

ABSTRACT
Leptomeningeal tertiary lymphoid structures (TLS) have emerged as a relatively common pathological feature of autoimmune disease, including multiple sclerosis (MS) and particularly in people with progressive and nonremitting MS. These ectopic lymphoid aggregates, observed in the leptomeninges adjacent to so‐called “Type 3” sub‐pial cortical lesions, are associated with more severe gray matter damage and worse clinical outcomes. Mouse models of MS that recapitulate TLS formation in the central nervous system (CNS) have provided critical insights into the mechanisms driving their development and maintenance. In these models of experimental autoimmune encephalomyelitis (EAE) initiation of TLS is facilitated by Th17 cells, which promote chronic inflammation via cytokines such as IL‐17 and GM‐CSF. The cell surface expression of lymphotoxin‐α and lymphotoxin‐β heterotrimers (LTαβ) on lymphocytes, including Th17 cells, elaborates the organization of ectopic lymphoid tissues via LTβR signaling on radio‐resistant stromal cells and resident fibroblasts. Ultimately a pro‐inflammatory environment characterized by cytokines such as IL‐17 and GM‐CSF promotes the recruitment of neutrophils which produce proteases and chemokines that sustain a pro‐inflammatory milieu. Emerging EAE data suggest that disrupting TLS organization or targeting key pathways involved in their maintenance could represent promising strategies for modulating chronic CNS inflammation in MS. Understanding the cellular and molecular mechanisms regulating TLS dynamics is therefore critical for the development of therapies aimed at halting or reversing nonremitting MS disease.
Keywords: autoimmunity, B cells, experimental autoimmune encephalomyelitis, lymphotoxin, multiple sclerosis, neutrophils, Th17
1. Introduction
Adaptive immune responses are initiated in secondary lymphoid organs, such as the spleen and regional lymph nodes. The organization of lymphocytes within secondary lymphoid tissues is key to generating an efficient adaptive immune response. Development of secondary lymphoid organs begins during embryogenesis, where fetal liver‐derived hematopoietic lymphoid tissue inducer (LTi) cells interact with mesenchymal lymphoid tissue organizer (LTo) cells at future sites of lymphoid organ development [1]. Lymphotoxin‐αβ (LTαβ), produced by LTi cells, promotes the production of chemokines and expression of adhesion molecules on LTos through LTβR signaling. Consequently, the LT pathway is necessary for secondary lymphoid organ development [1]. Such developmental LTβR‐dependent signals are echoed in the adult animal during homeostasis to maintain the chemokine networks that are essential for the organization of lymphocytes in secondary lymphoid organs [2, 3], and can drive the formation of so‐called tertiary lymphoid structure (TLS) in chronically inflamed tissues [4]. TLS, which resemble secondary lymphoid tissues insofar as they contain aggregates of T cells, B cells, and antigen‐presenting cells (APCs) supported by fibroblasts that produce a network of extracellular matrix (ECM), have been reported in the context of cancer [5], autoimmunity [6], transplantation [7, 8], and infection [9]. TLS is observed across multiple tissues, including a compartment adjacent to the central nervous system (CNS) called the leptomeninges [10]. In contrast to TLS in other tissues and pathologies, such as those observed in the salivary glands of Sjogren's disease patients [11] or the joints of rheumatoid arthritis patients [12], T cell and B cell zones in leptomeningeal TLS are less defined and evidence for bona fide GC reactions is lacking [13, 14]. Nevertheless, leptomeningeal TLS has garnered special attention in multiple sclerosis (MS) research as its presence has been correlated with clinical measures of disease progression [15]. For the purposes of this review, sites of leptomeningeal immune cell aggregates containing T cells and B cells will be considered TLS. We will highlight findings on leptomeningeal TLS formation, persistence, and function in MS and its animal model, experimental autoimmune encephalomyelitis (EAE), then discuss evidence that may rationalize the disruption of TLS as a therapeutic strategy for attenuating MS progression.
2. Anatomy of the CNS and Meningeal Layers
The meninges are composed of three distinct layers that envelop and protect the brain. The innermost layer, the pia mater, is in direct contact with the underlying brain parenchyma. Above it, the arachnoid mater is connected to the pia by fine connective tissue strands called trabeculae. The space between these two layers—the subarachnoid space (SAS), also referred to as the leptomeninges—is filled with cerebrospinal fluid (CSF). Overlying the arachnoid is the dura mater, a dense, fibrous layer that secures the meninges to the skull (Figure 1).
FIGURE 1.
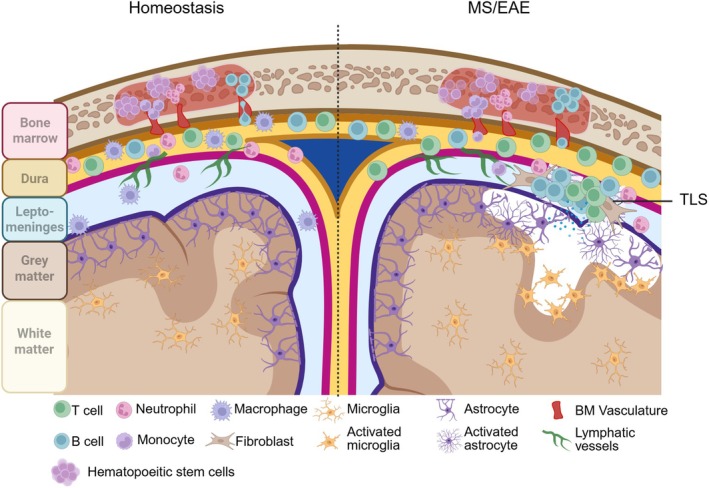
Structure of the steady‐state and inflamed meninges and brain. (Left) Homeostatic CNS and meninges: Reservoir of immune cells in the dura are continually supplied by the skull bone marrow, while leptomeninges remain relatively clear of immune cells and toxic factors. Lymphatic vessels in the leptomeninges drain solutes from CSF into dural sinuses. Underlying glia limitans maintained by tight junctions between astrocyte end feet are intact, and gray and white matter are healthy and myelinated. Microglia remain in resting state. (Right) Pathogenic T cells induce elaboration of fibroblast niche, recruitment of neutrophils and B cells from the periphery, and production of toxic factors that disrupt the glia limitans. A combination of noxious solutes from the leptomeninges and activated microglia leads to subpial gray matter damage (lesion). Persistent gray matter damage eventually results in neuronal death and cortical atrophy (not shown).
While small numbers of T lymphocytes reside in the CNS, presumably for the purpose of immunosurveillance [16, 17, 18], for the most part, the brain and spinal cord parenchyma are largely devoid of T cells and B cells at homeostasis. This paucity in lymphocytes is in part owing to tight endothelial junctions in the blood–brain barrier (BBB) which limits blood‐derived cells and proteins from entering the CNS [19]. However, unlike the tightly regulated vasculature of the blood–brain barrier, dural blood vessels are fenestrated and lack tight junctions, permitting the passage of large molecules and peripheral immune cells into this meningeal compartment [20, 21]. For this reason, the dura is densely populated by immune cells including lymphocytes, monocytes, and other myeloid cells, even under steady‐state conditions [20]. The anatomical and immunological features of the dura mater are quite unique. Adhered to the inner surface of the skull, the dura contains specialized structures known as dural venous sinuses, which facilitate the drainage of venous blood from the brain. In addition to venous channels, the dura is also traversed by arteries derived from the carotid circulation and veins that ultimately drain into the dural sinuses [20]. Dural blood vessels are also innervated and responsive to neurovascular signaling, allowing for dynamic interactions between the nervous and immune systems. Another unique feature of the dura is its connectivity to the skull bone marrow via diploic veins, which offer a direct migratory route for immune cells [20]. For example, in a mouse model of stroke, neutrophils were observed migrating from the skull bone marrow into the dura, underscoring the potential functional relevance of this tissue during injury and/or inflammation [22]. Furthermore, the calvarial bone marrow harbors distinct niches that communicate with both the dura and the SAS through osseous channels, providing a potential source of immune cells during CNS inflammation. A pivotal study by Marco Colonna's group showed that dural B cells originate from the skull bone marrow and migrate into the dura through skull vascular channels potentially in response to a gradient of CXCL12 chemokine [23]. Using an intrathecal injection of CD19‐tdTomato cells to track migration, Colonna and colleagues found tdTomato+ B cells in both dural lymphatic vessels and cervical lymph nodes 24 h following transfer, suggesting that the dura may be a migratory route for B cells [23]. Ongoing research continues to explore the role of skull bone marrow in regulating immune responses in the context of neuroinflammatory disease [23, 24].
The arachnoid mater at the base of the dura consists of squamous epithelial cells joined by tight junctions and is supported by a meshwork of collagenous trabeculae that span the SAS, forming the leptomeninges through which CSF circulates. The presence of tight junctions within the arachnoid epithelium establishes a physiological barrier between the dura mater and the CSF‐filled SAS. In addition to its structural role, the arachnoid epithelium expresses various efflux drug transporters and cytochrome p450 enzymes [25], suggesting it plays an active role in regulating molecular clearance from the CSF. Beneath the arachnoid lies the pia mater, a thin, delicate membrane that closely follows the contours of the brain and serves as a barrier between the parenchyma and penetrating blood vessels. Immediately below the pia is the glia limitans, a continuous layer of astrocyte end‐feet that forms the final boundary of the blood–meningeal barrier (BMB) [26, 27]. This multilayered system functions collectively to regulate immune cell access to the brain and maintain CNS immune privilege under homeostatic conditions.
In contrast to the fenestrated vasculature of the dura mater, blood vessels within the leptomeninges are nonfenestrated and sealed by tight junctions, forming a selective barrier that limits the extravasation of immune cells and macromolecules from the circulation into the CSF [26, 28]. This barrier is maintained by tightly connected endothelial cells and by bidirectional crosstalk with astrocytes of the glia limitans, which reinforces barrier integrity. A defining feature of these astrocytes is the expression of aquaporin‐4 (AQP4), a water channel protein enriched on astrocytic end‐feet surrounding cerebral blood vessels [29]. In vitro coculture models of the blood–brain barrier (BBB) have shown that astrocytes play a critical role in modulating endothelial expression of tight junction proteins, emphasizing the importance of astrocyte–endothelial cell cooperation in maintaining barrier function [30]. Despite the restrictive nature of the leptomeningeal vasculature, low‐level immune cell trafficking does occur under homeostatic conditions. Endothelial expression of adhesion molecules such as P‐selectin, E‐selectin, and intercellular adhesion molecule‐1 (ICAM‐1) supports limited leukocyte surveillance of the leptomeningeal compartment [31]. Additionally, C‐C chemokine ligand 19 (CCL19) is constitutively present in human CSF and may contribute to the basal recruitment of CCR7‐expressing leukocytes, even in the absence of inflammation [32].
There is debate in the literature regarding the presence and continuity of perivascular spaces within the SAS and cerebral cortex, and whether these are structurally or functionally continuous with Virchow‐Robin (VR) spaces, which envelop parenchymal penetrating blood vessels in the white matter. While VR spaces in the white matter are increasingly recognized as sites of immune surveillance and potential antigen presentation, the existence of analogous perivascular compartments surrounding leptomeningeal or cortical vessels is less well defined. However, if present, these perivascular spaces could offer a transient niche for immune cell trafficking and retention within the SAS, supporting localized immune surveillance even under noninflammatory conditions [33, 34, 35, 36]. In support of the concept that immune cells occupy the leptomeninges, flow cytometric studies of human CSF have revealed the presence of memory CD4+ T cells and occasional B cells under steady‐state conditions [31, 37]. Correspondingly, murine histological studies have identified a diverse population of immune cells within the meninges, including macrophages, dendritic cells, neutrophils, and mast cells [31, 38, 39, 40]. Recent transcriptomic analyses have further confirmed the presence of natural killer (NK) cells, T cells, and B cells in the leptomeninges of mice [38]. These findings suggest that, like the dura, the leptomeninges maintain a resident or semi‐resident immune cell population. However, the duration of immune cell residency within the SAS and their functional dynamics remains poorly defined. What is clear is that the density and diversity of immune cells in the leptomeninges are likely more limited than the dura, which appears to be more immunologically active at baseline.
Although relatively lymphocyte‐poor in steady state, the leptomeninges can alter dramatically during inflammation. This may be due to “hair‐trigger”‐like changes in resident fibroblast populations within the SAS. Fibroblasts are specialized stromal cells that are important for more than just architectural integrity of a tissue. In peripheral tissues there are a variety of fibroblasts that respond to inflammation, and lessons from these fibroblasts may give some insights into meningeal fibroblast remodeling [41, 42]. For example, Buechler et al. provided a framework for identifying and functionally characterizing fibroblasts, outlining that while there is heterogeneity of fibroblast populations within tissues, they adopt similar functional states in response to inflammation [42]. Inflammatory fibroblasts express conserved genes across tissues, including those involved in cytokine signaling (Il6, Ccl2, Cxcl1), ECM remodeling (Mmp3, Mmp13), and immune cell recruitment (Cxcl12, Ccl19, Ccl21) [42].
Although our understanding of fibroblasts in the leptomeninges is in its infancy, we previously characterized a network of CD31− podoplanin (PDPN)+ fibroblastic reticular cell (FRC)‐like stromal cells in the leptomeninges in mice [43, 44]. FRC organizes immune cell positioning and facilitates immune responses in secondary lymphoid organs via the production of various chemokines and survival factors [44, 45]. Experimental models of CNS infection—including viral and parasitic pathogens such as Toxoplasma gondii—demonstrate that leptomeningeal fibroblasts can upregulate chemokines such as CCL21, enabling the recruitment of CD8+ T cells into the SAS for pathogen clearance [46, 47]. As will be discussed in a later section, evidence from animal models of MS (EAE) has shown that fibroblasts are key orchestrators of leptomeningeal TLS.
