

Abstract
We introduce coherent infrared emission interferometry as a χ(2) vibrational spectroscopy technique and apply it to studying the initial dynamics upon photoactivation of myoglobin (Mb). By impulsive excitation (using 11-fs pulses) of a Mb crystal, vibrations that couple to the optical excitation are set in motion coherently. Because of the order in the crystal lattice the coherent oscillations of the different proteins in the crystal that are associated with charge motions give rise to a macroscopic burst of directional multi-teraHertz radiation. This radiation can be detected in a phase-sensitive way by heterodyning with a broad-band reference field. In this way both amplitude and phase of the different vibrations can be obtained. We detected radiation in the 1,000–1,500 cm−1 frequency region, which contains modes sensitive to the structure of the heme macrocycle, as well as peripheral protein modes. Both in carbonmonoxy-Mb and aquomet-Mb we observed emission from six modes, which were assigned to heme vibrations. The phase factors of the modes contributing to the protein electric field show a remarkable consistency, taking on values that indicate that the dipoles are created “emitting” at t = 0, as one would expect for impulsively activated modes. The few deviations from this behavior in Mb-CO we propose are the result of these modes being sensitive to the photodissociation process and severely disrupted by it.
The time scale of reaction dynamics in proteins and concomitant protein conformational changes spans many orders of magnitude. Small and highly directed structural changes occurring on the time scale of vibrations, i.e., tens of femtoseconds to picoseconds, often ultimately lead to a larger conformational change. Time-resolved optical measurements have been widely used to obtain information on the dynamics of the optically active cofactors on time scales ranging from tens of femtoseconds to seconds (1, 2). However, visualization of function-determining structural changes of the protein on the ultrafast time scale is still greatly missing. Specific structural changes may be monitored by vibrational spectroscopy (absorption or Raman scattering) in the mid-IR range, where the protein and cofactors have vibrational transitions, or by NMR and x-ray diffraction techniques. With time-resolved x-ray spectroscopy the structures of photointermediates of proteins have been resolved with nanosecond time resolution (3, 4). However, techniques with femtosecond resolution are necessary to obtain information on local structural changes and their coupling to a reaction on the time scale of molecular vibrations.
Information on the coherence time of cofactor–protein interactions with respect to reaction times in several protein complexes has been derived from three pulse-photon echo experiments performed either on electronic transitions of the cofactors (5–7) or vibrational transitions (8, 9). The relevance of low-frequency (<400 cm−1) heme–protein modes for the initial reactions in hemes has been studied by monitoring modulations of the pump-probe absorption spectra in this frequency range (10, 11).
We report here the application of a coherent heterodyne technique, in which we have used time-domain interferometry in the mid-IR to measure directly the electric field emitted by coherently oscillating vibrational dipoles in a protein complex. Special to this technique is that it is sensitive for both the phase and the direction of the different vibrations of the protein. This is made possible by performing the experiments on single crystals. Impulsive excitation of the proteins in the crystal results in coherent oscillation of the protein vibrations. The oscillations of the individual modes add up to a directional burst of THz radiation, because of the order of the crystal.** We impulsively set the vibrational dipoles in a coherent oscillation by electronic excitation of an optically active cofactor (in our case heme) with a short laser pulse. We have detected the macroscopic oscillating electric field by letting it interfere with a well-characterized IR pulse. The polarization of the oscillating dipoles can be determined relative to the orientation in the crystal, and a comparison to static x-ray structures can be made. Ultrafast kinetic information can be obtained because the time resolution is in principle subcycle, i.e., sub-30 fs. This is more than an order of magnitude higher than obtainable in time-resolved frequency-domain techniques as Raman or IR pump-probe. Earlier this technique was applied to detect the IR emission from coherent charge oscillations arising from electronic coherence in semiconductor quantum wells (12). Here we demonstrate its potential for measuring vibrational dynamics in a complex system as a protein.
Myoglobin (Mb) is a small, well-characterized heme protein, active in the storage of oxygen in muscle tissue. Within 50 fs of optical excitation of the heme (a protoporphyrin IX noncovalently bound to the protein backbone), carbon monoxide (CO), or other diatomic ligands dissociate from the heme, followed by formation of the unliganded heme ground state. In the latter state the heme has a domed configuration, i.e., the Fe has moved out of the heme plane (≈0.4 Å) toward the proximal helix (13). The early photophysics of Mb-CO can be summarized as follows: excitation leads to an excited photodissociated state in <50 fs, which decays to the unliganded ground state in 300 fs (14). The ultrafast time scale of this reaction probably explains the high (near-unity) quantum yield and may indicate that it is governed by coherent vibrational motions. In aquomet-Mb, which is formed when the Fe of the protoporphyrin IX is in the Fe3+ rather than in the Fe2+ state, no photodissociation takes place and only decay of the excited state to the liganded ground state occurs. We have performed our experiments on both crystals of Mb-CO and aquomet-Mb to be able to distinguish dissociation-induced structural changes from photophysics of the heme.
