

Abstract
The correlation functions of the fluctuations of vibrational frequencies of azide ions and carbon monoxide in proteins are determined directly from stimulated photon echoes generated with femtosecond infrared pulses. The asymmetric stretching vibration of azide bound to carbonic anhydrase II exhibits a pronounced evolution of its vibrational frequency distribution on the time scale of a few picoseconds, which is attributed to modifications of the ligand structure through interactions with the nearby Thr-199. When azide is bound in hemoglobin, a more complex evolution of the protein structure is required to interchange the different ligand configurations, as evidenced by the much slower relaxation of the frequency distribution in this case. The time evolution of the distribution of frequencies of carbon monoxide bound in hemoglobin occurs on the ≈10-ps time scale and is very nonexponential. The correlation functions of the frequency fluctuations determine the evolution of the protein structure local to the probe and the extent to which the probe can navigate those parts of the energy landscape where the structural configurations are able to modify the local potential energy function of the probe.
Chemical reactions in biology can be controlled by the fluctuations in the energies of the reactant states. When the reactions involve charge translocations, the motions of the charges in the surrounding medium cause changes in the electric fields in the neighborhood of the reacting species, which in turn cause fluctuations of the reactant energies. These fluctuations occur on the time scale of the nuclear motions of the medium. For chemical processes occurring in a protein environment, such as in enzyme catalysis, the dynamics of the protein nuclei can control the reaction. Therefore, it is very important to determine experimentally the essential properties of the fluctuations associated with various observables, such as their mean values, time-correlation functions, and structural origins.
The experimental approaches to determining time-correlation functions include spectroscopic line shape measurements and time-domain responses. In the vibrational frequency regime the line-shape approach is a common one (1), although it does not always expose the underlying physical origins of the fluctuations. Time-domain techniques were also developed some time ago that enabled investigations of the various contributions to the line width such as pure dephasing and population relaxation (2). The dephasing times of vibrational transitions having static inhomogeneous distributions can be measured by means of two-pulse photon echoes (3–5). Silvestri et al. (6) have shown that for optical transitions, a photon echo with three pulses can also yield the dynamics of the inhomogeneous distribution. There were many applications of this nonlinear technique in the electronic spectroscopy of dye molecules (refs. 6–8, and see ref. 9 for a review) and of cofactors in biological systems [light-harvesting complex II (10), reaction center (11), Zn–myoglobin (12)]. This experiment was only recently accomplished with IR pulses exciting vibrational modes (13, 14). The IR measurements report on the frequency-fluctuation correlation function for cases where the inhomogeneous distribution of vibrational frequencies changes over measurable time scales.
In the present work we introduce the idea of using molecular vibrations to probe time-dependent changes in the heterogeneity of proteins. Previous studies of vibrational dynamics in proteins were restricted to the determination of orientation, population, and dephasing relaxation times of ligand vibrations in heme proteins (15–21). In the present work the goal is to determine the dynamics of the inhomogeneous distribution of vibrational frequencies by measuring the time evolution of the vibrational-frequency correlation function. The vibrational frequency is modified when the interaction with the medium alters the functional form of the potential-energy surface of the vibrator. The idea is quite general, and the vibrational transitions could be intrinsic, corresponding to natural parts of the protein, or extrinsic, such as molecules or ions bound to protein or to enzyme active sites. This work is distinct both in philosophy and in methodology from the many studies of transient vibrational spectra of biological photoreactions and their intermediates (16, 22–24).
Correlation functions related to protein dynamics have been obtained by using a variety of fast optical methods. Fluorescent probes attached to proteins or nucleic acids have been used to determine orientational correlation functions through studies of their fluorescence anisotropy. These measurements yield properties of the fluctuations in the angular velocities of the probe-transition dipole and the coupled angular fluctuations of the protein (25). Studies of the time dependence of the Stokes shifts of charge-sensitive probes or cofactors by fluorescence lifetime, transient absorption, and photon echoes have all yielded information on the energy fluctuations experienced at the site of the probe (9). These experiments can measure the probe electronic-transition energy fluctuations induced by nuclear motions of the medium and of the probe chromophore itself.