3. Leptomeningeal TLS in MS
MS is widely regarded as an autoimmune disease directed against myelin that ensheathes axons, manifesting as neurological symptoms such as cognitive deficits and motor dysfunction [48]. Clinically, MS is classified into three subtypes: relapse–remitting MS (RRMS), primary progressive MS (PPMS), and secondary progressive MS (SPMS) [49]. Many people are initially diagnosed with relapsing–remitting MS, where acute symptoms are followed by a time of relative quiescence. With time, individuals may transition into progressive MS (PMS), where the influx of lymphocytes into the CNS has subsided and neurodegenerative processes dominate [50]. However, emerging evidence suggests that MS may invoke parallel disease processes that occur from disease onset [51]. MS diagnosis and monitoring of progression is achieved by longitudinal assessment of clinical deficits, using metrics such as the Expanded Disability Scoring System or white matter lesion burden using magnetic resonance imaging (MRI) [48]. Neither of these metrics clearly distinguishes between RRMS and PMS.
What triggers MS is unclear, but a genetic component is evident. Widescale genome mapping studies across different populations have consistently implicated a link between MS and HLA‐DRB1 risk alleles which encode for MHC‐II [52, 53]. Genetic variations in HLA genes determine the repertoire of peptides presented by MHC molecules, which in turn shapes CD4+ T cell reactivity and their capacity to provide help to cognate B cells. MS patient‐derived blood samples show evidence of elevated systemic T cell activation [54], impaired regulatory T cell function [55], and T cell autoreactivity against myelin‐derived peptides, highlighting the critical involvement of T cell dysregulation in disease. CD4+ T cells are thus thought to play a central role in MS pathogenesis, a concept also strongly supported by animal model data [56, 57]. The presence of oligoclonal antibody bands (OCBs) in MS CSF is evidence of abnormal intrathecal antibody production. This prompted the testing of B cell–depleting therapies in MS, such as anti‐CD20 antibodies (rituximab, ocrelizumab) [58]. These therapies have a profound impact on reducing relapsing biology in MS; however, this benefit occurs without altering levels of CSF oligoclonal bands [59]. One hypothesis is that the mechanism of action of anti‐CD20 antibodies in reducing relapsing biology may be linked to antibody‐independent B cell functions, such as production of anti‐inflammatory cytokines or T cell inhibitory ligands [60, 61].
The mechanisms underlying the transition from RRMS to PMS remain incompletely understood. One proposed explanation for this shift involves the gradual accumulation of gray matter (GM) injury [62, 63, 64]. Both imaging and histopathological studies have demonstrated the presence of GM abnormalities early in the disease course, with lesion burden intensifying as the disease advances [64, 65, 66]. Multiple cross‐sectional and longitudinal investigations have further established a correlation between GM pathology and the progression of physical and cognitive impairments [66, 67, 68]. Notably, the volume of cortical GM lesions has emerged as a robust predictor of disease progression and an indicator of the transition from RRMS to PMS. While various types of GM lesions exist, this review focuses on subpial cortical lesions, also called Type III lesions, which have been documented in postmortem MS brain tissue [65]. Type III lesions are localized to the surface of the brain, frequently forming ribbon‐like patterns across multiple gyri. Importantly, such lesions are not observed in other inflammatory CNS diseases such as Rasmussen's encephalitis [69] or neuromyelitis optica (NMO) [70], suggesting they are unique to MS [63]. Beyond demyelination, these lesions are also characterized by axonal, neuronal, and synaptic injury/loss [62, 63].
A hallmark of Type III lesions is their spatial association with leptomeningeal TLS [14, 15, 71]. Leptomeningeal TLS in postmortem MS brain tissues has been shown to contain a variety of immune cell types, including B cells, T cells, dendritic cells, macrophages, plasma cells, and stromal cells resembling follicular dendritic cells [14, 15, 72]. The presence of CXCL13 within these structures supports the recruitment and retention of CXCR5+ lymphocytes, including B cells and likely T follicular helper (Tfh) cells [73]. Moreover, RORγt+ Th17 cells, which can produce IL‐17, are preferentially enriched within immune aggregates [74] rather than diffusely infiltrated meningeal areas, suggesting that they play a role in the organized formation of these tertiary lymphoid‐like structures.
As mentioned, the glial limitans form a tight barrier reinforced by astrocyte end‐feet processes that separate the leptomeninges from the CNS parenchyma [45]. Due to this barrier, it is unlikely that leptomeningeal TLS‐resident immune cells directly penetrate the underlying cortex during MS/EAE. Instead, it is hypothesized that diffusible inflammatory mediators—such as cytokines, chemokines, and other toxic molecules—cross the glia limitans and induce localized subpial damage leading to Type III lesions. Supporting this model, CSF from people with MS with high cortical lesion burden exhibits elevated levels of proinflammatory cytokines (e.g., IFN‐γ, TNF, IL‐2, IL‐22), lymphoid‐organizing factors (e.g., CXCL13, LTα), B cell survival factors (e.g., BAFF), molecules associated with BBB dysfunction (e.g., fibrin, complement, coagulation proteins), and iron‐related oxidative stress indicators (e.g., hemoglobin, haptoglobin) [75, 76, 77].
Although subpial GM lesions are anatomically distinct from white matter (WM) lesions, there may be a connection [78]. Recent work revealed that postmortem tissues from people with MS that had high meningeal T and B cell infiltration exhibit not only more extensive subpial demyelination but also a higher frequency of active and mixed active‐inactive WM lesions relative to inactive or remyelinated WM lesions [78]. This observation aligns with longitudinal MRI studies linking WM lesion volume to GM atrophy [79, 80]. While it is not known how leptomeningeal inflammation is connected to WM lesions, these data suggest a broader interplay between CNS‐compartmentalized inflammation in the leptomeninges and widespread CNS pathology [69]. One possibility is that antigens released from chronically inflamed WM lesions drain via dural lymphatics to the cervical lymph nodes where they prime autoreactive T cells that home back to the leptomeninges [78]. Alternatively, axonal degeneration within subcortical WM may initiate retrograde damage to cortical neurons, leading to secondary GM injury [78].
While postmortem studies have been instrumental in characterizing the cellular and molecular composition of leptomeningeal TLS, translating these findings to living individuals remains difficult. Advanced imaging techniques, such as high‐resolution MRI with contrast enhancement, have provided some evidence of leptomeningeal contrast dye enhancement [81, 82, 83], yet these measurements are unable to confirm that these areas of contrast are due to resident lymphocytes versus residual fibrotic scarring. Moreover, the development of consistent and sensitive biomarkers to reflect leptomeningeal inflammation is hindered by the compartmentalized nature of the immune response, which may not be adequately captured by peripheral blood or even CSF analyses. Although certain cytokines and chemokines such as CXCL13, BAFF, and LTα have been associated with leptomeningeal TLS and cortical pathology [84], their levels can vary widely between individuals and disease stages, limiting their clinical utility. These technical and biological limitations underscore the need for novel, multimodal approaches to detect and monitor leptomeningeal TLS in vivo and to establish reliable biomarkers that can guide prognosis and therapeutic intervention, as well as animal models that replicate the relationship between leptomeningeal TLS and Type III sub‐pial lesions.
4. Mouse Models of Leptomeningeal TLS in MS
EAE is the collective term for animal models that mimic characteristics of MS, particularly CD4+ T cell‐mediated demyelination. The earliest deliberate induction of what is now known as EAE was established by Koritschoner and Schweinburg through the injection of human spinal cord homogenates into rabbits [85]. It was then shown in rhesus monkeys that the paralysis seen after repeated brain homogenate injections was accompanied by demyelinating brain lesions [86] Subsequently, EAE has been induced across a variety of model organisms and leveraged as a model for MS [87, 88, 89]. Amongst these, mouse models are the most widely used, benefiting from the availability of genetically modified strains that provide powerful tools for neuroimmunology research. Mouse models of EAE rely on sensitizing the adaptive immune system towards myelin proteins or peptides, such as myelin oligodendrocyte protein (MOG), proteolipid protein (PLP), and myelin basic protein (MBP), which can lead to different disease courses depending on the route of administration and the mouse strain used [90]. In active EAE models, mice are immunized towards myelin peptides or proteins, usually emulsified in Complete Freund's Adjuvant (CFA) which contains heat‐killed Mycobacterium tuberculosis [90]. This generates an encephalitogenic CD4+ T cell response towards a dominant myelin epitope and drives demyelination in a manner that, depending on the immunogen, may or may not depend on B cells [91, 92, 93]. Alternatively, passive EAE models involve the adoptive transfer of pre‐primed encephalitogenic CD4+ T cells into a naïve recipient animal, which provides an opportunity to study CD4+ T cell‐driven effector function independent of priming events [94, 95]. Spontaneous EAE models also exist—these are mice genetically engineered to express a T cell receptor and B cell receptor transgenes specific for myelin [94]. Ultimately, mouse EAE variations seek to recapitulate specific pathological features of disease, including a relapsing–remitting versus progressive disease course, and the localization of leptomeningeal TLS (Table 1).
TABLE 1.
Summary of EAE models featuring meningeal inflammation and/or gray matter demyelination.
| Strain | Induction | Disease features | TLS location and composition |
| C57BL/6 [90] | Subcutaneous injection of MOG35‐55 with CFA supplemented with pertussis toxin |
Monophasic EAE Spinal cord (white matter) demyelination |
Limited/none, rather diffuse meningeal inflammation with T cells, B cells, neutrophils, and monocytes |
| C57BL/6 [96] | Adoptive transfer (intraperitoneal or intravenous) of MOG35‐55‐primed T cells |
Monophasic EAE (young, < 6 months) Nonremitting EAE (old, > 8 months) Spinal cord (white matter) demyelination |
Limited/none, rather, diffuse meningeal inflammation with T cells, B cells, neutrophils, and monocytes |
| C57BL/6 [97] | Introduction of heat‐killed Mycobacterium in the piriform cortex after immunization against MOG35‐55 | Focal lesions in the brain, gray matter demyelination, microglia, and astrocyte activation adjacent to TLS | Delayed‐type hypersensitivity TLS, slow accumulation of T and B cells in the leptomeninges |
| C57BL/6, 2D2 T cells [98] | Spontaneous EAE driven by MOG‐reactive T cells, incidence increases with age and pertussis toxin |
Monophasic EAE Spinal cord demyelination |
Spinal cord meninges, clusters of T cells and B cells encapsulated by a reticulin+ ECM network |
| C57BL/6, IgHMOG B cells [99] | Subcutaneous injection of MOG35‐55 with CFA |
Chronic disease, nonremitting Spinal cord demyelination |
Spinal cord meninges, large clusters of B cells |
| NOD [100, 101, 102] | Immunization with MOG35‐55 with CFA supplemented with pertussis toxin | RR‐EAE with transition to chronic disease | TLS in the spinal cord meninges with CD4+ T cells, B220+ B cells, and CD21/CD35+ follicular dendritic cells |
| NOD IgHMOG [103] | Immunization with MOG35‐55 with CFA | Rapid and severe progressive EAE | Brain‐adjacent TLS with fibronectin‐rich, PDGFRα/β+ stromal cell network and prominent accumulation of B220+ B cells |
| Biozzi‐ABH [104, 105] | Immunization with spinal cord homogenate emulsified in CFA |
RR‐EAE (young, < 3 months), then progressive after 3 months Severe, progressive EAE (old, > 12 months) Cortical demyelination, axonal/synapse loss adjacent to TLS |
CD3+ T cell and B220+ B cell infiltration into the gray and white matter of the spinal cord, rather than the overlying leptomeninges. Microglia activation near regions of severe axonal damage. Damage tends to be perivascular |
| SJL/J [106, 107] | Subcutaneous injection of PLP139‐151 with CFA | RR‐EAE, cortical demyelination, axonal/synapse loss adjacent to TLS | Brain and spinal cord, B220+ B cells, CD4+ T cells, FDCs |
| SJL/J [43] | Adoptive transfer of PLP139‐151‐primed T cells |
Monophasic EAE (young, < 6 months) Nonremitting EAE (old, > 8 months) Cortical demyelination, axonal/synapse loss adjacent to TLS |
Brain and spinal cord, B220+ B cells, CD4+ T cells, CD11c+ myeloid cells |
4.1. C57Bl/6 EAE
MOG35‐55 EAE in C57Bl/6 mice is the most widely used EAE model for its robust CNS‐directed autoimmune CD4+ T cell response and the availability of genetically modified mice. Active MOG35‐55 EAE requires coadministration of pertussis toxin, which interrupts G protein‐coupled receptor signaling and has been reported to transiently loosen the BBB [108, 109]. C57Bl/6 mice typically develop a disease typified by acute onset followed by some extent of recovery and subsequent chronic disease [95, 108, 110]. Spinal cord inflammation dominates this model, with leptomeningeal TLS rarely reported in the brain [111]. One study has reported that stereotactic injection of heat‐killed Mycobacterium tuberculosis into the piriform cortex after immunization with MOG35‐55 is capable of inducing focal lesions in the brain, which are termed delayed‐type hypersensitivity (DTH)‐TLS [97]. In this model, T cell and B cell infiltration into the leptomeninges increases over time and is accompanied by demyelination, as well as microglial and astrocyte activation proximal to overlying leptomeningeal aggregates [97]. For these reasons, DTH‐TLS has been proposed as a model for recapitulating leptomeningeal TLS‐associated pathology observed in PMS.
