adv. 2021;11:29227–29246. doi: 10.1039/d1ra03820f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian C. Wei Y. Li J. Huang Z. Wang Q. Lin Y. Lv X. Chen Y. Fan Y. Sun P. Xiang R. Chang A. Yang S. Int. J. Mol. Sci. 2022;23:2892. doi: 10.3390/ijms23052892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H. Wei M. Xu J. Hua J. Liang C. Meng Q. Zhang Y. Liu J. Zhang B. Yu X. Shi S. Mol. Cancer. 2020;19:49. doi: 10.1186/s12943-020-01167-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwiatkowski N. Zhang T. Rahl P. B. Abraham B. J. Reddy J. Ficarro S. B. Dastur A. Amzallag A. Ramaswamy S. Tesar B. Jenkins C. E. Hannett N. M. McMillin D. Sanda T. Sim T. Kim N. D. Look T. Mitsiades C. S. Weng A. P. Brown J. R. Benes C. H. Marto J. A. Young R. A. Gray N. S. Nature. 2014;511:616–620. doi: 10.1038/nature13393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapenna S. Giordano A. Nat. Rev. Drug Discovery. 2009;8:547–566. doi: 10.1038/nrd2907. [DOI] [PubMed] [Google Scholar]
- Patel H. Abduljabbar R. Lai C.-F. Periyasamy M. Harrod A. Gemma C. Steel J. H. Patel N. Busonero C. Jerjees D. Clin. Cancer Res. 2016;22:5929–5938. doi: 10.1158/1078-0432.CCR-15-1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marineau J. J. Hamman K. B. Hu S. Alnemy S. Mihalich J. Kabro A. Whitmore K. M. Winter D. K. Roy S. Ciblat S. Ke N. Savinainen A. Wilsily A. Malojcic G. Zahler R. Schmidt D. Bradley M. J. Waters N. J. Chuaqui C. J. Med. Chem. 2022;65:1458–1480. doi: 10.1021/acs.jmedchem.1c01171. [DOI] [PubMed] [Google Scholar]
- Lu P. Geng J. Zhang L. Wang Y. Niu N. Fang Y. Liu F. Shi J. Zhang Z. G. Sun Y. W. Wang L. W. Tang Y. Xue J. Oncogene. 2019;38:3932–3945. doi: 10.1038/s41388-019-0701-1. [DOI] [PubMed] [Google Scholar]
- Zeng S. Lan B. Ren X. Zhang S. Schreyer D. Eckstein M. Yang H. Britzen-Laurent N. Dahl A. Mukhopadhyay D. Chang D. Kutschick I. Pfeffer S. Bailey P. Biankin A. Grützmann R. Pilarsky C. J. Exp. Clin. Cancer Res. 2022;41:241. doi: 10.1186/s13046-022-02443-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson C. M. Liang Y. Leggett A. Park W. D. Li L. Mills C. E. Elsarrag S. Z. Ficarro S. B. Zhang T. Düster R. Cell Chem. Biol. 2019;26:792–803. doi: 10.1016/j.chembiol.2019.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang A. Jiang J. Li Z. Kapner K. S. Feng H. Lowder K. E. Kuljanin M. Johnson W. Uribe G. Neggers J. E. Cancer Res. 2022;82:A057. [Google Scholar]
- Thode T. Li Z. Weston A. Soldi R. Kaadige M. R. Vankayalapti H. Sharma S. Cancer Res. 2019;79:LB107. [Google Scholar]
- Bashir B. Sharma M. Juric D. Papadopoulos K. P. Hamilton E. P. Richardson D. L. Shapiro G. Sahai V. Mettu N. B. Wainberg Z. A. Alese O. B. Dragovich T. Hodgson G. Henry S. Hall T. Paul S. Roth D. A. Kelly M. Abdul Malak T. Klimek V. J. Clin. Oncol. 2023;41:3080. [Google Scholar]
- Sunada S. Saito H. Zhang D. Xu Z. Miki Y. Biochem. Biophys. Res. Commun. 2021;550:56–61. doi: 10.1016/j.bbrc.2021.02.117. [DOI] [PubMed] [Google Scholar]