The experimental method used in the present work is the IR three-pulse or stimulated-photon echo. We employ femtosecond IR pulses in the first such experiments on vibrational transitions of probes associated with proteins. We will see that studies of vibrational dynamics using femtosecond nonlinear spectroscopy can expose features of protein dynamics that are not otherwise available.
OUTLINE OF THE FEMTOSECOND IR THREE-PULSE PHOTON-ECHO METHOD
In the experiment, three femtosecond IR pulses in a variable time sequence, having wave vectors k1, k2, and k3, interact with the sample and generate two new fields into directions kα and kβ as pictured in Fig. 1a. The magnitude of each of the generated fields in the nonlinear IR experiment is determined by the convolution of a probe-response function with the electric fields of the incident laser pulses (14, 26). The pulse sequence T, τ, t is depicted in Fig. 1a, where T is the time separation between the peaks of pulse 3 and pulse 2 for τ > 0, or between the peaks of pulse 3 and pulse 1 for τ < 0. The kα signal contains the same information at −τ as the kβ signal contains at τ. The detectors measure the square of each of the IR electric fields averaged over time t. When the vibrators change their frequencies very rapidly compared with the time scale of the observation the system is said to be homogeneous, and only then will the kα and kβ signals be identical. Any system will appear homogeneous when T becomes larger than all of the relaxation times, at which point there will be no longer any memory of the inhomogeneous distribution of frequencies present when the sample was first excited. It is the temporal evolution of the inhomogeneity that is measured in this work. The protein interaction with the probe is modeled by the correlation function of the vibrational frequency fluctuations, δω10(t) or δω21(t), about their mean values. These mean values, which are the transition frequencies connecting the probe vibrational quantum states v = 0 and v = 1 or v = 1 and v = 2 are separated by Δ, the anharmonicity. Both the linear and nonlinear IR spectra are characterized by the function g(t) defined as (see Appendix and ref. 14 for more details)
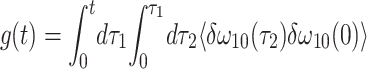 |
1 |
It is assumed here that the fluctuations Δω10(t) and Δω21(t) are strictly correlated, so that at this stage the analysis only considers the changes in the quadratic part of the probe potential energy arising from the probe–protein interaction. If the oscillator were actually harmonic, there would be no signal [apart from effects caused by variations of dephasing rates with quantum number (27)], so the anharmonicity assumes a vital role in this experiment.
Figure 1.

(a) The geometry and timing used in the stimulated-photon echo experiment. Two detectors collect the light emitted into the kα = −k1 + k2 + k3 and the kβ = k1 − k2 + k3 directions. (b) Stimulated-photon echo signal obtained for CA–N3− in the kα direction plotted against the delay time between pulse 1 and pulse 2 (τ) and pulse 2 and pulse 3 (T).
The value of T1 needed to interpret the experiment was measured from the exponential decay of the signal versus T at τ = 0, a situation that corresponds to a conventional transient-population grating experiment, or from a separate pump/probe experiment (19) with two IR pulses. The time over which the correlation function can be examined by the photon echo is limited by T1. For a free or a very loosely bound probe the orientational relaxation, which can also be obtained from separate polarized pump/probe experiments, would also need to be taken into account (14). The rotational diffusion of the protein is much slower than the echo time scales and therefore has negligible effect. Explicit, general forms of the response functions similar to those needed to interpret our experiments (14) have been given by Mukamel (26).
METHODS OF PROCEDURE
Tunable femtosecond IR pulses with pulse durations of 120 fs, energies of ≈1 μJ, bandwidths of ≈150 cm−1, and frequencies centered to the absorption lines of the N3− or CO in the different experiments were generated by mixing the outputs of a BBO-optical parametric amplifier in a AgGaS2 crystal (28). The output was split into three beams each with the same polarization and energy 300 nJ. The pulses with directions k1 and k3 traversed computer-controlled delay lines. The beams were focused at the sample (spot size 150 μm) in a box configuration (see Fig. 1) (29). A small third-order signal from the solvent (<3%) was observed when pulses 1 and 2 temporally overlap. Two mercury-cadmium-telluride detectors recorded the signals phase-matched in the two directions kβ = k1 − k2 + k3 and kα = −k1 + k2 + k3.