TLS in the spinal cord leptomeninges in C57Bl/6 mice has mostly been studied in the context of transgenic myelin‐specific TCR and BCR mice. T cells derived from mice that express a transgene encoding a MOG35‐55‐specific TCR composed of the Vα3.2 and Vβ11 chains (2D2 TCR) [98] are capable of inducing EAE in naïve WT C57Bl/6 recipient mice upon adoptive transfer. These T cells induce TLS in the spinal cord SAS that vary in size but are comprised of B cell clusters surrounded by T cells and encapsulated within reticulin+ ECM [112]. A MOG‐specific IgH (IgHMOG) knock‐in has also been generated [99]. Both 2D2 and IgHMOG mice are individually susceptible to EAE following immunization with MOG peptides [98, 113], or full‐length MOG protein [98]. When crossed together, 2D2 x IgHMOG mice develop spontaneous EAE characterized by B cell aggregates in the spinal cord leptomeninges [114, 115, 116]. These leptomeningeal TLS‐resident B cells are CD62Llow and CD80high, suggesting previous activation or priming. However, unlike secondary lymphoid tissues, these TLS do not contain distinct T cell and B cell zones nor do they support germinal centers [116]. Single‐cell transcriptomic analyses of leptomeningeal TLS from 2D2 × IgHMOG mice identified clusters of cells resembling follicular and marginal zone B cells, as well as various populations of CD4+ T cells and myeloid cells—lymphocyte populations reminiscent of their counterparts in the spleen and lymph nodes, albeit leptomeningeal lymphocytes exhibit a more proinflammatory phenotype [117].
4.2. Nonobese Diabetic (NOD) EAE
NOD mice have traditionally been used as a model for type 1 diabetes (T1D) for their spontaneous onset of disease driven by an autoimmune T cell response towards pancreatic beta cell antigens [100]. However, NOD mice are also susceptible to EAE upon immunization with MOG35‐55 [101]. Immunized mice initially develop relapse‐remitting EAE which transitions into progressive disease worsening [118], although there is some debate as to whether this resembles true progression as seen in people with MS [119]. Nonetheless, the model has been successfully used to study axonal injury [120], astrocyte activation [118, 121], and mechanisms leading to cortical demyelinating lesions [122]. One study reported the development of TLS in the spinal cord leptomeninges of chronic NOD‐EAE mice that were populated by CD4+ T cells, B220+ B cells, and CD21/CD35+ follicular dendritic cells (FDCs) [102]. Additionally, transcriptomic analysis of chronic NOD‐EAE spinal cords showed evidence of stromal cell remodeling, revealing a potential mechanism for TLS formation in this model [102].
To study the role of myelin‐reactive B cells, transgenic IgHMOG mice were backcrossed onto the NOD background [123]. Unlike NOD WT mice, which present initially with relapsing–remitting disease, NOD IgH[MOG] mice develop a rapid and severe form of progressive EAE after active immunization with MOG35‐55 peptide [103]. In this model, brain‐adjacent leptomeningeal TLS are observed and are colocalized with a fibronectin‐rich, PDGFRα/β+ stromal cell network [103]. These TLS show prominent accumulation of B220+ B cells, class‐switched CD138+ plasma cells, and T peripheral helper (Tph)‐like PD‐1+CXCR5− cells within the leptomeninges [103]. Interestingly, IgHMOG B cells may also exacerbate Th17‐driven passive EAE in the NOD background through a mechanism dependent on IL‐23 [103], a well‐established encephalitogenic cytokine [124, 125].
4.3. Biozzi AB/H EAE
Immunization with spinal cord homogenate emulsified in CFA in young Biozzi ABH mice (8–12 weeks old) induces a relapsing–remitting form of EAE, which transitions into a chronic, nonremitting disease approximately 3 months after immunization [104, 126]. In contrast, applying the same immunization protocol to aged Biozzi ABH mice (12 months or older) leads to the immediate onset of a progressive, nonremitting disease course, bypassing the initial relapsing–remitting phase entirely [105]. Aged mice not only exhibit this altered disease trajectory but also demonstrate exacerbated neuropathology compared to their younger counterparts. This includes more pronounced axonal damage, heightened microglial activation, and a significant increase in CD3+ T cell infiltration into the spinal cord. Additionally, both the incidence of EAE and the rate of disease‐associated mortality rise with age. Notably, juvenile mice (younger than 2 weeks) display a remarkable resistance to EAE induction, suggesting a potential age‐dependent vulnerability to CNS autoimmune responses. While this model provides valuable insights into the relationship between aging, microglial activation, peripheral T cell infiltration, and WM pathology within the spinal cord, it does not reproduce the formation of leptomeningeal TLS near regions of cortical GM injury, as observed in people with PMS [15, 71, 126, 127, 128]. Furthermore, as with all active models of EAE, the autoimmune response in this model is triggered using spinal cord homogenate and CFA. It remains possible that some aspects of CNS pathology may be influenced or exacerbated by the strong adjuvant‐induced inflammatory response, potentially confounding the interpretation of disease mechanisms.
4.4. SJL/J PLP139 ‐151 Active EAE
SJL/J mice are highly susceptible to both active and passive PLP139‐151‐driven EAE. Active immunization with PLP139‐151 in SJL/J mice induces a relapse‐remitting disease even without coadministration of pertussis toxin, making it a relevant model for understanding RRMS [106, 129]. On the other hand, adoptive transfer (A/T) of PLP139‐151‐primed T cells skewed ex vivo with IL‐23 induces a monophasic disease followed by a period of recovery in naïve recipient mice [43]. While disease course in SJL/J PLP139‐151 EAE can be influenced by method of EAE induction, sex [130] and age of mice [131, 132], and even substrain differences in gene copy numbers across vendors [107], the development of prominent leptomeningeal TLS in the brain remains consistent [43, 107, 132].
As mentioned, active immunization with PLP139‐151 induces a relapsing–remitting EAE (RR‐EAE) [13]. This disease course occurs with the appearance of TLS in the meninges and upregulation of Baff and Cxcl13 transcripts that increase with each relapse and wane with each remission. During the first relapse, there is an influx of CD4+ T cells and some B cell accumulation in the leptomeninges. However, during remission, leptomeningeal B cells become more clustered, and T cells are still visible at similar levels throughout the disease course [13], suggesting that leptomeningeal TLS persists even as clinical symptoms subside. These observations support the notion that TLS‐derived factors may contribute towards progression over time, making this model an appealing tool for dissecting the roles of TLS that ultimately lead to disease progression.
MRI studies in mice have been pivotal in elucidating pathologies associated with neuroinflammation and protective or detrimental effects of therapeutic interventions. Calabresi and colleagues utilized the active SJL/J EAE model to better understand the relationship between MRI studies and corresponding histopathology [133]. They demonstrated that areas of leptomeningeal enhancement visible by MRI matched areas that showed high infiltration of immune cells, including areas with features consistent with TLS, such as an accumulation of FDCs, B cells, T cells, macrophages, and CXCL13‐producing cells [13, 133]. In adjacent cortical gray matter, they identified regions of damage, including demyelination, astrocytosis, and microgliosis. Utilizing the power of this model, Calabresi and colleagues showed that treatment with Bruton's tyrosine kinase (BTK) inhibitor reduced leptomeningeal enhancement in the treated mice compared to the vehicle group [133].
4.5. SJL/J PLP139 ‐151 Passive EAE
The consistent induction of leptomeningeal TLS in the brain in SJL/J A/T EAE mice has established this model as a valuable tool for probing the well‐documented association between leptomeningeal inflammation and GM injury in MS [15, 69, 72, 134], a process that remains poorly understood. In the SJL/J A/T EAE model, recipient mice (8–10 weeks old) develop brain leptomeningeal TLS underpinned by a fibronectin+ and PDGFRα/β+ stromal cell network, exhibiting various degrees of lymphocyte infiltration across the leptomeninges overlying different anatomical locations in the brain, including the hippocampal, cerebellar, and brainstem sulci, and periventricular regions [43]. Notably, these TLS are associated with a gradient of cortical gray matter demyelination and increased microglial activation in TLS‐proximal regions [135]. While EAE and leptomeningeal TLS are initiated by Th17 cells in this model [43], the age of the recipient can alter the persistence of TLS. In aged recipient mice (> 8 months) that receive PLP139‐151‐primed Th17 cells from young donor animals, TLS is sustained in the brain leptomeninges, correlating with a progressive disease phenotype including loss of brain volume [132]. While CD4+ T cell numbers and cytokine secretion in the leptomeninges are relatively consistent between young and aged recipient mice, TLS in aged mice contained more neutrophils and class‐switched B cells [136]. This is also seen in postmortem brain tissues in people with PMS [132]. Using this A/T EAE model, we and others have been able to further interrogate the mechanisms of MS therapies on modulating leptomeningeal TLS and the role of aging in TLS persistence.
5. Formation and Persistence of Leptomeningeal TLS in MS/EAE
5.1. TLS Elaboration—The Lymphotoxin Pathway
As mentioned, LTβR signaling is critical for the formation of lymph nodes in utero and the homeostatic maintenance of secondary lymphoid tissue organization in the adult. Accordingly, the disruption of the LT pathway in mice via genetic deletion of Lta, Ltb, or Ltbr results in the absence of lymph nodes and Peyer's patches [4, 137], as well as architectural disorganization of the spleen and thymus [137]. Although LTαβ is expressed on LTi during development, in adults, LTαβ expression is found on T cells, B cells, innate lymphoid cells, and NK cells. Additionally, LIGHT (TNFSF14)—another ligand for LTβR—is expressed by T cells and myeloid cells, including neutrophils, macrophages, and DCs [4, 84].
It has been proposed that Th17 cells—a T helper subset implicated in MS and EAE—may mimic the in utero function of LTi cells to initiate TLS formation. In support of this, Pikor et al. adoptively transferred PLP139–151‐specific Th17 cells into SJL/J mice to induce EAE and examined the leptomeninges for TLS development [43]. They observed an expansion of gp38+CD31−PDGFRα+PDGFRβ+ stromal cells, characteristic of FRCs, in the leptomeninges of EAE mice. In vitro studies further demonstrated that the addition of recombinant IL‐17 and IL‐22 to meningeal fibroblasts could induce the expression of ECM components, such as fibronectin and collagen, implicating Th17‐derived cytokines in the earliest steps of fibroblast remodeling and the establishment of TLS. Immunofluorescence microscopy revealed the accumulation of CD4+ T cells and B220+ B cells within a fibronectin+ERTR7+ ECM network, forming organized structures. These TLS were situated near areas of myelin rarefaction, paralleling findings from postmortem MS tissue. To dissect the role of LTβR signaling, the authors administered LTβR‐Ig intrathecally to block LTβR signaling. While this did not significantly alter the number or spatial organization of gp38+ stromal cells, it led to a reduction in B220+ B cells within the TLS, suggesting that LTβR signaling contributes to the maintenance or recruitment of B cells within the meningeal niche. Further, meningeal fibroblasts expressed transcripts for the B cell‐attracting chemokine Cxcl13, and Cxcl13 levels were nearly undetectable when EAE was induced in Ltbr −/− mice. Moreover, the absence of LTβR signaling in radio‐resistant, but not radio‐sensitive, cells impaired CD4+ T cell IL‐17 and IFN‐γ production.
Further work investigating the link between the LT pathway and TLS development is underway. In a preprint by Naouar et al., researchers used the SJL/J model to investigate whether inhibition of BTK impacts TLS formation and associated gray matter pathology [138]. Small molecule BTK inhibitors such as Tolebrutinib have been tested in PMS (HERCULES trial) and were found to slow disability progression compared to placebo [139]. Naouar and colleagues found that TLS development was abrogated in SJL/J A/T EAE mice following treatment with the BTK inhibitor remibrutinib. Further, treatment with remibrutinib protected against subpial cortical GM demyelination, microglial activation, and disruption of the glia limitans. By immunofluorescence, CXCL13 was found to be significantly reduced in EAE mice treated with remibrutinib within the leptomeninges. Whole tissue qPCR for lymphotoxin ligand transcripts revealed that remibrutinib‐treated mice downregulated leptomeningeal Ltb expression. These data suggest that BTK inhibition may influence the production of both CXCL13 and Ltb, and administration of an LTBR agonist to remibrutinib‐treated SJL/J A/T EAE mice reversed the protective effects of BTK inhibition and normalized CXCL13 expression in the leptomeninges to untreated levels [138]. Collectively, these results suggest a link between BTK, the LTBR pathway, and CXCL13 in the formation of brain leptomeningeal TLS and associated cortical pathology.
5.2. Expansion of the TLS: Th17 Cells and Neutrophils
In addition to inducing the formation of TLS, Th17 cells contribute to meningeal inflammation by recruiting other immune cells, particularly (Table 1) neutrophils (Figure 2). Neutrophils can be recruited by Th17 cells either directly or indirectly. Activated Th17 cells secrete IL‐17A, which can induce stromal cells (endothelial cells, fibroblasts) and glia to produce neutrophil chemoattractants CXCL1, CXCL5, CXCL6, and CXCL8 [140, 141]. Additionally, IL‐17A induces upregulation of G‐CSF, which leads to increased production of granulocyte progenitor cells in the bone marrow, thereby modulating neutrophil granulopoiesis [142]. In a mouse model of Staphylococcus aureus infection, Cavagnero et al. demonstrated that IL‐17A promotes fibroblast‐mediated neutrophil recruitment. Using scRNA‐seq 1 day postinfection, they identified CXCL12+ fibroblasts as major producers of neutrophil chemokines (Cxcl1, Cxcl2, Cxcl3, Cxcl5), which were upregulated in response to IL‐17A [143]. Time‐course analysis showed early induction of Cxcl1 and Cxcl5 (within 3 h), followed by Cxcl12 upregulation (by 12 h), with chemokine expression declining by Day 2 and returning to baseline by Day 10. The authors suggest Cxcl1/Cxcl5 drive early neutrophil recruitment, while Cxcl12 may mediate later recruitment. These findings raise the possibility that fibroblasts in the leptomeninges and brain may similarly coordinate neutrophil recruitment in IL‐17–driven neuroinflammation.