Materials and Methods
In our experiments, to obtain constructive interference of the emission of an ensemble of molecules, a macroscopic orientation of dipoles is required; therefore we used crystals of horse heart Mb. Crystals of space group P21 containing two proteins per unit cell were grown as described (15). Fig. 1 schematically depicts the orientation of the two proteins per unit cell in the rectangular face of the crystal. Typical crystal dimensions were 50 × 200 × 8 μm. The crystals were kept in the mother liquor, a 3.6 M (NH4)2SO4, 50 mM Tris, pH 7.4 buffer. For met-Mb, (Fe3+-H2O), the crystals were used as prepared. To obtain Mb-CO, the crystals were soaked in an anaerobic, CO-saturated buffer containing 100 mM dithionite for 30 min, in which they turned bright red. The crystals were mounted between CaF2 windows, separated by a spacer of 8 μm. CaF2 (transmission cuts off at ω <900 cm−1) windows were used, because materials with a broader mid-IR transmission range either are not transparent around 560 nm, or, as BaF2, reacted with the ammonium sulfate. The result of the procedure to bind CO to Mb was checked by measuring the transmission spectrum (16).
Figure 1.

Schematic view of the orientation of the horse heart Mb molecules in the unit cell for crystals of the space group P21, which contains two Mb molecules per unit cell. The protein complexes are indicated by their heme group (Left). (Right) A crystal is shown with the crystal axis indicated: b lies in the plane of the crystal, c is perpendicular to the plane, and a is tilted in the plane. The angle over which the crystal is rotated in the experiments is marked.
The experimental setup contained a home-built, Ar+ laser-pumped, Ti:sapphire oscillator (centered at 810 nm, bandwidth 40 nm) operating at 100 MHz. The oscillator output was seeded into a 1-kHz regenerative amplifier based on chirped-pulse amplification (Spitfire, Spectra-Physics) and amplified to 0.6 mJ with a pulse duration of ≈50 fs. The amplified output was used to pump a noncollinear optical parametric amplifier (NOPA) built according to refs. 17–20. Care was taken to avoid dispersive optics after the white-light generation. The output, tuned to 560 nm, was typically 1–2 μJ, using 15 μJ of 400-nm pump light. The pulses were compressed to 10–15 fs with a double pair of small apex angle prisms with a broadband AR coating (18).
The reference IR fields were generated and characterized as shown in Fig. 2 Left, using a diffracting Fourier transform (FT) IR interferometer as described (21). Broadband mid-IR was created by optical rectification of 100 nJ of the NOPA output in a GaAs crystal (type <1 1̄ 0>, thickness 110 μm) (22). In short, optical rectification is the effect that when an electric field in the form of a short pulse is applied to a nonlinear material a second-order polarization is induced that follows the envelope of the field. The induced polarization is the source of a single-cycle IR pulse. The efficiency of the optical rectification process was similar as reported earlier by using the oscillator output (22), i.e., 10−6.
Figure 2.

Layout of the experimental setup in autocorrelation configuration (Left) and coherent emission configuration (Right). (Left) The NOPA output is split in two equal parts that are focused on the GaAs to create mid-IR single-cycle radiation by means of optical rectification. (Right) One of the two NOPA beams is focused on the Mb crystal to induce coherent mid-IR vibrational radiation. In both configurations one of the two beams travels over a variable delay line that is scanned to obtain the interferogram.