- Neganova I. Tilgner K. Buskin A. Paraskevopoulou I. Atkinson S. P. Peberdy D. Passos J. F. Lako M. Cell Death Dis. 2014;5:e1508. doi: 10.1038/cddis.2014.464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wijnen R. Pecoraro C. Carbone D. Fiuji H. Avan A. Peters G. J. Giovannetti E. Diana P. Cancers. 2021;13:4389. doi: 10.3390/cancers13174389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbone D. Pecoraro C. Panzeca G. Xu G. Roeten M. S. F. Cascioferro S. Giovannetti E. Diana P. Parrino B. Mar. Drugs. 2023;21:412. doi: 10.3390/md21070412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Matre S. Huq S. Akana L. Eldridge D. E. Zuniga O. Rodrigues H. Wolfe A. R. Radiother. Oncol. 2024;200:110531. doi: 10.1016/j.radonc.2024.110531. [DOI] [PubMed] [Google Scholar]
- Bubin R. Uljanovs R. Strumfa I. Int. J. Mol. Sci. 2023;24:1–23. doi: 10.3390/ijms24087030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brasca M. G. Amboldi N. Ballinari D. Cameron A. Casale E. Cervi G. Colombo M. Colotta F. Croci V. D'Alessio R. Fiorentini F. Isacchi A. Mercurio C. Moretti W. Panzeri A. Pastori W. Pevarello P. Quartieri F. Roletto F. Traquandi G. Vianello P. Vulpetti A. Ciomei M. J. Med. Chem. 2009;52:5152–5163. doi: 10.1021/jm9006559. [DOI] [PubMed] [Google Scholar]
- Abdullah M. Guruprasad L. Struct. Chem. 2019;30:213–226. [Google Scholar]
- Aspeslagh S. Shailubhai K. Bahleda R. Gazzah A. Varga A. Hollebecque A. Massard C. Spreafico A. Reni M. Soria J.-C. Cancer Chemother. Pharmacol. 2017;79:1257–1265. doi: 10.1007/s00280-017-3303-z. [DOI] [PubMed] [Google Scholar]
- Parry D. Guzi T. Shanahan F. Davis N. Prabhavalkar D. Wiswell D. Seghezzi W. Paruch K. Dwyer M. P. Doll R. Nomeir A. Windsor W. Fischmann T. Wang Y. Oft M. Chen T. Kirschmeier P. Lees E. M. Mol. Cancer Ther. 2010;9:2344–2353. doi: 10.1158/1535-7163.MCT-10-0324. [DOI] [PubMed] [Google Scholar]
- Martin M. P. Olesen S. H. Georg G. I. Schönbrunn E. ACS Chem. Biol. 2013;8:2360–2365. doi: 10.1021/cb4003283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Z. Sun Y. Liang L. Lu W. Luo B. Wu Z. Huo B. Hu Y. Huang P. Wu Q. Wen S. J. Med. Chem. 2022;65:6573–6592. doi: 10.1021/acs.jmedchem.1c01876. [DOI] [PubMed] [Google Scholar]
- Almehmadi S. J. Alsaedi A. M. R. Harras M. F. Farghaly T. A. Bioorg. Chem. 2021;117:105431. doi: 10.1016/j.bioorg.2021.105431. [DOI] [PubMed] [Google Scholar]
- Feldmann G. Mishra A. Bisht S. Karikari C. Garrido-Laguna I. Rasheed Z. Ottenhof N. A. Dadon T. Alvarez H. Fendrich V. Rajeshkumar N. V. Matsui W. Brossart P. Hidalgo M. Bannerji R. Maitra A. Nelkin B. D. Cancer Biol. Ther. 2011;12:598–609. doi: 10.4161/cbt.12.7.16475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy A. G. Zahurak M. Shah M. Weekes C. D. Hansen A. Siu L. L. Spreafico A. LoConte N. Anders N. M. Miles T. Clin. Transl. Sci. 2020;13:1178–1188. doi: 10.1111/cts.12802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y. Zheng Z. Xiao L. Chen Y. Liu X. Long D. Chai L. Li Y. Tan C. Anti-Cancer Drugs. 2024;35:140–154. doi: 10.1097/CAD.0000000000001545. [DOI] [PubMed] [Google Scholar]
- Wang Q. Bode A. M. Zhang T. npj Precis. Oncol. 2023;7:58. doi: 10.1038/s41698-023-00407-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Y. Chen Y. Chen J. Dong J. Cancers. 2023;15:5424. [Google Scholar]