Three different samples have been investigated in this work: N3− bound to bovine carbonic anhydrase II (CA–N3−), N3− bound to hemoglobin A (Hb–N3−), and CO bound to hemoglobin (Hb–CO). The protein samples were purchased from Sigma and used without further purification. The CA–N3− sample (8 mM) was prepared in D2O containing 7 mM NaN3 and 100 mM Mes buffer (pH 5.7). Under these conditions, 91% of the total azide was bound to protein. Its absorption peak shifts from 2,043 cm−1 in D2O to 2,094 cm−1 in the protein. The Hb–N3− sample (≈13 mM Hb) was dissolved in D2O containing 100 mM potassium phosphate buffer (pH 6.8) and 7 mM NaN3. In that case, ≈90% of the ions also are bound, and the vibrational frequency shifts to 2,023 cm−1. The samples were filtered through a 5-μm membrane filter before being loaded into a gas-tight 50-μm pathlength sample cell. The Hb–CO sample (16 mM) in 0.1 M phosphate buffer (pH 7.6) was reduced with 0.1 M dithionate, stirred under 1 atmosphere (= 101.3 kPa) of CO for several hours and again filtered through a 5-μm membrane filter. The main absorption peak appears at 1,951 cm−1. However, a small band (10%) is also observed at ≈1,970 cm−1 (i.e., the A0 band).
The continual irradiation of protein solutions with IR pulses has no known influence on the integrity of the samples, which can therefore be used repeatedly with little if any denaturation.
RESULTS
An example of a complete set of stimulated-photon echo data as a function of both delay times T and τ is shown in Fig. 1b for CA–N3− (kα direction only). In Fig. 2, the signal as a function of τ for some selected times T is shown for both the kα and the kβ directions. Neither the kα nor the kβ signals are symmetric in τ, proving that an inhomogeneous distribution is present in all of the cases studied and for all times T. However, a careful inspection indicates that the asymmetry of each of the signals with respect to τ decreases with T. This is most evident for Hb–CO, in which the longer vibrational lifetime permits the process to be followed for up to ≈40 ps. The temporal evolution of the asymmetry is evident from first-moment M1(T) of the signals† (see Fig. 3). The first moments have no fundamental significance other than they are convenient measures of the asymmetry of the signal versus τ at a given value of T. They assess qualitatively whether an inhomogeneous distribution still exists after time T, so the approach of the photon-echo signals to a symmetric form centered at τ = 0, at which point M1 = 0, mimics in some respects the evolution of the spectral diffusion. They also give an idea of the time scales of the spectral-diffusion processes. In the case of CA–N3−, M1(T) decays over the first 4 ps, while for Hb–N3−, the first moment M1(T) stays constant during this period after a small initial decay. The inhomogeneity of Hb–CO decays on at least two time scales within the observation window of 40 ps.
Figure 2.

Stimulated-photon echo signal of CA–N3−, Hb–N3−, and Hb–CO plotted against delay time τ for selected delay times T together with global fits (solid lines). The signals in the kα (solid symbols) and the kβ (shaded symbols) directions are both shown. The kα signal at +τ is identical to the kβ signal at −τ, and the shift between both signals is a measure of inhomogeneity at time T. The asymmetry of the signals with respect to τ decays with time T.
Figure 3.

The normalized first moments of CA–N3−, Hb–N3−, and Hb–CO as a function of delay time T. The Inset shows the Hb–CO data with an extended time axis. The inhomogeneity decays with time T as a result of conformational fluctuations of the proteins.