In the coherent emission experiment (Fig. 2 Right) the radiation induced by the beam traveling the variable delay line was used as a reference electric field. Another part of the NOPA output, ≈75 nJ, was focused on a Mb crystal that could be rotated in the crystal plane. The diameter of the focus was ≈50 μm. To facilitate overlapping the reference beam and the protein emission on the HgCdTe detector, an alignment was used where the reference beam also traveled through the Mb crystal. To this end a second pair of parabolic mirrors was used to focus the reference beam to ≈50 μm on the Mb crystal and to collimate the reference and protein emission beams before focusing them on the detector. Signal was found with a second GaAs crystal at the sample position, in which a pump-probe signal was used to find temporal overlap. Subsequently the pump was moved slightly to optimize the emission signal from the GaAs on the detector, typically reducing the pump-probe signal to 20–30%. The output of the HgCdTe detector was measured every shot (at 1 kHz). An interferogram was measured by scanning the reference beam delay. One scan took about 1 min and consisted of 256 time points; the scan length was 2 ps, corresponding to a spectral resolution of about 8 cm−1. Correction for slow fluctuations in the reference intensity was achieved by chopping the excitation beam of the protein emission at a rate of ≈200 Hz. To avoid irreversible crystal degradation, the crystal was translated after each scan. The resulting small variations in signal intensity did not affect the results, as the emission was detected background free; we made sure that the shape of the FT spectrum of the interferograms did not change during accumulation. Interferograms were measured as a function of the angle between the polarization of both reference beam and visible excitation beam, which were vertical, and the long axis of the crystal rectangular face. Because the modulation depth of the interferogram depends on the angle between the polarization of reference and the protein electric fields, the contributions of differently oriented dipoles in the interferogram will vary when the crystal is rotated. Because of the particular orientation of the two proteins in the unit cell (see Fig. 1) a 0–90o angle covers the whole orientation plane of the dipoles. Measurements were performed at one small tilt angle, corresponding to an internal angle of 15o.
Results
The Reference E-Field.
In the coherent emission configuration, an interferogram of the temporal profiles of the E-field emitted by the protein crystal and a reference E-field is measured. Determination of the true E-field of the protein emission requires deconvolution of the interferogram and hence the temporal shape of the reference E-field itself needs to be determined. Fig. 3 Upper shows the interferogram ISingleCycle(t) of two single-cycle (i.e., ≈30 fs) mid-IR pulses created by optical rectification of the 560-nm NOPA output in GaAs, using the experimental configuration of Fig. 2 Left. The small amplitude oscillations on both sides of the interferogram are caused by the sharp cut-off of the detector at ω <800 cm−1. In Fig. 3 Lower the FT spectrum of the interferogram is shown (solid black line); the radiation extends up to 2,000 cm−1 (60 THz). The spectrum corrected for the detector response for ω >800 cm−1 (blue line) corresponds very well with the spectrum calculated for radiation induced by optical rectification of an 11-fs Gaussian pulse (red line).
Figure 3.

(Upper) An interferogram of two single-cycle pulses created by optical rectification in GaAs. The small amplitude oscillations on either side of the interferogram are caused by the cut-off of the detector response. (Lower) The corresponding FT spectrum (black line), the spectrum after correction for the detector response (blue line), and the calculated spectrum for optical rectification of an 11-fs Gaussian pulse.
The reference E-field will be both spectrally and temporally altered by its passage through the CaF2 window and the Mb crystal. We determined this by recording the interferogram IDispersed(t) of two reference beams (see Fig. 2 Left), one of which was passed through one CaF2 window and a Mb crystal, i.e., the difference in dispersive material between reference and protein E-field. We found that the spectral content in the reference electric field was limited to a ≈1,050–1,450 cm−1 region because of the cut-off of the CaF2 window and a strong sulfate absorption band of the buffer solution on the low-frequency side, and the absorption from the amide bands of the protein and water on the high-frequency side. The dispersion in the reference E-field is negative, i.e., high frequencies travel in front of the pulse envelope and amount to about 150 fs between 1,100 and 1,450 cm−1 and to 700 fs between 930 and 1,100 cm−1. The corresponding variation in the refraction index calculated from the phase spectrum of the FT of the interferogram shows very good agreement with literature values for the dispersion of CaF2 (not shown). The reference electric field can now be calculated by factoring out the contribution of the single-cycle electric field, obtained by Fourier transforming the single-cycle interferogram:
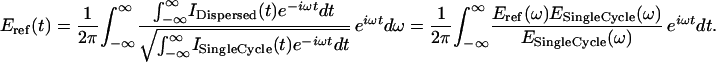 |
1 |
Coherent Emission.
As stated above, in the coherent emission experiment (see Fig. 2 Right), an interferogram of the temporal profiles of the E-field emitted by the sample and the reference E-field is measured. The temporal profiles in Fig. 4 demonstrate that macroscopic mid-IR emission can indeed be obtained from Mb crystals upon electronic excitation of the heme by a visible pulse. We stress that this process is a χ(2) process (or three-wave mixing experiment): the sample has two interactions with the visible field creating a vibrational coherence that leads (in an anisotropic medium) to the creation of a third field emitted in the IR. Higher-order χ(3) processes involving an interaction of the reference IR beam with the sample give rise to (long-lived) population terms (12), which do not contribute to a detectable level in the present experiments.