- Kretz A. L. Schaum M. Richter J. Kitzig E. F. Engler C. C. Leithäuser F. Henne-Bruns D. Knippschild U. Lemke J. Tumour Biol. 2017;39:1010428317694304. doi: 10.1177/1010428317694304. [DOI] [PubMed] [Google Scholar]
- Lücking U. Scholz A. Lienau P. Siemeister G. Kosemund D. Bohlmann R. Briem H. Terebesi I. Meyer K. Prelle K. Denner K. Bömer U. Schäfer M. Eis K. Valencia R. Ince S. von Nussbaum F. Mumberg D. Ziegelbauer K. Klebl B. Choidas A. Nussbaumer P. Baumann M. Schultz-Fademrecht C. Rühter G. Eickhoff J. Brands M. ChemMedChem. 2017;12:1776–1793. doi: 10.1002/cmdc.201700447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lücking U. Org. Chem. Front. 2019;6:1319–1324. [Google Scholar]
- Ruff J. P. Kretz A. L. Kornmann M. Henne-Bruns D. Lemke J. Traub B. Anticancer Res. 2021;41:5973–5985. doi: 10.21873/anticanres.15416. [DOI] [PubMed] [Google Scholar]
- Lücking U. Kosemund D. Böhnke N. Lienau P. Siemeister G. Denner K. Bohlmann R. Briem H. Terebesi I. Bömer U. Schäfer M. Ince S. Mumberg D. Scholz A. Izumi R. Hwang S. von Nussbaum F. J. Med. Chem. 2021;64:11651–11674. doi: 10.1021/acs.jmedchem.1c01000. [DOI] [PubMed] [Google Scholar]
- Frame S. Saladino C. MacKay C. Atrash B. Sheldrake P. McDonald E. Clarke P. A. Workman P. Blake D. Zheleva D. PLoS One. 2020;15:e0234103. doi: 10.1371/journal.pone.0234103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt M. Rohe A. Platzer C. Najjar A. Erdmann F. Sippl W. Molecules. 2017;22:2045. doi: 10.3390/molecules22122045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghelli Luserna di Rorà A. Cerchione C. Martinelli G. Simonetti G. J. Hematol. Oncol. 2020;13:126. doi: 10.1186/s13045-020-00959-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geenen J. J. Schellens J. H. Clin. Cancer Res. 2017;23:4540–4544. doi: 10.1158/1078-0432.CCR-17-0520. [DOI] [PubMed] [Google Scholar]
- Diehl J. N. Klomp J. E. Snare K. R. Hibshman P. S. Blake D. R. Kaiser Z. D. Gilbert T. S. K. Baldelli E. Pierobon M. Papke B. Yang R. Hodge R. G. Rashid N. U. Petricoin, 3rd E. F. Herring L. E. Graves L. M. Cox A. D. Der C. J. J. Biol. Chem. 2021;297:101335. doi: 10.1016/j.jbc.2021.101335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L. Moore E. Berman R. Clavijo P. E. Saleh A. Chen Z. Van Waes C. Davies J. Friedman J. Allen C. T. Onco Targets Ther. 2018;7:e1488359. doi: 10.1080/2162402X.2018.1488359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartman S. J. Bagby S. M. Yacob B. W. Simmons D. M. MacBeth M. Lieu C. H. Davis S. L. Leal A. D. Tentler J. J. Diamond J. R. Front. Oncol. 2021;11:642328. doi: 10.3389/fonc.2021.642328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirai H. Iwasawa Y. Okada M. Arai T. Nishibata T. Kobayashi M. Kimura T. Kaneko N. Ohtani J. Yamanaka K. Itadani H. Takahashi-Suzuki I. Fukasawa K. Oki H. Nambu T. Jiang J. Sakai T. Arakawa H. Sakamoto T. Sagara T. Yoshizumi T. Mizuarai S. Kotani H. Mol. Cancer Ther. 2009;8:2992–3000. doi: 10.1158/1535-7163.MCT-09-0463. [DOI] [PubMed] [Google Scholar]