The fitting procedure consisted of first estimating a correlation function from the first moments M1(T) and then refining the parameters of this function by least-squares fitting to the complete set of data points using the formalism outlined in the Appendix. Model-correlation functions contained sum of exponentials and a constant:
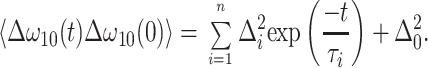 |
2 |
Two terms were needed for CA–N3− and Hb–CO and one for Hb–N3−. The fits are included in Fig. 2, showing a reasonable overall agreement.‡ The fit parameters are summarized in Table 1, and the resulting correlation functions are plotted in Fig. 4. The parameter τ2⋅Δ2 is <1 in all three samples, indicating that the faster process is close to the motional narrowing limit that would be describe by a pure dephasing time T2 = (τ2⋅Δ22)−1. The vibrational relaxation times, T1, were deduced from the transient grating signal (i.e., τ = 0) and are included in Table 1.
Table 1.
The parameter of the energy fluctuation correlation function (Eq. 2) obtained from global fits and vibrational relaxation time T1 obtained from the transient grating signal (i.e., τ = 0)
| Species | Δ0/ps−1 | Δ1/ps−1 | τ1/ps | Δ2/ps−1 | τ2/ps | T1/ps |
|---|---|---|---|---|---|---|
| CA–N3− | — | 0.95 | 17 | 1.6 | 0.2 | 2.8 |
| Hb–N3− | 0.56 | — | — | 1.2 | 0.3 | 2.5 |
| Hb–Co | 0.35 | 0.41 | 12 | 0.71 | 0.5 | 25 |
Figure 4.

The frequency-fluctuation correlation functions (Eq. 2) obtained from global fits of the complete data sets (such as the one shown in Fig. 1) of CA–N3− (solid line), Hb–N3− (dashed line), and Hb–CO (dashed-dotted line). The Inset shows the Hb–CO results with the time axis extended to 40 ps.
DISCUSSION
The data show that a time-dependent inhomogeneous distribution or a spectral-diffusion process is present in all of the cases studied. The transition-frequency correlation functions (see Table 1), determined from the complete data sets, provide quantitative proof of this conclusion. They indicate the time scales over which the memory of the initial vibrational frequency distribution is lost by structural rearrangements. Qualitatively, the existence of spectral diffusion is easily seen from the first-moment plots. The finite values of M1(T) indicate that the kα and kβ signals are different even at the largest values of T reached in each of the experiments. Technically, the results show that the vibrational coherence introduced by pulse 1, and the corresponding phase information stored in the population created by pulse 2, continues to be detectable after rephasing is induced by pulse 3, albeit in an ever-decreasing manner, as T increases. The rephasing process that generates the echo can only occur when there is some memory of the original inhomogeneous distribution of frequencies. If this memory was obliterated, the kα and kβ signals would be identical at each value of τ. Therefore in the examples given, an inhomogeneous distribution of vibrational frequencies must initially exist and it must be time-dependent.
The time-dependence of the inhomogeneous distribution has a special significance in the case of proteins because it measures the changes in the structure of those parts of the protein that influence the probe vibrational spectrum. The potential-energy surface of the probe molecule is changing as a result of the interaction with the protein. The potential will be sensitive to forces that can influence the charge distribution in the probe. Therefore, this method is a probe of the local structure.
Long-range interactions can also cause frequency changes by the shifting of vibrational transition in response to the fields from the fluctuating charges of the medium. However, the dipole or quadrupole moments needed to couple to those fields vary only slightly with the quantum number of vibrational states because of the small anharmonicity of the oscillators, so these perturbations will be small. However, an important effect of longer range interactions on the spectral diffusion would be to cause energy and nuclear-position fluctuations of those parts of the protein that are involved in direct coupling to the probe atoms. By this mechanism, the local interactions can sense the bulk fluctuations of the protein. The local forces can be changed either by fluctuations in the local structure or by the local structure responding to changes in other parts of the protein. This picture suggests a plausible interpretation of the time sequence of events in the evolution of the inhomogeneous distribution around a local region.
The stimulated-echo results for the proteins are to be contrasted with those for isolated ions (13, 14) in water, where the inhomogeneity disappears after only few ps. The time scale for the loss of the memory of the inhomogeneous distributions is much shorter than found in the proteins. This provides additional support for the conclusion that the protein dynamics are directly responsible for the effects reported here. For the case of azide in water, the relaxation time of 1.5 ps is not that reported for water (30); rather, it corresponds to the making and breaking of the hydrogen bonds between azide ions and water molecules, establishing that it is the local-structure fluctuations that dominate the spectral diffusion for that case (14). This conclusion forms the basis of each of the following specific examples.