Figure 4.

Interferograms of Mb-CO (Upper) and aquomet-Mb (Lower) emission as a function of rotation angle of the crystal (black lines) and fitted interferograms (see text, red and blue lines, respectively). The upper black line marked 30o (Upper) is the calculated oscillating electric field of the Mb-CO radiation at 30o as obtained from these data.
In Fig. 4 (Upper, black lines) emission interferograms of Mb-CO crystals measured at different rotation angles are shown. The interferograms show a set of beats, indicating that several different oscillators contribute to the protein electric field. The interference pattern depends on the rotation angle.
The interferograms of aquomet-Mb (Fig. 4 Lower) show a clearly different modulation pattern (one beat is much larger than the subsequent beats), and again a dependence on the rotation angle. From the relative modulation of the interferogram (≈1%), and the intensity in the reference (about 100 fJ), we estimate the number of detected protein–emission photons to be ≈300 per laser shot.
Analysis.
The interferograms are a convolution of the protein electric field with the ≈15-fs excitation pulse‡‡ and the reference electric field. A Fourier transformation yields the product of the spectra of the protein radiation field and the reference field and the difference of their phases. However, because of the dispersed reference, this procedure does not directly yield the phase of the protein electric field at t = 0. Therefore, and also to avoid artifacts caused by the asymmetric nature of the interferograms, we fitted the interferogram directly with a protein electric field constructed from a sum of damped sine functions, E(ω,φ,t) = ΣiAisin(ωit + φi)e−t/τ, convoluted with a 15-fs Gaussian excitation pulse (see Discussion) and with the experimentally determined electric field of the reference. This procedure in principle also enables the use of a kinetic model to fit the data. The calculated interferogram was fitted to the data by adjusting the amplitudes, frequencies, and phases of the protein field. The dephasing time and t = 0 were varied manually to obtain a minimal sum-squared value (see below). A fit was considered satisfactory when the FT of the residuals contained no peaks above the noise level; the fitted spectra were also compared with the FT of the interferograms for consistency. For each data set a unique fit was found, which was checked by starting the fit procedure with different starting values.
The colored lines in Fig. 4 represent the fitted interferograms. Satisfactory results are obtained, including a good fit of the apparent rising part of the interferogram, at early times, caused by the trailing of the low frequencies in the reference field. The spectra of the oscillators of Mb-CO and aquomet-Mb are shown in Fig. 5. The aquomet-Mb spectra differ in some respects from the Mb-CO spectra (see below). The dephasing time (T2) of all oscillators was 0.7 ± 0.1 ps, which corresponds to a spectral width of ≈25–30 cm−1. This corresponds well with the excited-state lifetime (T1) of ≈300 fs of Mb-CO and aquomet-Mb (ref. 14 and unpublished data) because the E-field is related to the square root of the population and therefore T2 ≤ 2T1.
Figure 5.

Spectra of the oscillators used to fit the interferograms of aquomet-Mb (blue lines) and Mb-CO (red lines), at each rotation angle of the crystal.
In Fig. 6 Upper we have plotted the phase factor sin(φi) for each frequency ωi contributing to the protein electric field at t = 0 for the Mb-CO data. As shown in the Inset, the fit quality depends critically on the choice T of t = 0, the sum of squares of the residuals showing a clear global minimum, as well as periodic local minima. As the choice of t = 0 at the minimum also corresponds to absence of linear, dispersion-like behavior of the phases (with φ = ωt), this finding indicates that we can indeed satisfactorily determine t = 0. The data at different rotation angles show a strong correlation; for most bands the phase factor is close to either +1 or −1. In contrast, the band around ≈1,230 cm−1 has a phase factor that varies between 0 and 1 and the band at 1,400 cm−1 has a very low phase factor. We verified that the phase factors of the FT of the raw data, both with and without correcting for the dispersion of the reference, behaved similarly. In fact, the difference in the oscillation pattern in the interferograms of Mb-CO and aquomet-Mb is largely caused by bands 4 and 5 being in phase at t = 0 in aquomet-Mb but ½π to π out of phase in Mb-CO.
Figure 6.