FIGURE 2.
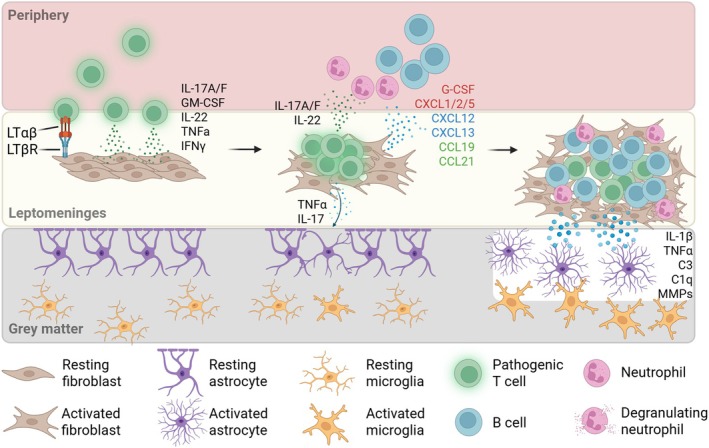
Elaboration of a leptomeningeal immune cell niche. (Left) Invasion of the leptomeninges by pathogenic Th17 cells. Engagement of LTβR on fibroblasts and secretion of inflammatory factors such as IL‐17 and IL‐22 induces fibroblast remodeling and activation. Glia limitans remains intact and gray matter is undisturbed. (Middle) Activated fibroblasts produce factors that induce granulopoiesis in the periphery (G‐CSF), as well as chemokines for neutrophil (in red), B cell (in blue), and T cell (in green) recruitment to the inflammatory milieu. Secretion of IL‐17 and TNFα by Th17 cells disrupts the subpial glia limitans. (Right) Prolonged inflammation and immune cell recruitment result in an elaborated lymphoid niche supported by activated fibroblasts (TLS). Inflammatory factors secreted by immune cells in the TLS further disrupt glia limitans (MMPs) and cause activation of glial cells (IL‐1β, TNFα, C3, C1q), resulting in damage to gray matter.
Given that neutrophil granules are packed with ECM‐degrading enzymes such as MMPs, neutrophils may further contribute to the remodeling of the leptomeningeal space, potentially to promote TLS expansion and/or to erode the underlying glia limitans and allow infiltration of other immune cells. Indeed, Mmp2 and Mmp9 double knockout mice show resistance to EAE and reduced leukocyte penetration of the CNS [144], as well as persistence of an intact BBB [144].
Lastly, a study by Harp et al. has established a putative role for neutrophils in guiding B cell trafficking to the spinal cord leptomeninges [145]. Using a model that ensures B cell‐restricted antigen presentation to T cells, these mice develop B cell‐rich follicles in the leptomeninges overlying the spinal cord. Blockade of CXCR2, a neutrophil‐specific chemokine receptor, resulted in the disappearance of B cell follicles, suggesting that neutrophil infiltration into the spinal cord leptomeninges is required for B cell recruitment. These data suggest that leptomeningeal neutrophil infiltration influences B cell recruitment. B cell infiltration into the CNS and the role of B cells in perpetuating the TLS will be discussed in the next section.
5.3. Expansion of the TLS: B Cells
Our group has recently profiled B cell subtypes that infiltrate the CNS during SJL/J A/T EAE, and the effect of age on the types of B cells that appear in the LM and dura [136]. While young mice have an accumulation of B cells in the leptomeninges at peak disease, old mice have a paucity of B cells [132]. Single‐cell RNA sequencing of these cells revealed that many of the B cells infiltrating the young LM are developing B cells based on the expression of VpreB, Ebf1, Cd79b, and Ighm [132]. These cells are partially resistant to anti‐CD20 treatment and may potentially be sustained by the increase in CNS BAFF concentrations after B cell depletion [136, 146]. Florescu et al. recently explored this phenotype further, finding that while young mice exhibit an accumulation of B220low developing B cells that are IgM+ and IgD+, old mice harbored more class‐switched (IgM−IgD−) B220high mature B cells [136]. Moreover, the ratio of B220high‐to‐B220low B cells decreases with age [136]. Therefore, class‐switched B cells correlate with a nonremitting clinical phenotype and persistent LM aggregates, whereas B220low B cells are associated with disease remission. In humans, aging is associated with a shift in B cell phenotype, including the expansion of age‐associated B cells (ABCs)—a subset implicated in chronic inflammation and autoimmunity [147]. Notably, ABCs accumulate within the dura mater over time and may displace other regulatory B cell subsets, such as IgA‐producing plasma cells, which have been shown by our group to deliver anti‐inflammatory cytokines like IL‐10 to the CNS during inflammation [148]. These findings suggest that the aging meningeal immune environment may influence B cell composition and function in ways that impact disease progression and regulation.
5.4. Regulation of the TLS: DCs, Monocytes and Macrophages
Monocytes, macrophages, and dendritic cells (DCs) may play important roles in CNS autoimmunity, but how they contribute to the formation and maintenance of TLS in the brain is unclear. Infiltrating monocytes are recruited to the CNS during inflammation via CCL2–CCR2 signaling and differentiate into macrophages within active lesions where they produce pro‐inflammatory cytokines (e.g., TNF, IL‐1β, IL‐6) and reactive oxygen species, contributing to demyelination and axonal damage [149, 150, 151]. These monocyte‐derived macrophages often adopt an M1‐like phenotype in active MS lesions, while a shift toward M2‐like profiles may be associated with tissue repair in resolving lesions [149, 152]. Macrophages and meningeal‐resident myeloid cells also play a structural and immunoregulatory role in TLS formation. They can interact with FRCs to remodel the ECM and produce lymphoid chemokines such as CXCL13, CCL19, and CCL21, which are critical for lymphocyte recruitment and organization within TLS [14, 153, 154, 155]. These chemokines attract CXCR5+ B cells and CCR7+ T cells, promoting compartmentalized adaptive immune responses. Notably, young SJL/J mice also have an accumulation of Ly6C+ monocytes in the leptomeninges during the acute phase of A/T EAE, although most of these monocytes disappear during remission. In aged mice, monocytes are continually present in the leptomeninges. Emerging studies have shown that monocytes from the skull and vertebral bone marrow can differentiate into macrophages (monocyte‐derived macrophages; MDMs) after infiltrating the brain parenchyma and brain borders [22, 24, 156]. These MDMs, although transcriptionally distinct from yolk‐sac derived microglia, can repopulate microglia‐depleted brains at steady‐state [24, 156], and invade the meninges and parenchyma during inflammation and injury [156]. Moreover, MDMs from the bone marrow were found to express genes associated with wound healing while blood‐derived myeloid cells expressed genes involved in propagating inflammation [24]. Follow‐up studies are required to determine whether the persistent monocyte signature in the leptomeninges of aged mice is pathogenic or immunoregulatory, and elucidating their origin may give some insights into their function.
Dendritic cells (DCs) are found within the leptomeninges, perivascular spaces, and SAS of both people with MS and EAE models, where they function as professional antigen‐presenting cells (APCs). They present CNS‐derived antigens to T cells via MHC class II and costimulatory molecules such as CD80/86 and CD40, supporting the activation and expansion of autoreactive T cells [157, 158]. Moreover, conventional DCs (cDCs) can produce IL‐12 and IL‐23, promoting the differentiation of Th1 and Th17 cells, both of which contribute to disease pathology and potentially to TLS induction [158, 159, 160, 161, 162]. Pikor et al. demonstrated the presence of leptomeningeal CD11c+ cells interspersed amongst CD4+ and B220+ cells in the SJL/J A/T model of EAE [43], although whether these myeloid cells were actively presenting antigen to lymphocytes is unknown. At steady‐state DCs have been found to populate brain regions where lesions tend to form, such as periventricular areas, and it has been theorized that these cells may act as gatekeepers for CD4+ T cells entering the brain [163, 164]. The Th17‐polarizing capability of DCs, coupled with their presence near or within TLS‐like structures, suggests a role for these cells in sustaining local immune activation [43, 157, 158, 165].
Collectively, myeloid cells may contribute to TLS persistence through chemokine‐driven recruitment of lymphocytes and polarization of Th17 differentiation while also driving pathogenic inflammation via antigen presentation and cytokine production.
5.5. Resolution of TLS
The resolution of TLS is a complex and poorly understood process, especially in the context of chronic diseases like MS, where we only have cross‐sectional (postmortem) tissue to study their composition. In peripheral tissues, TLS resolution is thought to occur via a combination of mechanisms, including the cessation of pro‐inflammatory cytokine and chemokine signaling, loss of stromal cell activation, and re‐entry of immune cells into circulation or draining lymphatics. Anti‐inflammatory cytokines (e.g., IL‐10 [121, 148], TGF‐β), regulatory T cells [166, 167], and immunosuppressive macrophages and B cells or plasma cells [60] may also play active roles in suppressing lymphoid neogenesis and promoting tissue remodeling [167].
Aging appears to negatively impact the resolution of TLS. As mentioned earlier, SJL/J mice over 8 months of age develop TLS following adoptive transfer of encephalitogenic T cells, and unlike young mice, these TLS persist in the CNS concomitant with a nonremitting clinical phenotype. This suggests that age is a predictor of impaired TLS resolution. One possible reason that has been explored in other settings is due to dysfunctional clearance mechanisms in the aged brain. For many years, the CNS was thought to lack classical lymphatic drainage. However, recent discoveries have overturned this view, revealing that the brain has both lymphatic and glymphatic mechanisms for fluid and solute clearance. Studies investigating lymphatic drainage have shown that this could be mediated by lymphocytes egressing from the leptomeninges through arachnoid cuff exit (ACE) points that lead into the dura, as molecules for retention are slowly outweighed by chemotactic signals from elsewhere in the periphery, such as the dura or cervical lymph nodes.
Rustenhoven and colleagues recently found that an aged dural lymphatic system exhibits changes in ECM remodeling [168]. For example, using immunostaining for type I collagen fibers, they found thicker bands of collagen‐positive staining near the dural sinuses of aged mice compared to young. This was supported by an increase in Col1a1 expression in the aged dura compared to the young dura. Furthermore, ECM remodeling driven by constitutive expression of Tgfbr1 using an adeno‐associated viral (AAV) vector resulted in an impaired drainage of intrathecal OVA to the cervical lymph nodes as well as impaired accumulation of OVA in dural macrophages and DCs. These data suggest ECM remodeling results in impaired clearance of CSF proteins, as well as a deficit in immunosurveillance by dural myeloid cells. Other studies in the context of subarachnoid hemorrhage and Alzheimer's disease have shown that promoting T cell egress via the CCR7‐CCL21 pathway is crucial for neuroprotection [169, 170]. It is tempting to speculate that impaired lymphatic drainage due to ECM remodeling may also cause lymphocytes to become trapped in the leptomeninges during neuroinflammation and may act as a roadblock to resolving inflammation in aged SJL/J A/T EAE mice.
6. Concluding Thoughts
Much progress has been made in our understanding of how TLS forms in brain‐adjacent regions, specifically the leptomeninges. This has tremendous impact not only on how we think about the neuro‐immune axis but also on how we may treat PMS, which is characterized by persistent leptomeningeal TLS that correlates with GM pathology. However, many key questions remain unresolved. For example, what is happening in the dura that overlies leptomeningeal TLS? Of note, Florescu et al. compared the kinetics of B cell infiltration into the brain, leptomeninges, and dura during neuroinflammation and observed a conspicuous loss of immature dural B cells during EAE [136] followed by their accumulation in the leptomeninges and brain parenchyma in young but not old mice. Whether these immature dural B cells contribute to disease remission remains to be determined. Another black box is the identity of leptomeningeal fibroblasts and determining if they change during steady state versus during MS/EAE [171]. Do fibroblasts perpetuate neuroinflammation by creating a self‐sufficient niche in the CNS? In addition, we know that aging alters fibroblast phenotype in the context of traumatic brain injury [172, 173]; is the same true for EAE, and if so, could that be another reason why TLS does not resolve in aged mice? Answers to these questions will allow us to better understand the dynamics of TLS formation and resolution and may provide a therapeutic window of opportunity for the treatment of PMS.
Author Contributions
M.Z., A.A.W., and J.L.G. worked together in writing the manuscript. M.Z. compiled the sections and generated figures. M.Z., A.A.W., and J.L.G. reviewed and edited the study.
Conflicts of Interest
The authors declare no conflicts of interest.
Zuo M., Wang A. A., and Gommerman J. L., “Follicle on the Roof: Tertiary Lymphoid Structures in Central Nervous System Autoimmunity,” Immunological Reviews 332, no. 1 (2025): e70045, 10.1111/imr.70045.
Funding: This work was supported by the Multiple Sclerosis Society of Canada and the National Multiple Sclerosis Society, RFA‐2203‐39259.
Michelle Zuo and Angela A. Wang contributed equally to this study.
Data Availability Statement
Data sharing not applicable to this article as no datasets were generated or analysed during the current study.