Fluctuations in the Active Site of Carbonic Anhydrase II.
Carbonic anhydrase (CA-II) is a zinc enzyme that catalyzes the interconversion of CO2 and bicarbonate. The azide ion, which is a competitive inhibitor of bicarbonate dehydration in CA-II, binds at Zn2+ without compromising the three-dimensional structure of the active site of the enzyme (Fig. 5; refs. 31 and 32). The active site is a Zn2+ ion buried ≈10 Å from the protein surface and coordinated in an approximately tetrahedral geometry to four ligands (31, 32): azide, His-94, His-96, and His-119. The azide nitrogen (N(1)) closest to the metal and the central nitrogen (N(2)) have short contacts (3.3 Å) to the hydroxyl oxygen of Thr-199. In addition, N(2) (at 3.7 Å) and N(3) (at 3.5 Å) must sense the amide nitrogen of Thr-199. Therefore, it is natural to invoke the nearby Thr-199 as the group that modifies the potential-energy function and controls the charges of Zn-bound azide. The charge on N(2) in isolated azide is ≈+1 electron (e), whereas the end atoms each have a ≈−1 e charge (33). Recent calculations (34) have shown that the Zn-bound azide N(2) becomes more positive by 0.11 e, N(1) becomes less negative by 0.03 e, and N(3) becomes more negative by 0.24 e when the effect of the enzyme environment, mainly the Thr-199, is taken into account. These simulations and quantum chemical calculations suggest that protein motions should cause fluctuations in the admixtures of the dominant valence bond structures N−1⩵N+1⩵N−1 and N N+1—N−2 of the bound azide. Each admixture corresponds to a different potential-energy function for the ground state and hence a different vibrational frequency. The solvent-coupling of valence-bond structures provides a simple model for dephasing and spectral diffusion. Such an interpretation is consistent with the azide vibrational frequency in the enzyme being increased by ≈50 cm−1 compared with azide in water and by 110 cm−1 compared with azide in the gas phase (35). The presence of a small admixture of the triple-bonded structure causes the averaged frequency to increase significantly.
Figure 5.

The structure of the binding pocket of CA–N3− (32). The atoms of the azide ion, which is bound to the active site (Zn2+), is in close contact with Thr-199 (indicated by the dotted lines).
Fluctuations of the CO Ligands of Hemoglobin.
The IR transition of a bound carbon monoxide molecule has been used before to probe hemoglobin dynamics (15–21). Both x-ray diffraction (36) and IR polarized absorption (17, 19) find the CO to be oriented essentially perpendicular to the heme plane and angularly immobilized on the time scale of tens of picoseconds (17). The vibrational-energy relaxation time (19) in this case is long enough to open a significant time window—in excess of 40 ps—for studies of the stimulated photon echo. Fig. 2 shows the stimulated echo arising from the CO vibration at 1,951 cm−1. Again, there is evidence for a significant local-structure evolution over the time scale of the experiment. The correlation function of the frequency fluctuations does not decay exponentially but can be fitted to a sum of two exponential decays plus a part that is stationary over the time scale of the observations (see Table 1). The variations in the CO potential can be caused by its interactions through the iron with the heme and its proximal ligand, and by forces exerted on the CO by the amino acids in the distal heme pocket. The fast component of the correlation function was measured previously for Mb–CO by two-pulse photon-echo experiments (4, 5). This essentially homogeneous bandwidth was attributed to energy fluctuations brought about by motions of the distal amino acids. The slower components of the correlation function, responsible for the major part of the decay of the first moment, correspond to the relaxation of the inhomogeneous distribution of CO frequencies. This is suggested to arise from the interaction of the CO with the distal histidine. It is known that the CO vibrational frequency and hence the potential energy for the CO stretching motion is extremely sensitive to the presence and positioning of His-E7 in hemoglobin (37) and myoglobin (38, 39). On the other hand, the CO frequency is less sensitive to mutations of Val-E11, another heme-pocket residue. Variations in the relative positioning of the polar His-E7 and the CO can cause mixing of the valence bond structures Fe⩵C⩵O and Fe—C O (41). The latter structure is stabilized by the hydrogen bond to E7 causing an increase in the vibrational frequency. Again, we invoke medium-induced mixing of valence bond structures as a model for fluctuating the vibrational frequency. We suggest that the 12-ps process, which is a main part of the correlation function, corresponds to relaxation of these distal pocket structures.