The Mb-CO (Upper) and aquomet (Lower) phase factor sin φ at t = 0, at different signal frequencies and at five rotational angles of the crystal. (Insets) Plots of the fit error (sum of squares of the fit residuals) as a function of the choice T of t = 0 calculated for one of the interferograms (Mb-CO, 70°). This illustrates that variation of T in the fit leads by means of local minima to a global minimum, thus allowing accurate determination of the experimental t = 0.
The phase factors of the aquomet-Mb electric field also show a high degree of correlation (Fig. 6 Lower): the bands at ω >1,200 cm−1 have a phase factor of either +1 or −1 where it may be noted that the 0o measurement closely overlaps with the other two measurements if it were multiplied by −1. A correlation between the phase factors at different rotation angles is expected if a frequency corresponds to the same dipole. The plus or minus sign then depends on the relative net orientation (up or down) of the emission field. Because the emitted field is the sum of the fields emitted by two protein complexes having different orientation within the unit cell, the emission can change sign for those dipoles polarized nearly antiparallel in the unit cell (compare Fig. 1). The ≈1,100 cm−1 band has a lower phase factor.
Discussion
Origin of Emission.
We have observed coherent IR radiation from Mb crystals induced by electronic excitation of the heme with a short laser pulse. In principle, this emission can arise from (i) oscillating vibrational dipoles of the heme and/or the nearby protein residues, (ii) oscillating electronic wave packets, i.e., quantum beats between different coherently excited electronic states (cf. ref. 12), or (iii) a nonlinear, resonant or nonresonant, electronic χ(2) process of the heme itself. The second possibility can be excluded, because the two electronic QX and QY transitions at 580 nm (560 nm for aquomet-Mb) are oriented at a right angle in the molecular plane and are near-degenerate; i.e., separated by an energy difference of only ≈300 cm−1 (23). The third possibility is also not likely; the emission clearly arises from a long-lived process and is sufficiently different from the IR absorption spectrum (not shown) to conclude that it is not caused by an optical rectification process in the crystal itself, modulated by the absorption of the heme-protein complex. Therefore we conclude that the emission is caused by two interactions with the visible field creating coherently oscillating vibrational wave packets, either on the excited electronic state, by means of coupling with the π-electron system of the heme electronic transition, or on the ground state by means of impulsive stimulated Raman scattering. Formally the emission process is a heterodyne-detected, doubly resonant, second-order (χ(2)) process, with χ(2) strongly frequency-dependent (see below). According to the classification of Albrecht and coworkers (24) it can be regarded as a second-order class II spectroscopy.
Formally, the second-order polarization tensor P(2) corresponding to difference frequency generation has the form
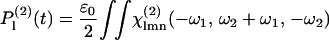 |
2 |
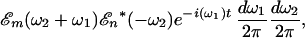 |
in which ℰ and ℰ* represent, respectively, the complex electric field and its conjugate. If χ(2) is frequency-independent, as is the case for the electronic nonresonant optical rectification used to generate the reference pulse, the polarization elements reduce to the form (22) P(2)(t) = ½ɛ0χ(2)I(t), in which I(t) is the intensity of the generating pulse. For our case, where the elements χ(2) varies slowly with the excitation (visible) frequency ω2 and strongly with the generated (IR) difference frequency ω1, the usual expression for χ(2) within the Bloch formalism (25) can be simply approximated with
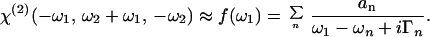 |
3 |
This yields polarization elements of the form P(2)(t) = ½ɛ0/2f(t)⊗I(t), in which f(t) is the FT of f(ω1). With Eq. 3 this implies that the emitted electric field, which is proportional to dP/dt, has the form of damped sine functions, as assumed in the analysis. Vibrational wavepackets may be efficiently excited if their period is longer than the pump pulse. The signals that we observe may originate from motions on the excited state or on the ground-state potential energy surface. For vibrational excitation on the ground state by means of impulsive stimulated Raman scattering there is an optimum pulse length (26): the pulse should be long enough to ensure that the second interaction with the pump field occurs after the wave packet, created by the first interaction with the pump, has had sufficient time to evolve on the excited-state surface to allow displacement upon return to the ground-state surface. A too long excitation pulse would smear out the wave packets on the ground state and result in a stationary state. If we scale the calculations of Pollard et al. (26) to our case, a 12- to 15-fs pulse would be able to efficiently excite ground-state modes between 1,000 and 2,000 cm−1, hence some of the vibrations that we visualize may come from ground-state vibrational modes (see also below).
Band Assignments.