- Zhu J. Y. Cuellar R. A. Berndt N. Lee H. E. Olesen S. H. Martin M. P. Jensen J. T. Georg G. I. Schönbrunn E. J. Med. Chem. 2017;60:7863–7875. doi: 10.1021/acs.jmedchem.7b00996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matheson C. J. Casalvieri K. A. Backos D. S. Reigan P. ChemMedChem. 2018;13:1681–1694. doi: 10.1002/cmdc.201800188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serpico A. F. D'Alterio G. Vetrei C. Della Monica R. Nardella L. Visconti R. Grieco D. Cancers. 2019;11:819. doi: 10.3390/cancers11060819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guler S. DiPoto M. C. Crespo A. Caldwell R. Doerfel B. Grossmann N. Ho K. Huck B. Jones C. C. V. Lan R. Musil D. Potnick J. Schilke H. Sherer B. Simon S. Sirrenberg C. Zhang Z. Liu-Bujalski L. ACS Med. Chem. Lett. 2023;14:566–576. doi: 10.1021/acsmedchemlett.2c00481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alli V. J. Yadav P. Suresh V. Jadav S. S. ACS Omega. 2023;8:20196–20233. doi: 10.1021/acsomega.3c01558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C. Li Z. Li Q. Xia Y. Chan C.-C. Yuan X. Wang Y. Chen S. Qian W. J. Clin. Oncol. 2020;38:e15637. [Google Scholar]
- Huang P. Q. Boren B. C. Hegde S. G. Liu H. Unni A. K. Abraham S. Hopkins C. D. Paliwal S. Samatar A. A. Li J. Bunker K. D. J. Med. Chem. 2021;64:13004–13024. doi: 10.1021/acs.jmedchem.1c01121. [DOI] [PubMed] [Google Scholar]
- Matheson C. J. Venkataraman S. Amani V. Harris P. S. Backos D. S. Donson A. M. Wempe M. F. Foreman N. K. Vibhakar R. Reigan P. ACS Chem. Biol. 2016;11:921–930. doi: 10.1021/acschembio.5b00725. [DOI] [PubMed] [Google Scholar]
- Lee S. Y. Jang C. Lee K. A. Dev. Reprod. 2014;18:65–71. doi: 10.12717/DR.2014.18.1.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J. Wang R. Schweickert P. G. Karki A. Yang Y. Kong Y. Ahmad N. Konieczny S. F. Liu X. Cell Cycle. 2016;15:711–719. doi: 10.1080/15384101.2016.1148838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kargbo R. B. ACS Med. Chem. Lett. 2021;12:1514–1516. doi: 10.1021/acsmedchemlett.1c00442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin X. Lin F. Wang Q. Yin L. Mahato R. I. ACS Appl. Mater. Interfaces. 2019;11:14647–14659. doi: 10.1021/acsami.9b02756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutteridge R. E. A. Ndiaye M. A. Liu X. Ahmad N. Mol. Cancer Ther. 2016;15:1427–1435. doi: 10.1158/1535-7163.MCT-15-0897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright G. Golubeva V. Remsing Rix L. L. Berndt N. Luo Y. Ward G. A. Gray J. E. Schonbrunn E. Lawrence H. R. Monteiro A. N. A. Rix U. ACS Chem. Biol. 2017;12:1883–1892. doi: 10.1021/acschembio.7b00147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beria I. Bossi R. T. Brasca M. G. Caruso M. Ceccarelli W. Fachin G. Fasolini M. Forte B. Fiorentini F. Pesenti E. Pezzetta D. Posteri H. Scolaro A. Depaolini S. R. Valsasina B. Bioorg. Med. Chem. Lett. 2011;21:2969–2974. doi: 10.1016/j.bmcl.2011.03.054. [DOI] [PubMed] [Google Scholar]
- Lu J. Lei H. Bai X. Wang W. Liu C. Yang Y. Zou F. Wang L. Wang Y. Du G. Bioorg. Chem. 2023;139:106711. doi: 10.1016/j.bioorg.2023.106711. [DOI] [PubMed] [Google Scholar]
- Li Z. Mei S. Liu J. Huang J. Yue H. Ge T. Wang K. He X. Gu Y. C. Hu C. Tong M. Shi X. Zhao Y. Liu Y. Qin M. Gong P. Hou Y. Eur. J. Med. Chem. 2023;251:115242. doi: 10.1016/j.ejmech.2023.115242. [DOI] [PubMed] [Google Scholar]