References
- 1. Randall T. D., Carragher D. M., and Rangel‐Moreno J., “Development of Secondary Lymphoid Organs,” Annual Review of Immunology 26 (2008): 627–650, 10.1146/ANNUREV.IMMUNOL.26.021607.090257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ansel K. M., Ngo V. N., Hyman P. L., et al., “A Chemokine‐Driven Positive Feedback Loop Organizes Lymphoid Follicles,” Nature 406, no. 6793 (2000): 309–314, 10.1038/35018581. [DOI] [PubMed] [Google Scholar]
- 3. Ansel K. M., Harris R. B. S., and Cyster J. G., “CXCL13 is Required for B1 Cell Homing, Natural Antibody Production, and Body Cavity Immunity,” Immunity 16, no. 1 (2002): 67–76, 10.1016/S1074-7613(01)00257-6. [DOI] [PubMed] [Google Scholar]
- 4. Gommerman J. L., Browning J. L., and Ware C. F., “The Lymphotoxin Network: Orchestrating a Type I Interferon Response to Optimize Adaptive Immunity,” Cytokine & Growth Factor Reviews 25, no. 2 (2014): 139–145, 10.1016/J.CYTOGFR.2014.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sautès‐Fridman C., Petitprez F., Calderaro J., and Fridman W. H., “Tertiary Lymphoid Structures in the Era of Cancer Immunotherapy,” Nature Reviews. Cancer 19, no. 6 (2019): 307–325, 10.1038/S41568-019-0144-6. [DOI] [PubMed] [Google Scholar]
- 6. Pipi E., Nayar S., Gardner D. H., Colafrancesco S., Smith C., and Barone F., “Tertiary Lymphoid Structures: Autoimmunity Goes Local,” Frontiers in Immunology 9 (2018): 9(SEP), 10.3389/FIMMU.2018.01952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhang G., Feizi N., Zhao D., et al., “Lymphotoxin β Receptor and Tertiary Lymphoid Organs Shape Acute and Chronic Allograft Rejection,” JCI Insight 9, no. 15 (2024): 177555, 10.1172/JCI.INSIGHT.177555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ruddle N. H., “Posttransplant Tertiary Lymphoid Organs,” Transplantation 108, no. 5 (2024): 1090–1099, 10.1097/TP.0000000000004812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Neyt K., Perros F., GeurtsvanKessel C. H., Hammad H., and Lambrecht B. N., “Tertiary Lymphoid Organs in Infection and Autoimmunity,” Trends in Immunology 33, no. 6 (2012): 297–305, 10.1016/j.it.2012.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pikor N. B., Prat A., Bar‐Or A., and Gommerman J. L., “Meningeal Tertiary Lymphoid Tissues and Multiple Sclerosis: A Gathering Place for Diverse Types of Immune Cells During CNS Autoimmunity,” Frontiers in Immunology 6 (2016): 657, 10.3389/FIMMU.2015.00657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bombardieri M., Barone F., Humby F., et al., “Activation‐Induced Cytidine Deaminase Expression in Follicular Dendritic Cell Networks and Interfollicular Large B Cells Supports Functionality of Ectopic Lymphoid Neogenesis in Autoimmune Sialoadenitis and MALT Lymphoma in Sjögren's Syndrome,” Journal of Immunology 179, no. 7 (2007): 4929–4938, 10.4049/JIMMUNOL.179.7.4929. [DOI] [PubMed] [Google Scholar]
- 12. Bombardieri M., Lewis M., and Pitzalis C., “Ectopic Lymphoid Neogenesis in Rheumatic Autoimmune Diseases,” Nature Reviews Rheumatology 13, no. 3 (2017): 141–154, 10.1038/NRRHEUM.2016.217. [DOI] [PubMed] [Google Scholar]
- 13. Magliozzi R., Columba‐Cabezas S., Serafini B., and Aloisi F., “Intracerebral Expression of CXCL13 and BAFF is Accompanied by Formation of Lymphoid Follicle‐Like Structures in the Meninges of Mice With Relapsing Experimental Autoimmune Encephalomyelitis,” Journal of Neuroimmunology 148, no. 1–2 (2004): 11–23, 10.1016/j.jneuroim.2003.10.056. [DOI] [PubMed] [Google Scholar]
- 14. Serafini B., Rosicarelli B., Magliozzi R., Stigliano E., and Aloisi F., “Detection of Ectopic B‐Cell Follicles With Germinal Centers in the Meninges of Patients With Secondary Progressive Multiple Sclerosis,” Brain Pathology 14, no. 2 (2004): 164–174, 10.1111/J.1750-3639.2004.TB00049.X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Magliozzi R., Howell O., Vora A., et al., “Meningeal B‐Cell Follicles in Secondary Progressive Multiple Sclerosis Associate With Early Onset of Disease and Severe Cortical Pathology,” Brain 130, no. Pt 4 (2007): 1089–1104, 10.1093/BRAIN/AWM038. [DOI] [PubMed] [Google Scholar]
- 16. Ellwardt E., Walsh J. T., Kipnis J., and Zipp F., “Understanding the Role of T Cells in CNS Homeostasis,” Trends in Immunology 37, no. 2 (2016): 154–165, 10.1016/j.it.2015.12.008. [DOI] [PubMed] [Google Scholar]
- 17. Smolders J., Heutinck K. M., Fransen N. L., et al., “Tissue‐Resident Memory T Cells Populate the Human Brain,” Nature Communications 9, no. 1 (2018): 4593, 10.1038/S41467-018-07053-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yoshida T. M., Nguyen M., Zhang L., et al., “The Subfornical Organ is a Nucleus for Gut‐Derived T Cells That Regulate Behaviour,” Nature (2025): 1–10, 10.1038/S41586-025-09050-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kadry H., Noorani B., and Cucullo L., “A Blood–Brain Barrier Overview on Structure, Function, Impairment, and Biomarkers of Integrity,” Fluids and Barriers of the CNS 17, no. 1 (2020): 69, 10.1186/S12987-020-00230-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rustenhoven J., Drieu A., Mamuladze T., et al., “Functional Characterization of the Dural Sinuses as a Neuroimmune Interface,” Cell 184, no. 4 (2021): 1000–1016.e27, 10.1016/j.cell.2020.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Balin B. J., Broadwell R. D., Salcman M., and El‐Kalliny M., “Avenues for Entry of Peripherally Administered Protein to the Central Nervous System in Mouse, Rat, and Squirrel Monkey,” Journal of Comparative Neurology 251, no. 2 (1986): 260–280, 10.1002/CNE.902510209. [DOI] [PubMed] [Google Scholar]
- 22. Herisson F., Frodermann V., Courties G., et al., “Direct Vascular Channels Connect Skull Bone Marrow and the Brain Surface Enabling Myeloid Cell Migration,” Nature Neuroscience 21, no. 9 (2018): 1209–1217, 10.1038/s41593-018-0213-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Brioschi S., Le Wang W., Peng V., et al., “Heterogeneity of Meningeal B Cells Reveals a Lymphopoietic Niche at the CNS Borders,” Science 373, no. 6553 (2021): 9277, 10.1126/SCIENCE.ABF9277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cugurra A., Mamuladze T., Rustenhoven J., et al., “Skull and Vertebral Bone Marrow Are Myeloid Cell Reservoirs for the Meninges and CNS Parenchyma,” Science 373, no. 6553 (2021): 7844, 10.1126/SCIENCE.ABF7844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yasuda K., Cline C., Vogel P., et al., “Drug Transporters on Arachnoid Barrier Cells Contribute to the Blood–Cerebrospinal Fluid Barrier,” Drug Metabolism and Disposition 41, no. 4 (2013): 923–931, 10.1124/DMD.112.050344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hutchings M. and Weller R. O., “Anatomical Relationships of the Pia Mater to Cerebral Blood Vessels in Man,” Journal of Neurosurgery 65, no. 3 (1986): 316–325, 10.3171/JNS.1986.65.3.0316. [DOI] [PubMed] [Google Scholar]
- 27. Zhang E. T., Inmant C. B. E., and Wellert R., “Interrelationships of the Pia Mater and the Perivascular (Virchow‐Robin) Spaces in the Human Cerebrum,” Journal of Anatomy 170 (1990): 111. [PMC free article] [PubMed] [Google Scholar]
- 28. Nag S., “Pathophysiology of Blood‐Brain Barrier Breakdown,” Methods in Molecular Medicine 89 (2003): 97–119, 10.1385/1-59259-419-0:97/COVER. [DOI] [PubMed] [Google Scholar]
- 29. Nicchia G. P., Nico B., Camassa L. M. A., et al., “The Role of Aquaporin‐4 in the Blood‐Brain Barrier Development and Integrity: Studies in Animal and Cell Culture Models,” Neuroscience 129, no. 4 (2004): 935–944, 10.1016/j.neuroscience.2004.07.055. [DOI] [PubMed] [Google Scholar]
- 30. Park J. S., Choe K., Khan A., et al., “Establishing Co‐Culture Blood‐Brain Barrier Models for Different Neurodegeneration Conditions to Understand Its Effect on BBB Integrity,” International Journal of Molecular Sciences 24, no. 6 (2023): 5283, 10.3390/IJMS24065283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kivisäkk P., Mahad D. J., Callahan M. K., et al., “Human Cerebrospinal Fluid Central Memory CD4+ T Cells: Evidence for Trafficking Through Choroid Plexus and Meninges via P‐Selectin,” Proceedings of the National Academy of Sciences of the United States of America 100, no. 14 (2003): 8389–8394, 10.1073/PNAS.1433000100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Krumbholz M., Theil D., Steinmeyer F., et al., “CCL19 is Constitutively Expressed in the CNS, Up‐Regulated in Neuroinflammation, Active and Also Inactive Multiple Sclerosis Lesions,” Journal of Neuroimmunology 190, no. 1–2 (2007): 72–79, 10.1016/j.jneuroim.2007.07.024. [DOI] [PubMed] [Google Scholar]
- 33. Morris A. W. J., Sharp M. M. G., Albargothy N. J., et al., “Vascular Basement Membranes as Pathways for the Passage of Fluid Into and out of the Brain,” Acta Neuropathologica 131, no. 5 (2016): 725–736, 10.1007/S00401-016-1555-Z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Engelhardt B., Carare R. O., Bechmann I., Flügel A., Laman J. D., and Weller R. O., “Vascular, Glial, and Lymphatic Immune Gateways of the Central Nervous System,” Acta Neuropathologica 132, no. 3 (2016): 317–338, 10.1007/S00401-016-1606-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mastorakos P. and McGavern D., “The Anatomy and Immunology of Vasculature in the Central Nervous System,” Science Immunology 4, no. 37 (2019): 492, 10.1126/sciimmunol.aav0492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Goldmann T., Wieghofer P., Jordão M. J. C., et al., “Origin, Fate and Dynamics of Macrophages at Central Nervous System Interfaces,” Nature Immunology 17, no. 7 (2016): 797–805, 10.1038/ni.3423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Spector R., Robert Snodgrass S., and Johanson C. E., “A Balanced View of the Cerebrospinal Fluid Composition and Functions: Focus on Adult Humans,” Experimental Neurology 273 (2015): 57–68, 10.1016/j.expneurol.2015.07.027. [DOI] [PubMed] [Google Scholar]
- 38. van Hove H., Martens L., Scheyltjens I., et al., “A Single‐Cell Atlas of Mouse Brain Macrophages Reveals Unique Transcriptional Identities Shaped by Ontogeny and Tissue Environment,” Nature Neuroscience 22, no. 6 (2019): 1021–1035, 10.1038/s41593-019-0393-4. [DOI] [PubMed] [Google Scholar]
- 39. Mrdjen D., Pavlovic A., Hartmann F. J., Merkler D., Greter M., and Correspondence B. B., “High‐Dimensional Single‐Cell Mapping of Central Nervous System Immune Cells Reveals Distinct Myeloid Subsets in Health, Aging, and Disease,” Immunity 48, no. 2 (2018): 380–395, 10.1016/j.immuni.2018.01.011. [DOI] [PubMed] [Google Scholar]
- 40. Arac A., Grimbaldeston M. A., Nepomuceno A. R. B., et al., “Evidence That Meningeal Mast Cells Can Worsen Stroke Pathology in Mice,” American Journal of Pathology 184, no. 9 (2014): 2493–2504, 10.1016/j.ajpath.2014.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yan M., Komatsu N., Muro R., et al., “ETS1 Governs Pathological Tissue‐Remodeling Programs in Disease‐Associated Fibroblasts,” Nature Immunology 23, no. 9 (2022): 1330–1341, 10.1038/s41590-022-01285-0. [DOI] [PubMed] [Google Scholar]
- 42. Buechler M. B., Pradhan R. N., Krishnamurty A. T., et al., “Cross‐Tissue Organization of the Fibroblast Lineage,” Nature 593, no. 7860 (2021): 575–579, 10.1038/S41586-021-03549-5. [DOI] [PubMed] [Google Scholar]
- 43. Pikor N. B., Astarita J. L., Summers‐Deluca L., et al., “Integration of Th17‐ and Lymphotoxin‐Derived Signals Initiates Meningeal‐Resident Stromal Cell Remodeling to Propagate Neuroinflammation,” Immunity 43, no. 6 (2015): 1160–1173, 10.1016/j.immuni.2015.11.010. [DOI] [PubMed] [Google Scholar]
- 44. Pikor N. B., Cupovic J., Onder L., Gommerman J. L., and Ludewig B., “Stromal Cell Niches in the Inflamed Central Nervous System,” Journal of Immunology 198, no. 5 (2017): 1775–1781, 10.4049/JIMMUNOL.1601566. [DOI] [PubMed] [Google Scholar]