The bands in our emission spectra are tentatively assigned (see Table 1) to IR active modes of the Fe-protoporphyrin IX (in some cases assignment to histidine residues also is possible) following refs. 27–33. As can be seen in Fig. 5, the peak position of the bands varies somewhat as a function of rotation angle (with the same number of bands satisfactory fits could not be obtained with the same peak positions for all angles). This finding can be explained by the appearance of modes of similar origin but on different groups when the crystal is rotated, as for example modes involving either the two- or four-vinyl groups; the different redox state and possibly differing protein contacts are also likely to induce band shifts in Mb-CO with respect to aquomet-Mb modes.
Table 1.
| Band | Frequency in cm−1 | Assignment |
|---|---|---|
| 1 | 1,070 (in MbCO) | Vinyl⩵CH2 deformation |
| 2 | 1,090–1,105 (in Aq-Mb) | Vinyl⩵CH2 deformation or proximal histidine C—H bending |
| 3 | 1,130–1,135 | ν43(pyr half-ring)asym |
| 1,155 | ν44(CβC1)sym | |
| 4 | 1,205–1,213 | Peripheral vinyl CH2 twist mode |
| 5 | 1,225–1,240 | ν42δ(CmH) core mode |
| 6 | 1,325 | 2- and 4-vinyl group [δ(CaH)], in-plane bending or ν41(pyr half-ring)sym |
| 7 | 1,400 | ν40(pyr quarter-ring)asym |
Dynamics.
A phase factor sin φ of ≈±1, as observed for most modes, is expected for emission from a vibrational wave packet on the excited state: at t = 0 the mode is driven by the optical light field from its rest position into an off-equilibrium position on the excited state. The wave packet is created with maximum acceleration, i.e., is created emitting and will give rise to emission with a phase factor of ±1 at t = 0. For modes excited on the ground state by the impulsive stimulated Raman mechanism, a phase delay is expected. Full understanding of the phases on the ground state and the relative intensity of population in the ground and excited state requires more theoretical modeling. At present, we limit ourselves to comparing Mb-CO data with the nonphotodissociating aquomet-Mb data.
In Mb-CO, two bands at 1,225–1,240 cm−1 (band 5) and 1,405 cm−1 (band 7) show a phase factor significantly deviating from ±1. As remarked earlier, the difference in the oscillation pattern in the interferograms of Mb-CO and aquomet-Mb is largely because of bands 4 and 5 being in phase at t = 0 in aquomet-Mb but ½π to π out of phase in Mb-CO. In aquomet-Mb the phase of bands 4, 5, and 7 is always ≈±1, therefore the low phase factor of these bands in Mb-CO cannot be explained by “normal” excited- or ground-state vibrational dynamics.
Another possible cause of a phase factor deviating from ±1 is delayed alteration or activation of the mode by a reaction or structural change of the heme, causing the mode to be shifted slightly in frequency or equilibrium position or to gain oscillator strength. The latter possibility is supported by the observation of a large positive band at 1,230 cm−1 in the nanosecond-FTIR absorption-difference spectrum of Mb-CO (F. Siebert, private communication), which would be consistent with our result that at t = 0 the bands 4 and 5 are almost in counter-phase, as if they are not activated at t = 0 by direct excitation, but with a time delay. For the emission to be observed such a reaction would have to be impulsive, i.e., faster than the vibrational period, or at least take place during a well-defined time span during which the phase relationships of the mode are maintained. Band 5 as well as band 7 arise from vibrational modes closely associated with the core of the heme (see Table 1) and they have been shown to be markers for the heme macrocycle structure. Asher and Schuster (30) found a correlation between a frequency shift from 1,232 to 1,217 cm−1 and a decreased heme core size as well as an out-of-plane movement of the iron toward the proximal histidine, for the hemes of α and β hemoglobin subunits in Raman measurements. Berthomieu et al. (32) showed that upon oxidation a band shift from 1,237 to 1,223 cm−1 occurred in IR-absorption spectra of double-coordinated Fe-protoporphyrin IX. Therefore we suggest that the phase behavior of the 1,230 and the 1,405 cm−1 (periods 27 fs and 24 fs, respectively) emission bands reflect the photodissociation reaction itself or a structural change of the heme related to the ultrafast CO dissociation, such as a redistribution of electron densities, or change in the local electric field.