- Rudolph D. Steegmaier M. Hoffmann M. Grauert M. Baum A. Quant J. Haslinger C. Garin-Chesa P. Adolf G. n. R. Clin. Cancer Res. 2009;15:3094–3102. doi: 10.1158/1078-0432.CCR-08-2445. [DOI] [PubMed] [Google Scholar]
- Kuntz K. W. and Emmitte K. A., Kinase Inhib. Drugs, 2009, pp. 351–364 [Google Scholar]
- Li J. Wang R. Schweickert P. G. Karki A. Yang Y. Kong Y. Ahmad N. Konieczny S. F. Liu X. Cell Cycle. 2016;15:711–719. doi: 10.1080/15384101.2016.1148838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z. Cheng L. Li J. Qiao Q. Karki A. Allison D. B. Shaker N. Li K. Utturkar S. M. Atallah Lanman N. M. Rao X. Rychahou P. He D. Konieczny S. F. Wang C. Shao Q. Evers B. M. Liu X. Cancer Res. 2022;82:3532–3548. doi: 10.1158/0008-5472.CAN-22-0018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Infante J. R. Hollebecque A. Postel-Vinay S. Bauer T. M. Blackwood E. M. Evangelista M. Mahrus S. Peale F. V. Lu X. Sahasranaman S. Zhu R. Chen Y. Ding X. Murray E. R. Schutzman J. L. Lauchle J. O. Soria J.-C. LoRusso P. M. Clin. Cancer Res. 2017;23:2423–2432. doi: 10.1158/1078-0432.CCR-16-1782. [DOI] [PubMed] [Google Scholar]
- Deng M. Wang P. Long X. Xu G. Wang C. Li J. Zhou Y. Liu T. ChemMedChem. 2023:e202200664. doi: 10.1002/cmdc.202200664. [DOI] [PubMed] [Google Scholar]
- Dent P. Expert Opin. Invest. Drugs. 2019;28:1095–1100. doi: 10.1080/13543784.2019.1694661. [DOI] [PubMed] [Google Scholar]
- Klomp J. E. Lee Y. S. Goodwin C. M. Papke B. Klomp J. A. Waters A. M. Stalnecker C. A. DeLiberty J. M. Drizyte-Miller K. Yang R. Diehl J. N. Yin H. H. Pierobon M. Baldelli E. Ryan M. B. Li S. Peterson J. Smith A. R. Neal J. T. McCormick A. K. Kuo C. J. Counter C. M. Petricoin, 3rd E. F. Cox A. D. Bryant K. L. Der C. J. Cell Rep. 2021;37:110060. doi: 10.1016/j.celrep.2021.110060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu Q. S. Jonker D. J. Provencher D. M. Miller W. H. Bouganim N. Shields A. F. Shapiro G. Sawyer M. B. Lheureux S. Samouelian V. Cancers. 2023;10:448. [Google Scholar]
- Huffman B. M. Feng H. Parmar K. Wang J. Kapner K. S. Kochupurakkal B. Martignetti D. B. Sadatrezaei G. Abrams T. A. Biller L. H. Giannakis M. Ng K. Patel A. K. Perez K. J. Singh H. Rubinson D. A. Schlechter B. L. Andrews E. Hannigan A. M. Dunwell S. Getchell Z. Raghavan S. Wolpin B. M. Fortier C. D'Andrea A. D. Aguirre A. J. Shapiro G. I. Cleary J. M. Clin. Cancer Res. 2023;29:5047–5056. doi: 10.1158/1078-0432.CCR-23-2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laquente B. Lopez-Martin J. Richards D. Illerhaus G. Chang D. Z. Kim G. Stella P. Richel D. Szcylik C. Cascinu S. Frassineti G. L. Ciuleanu T. Hurt K. Hynes S. Lin J. Lin A. B. Von Hoff D. Calvo E. BMC Cancer. 2017;17:137. doi: 10.1186/s12885-017-3131-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaaks P. Coker E. A. Vis D. J. Edwards O. Carpenter E. F. Leto S. M. Dwane L. Sassi F. Lightfoot H. Barthorpe S. van der Meer D. Yang W. Beck A. Mironenko T. Hall C. Hall J. Mali I. Richardson L. Tolley C. Morris J. Thomas F. Lleshi E. Aben N. Benes C. H. Bertotti A. Trusolino L. Wessels L. Garnett M. J. Nature. 2022;603:166–173. doi: 10.1038/s41586-022-04437-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiechmann S. Saupp E. Schilling D. Heinzlmeir S. Schneider G. Schmid R. M. Combs S. E. Kuster B. Dobiasch S. Mol. Cell. Proteomics. 2020;19:1649–1663. doi: 10.1074/mcp.RA120.002046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin T. Xu L. Wang P. Hu X. Zhang R. Wu Z. Du W. Kan W. Li K. Wang C. Zhou Y. Li J. Liu T. J. Med. Chem. 2021;64:15069–15090. doi: 10.1021/acs.jmedchem.1c00994. [DOI] [PubMed] [Google Scholar]