- 45. Ramaglia V., Florescu A., Zuo M., Sheikh‐Mohamed S., and Gommerman J. L., “Stromal Cell‐Mediated Coordination of Immune Cell Recruitment, Retention, and Function in Brain‐Adjacent Regions,” Journal of Immunology 206, no. 2 (2021): 282–291, 10.4049/JIMMUNOL.2000833. [DOI] [PubMed] [Google Scholar]
- 46. Noor S., Habashy A. S., Nance J. P., et al., “CCR7‐Dependent Immunity During Acute Toxoplasma Gondii Infection,” Infection and Immunity 78, no. 5 (2010): 2257–2263, 10.1128/IAI.01314-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wilson E. H., Harris T. H., Mrass P., et al., “Behavior of Parasite‐Specific Effector CD8+ T Cells in the Brain and Visualization of a Kinesis‐Associated System of Reticular Fibers,” Immunity 30, no. 2 (2009): 300–311, 10.1016/J.IMMUNI.2008.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Jakimovski D., Bittner S., Zivadinov R., et al., “Multiple Sclerosis,” Lancet 403, no. 10422 (2024): 183–202, 10.1016/S0140-6736(23)01473-3. [DOI] [PubMed] [Google Scholar]
- 49. Klineova S. and Lublin F. D., “Clinical Course of Multiple Sclerosis,” Cold Spring Harbor Perspectives in Medicine 8, no. 9 (2018): a028928, 10.1101/CSHPERSPECT.A028928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Frischer J. M., Bramow S., Dal‐Bianco A., et al., “The Relation Between Inflammation and Neurodegeneration in Multiple Sclerosis Brains,” Brain 132, no. Pt 5 (2009): 1175–1189, 10.1093/BRAIN/AWP070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Makhani N. and Tremlett H., “The Multiple Sclerosis Prodrome,” Nature Reviews. Neurology 17, no. 8 (2021): 515–521, 10.1038/S41582-021-00519-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hollenbach J. A. and Oksenberg J. R., “The Immunogenetics of Multiple Sclerosis: A Comprehensive Review,” Journal of Autoimmunity 64 (2015): 13–25, 10.1016/J.JAUT.2015.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Naito S., Namerow N., Mickey M. R., and Terasaki P. I., “Multiple Sclerosis: Association With HL—A3,” Tissue Antigens 2, no. 1 (1972): 1–4, 10.1111/J.1399-0039.1972.TB00111.X. [DOI] [PubMed] [Google Scholar]
- 54. Hafler D. A., Hemler M. E., Christenson L., et al., “Investigation of In Vivo Activated T Cells in Multiple Sclerosis and Inflammatory Central Nervous System Diseases,” Clinical Immunology and Immunopathology 37, no. 2 (1985): 163–171, 10.1016/0090-1229(85)90147-3. [DOI] [PubMed] [Google Scholar]
- 55. Khoury S. J., Guttmann C. R. G., Orav E. J., Kikinis R., Jolesz F. A., and Weiner H. L., “Changes in Activated T Cells in the Blood Correlate With Disease Activity in Multiple Sclerosis,” Archives of Neurology 57, no. 8 (2000): 1183–1189, 10.1001/ARCHNEUR.57.8.1183. [DOI] [PubMed] [Google Scholar]
- 56. Gonatas N. K. and Howard J. C., “Inhibition of Experimental Allergic Encephalomyelitis in Rats Severely Depleted of T Cells,” Science 186, no. 4166 (1974): 839–841, 10.1126/SCIENCE.186.4166.839. [DOI] [PubMed] [Google Scholar]
- 57. Ortiz‐Ortiz L. and Weigle W. O., “Cellular Events in the Induction of Experimental Allergic Encephalomyelitis in Rats,” Journal of Experimental Medicine 144, no. 3 (1976): 604–616, 10.1084/JEM.144.3.604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Lee D. S. W., Rojas O. L., and Gommerman J. L., “B Cell Depletion Therapies in Autoimmune Disease: Advances and Mechanistic Insights,” Nature Reviews. Drug Discovery 20, no. 3 (2021): 179–199, 10.1038/S41573-020-00092-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Piccio L., Naismith R. T., Trinkaus K., et al., “Changes in B‐ and T‐Lymphocyte and Chemokine Levels With Rituximab Treatment in Multiple Sclerosis,” Archives of Neurology 67, no. 6 (2010): 707–714, 10.1001/ARCHNEUROL.2010.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wang A., Rojas O., Lee D., and Gommerman J. L., “Regulation of Neuroinflammation by B Cells and Plasma Cells,” Immunological Reviews 299, no. 1 (2021): 45–60, 10.1111/IMR.12929. [DOI] [PubMed] [Google Scholar]
- 61. Shen P. and Fillatreau S., “Antibody‐Independent Functions of B Cells: A Focus on Cytokines,” Nature Reviews. Immunology 15, no. 7 (2015): 441–451, 10.1038/NRI3857. [DOI] [PubMed] [Google Scholar]
- 62. Eshaghi A., Marinescu R. V., Young A. L., et al., “Progression of Regional Grey Matter Atrophy in Multiple Sclerosis,” Brain 141, no. 6 (2018): 1665–1677, 10.1093/BRAIN/AWY088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Calabrese M., Magliozzi R., Ciccarelli O., Geurts J. J. G., Reynolds R., and Martin R., “Exploring the Origins of Grey Matter Damage in Multiple Sclerosis,” Nature Reviews. Neuroscience 16, no. 3 (2015): 147–158, 10.1038/nrn3900. [DOI] [PubMed] [Google Scholar]
- 64. Geurts J. J. G., Calabrese M., Fisher E., and Rudick R. A., “Measurement and Clinical Effect of Grey Matter Pathology in Multiple Sclerosis,” Lancet Neurology 11, no. 12 (2012): 1082–1092, 10.1016/S1474-4422(12)70230-2. [DOI] [PubMed] [Google Scholar]
- 65. Chard D. T., Griffin C. M., Rashid W., et al., “Progressive Grey Matter Atrophy in Clinically Early Relapsing‐Remitting Multiple Sclerosis,” Multiple Sclerosis 10, no. 4 (2004): 387–391, 10.1191/1352458504MS1050OA. [DOI] [PubMed] [Google Scholar]
- 66. Khaleeli Z., Cercignani M., Audoin B., Ciccarelli O., Miller D. H., and Thompson A. J., “Localized Grey Matter Damage in Early Primary Progressive Multiple Sclerosis Contributes to Disability,” NeuroImage 37, no. 1 (2007): 253–261, 10.1016/J.NEUROIMAGE.2007.04.056. [DOI] [PubMed] [Google Scholar]
- 67. Benedict R. H. B., Ramasamy D., Munschauer F., Weinstock‐Guttman B., and Zivadinov R., “Memory Impairment in Multiple Sclerosis: Correlation With Deep Grey Matter and Mesial Temporal Atrophy,” Journal of Neurology, Neurosurgery, and Psychiatry 80, no. 2 (2009): 201–206, 10.1136/JNNP.2008.148403. [DOI] [PubMed] [Google Scholar]
- 68. Chard D. and Miller D., “Grey Matter Pathology in Clinically Early Multiple Sclerosis: Evidence From Magnetic Resonance Imaging,” Journal of the Neurological Sciences 282, no. 1–2 (2009): 5–11, 10.1016/J.JNS.2009.01.012. [DOI] [PubMed] [Google Scholar]
- 69. Howell O. W., Reeves C. A., Nicholas R., et al., “Meningeal Inflammation is Widespread and Linked to Cortical Pathology in Multiple Sclerosis,” Brain 134, no. 9 (2011): 2755–2771, 10.1093/BRAIN/AWR182. [DOI] [PubMed] [Google Scholar]
- 70. Popescu B. F. G., Parisi J. E., Cabrera‐Gómez J. A., et al., “Absence of Cortical Demyelination in Neuromyelitis Optica,” Neurology 75, no. 23 (2010): 2103–2109, 10.1212/WNL.0B013E318200D80C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Magliozzi R., Howell O. W., Reeves C., et al., “A Gradient of Neuronal Loss and Meningeal Inflammation in Multiple Sclerosis,” Annals of Neurology 68, no. 4 (2010): 477–493, 10.1002/ANA.22230. [DOI] [PubMed] [Google Scholar]
- 72. Reali C., Magliozzi R., Roncaroli F., Nicholas R., Howell O. W., and Reynolds R., “B Cell Rich Meningeal Inflammation Associates With Increased Spinal Cord Pathology in Multiple Sclerosis,” Brain Pathology 30, no. 4 (2020): 779–793, 10.1111/BPA.12841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Schafflick D., Xu C. A., Hartlehnert M., et al., “Integrated Single Cell Analysis of Blood and Cerebrospinal Fluid Leukocytes in Multiple Sclerosis,” Nature Communications 11, no. 1 (2020): 1–14, 10.1038/s41467-019-14118-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Serafini B., Rosicarelli B., Veroni C., Zhou L., Reali C., and Aloisi F., “RORγt Expression and Lymphoid Neogenesis in the Brain of Patients With Secondary Progressive Multiple Sclerosis,” Journal of Neuropathology and Experimental Neurology 75, no. 9 (2016): 877–888, 10.1093/JNEN/NLW063. [DOI] [PubMed] [Google Scholar]
- 75. Calabrese M., Poretto V., Favaretto A., et al., “Cortical Lesion Load Associates With Progression of Disability in Multiple Sclerosis,” Brain 135, no. Pt 10 (2012): 2952–2961, 10.1093/BRAIN/AWS246. [DOI] [PubMed] [Google Scholar]
- 76. Fischer M. T., Wimmer I., Höftberger R., et al., “Disease‐Specific Molecular Events in Cortical Multiple Sclerosis Lesions,” Brain 136, no. Pt 6 (2013): 1799–1815, 10.1093/BRAIN/AWT110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Magliozzi R., Hametner S., Facchiano F., et al., “Iron Homeostasis, Complement, and Coagulation Cascade as CSF Signature of Cortical Lesions in Early Multiple Sclerosis,” Annals of Clinical Translational Neurology 6, no. 11 (2019): 2150–2163, 10.1002/ACN3.50893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Ahmed S. M., Fransen N. L., Touil H., et al., “Accumulation of Meningeal Lymphocytes Correlates With White Matter Lesion Activity in Progressive Multiple Sclerosis,” JCI Insight 7, no. 5 (2022): 151683, 10.1172/JCI.INSIGHT.151683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Amato M. P., Portaccio E., Goretti B., et al., “Association of Neocortical Volume Changes With Cognitive Deterioration in Relapsing‐Remitting Multiple Sclerosis,” Archives of Neurology 64, no. 8 (2007): 1157–1161, 10.1001/ARCHNEUR.64.8.1157. [DOI] [PubMed] [Google Scholar]
- 80. Harrison D. M., Roy S., Oh J., et al., “Association of Cortical Lesion Burden on 7‐T Magnetic Resonance Imaging With Cognition and Disability in Multiple Sclerosis,” JAMA Neurology 72, no. 9 (2015): 1004–1012, 10.1001/JAMANEUROL.2015.1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Makshakov G., Magonov E., Totolyan N., et al., “Leptomeningeal Contrast Enhancement is Associated With Disability Progression and Grey Matter Atrophy in Multiple Sclerosis,” Neurological Research International 2017, no. 1 (2017): 8652463, 10.1155/2017/8652463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Absinta M., Vuolo L., Rao A., et al., “Gadolinium‐Based MRI Characterization of Leptomeningeal Inflammation in Multiple Sclerosis,” Neurology 85, no. 1 (2015): 18–28, 10.1212/WNL.0000000000001587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Kappos L., Moeri D., Radue E. W., et al., “Predictive Value of Gadolinium‐Enhanced Magnetic Resonance Imaging for Relapse Rate and Changes in Disability or Impairment in Multiple Sclerosis: A Meta‐Analysis,” Gadolinium MRI Meta‐analysis Group, Lancet 353, no. 9157 (1999): 964–969, 10.1016/S0140-6736(98)03053-0. [DOI] [PubMed] [Google Scholar]
- 84. James Bates R. E., Browne E., Schalks R., et al., “Lymphotoxin‐Alpha Expression in the Meninges Causes Lymphoid Tissue Formation and Neurodegeneration,” Brain 145, no. 12 (2022): 4287–4307, 10.1093/BRAIN/AWAC232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Koritschoner R. S. and Schweinburg F., “Induktion von Paralyse und Rückenmarksentzündung Durch Immunisierung von Kaninchen mit Menschlichem Rückenmarksgewebe,” Zeitschrift Für Immunitätsforschung Experimentelle Therapie 42 (1925): 217–283. [Google Scholar]
- 86. Rivers T. M. and Schwentke F. F., “Encephalomyelitis Accompanied by Myelin Destruction Experimentally Produced in Monkeys,” Journal of Experimental Medicine 61, no. 5 (1935): 689–702, 10.1084/JEM.61.5.689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Constantinescu C. S., Farooqi N., O'Brien K., and Gran B., “Experimental Autoimmune Encephalomyelitis (EAE) as a Model for Multiple Sclerosis (MS),” British Journal of Pharmacology 164, no. 4 (2011): 1079–1106, 10.1111/J.1476-5381.2011.01302.X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Stimmer L., Fovet C. M., and Serguera C., “Experimental Models of Autoimmune Demyelinating Diseases in Nonhuman Primates,” Veterinary Pathology 55, no. 1 (2018): 27–41, 10.1177/0300985817712794. [DOI] [PubMed] [Google Scholar]