We also observed an anomalous phase factor for the modes at ≈1,100 cm−1 in the aquomet-Mb emission. Vibrational modes at this frequency have been ascribed to histidine C⩵H bending modes (29) and have been reported to gain IR intensity in the reduced state (32). This finding may explain the spectral differences between Mb-CO and aquomet-Mb in this region. The different fate of the excited states in aquomet-Mb, Mb-CO, and MbO2, apart from being determined by redox state, has been suggested to be caused by different conformational states of the heme-ligand-distal histidine complex observed in high-resolution x-ray structures (13). In some of the conformations a slightly different orientation of both ligand and distal histidine results in the formation of a hydrogen bond. It was suggested that this hydrogen bond prevents the photodissociation of the ligand and the subsequent escape from the pocket. We might speculate that the 1,100 cm−1 mode-phase shift reflects an “attempted” dissociation.
Conclusions
We have succeeded in measuring mid-IR radiation emitted by oscillating dipoles in a protein complex. Based on the observed frequencies, the dipoles can be associated with heme vibrational modes and, in some cases, to histidine modes. The phases at t = 0 of the modes contributing to the protein electric field show a remarkable consistency, taking on values that indicate that the dipoles are created emitting at t = 0 as one would expect from simple first principles. We suggest that the few deviations from this behavior arise from the fact that, after a delay, these modes are set in motion or altered by the photodissociation process. The consequence of the fact that these modes are observed is that the photodissociation process is impulsive on the time scale of the vibrational periods.
In this work we report an exploration of the coherent emission technique for studying ultrafast charge displacements in proteins. The method can be extended to reconstruct the direction of the motions in the protein frame by rotation of the crystals in three dimensions and combining the information with the crystal structure and packing. In principle, technical developments should be able to diminish at least part of the spectral limitations of the present work. Finally, a challenging development is the possibility to map out the second-order susceptibility χ(2) in two frequency dimensions (34) (the visible generator and the emitted IR), which would allow to get a detailed description of the molecular response to electronic perturbation.
Acknowledgments
M.-L.G. was supported by a Marie Curie Fellowship from the European Community.
Abbreviations
- Mb
myoglobin
- NOPA
noncollinear optical parametric amplifier
- FT
Fourier transform
Footnotes
Note that these experiments cannot be performed on solutions of Mb, because the process of photoselection, using a single excitation beam, excites as many dipoles oriented up as down and would not result in net emission.
Because the compression of the NOPA output was optimized for the optical rectification process, and the beam paths for OR generation and protein emission generation differed slightly, we estimate the excitation pulse length to be 15 fs, i.e., slightly longer than the pulse at the position of the GaAs (see above).
References
- 1.Thorn Leeson D, Wiersma D A. Nat Struct Biol. 1995;2:848–851. doi: 10.1038/nsb1095-848. [DOI] [PubMed] [Google Scholar]
- 2.Austin R H, Beeson K W, Eisenstein L, Frauenfelder H, Gunsalus I C. Biochemistry. 1975;14:5355–5373. doi: 10.1021/bi00695a021. [DOI] [PubMed] [Google Scholar]
- 3.Srajer V, Teng T Y, Ursby T, Pradervand C, Ren Z, Adachi S, Schildkamp W, Bourgeois D, Wulff M, Moffat K. Science. 1996;274:1726–1729. doi: 10.1126/science.274.5293.1726. [DOI] [PubMed] [Google Scholar]