- Guo N. Li M. Z. Wang L. M. Chen H. D. Song S. S. Miao Z. H. He J. X. Cancer Biol. Ther. 2022;23:69–82. doi: 10.1080/15384047.2021.2024414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang A. Gao K. Chu L. Zhang R. Yang J. Zheng J. Oncotarget. 2017;8:23937–23954. doi: 10.18632/oncotarget.14893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D. Zhu J. Firozi P. F. Abbruzzese J. L. Evans D. B. Cleary K. Friess H. Sen S. Clin. Cancer Res. 2003;9:991–997. [PubMed] [Google Scholar]
- Pollard J. R. Mortimore M. J. Med. Chem. 2009;52:2629–2651. doi: 10.1021/jm8012129. [DOI] [PubMed] [Google Scholar]
- dos Santos E. O. Carneiro-Lobo T. C. Aoki M. N. Levantini E. Bassères D. S. Mol. Cancer. 2016;15:12. doi: 10.1186/s12943-016-0494-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes-Filho S. M. Dos Santos E. O. Bertoldi E. R. M. Scalabrini L. C. Heidrich V. Dazzani B. Levantini E. Reis E. M. Bassères D. S. Cell. Oncol. 2020;43:445–460. doi: 10.1007/s13402-020-00498-5. [DOI] [PubMed] [Google Scholar]
- Sells T. B. Chau R. Ecsedy J. A. Gershman R. E. Hoar K. Huck J. Janowick D. A. Kadambi V. J. LeRoy P. J. Stirling M. Stroud S. G. Vos T. J. Weatherhead G. S. Wysong D. R. Zhang M. Balani S. K. Bolen J. B. Manfredi M. G. Claiborne C. F. ACS Med. Chem. Lett. 2015;6:630–634. doi: 10.1021/ml500409n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Y. Zhu S. Zhong M. Yang M. Sun X. Liu J. Kroemer G. Lotze M. Zeh, 3rd H. J. Kang R. Tang D. Gastroenterology. 2017;153:1429–1443. doi: 10.1053/j.gastro.2017.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J. A. Huynh J. C. Wu C. Y. Yu A. M. Matsukuma K. Semrad T. J. Gandara D. R. Li T. Riess J. W. Tam K. Mack P. C. Martinez A. Mahaffey N. Kelly K. L. Kim E. J. Cancer Chemother. Pharmacol. 2022;90:217–228. doi: 10.1007/s00280-022-04457-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fancelli D. Moll J. Varasi M. Bravo R. Artico R. Berta D. Bindi S. Cameron A. Candiani I. Cappella P. Carpinelli P. Croci W. Forte B. Giorgini M. L. Klapwijk J. Marsiglio A. Pesenti E. Rocchetti M. Roletto F. Severino D. Soncini C. Storici P. Tonani R. Zugnoni P. Vianello P. J. Med. Chem. 2006;49:7247–7251. doi: 10.1021/jm060897w. [DOI] [PubMed] [Google Scholar]
- Kirbiyik I. A. Ozcimen A. A. J. Cancer Res. Ther. 2021;17:1419–1424. doi: 10.4103/jcrt.JCRT_827_19. [DOI] [PubMed] [Google Scholar]
- Carlino M. S. Larkin J. Long G. V. Lancet. 2021;398:1002–1014. doi: 10.1016/S0140-6736(21)01206-X. [DOI] [PubMed] [Google Scholar]