- 89. Kulkarni P., Yellanki S., Medishetti R., Sriram D., Saxena U., and Yogeeswari P., “Novel Zebrafish EAE Model: A Quick In Vivo Screen for Multiple Sclerosis,” Multiple Sclerosis and Related Disorders 11 (2017): 32–39, 10.1016/j.msard.2016.11.010. [DOI] [PubMed] [Google Scholar]
- 90. Stromnes I. M. and Goverman J. M., “Active Induction of Experimental Allergic Encephalomyelitis,” Nature Protocols 1, no. 4 (2006): 1810–1819, 10.1038/NPROT.2006.285. [DOI] [PubMed] [Google Scholar]
- 91. Wolf S. D., Dittel B. N., Hardardottir F., and Janeway C. A., “Experimental Autoimmune Encephalomyelitis Induction in Genetically B Cell‐Deficient Mice,” Journal of Experimental Medicine 184, no. 6 (1996): 2271–2278, 10.1084/JEM.184.6.2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Lyons J. A., San M., Happ M. P., and Cross A. H., “B Cells Are Critical to Induction of Experimental Allergic Encephalomyelitis by Protein but Not by a Short Encephalitogenic Peptide,” European Journal of Immunology 29, no. 11 (1999): 3432–3439, 10.1002/(SICI)1521-4141(199911)29:11<3432::AID-IMMU3432>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 93. Pierson E. R., Stromnes I. M., and Goverman J. M., “B Cells Promote Induction of Experimental Autoimmune Encephalomyelitis by Facilitating Reactivation of T Cells in the Central Nervous System,” Journal of Immunology 192, no. 3 (2014): 929–939, 10.4049/JIMMUNOL.1302171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Rangachari M. and Kuchroo V. K., “Using EAE to Better Understand Principles of Immune Function and Autoimmune Pathology,” Journal of Autoimmunity 45 (2013): 31–39, 10.1016/j.jaut.2013.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Miller S. D., Karpus W. J., and Davidson T. S., “Experimental Autoimmune Encephalomyelitis in the Mouse,” Current Protocols in Immunology 88, no. 1 (2010): 15, 10.1002/0471142735.im1501s77. [DOI] [PubMed] [Google Scholar]
- 96. Stromnes I. M. and Goverman J. M., “Passive Induction of Experimental Allergic Encephalomyelitis,” Nature Protocols 1, no. 4 (2006): 1952–1960, 10.1038/nprot.2006.284. [DOI] [PubMed] [Google Scholar]
- 97. Roodselaar J., Zhou Y., Leppert D., Hauser A. E., Urich E., and Anthony D. C., “Anti‐CD20 Disrupts Meningeal B‐Cell Aggregates in a Model of Secondary Progressive Multiple Sclerosis,” Neurology Neuroimmunology & Neuroinflammation 8, no. 3 (2021): 975, 10.1212/NXI.0000000000000975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Bettelli E., Pagany M., Weiner H. L., Linington C., Sobel R. A., and Kuchroo V. K., “Myelin Oligodendrocyte Glycoprotein‐Specific T Cell Receptor Transgenic Mice Develop Spontaneous Autoimmune Optic Neuritis,” Journal of Experimental Medicine 197, no. 9 (2003): 1073–1081, 10.1084/JEM.20021603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Litzenburger T., Fässler R., Bauer J., et al., “B Lymphocytes Producing Demyelinating Autoantibodies: Development and Function in Gene‐Targeted Transgenic Mice,” Journal of Experimental Medicine 188, no. 1 (1998): 169–180, 10.1084/JEM.188.1.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Pearson J. A., Wong F. S., and Wen L., “The Importance of the Non Obese Diabetic (NOD) Mouse Model in Autoimmune Diabetes,” Journal of Autoimmunity 66 (2016): 76–88, 10.1016/j.jaut.2015.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Ignatius Arokia Doss P. M., Roy A. P., Wang A. L., Anderson A. C., and Rangachari M., “The Non‐Obese Diabetic Mouse Strain as a Model to Study CD8+ T Cell Function in Relapsing and Progressive Multiple Sclerosis,” Frontiers in Immunology 6 (2015): 6(OCT), 10.3389/FIMMU.2015.00541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Hamano S., Yoshimizu T., Mori M., Iida A., and Yamashita T., “Characterization of Pathological Stages in a Mouse Model of Progressive Multiple Sclerosis,” Neuroscience Research 204 (2024): 46–57, 10.1016/j.neures.2024.01.009. [DOI] [PubMed] [Google Scholar]
- 103. Fazazi M. R., Doss P. M. I. A., Pereira R., et al., “Myelin‐Reactive B Cells Exacerbate CD4+ T Cell‐Driven CNS Autoimmunity in an IL‐23‐Dependent Manner,” Nature Communications 15, no. 1 (2024): 5404, 10.1038/S41467-024-49259-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Al‐Izki S., Pryce G., O'Neill J. K., et al., “Practical Guide to the Induction of Relapsing Progressive Experimental Autoimmune Encephalomyelitis in the Biozzi ABH Mouse,” Multiple Sclerosis and Related Disorders 1, no. 1 (2012): 29–38, 10.1016/J.MSARD.2011.09.001. [DOI] [PubMed] [Google Scholar]
- 105. Peferoen L. A. N., Breur M., van de Berg S., et al., “Ageing and Recurrent Episodes of Neuroinflammation Promote Progressive Experimental Autoimmune Encephalomyelitis in Biozzi ABH Mice,” Immunology 149, no. 2 (2016): 146–156, 10.1111/IMM.12644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. McRae B. L., Kennedy M. K., Tan L. J., Dal Canto M. C., Picha K. S., and Miller S. D., “Induction of Active and Adoptive Relapsing Experimental Autoimmune Encephalomyelitis (EAE) Using an Encephalitogenic Epitope of Proteolipid Protein,” Journal of Neuroimmunology 38, no. 3 (1992): 229–240, 10.1016/0165-5728(92)90016-E. [DOI] [PubMed] [Google Scholar]
- 107. deLuca L. E., Pikor N. B., O'Leary J., et al., “Substrain Differences Reveal Novel Disease‐Modifying Gene Candidates That Alter the Clinical Course of a Rodent Model of Multiple Sclerosis,” Journal of Immunology 184, no. 6 (2010): 3174–3185, 10.4049/jimmunol.0902881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Kügler S., Böcker K., Heusipp G., Greune L., Kim K. S., and Schmidt M. A., “Pertussis Toxin Transiently Affects Barrier Integrity, Organelle Organization and Transmigration of Monocytes in a Human Brain Microvascular Endothelial Cell Barrier Model,” Cellular Microbiology 9, no. 3 (2007): 619–632, 10.1111/J.1462-5822.2006.00813.X. [DOI] [PubMed] [Google Scholar]
- 109. Fields T. A. and Casey P. J., “Signalling Functions and Biochemical Properties of Pertussis Toxin‐Resistant G‐Proteins,” Biochemical Journal 321, no. 3 (1997): 561–571, 10.1042/BJ3210561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Mendel I., de Rosbo N. K., and Ben‐Nun A., “A Myelin Oligodendrocyte Glycoprotein Peptide Induces Typical Chronic Experimental Autoimmune Encephalomyelitis in H‐2b Mice: Fine Specificity and T Cell Receptor Vβ Expression of Encephalitogenic T Cells,” European Journal of Immunology 25, no. 7 (1995): 1951–1959, 10.1002/EJI.1830250723. [DOI] [PubMed] [Google Scholar]
- 111. Pol S., Schweser F., Bertolino N., et al., “Characterization of Leptomeningeal Inflammation in Rodent Experimental Autoimmune Encephalomyelitis (EAE) Model of Multiple Sclerosis,” Experimental Neurology 314 (2019): 82–90, 10.1016/j.expneurol.2019.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Peters A., Pitcher L. A., Sullivan J. M., et al., “Th17 Cells Induce Ectopic Lymphoid Follicles in Central Nervous System Tissue Inflammation,” Immunity 35, no. 6 (2011): 986–996, 10.1016/j.immuni.2011.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Dang A. K., Jain R. W., Craig H. C., and Kerfoot S. M., “B Cell Recognition of Myelin Oligodendrocyte Glycoprotein Autoantigen Depends on Immunization With Protein Rather Than Short Peptide, While B Cell Invasion of the CNS in Autoimmunity Does Not,” Journal of Neuroimmunology 278 (2015): 73–84, 10.1016/j.jneuroim.2014.12.008. [DOI] [PubMed] [Google Scholar]
- 114. Krishnamoorthy G., Lassmann H., Wekerle H., and Holz A., “Spontaneous Opticospinal Encephalomyelitis in a Double‐Transgenic Mouse Model of Autoimmune T Cell/B Cell Cooperation,” Journal of Clinical Investigation 116, no. 9 (2006): 2385–2392, 10.1172/JCI28330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Bettelli E., Baeten D., Jäger A., Sobel R. A., and Kuchroo V. K., “Myelin Oligodendrocyte Glycoprotein‐Specific T and B Cells Cooperate to Induce a Devic‐Like Disease in Mice,” Journal of Clinical Investigation 116, no. 9 (2006): 2393–2402, 10.1172/JCI28334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Dang A. K., Tesfagiorgis Y., Jain R. W., Craig H. C., and Kerfoot S. M., “Meningeal Infiltration of the Spinal Cord by Non‐Classically Activated B Cells is Associated With Chronic Disease Course in a Spontaneous B Cell‐Dependent Model of CNS Autoimmune Disease,” Frontiers in Immunology 6 (2015): 6(SEP), 10.3389/FIMMU.2015.00470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Diddens J., Lepennetier G., Friedrich V., et al., “Single‐Cell Profiling Indicates a Proinflammatory Role of Meningeal Ectopic Lymphoid Tissue in Experimental Autoimmune Encephalomyelitis,” Neurology(R) Neuroimmunology & Neuroinflammation 11, no. 1 (2024): e200185, 10.1212/NXI.0000000000200185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Encinas J. A., Wicker L. S., Peterson L. B., et al., “QTL Influencing Autoimmune Diabetes and Encephalomyelitis Map to a 0.15‐cM Region Containing 1/2 [3],” Nature Genetics 21, no. 2 (1999): 158–160, 10.1038/5941. [DOI] [PubMed] [Google Scholar]
- 119. Baker D., Nutma E., O'Shea H., Cooke A., Orian J. M., and Amor S., “Autoimmune Encephalomyelitis in NOD Mice is Not Initially a Progressive Multiple Sclerosis Model,” Annals of Clinical Translational Neurology 6, no. 8 (2019): 1362–1372, 10.1002/ACN3.792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Basso A. S., Frenkel D., Quintana F. J., et al., “Reversal of Axonal Loss and Disability in a Mouse Model of Progressive Multiple Sclerosis,” Journal of Clinical Investigation 118, no. 4 (2008): 1532–1543, 10.1172/JCI33464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Mayo L., Da Cunha A. P., Madi A., et al., “IL‐10‐Dependent Tr1 Cells Attenuate Astrocyte Activation and Ameliorate Chronic Central Nervous System Inflammation,” Brain 139, no. 7 (2016): 1939–1957, 10.1093/BRAIN/AWW113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Levy H., Assaf Y., and Frenkel D., “Characterization of Brain Lesions in a Mouse Model of Progressive Multiple Sclerosis,” Experimental Neurology 226, no. 1 (2010): 148–158, 10.1016/j.expneurol.2010.08.017. [DOI] [PubMed] [Google Scholar]
- 123. Anderson A. C., Chandwaskar R., Lee D. H., et al., “A Transgenic Model of Central Nervous System Autoimmunity Mediated by CD4+ and CD8+ T and B Cells,” Journal of Immunology 188, no. 5 (2012): 2084–2092, 10.4049/JIMMUNOL.1102186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Langrish C. L., Chen Y., Blumenschein W. M., et al., “IL‐23 Drives a Pathogenic T Cell Population That Induces Autoimmune Inflammation,” Journal of Experimental Medicine 201, no. 2 (2005): 233–240, 10.1084/JEM.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. McGeachy M. J., Chen Y., Tato C. M., et al., “The Interleukin 23 Receptor is Essential for the Terminal Differentiation of Interleukin 17‐Producing Effector T Helper Cells In Vivo,” Nature Immunology 10, no. 3 (2009): 314–324, 10.1038/NI.1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Nishri Y., Fainstein N., Goldfarb S., et al., “Modeling Compartmentalized Chronic Immune‐Mediated Demyelinating CNS Disease in the Biozzi ABH Mouse,” Journal of Neuroimmunology 356 (2021): 577582, 10.1016/J.JNEUROIM.2021.577582. [DOI] [PubMed] [Google Scholar]
- 127. van Olst L., Rodriguez‐Mogeda C., Picon C., et al., “Meningeal Inflammation in Multiple Sclerosis Induces Phenotypic Changes in Cortical Microglia That Differentially Associate With Neurodegeneration,” Acta Neuropathologica 141, no. 6 (2021): 881–899, 10.1007/S00401-021-02293-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Bevan R. J., Evans R., Griffiths L., et al., “Meningeal Inflammation and Cortical Demyelination in Acute Multiple Sclerosis,” Annals of Neurology 84, no. 6 (2018): 829–842, 10.1002/ANA.25365. [DOI] [PubMed] [Google Scholar]