Measurements in the optical regime below 20 K (12) have shown that the interchange of structural configurations or protein substates that cause fluctuations in the heme transition energy stretch over a very wide time scale from picoseconds to microseconds. This evolution would be speeded up at 300 K. According to our results, motion within the distribution of substates that modify the CO vibrational frequency begins significantly on the time scale of 12 ps. This implies that the CO oscillator potential in Hb–CO varies significantly over this time scale. However, even after the experimental limit of 40 ps, the inhomogeneity is not yet equilibrated, and its complete relaxation must be stretched over an even longer time scale. We have suggested that the relaxation of the probe frequency distribution caused by only the local motions (e.g., His-E7 of Hb–CO) is the main contributor to the ≈12-ps dynamics. The slower changes in the CO potential must require more complex reorganizations of the protein, presumably involving many more protein atoms.§ The residual inhomogeneity is therefore attributed to more global structural changes. Forces on the iron, transmitted by motions of the proximal structure and heme, would fall into this category and would cause changes in the CO frequency, but in a less direct manner.
Fluctuations of the N3− Ligands of Hemoglobin.
Myoglobin azide has a structure with the linear azido group forming an angle of ≈60° with the heme plane normal (41). A close polar contact with the ligand is the distal histidine, His-64, but the ligand is in van der Waals contact with numerous other amino acids in the heme pocket. The lifetime of the azide transition limits the data collection to ≈4 ps. It is apparent, however, that the correlation function is different from that obtained from CO in the same environment; it decays significantly more slowly at early times. This suggests that the inhomogeneous distribution is not relaxed by local fluctuations on a fast time scale. Either the relaxation requires a more complex structural reorganization of the protein or the azide is significantly more tightly bound to sites in the heme pocket than is CO.
CONCLUSIONS
Protein motions are expected to stretch over many orders of magnitude in time at low temperatures (12, 42) and even at ambient temperatures (43, 44). It is evident that different methods of examining these motions will give results that depend on what is probed. In this work we have introduced a method that is sensitive to the microscopic protein structure changes around a small molecule probe, inasmuch as they are able to modify the potential energy determining the probe vibrational spectrum.
A unique feature of the vibrational approach is that small molecular or ionic probes can be employed, thereby allowing information about local structural fluctuations to be obtained while minimizing deleterious effects on the protein function. Optical probes are usually fairly large cofactors or dye molecules. Their responses are generally assumed to provide properties of the charge fluctuations of the whole medium. It is the fluctuations of the parts of the protein that are directly coupled to the probe—and are therefore the main perturbers of its potential-energy surface—that are important for small molecule vibrational probes. Accurate calculations of these fluctuations seem to be achievable with the help of molecular dynamic simulations and quantum-chemistry calculations, providing a direct link between theory and experiment.
In experimental and theoretical work on electronic transitions of two-level systems, the essential feature of the dynamics revealed by the three-pulse echo is the correlation function of the fluctuations of the electronic transition energy. The measurement can expose solvent-induced energy changes that occur when the charge distribution is modified by electronic excitation. As mentioned above, the practical situation is qualitatively different for vibrational transitions. For example, there are always more than two vibrational levels in the nonlinear IR response because the vibrator consists of a set of roughly equally spaced levels. Another property unique to vibrations is that the fluctuations of the transition frequencies of nearly harmonic potentials, inasmuch as they derive from solvent-induced changes in the whole potential-energy function of the probe molecule, can be highly correlated. In this work we assumed they were fully correlated, but further work is needed determine the actual correlation coefficients.