- 4.Perman B, Srajer V, Ren Z, Teng T, Pradervand C, Ursby T, Bourgeois D, Schotte F, Wulff M, Kort R, et al. Science. 1998;279:1940–1945. doi: 10.1126/science.279.5358.1946. [DOI] [PubMed] [Google Scholar]
- 5.Jimenez R, van Mourik F, Yu J-Y, Fleming G R. J Phys Chem. 1997;101:7350–7359. [Google Scholar]
- 6.Groot M-L, Yu J-Y, Agarwal R, Norris J R, Fleming G R. J Phys Chem. 1998;102:5923–5931. [Google Scholar]
- 7.Thorn Leeson D, Wiersma D A, Fritsch K, Friedrich J. J Phys Chem. 1997;101:6331–6340. [Google Scholar]
- 8.Rector K D, Rella C W, Hill J R, Kwok A S, Sligar S G, Chien E Y T, Dlott D D, Fayer M D. J Phys Chem. 1997;101:1468–1475. [Google Scholar]
- 9.Lim M, Hamm P, Hochstrasser R M. Proc Natl Acad Sci USA. 1998;95:15315–15320. doi: 10.1073/pnas.95.26.15315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liebl U, Lipowski G, Negrerie M, Lambry J C, Martin J L, Vos M H. Nature (London) 1999;401:181–184. doi: 10.1038/43699. [DOI] [PubMed] [Google Scholar]
- 11.Rosca F, Kumar A T N, Ye X, Sjodin T, Demidov A A, Champion P M. J Phys Chem. 2000;104:4280–4290. [Google Scholar]
- 12.Bonvalet A, Nagle J, Berger V, Migus A, Martin J-L, Joffre M. Phys Rev Lett. 1996;76:4392–4395. doi: 10.1103/PhysRevLett.76.4392. [DOI] [PubMed] [Google Scholar]
- 13.Vojtechovsky J, Chu K, Berendzen J, Sweet R M, Schlichting I. Biophys J. 1999;77:2153–2174. doi: 10.1016/S0006-3495(99)77056-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Petrich J W, Poyart C, Martin J-L. Biochemistry. 1988;27:4049–4060. doi: 10.1021/bi00411a022. [DOI] [PubMed] [Google Scholar]
- 15.Chu K, Vojtchosky J, McMahon B H, Sweet R M, Berendzen K, Schlichting I. Nature (London) 2000;403:921–923. doi: 10.1038/35002641. [DOI] [PubMed] [Google Scholar]
- 16.Eaton W A, Hofrichter J. Methods Enzymol. 1981;76:175–261. doi: 10.1016/0076-6879(81)76126-3. [DOI] [PubMed] [Google Scholar]
- 17.Cerullo G, Nisoli M, Stagira S, De Silvestri S. Opt Lett. 1998;23:1283–1285. doi: 10.1364/ol.23.001283. [DOI] [PubMed] [Google Scholar]
- 18.Cerullo G, Nisoli M, Stagira S, De Silvestri S. Appl Phys Lett. 1997;71:3616–3618. [Google Scholar]
- 19.Shirakawa A, Sakane I, Kobayashi T. Opt Lett. 1998;23:1292–1294. doi: 10.1364/ol.23.001292. [DOI] [PubMed] [Google Scholar]
- 20.Wilhelm T, Piedl J, Riedle E. Opt Lett. 1997;22:1494–1496. doi: 10.1364/ol.22.001494. [DOI] [PubMed] [Google Scholar]
- 21.Joffre M, Bonvalet A, Migus A, Martin J-L. Opt Lett. 1996;21:964–966. doi: 10.1364/ol.21.000964. [DOI] [PubMed] [Google Scholar]
- 22.Bonvalet A, Joffre M, Martin J-L, Migus A. Appl Phys Lett. 1995;67:2907–2909. [Google Scholar]
- 23.Shibata Y, Kushida T. Chem Phys Lett. 1998;284:115–120. [Google Scholar]
- 24.Kirkwood J C, Ulness D J, Albrecht A C. J Phys Chem. 2000;104:4167–4173. [Google Scholar]
- 25.Butcher P N, Cotter D. The Elements of Nonlinear Optics. Cambridge, U.K.: Cambridge Univ. Press; 1990. [Google Scholar]
- 26.Pollard W T, Dexheimer S L, Wang Q, Peteanu L A, Shank C V, Mathies R A. J Phys Chem. 1992;96:6147–6158. [Google Scholar]
- 27.Choi S, Spiro T G, Langry K C, Smith K M. J Am Chem Soc. 1982;104:4337–4344. [Google Scholar]
- 28.Hu S, Smith K M, Spiro T G. J Am Chem Soc. 1996;118:12638–12646. [Google Scholar]
- 29.Miller L M, Chance M R. J Am Chem Soc. 1994;116:9662–9669. [Google Scholar]
- 30.Asher S A, Schuster T M. Biochemistry. 1981;20:1866–1873. doi: 10.1021/bi00510a023. [DOI] [PubMed] [Google Scholar]
- 31.Gregoriou V G, Jayaraman V, Hu X, Spiro T G. Biochemistry. 1995;34:6876–6882. doi: 10.1021/bi00020a035. [DOI] [PubMed] [Google Scholar]
- 32.Bertomieu C, Boussac A, Mantele W, Breton J, Nabedryk E. Biochemistry. 1992;31:11460–11471. doi: 10.1021/bi00161a026. [DOI] [PubMed] [Google Scholar]
- 33.Li X Y, Czernuszewicz R S, Kincaid J R, Stein P, Spiro T G. J Phys Chem. 1990;94:31–47. [Google Scholar]
- 34.Lepetit L, Joffre M. Opt Lett. 1996;21:564–566. doi: 10.1364/ol.21.000564. [DOI] [PubMed] [Google Scholar]