- 129. Kozlowski P. B., Schuller‐Levis G. B., and Wisniewski H. M., “Induction of Synchronized Relapses in SJL/J Mice With Chronic Relapsing Experimental Allergic Encephalomyelitis,” Acta Neuropathologica 74, no. 2 (1987): 163–168, 10.1007/BF00692847. [DOI] [PubMed] [Google Scholar]
- 130. Bebo B. F., Vandenbark A. A., and Offner H., “Male SJL Mice Do Not Relapse After Induction of EAE With PLP 139‐151,” Journal of Neuroscience Research 45, no. 6 (1996): 680–689, 10.1002/(SICI)1097-4547(19960915)45:6<680::AID-JNR4>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 131. Spach K. M., Blake M., Bunn J. Y., et al., “Cutting Edge: The Y Chromosome Controls the Age‐Dependent Experimental Allergic Encephalomyelitis Sexual Dimorphism in SJL/J Mice,” Journal of Immunology 182, no. 4 (2009): 1789–1793, 10.4049/JIMMUNOL.0803200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Zuo M., Fettig N., Bernier L. P., et al., “Age‐Dependent Gray Matter Demyelination is Associated With Leptomeningeal Neutrophil Accumulation,” JCI Insight 7 (2022): 158144, 10.1172/JCI.INSIGHT.158144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Bhargava P., Kim S., Reyes A. A., et al., “Imaging Meningeal Inflammation in CNS Autoimmunity Identifies a Therapeutic Role for BTK Inhibition,” Brain 144, no. 5 (2021): 1396–1408, 10.1093/BRAIN/AWAB045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Choi S. R., Howell O. W., Carassiti D., et al., “Meningeal Inflammation Plays a Role in the Pathology of Primary Progressive Multiple Sclerosis,” Brain 135, no. Pt 10 (2012): 2925–2937, 10.1093/BRAIN/AWS189. [DOI] [PubMed] [Google Scholar]
- 135. Ward L. A., Lee D. S. W., Sharma A., et al., “Siponimod Therapy Implicates Th17 Cells in a Preclinical Model of Subpial Cortical Injury,” JCI Insight 5, no. 1 (2020): 132522, 10.1172/jci.insight.132522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Florescu A., Zuo M., Wang A. A., et al., “Dynamic Alterations of Dural and Bone Marrow B Cells in an Animal Model of Progressive Multiple Sclerosis,” bioRxiv 8, no. 23 (2024): 609437, 10.1101/2024.08.23.609437. [DOI] [Google Scholar]
- 137. Mebius R. E., “Organogenesis of Lymphoid Tissues,” Nature Reviews Immunology 3, no. 4 (2003): 292–303, 10.1038/nri1054. [DOI] [PubMed] [Google Scholar]
- 138. Ramaglia V., Naouar I., Pangan A., et al., “Lymphotoxin‐Driven Meningeal BAFF/CXCL13 Imbalance and Grey Matter Injury,” Research Square (2024), 10.21203/RS.3.RS-5118485/V1. [DOI] [Google Scholar]
- 139. Fox R. J., Bar‐Or A., Traboulsee A., et al., “Tolebrutinib in Nonrelapsing Secondary Progressive Multiple Sclerosis,” New England Journal of Medicine 392 (2025): 5988, 10.1056/NEJMoa2415988. [DOI] [PubMed] [Google Scholar]
- 140. Wu L., Awaji M., Saxena S., Varney M. L., Sharma B., and Singh R. K., “IL‐17–CXC Chemokine Receptor 2 Axis Facilitates Breast Cancer Progression by Up‐Regulating Neutrophil Recruitment,” American Journal of Pathology 190, no. 1 (2020): 222–233, 10.1016/j.ajpath.2019.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Fan X., Shu P., Wang Y., Ji N., and Zhang D., “Interactions Between Neutrophils and T‐Helper 17 Cells,” Frontiers in Immunology 14 (2023): 1279837, 10.3389/FIMMU.2023.1279837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Witowski J., Ksia̧zek K., Warnecke C., et al., “Role of Mesothelial Cell‐Derived Granulocyte Colony‐Stimulating Factor in Interleukin‐17‐Induced Neutrophil Accumulation in the Peritoneum,” Kidney International 71, no. 6 (2007): 514–525, 10.1038/SJ.KI.5002082. [DOI] [PubMed] [Google Scholar]
- 143. Cavagnero K. J., Li F., Dokoshi T., et al., “CXCL12+ Dermal Fibroblasts Promote Neutrophil Recruitment and Host Defense by Recognition of IL‐17,” Journal of Experimental Medicine 221, no. 4 (2024): 276576, 10.1084/JEM.20231425/276576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Wu F., Cao W., Yang Y., and Liu A., “Extensive Infiltration of Neutrophils in the Acute Phase of Experimental Autoimmune Encephalomyelitis in C57BL/6 Mice,” Histochemistry and Cell Biology 133, no. 3 (2010): 313–322, 10.1007/S00418-009-0673-2. [DOI] [PubMed] [Google Scholar]
- 145. Parker Harp C. R., Archambault A. S., Cheung M., et al., “Neutrophils Promote VLA‐4–Dependent B Cell Antigen Presentation and Accumulation Within the Meninges During Neuroinflammation,” Proceedings of the National Academy of Sciences of the United States of America 116, no. 48 (2019): 24221–24230, 10.1073/PNAS.1909098116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Wang A. A., Luessi F., Neziraj T., et al., “B Cell Depletion With Anti‐CD20 Promotes Neuroprotection in a BAFF‐Dependent Manner in Mice and Humans,” Science Translational Medicine 16, no. 737 (2024): eadi0295, 10.1126/SCITRANSLMED.ADI0295. [DOI] [PubMed] [Google Scholar]
- 147. Cancro M. P., “Age‐Associated B Cells,” Annual Review of Immunology 38 (2020): 315–340, 10.1146/ANNUREV-IMMUNOL-092419-031130. [DOI] [PubMed] [Google Scholar]
- 148. Rojas O. L., Pröbstel A. K., Porfilio E. A., et al., “Recirculating Intestinal IgA‐Producing Cells Regulate Neuroinflammation via IL‐10,” Cell 176 (2019): 35, 10.1016/j.cell.2018.11.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Ajami B., Bennett J. L., Krieger C., McNagny K. M., and Rossi F. M. V., “Infiltrating Monocytes Trigger EAE Progression, but Do Not Contribute to the Resident Microglia Pool,” Nature Neuroscience 14, no. 9 (2011): 1142–1149, 10.1038/nn.2887. [DOI] [PubMed] [Google Scholar]
- 150. Mildner A., Schmidt H., Nitsche M., et al., “Microglia in the Adult Brain Arise From Ly‐6ChiCCR2+ Monocytes Only Under Defined Host Conditions,” Nature Neuroscience 10, no. 12 (2007): 1544–1553, 10.1038/NN2015. [DOI] [PubMed] [Google Scholar]
- 151. Mildner A., MacK M., Schmidt H., et al., “CCR2+Ly‐6Chi Monocytes are Crucial for the Effector Phase of Autoimmunity in the Central Nervous System,” Brain 132, no. 9 (2009): 2487–2500, 10.1093/BRAIN/AWP144. [DOI] [PubMed] [Google Scholar]
- 152. Yamasaki R., Lu H., Butovsky O., et al., “Differential Roles of Microglia and Monocytes in the Inflamed Central Nervous System,” Journal of Experimental Medicine 211, no. 8 (2014): 1533–1549, 10.1084/JEM.20132477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Carragher D. M., Rangel‐Moreno J., and Randall T. D., “Ectopic Lymphoid Tissues and Local Immunity,” Seminars in Immunology 20, no. 1 (2008): 26–42, 10.1016/j.smim.2007.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Miron V. E., Boyd A., Zhao J. W., et al., “M2 Microglia and Macrophages Drive Oligodendrocyte Differentiation During CNS Remyelination,” Nature Neuroscience 16, no. 9 (2013): 1211–1218, 10.1038/NN.3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Prinz M. and Priller J., “Microglia and Brain Macrophages in the Molecular Age: From Origin to Neuropsychiatric Disease,” Nature Reviews Neuroscience 15, no. 5 (2014): 300–312, 10.1038/nrn3722. [DOI] [PubMed] [Google Scholar]
- 156. Du S., Drieu A., Cheng Y., et al., “Brain‐Engrafted Monocyte‐Derived Macrophages From Blood and Skull‐Bone Marrow Exhibit Distinct Identities From Microglia,” bioRxiv 8, no. 8 (2024): 606900, 10.1101/2024.08.08.606900. [DOI] [Google Scholar]
- 157. Greter M., Heppner F. L., Lemos M. P., et al., “Dendritic Cells Permit Immune Invasion of the CNS in an Animal Model of Multiple Sclerosis,” Nature Medicine 11, no. 3 (2005): 328–334, 10.1038/NM1197. [DOI] [PubMed] [Google Scholar]
- 158. Brown J. A., Dorfman D. M., Ma F. R., et al., “Blockade of Programmed Death‐1 Ligands on Dendritic Cells Enhances T Cell Activation and Cytokine Production,” Journal of Immunology 170, no. 3 (2003): 1257–1266, 10.4049/JIMMUNOL.170.3.1257. [DOI] [PubMed] [Google Scholar]
- 159. Carlson T., Kroenke M., Rao P., Lane T. E., and Segal B., “The Th17–ELR+ CXC Chemokine Pathway is Essential for the Development of Central Nervous System Autoimmune Disease,” Journal of Experimental Medicine 205, no. 4 (2008): 811–823, 10.1084/JEM.20072404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Huber A. K., Wang L., Han P., et al., “Dysregulation of the IL‐23/IL‐17 Axis and Myeloid Factors in Secondary Progressive MS,” Neurology 83, no. 17 (2014): 1500–1507, 10.1212/WNL.0000000000000908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Bailey S. L., Schreiner B., McMahon E. J., and Miller S. D., “CNS Myeloid DCs Presenting Endogenous Myelin Peptides “Preferentially” Polarize CD4+ TH‐17 Cells in Relapsing EAE,” Nature Immunology 8, no. 2 (2007): 172–180, 10.1038/NI1430. [DOI] [PubMed] [Google Scholar]
- 162. Huang G., Wang Y., Vogel P., Kanneganti T. D., Otsu K., and Chi H., “Signaling via the Kinase p38α Programs Dendritic Cells to Drive T H17 Differentiation and Autoimmune Inflammation,” Nature Immunology 13, no. 2 (2012): 152–161, 10.1038/NI.2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Prodinger C., Bunse J., Krüger M., et al., “CD11c‐Expressing Cells Reside in the Juxtavascular Parenchyma and Extend Processes Into the Glia Limitans of the Mouse Nervous System,” Acta Neuropathologica 121, no. 4 (2011): 445–458, 10.1007/S00401-010-0774-Y. [DOI] [PubMed] [Google Scholar]
- 164. Anandasabapathy N., Victora G. D., Meredith M., et al., “Flt3L Controls the Development of Radiosensitive Dendritic Cells in the Meninges and Choroid Plexus of the Steady‐State Mouse Brain,” Journal of Experimental Medicine 208 (2011): 1695–1705, 10.1084/JEM.20102657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Reis E. and Sousa C., “Activation of Dendritic Cells: Translating Innate Into Adaptive Immunity,” Current Opinion in Immunology 16, no. 1 (2004): 21–25, 10.1016/j.coi.2003.11.007. [DOI] [PubMed] [Google Scholar]
- 166. Ito M., Komai K., Mise‐Omata S., et al., “Brain Regulatory T Cells Suppress Astrogliosis and Potentiate Neurological Recovery,” Nature 565, no. 7738 (2019): 246–250, 10.1038/s41586-018-0824-5. [DOI] [PubMed] [Google Scholar]
- 167. Gagliani N., Amezcua Vesely M. C., Iseppon A., et al., “Th17 Cells Transdifferentiate Into Regulatory T Cells During Resolution of Inflammation,” Nature 523, no. 7559 (2015): 221–225, 10.1038/nature14452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Hitpass Romero K., Stevenson T. J., Smyth L. C. D., et al., “Age‐Related Meningeal Extracellular Matrix Remodeling Compromises CNS Lymphatic Function,” Journal of Neuroinflammation 22, no. 1 (2025): 1–21, 10.1186/S12974-025-03436-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Gao D., Zou B., Zhu K., et al., “Enhancing Th17 Cells Drainage Through Meningeal Lymphatic Vessels Alleviate Neuroinflammation After Subarachnoid Hemorrhage,” Journal of Neuroinflammation 21, no. 1 (2024): 269, 10.1186/s12974-024-03252-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Da Mesquita S., Herz J., Wall M., et al., “Aging‐Associated Deficit in CCR7 is Linked to Worsened Glymphatic Function, Cognition, Neuroinflammation, and β‐Amyloid Pathology,” Science Advances 7, no. 21 (2021): 4601–4622, 10.1126/SCIADV.ABE4601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Ewing‐Crystal N. A., Mroz N. M., Chang A. A., et al., “Dynamic Fibroblast‐Immune Interactions Shape Wound Healing After Brain Injury,” bioRxiv 3, no. 13 (2024): 584873, 10.1101/2024.03.13.584873. [DOI] [Google Scholar]
- 172. Bolte A. C., Dutta A. B., Hurt M. E., et al., “Meningeal Lymphatic Dysfunction Exacerbates Traumatic Brain Injury Pathogenesis,” Nature Communications 11, no. 1 (2020): 4524, 10.1038/S41467-020-18113-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Bolte A. C., Shapiro D. A., Dutta A. B., et al., “The Meningeal Transcriptional Response to Traumatic Brain Injury and Aging,” eLife 12 (2023): 12, 10.7554/ELIFE.81154. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing not applicable to this article as no datasets were generated or analysed during the current study.