- Petroni G. Formenti S. C. Chen-Kiang S. Galluzzi L. Nat. Rev. Immunol. 2020;20:669–679. doi: 10.1038/s41577-020-0300-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lelliott E. J. Sheppard K. E. McArthur G. A. npj Precis. Oncol. 2022;6:26. doi: 10.1038/s41698-022-00273-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goel S. DeCristo M. J. Watt A. C. BrinJones H. Sceneay J. Li B. B. Khan N. Ubellacker J. M. Xie S. Metzger-Filho O. Hoog J. Ellis M. J. Ma C. X. Ramm S. Krop I. E. Winer E. P. Roberts T. M. Kim H. J. McAllister S. S. Zhao J. J. Nature. 2017;548:471–475. doi: 10.1038/nature23465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J. Bu X. Wang H. Zhu Y. Geng Y. Nihira N. T. Tan Y. Ci Y. Wu F. Dai X. Guo J. Huang Y. H. Fan C. Ren S. Sun Y. Freeman G. J. Sicinski P. Wei W. Nature. 2018;553:91–95. doi: 10.1038/nature25015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M. Zhang L. Hei R. Li X. Cai H. Wu X. Zheng Q. Cai C. Am. J. Cancer Res. 2021;11:1913–1935. [PMC free article] [PubMed] [Google Scholar]
- Hossain D. M. S. Javaid S. Cai M. Zhang C. Sawant A. Hinton M. Sathe M. Grein J. Blumenschein W. Pinheiro E. M. J. Clin. Invest. 2018;128:644–654. doi: 10.1172/JCI94586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z. Cheng L. Li J. Qiao Q. Karki A. Allison D. B. Shaker N. Li K. Utturkar S. M. Atallah Lanman N. M. Rao X. Rychahou P. He D. Konieczny S. F. Wang C. Shao Q. Evers B. M. Liu X. Cancer Res. 2022;82:3532–3548. doi: 10.1158/0008-5472.CAN-22-0018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel M. R. Falchook G. S. Wang J. S.-Z. Rodrigo Imedio E. Kumar S. Motlagh P. Miah K. Mugundu G. M. Jones S. F. Spigel D. R. Hamilton E. P. J. Clin. Oncol. 2019;37:2562. [Google Scholar]
- Punt S. Malu S. McKenzie J. A. Manrique S. Z. Doorduijn E. M. Mbofung R. M. Williams L. Silverman D. A. Ashkin E. L. Dominguez A. L. Wang Z. Chen J. Q. Maiti S. N. Tieu T. N. Liu C. Xu C. Forget M. A. Haymaker C. Khalili J. S. Satani N. Muller F. Cooper L. J. N. Overwijk W. W. Amaria R. N. Bernatchez C. Heffernan T. P. Peng W. Roszik J. Hwu P. Cancer Immunol., Immunother. 2021;70:1101–1113. doi: 10.1007/s00262-020-02748-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rampioni Vinciguerra G. L. Sonego M. Segatto I. Dall'Acqua A. Vecchione A. Baldassarre G. Belletti B. Front. Oncol. 2022;12:891580. doi: 10.3389/fonc.2022.891580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wander S. A. Cohen O. Gong X. Johnson G. N. Buendia-Buendia J. E. Lloyd M. R. Kim D. Luo F. Mao P. Helvie K. Kowalski K. J. Nayar U. Waks A. G. Parsons S. H. Martinez R. Litchfield L. M. Ye X. S. Yu C. Jansen V. M. Stille J. R. Smith P. S. Oakley G. J. Chu Q. S. Batist G. Hughes M. E. Kremer J. D. Garraway L. A. Winer E. P. Tolaney S. M. Lin N. U. Buchanan S. G. Wagle N. Cancer Discovery. 2020;10:1174–1193. doi: 10.1158/2159-8290.CD-19-1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodwin C. M. Waters A. M. Klomp J. E. Javaid S. Bryant K. L. Stalnecker C. A. Drizyte-Miller K. Papke B. Yang R. Amparo A. M. Ozkan-Dagliyan I. Baldelli E. Calvert V. Pierobon M. Sorrentino J. A. Beelen A. P. Bublitz N. Lüthen M. Wood K. C. Petricoin E. F. Sers C. McRee A. J. Cox A. D. Der C. J. Cancer Res. 2023;83:141–157. doi: 10.1158/0008-5472.CAN-22-0391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto G. Tanaka K. Kamata R. Mori S. Liu J. Yamauchi T. Sakae Y. Ohashi A. Kobayashi S. S. Cancer Res. 2023;83:5739. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.