The time scale over which the correlation function can be measured by vibrational probing is limited by the energy relaxation time, T1, of the probed vibrational states. Although there is considerable information on these relaxation times (2, 45, 46), this is not the case for molecules in protein environments or for small molecules or ions in liquids (46). Some molecular vibrations relax by transferring the energy excess directly to the environment, whereas others first undergo intramolecular vibrational-energy redistribution, which can be less sensitive to the environment. Some examples are the vibrational excitations of ions such as CN−, which live for ≈30 ps in water (28), the CH vibration of neutral HCN, which lives for hundreds of picoseconds in nonpolar solvents (47), and CO2, which relaxes within 10 ps in water (13) but can live much longer in less polar environments. As an example, in a non-hydrogen bonding environment the azide-ion relaxation time is extended to 15 ps (48). In these and other cases in which a large amount of energy relaxes directly into the surroundings in a multiphonon process, the relaxation times themselves are sensitive indicators of the local environment of the probe molecules and hence of the local fluctuations of the protein structure.
Acknowledgments
We thank J. T. Hynes for valuable discussion. P.H. is grateful to the Deutsche Forschungsgemeinschaft for a Fellowship. The research was supported by the National Institutes of Health and the National Science Foundation with instrumentation developed under National Institutes of Health Grant RR13456.
ABBREVIATIONS
- CA–N3−
azide carbonic anhydrase
- Hb–N3−
azide hemoglobin
- Hb–CO
carbon monoxide hemoglobin
- e
electron
Appendix
With the help of the function g(t), defined by Eq. 1, the total response function, R(T,τ,t), is assumed to be
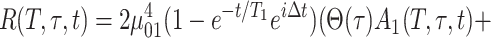 |
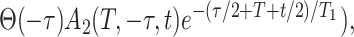 |
where Θ(t) is the Heaviside function. A1 and A2 are given by
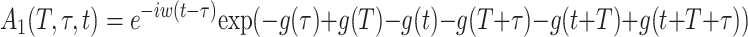 |
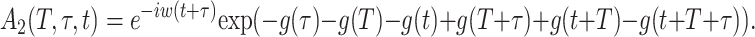 |
The detector measures the integrated four-wave mixing signal to which our data are fit:
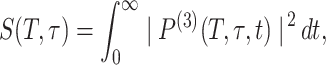 |
where the third-order polarization P(3) is evaluated by convoluting the total response function R(T,τ,t) with the electric fields of the IR pulses. A harmonic oscillator approximation implies 2μ102 = μ212, where μij is the transition moment from state i to state j in the vibrator.
Footnotes
A Commentary on this article begins on page 15161.
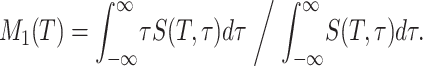 |
The fits are not expected to be perfect. There is a small fifth-order contribution to the signal that is not included in the fitting. This causes the oscillations at the frequency of the anharmonic shift between adjacent levels, which are most evident in the Hb−CO signal in Fig. 2. Furthermore, the fits assume that only a single IR absorption band generates the signal. Actually, in each example there is a weak satellite band with ≈10–20% of the strength of the main band (see Methods of Procedure) that contributes a small amount to the signal.
The CO absorption is composed of transitions from the α- and β-subunits of hemoglobin that have similar heme pockets (36). Although these transitions are not separated in the IR spectrum, the correlation function can be influenced by this inhomogeneity as well.
References
- 1.Gordon R G. Adv Magn Reson. 1968;3:1–42. [Google Scholar]
- 2.Laubereau A, Kaiser W. Rev Mod Phys. 1978;50:607–665. [Google Scholar]
- 3.Zimdars D, Tokmakoff A, Chen S, Greenfield S R, Fayer M D, Smith T I, Schwettman H A. Phys Rev Lett. 1993;70:2718–2721. doi: 10.1103/PhysRevLett.70.2718. [DOI] [PubMed] [Google Scholar]
- 4.Rector K D, Rella C W, Jeffrey R H, Kwok A S, Sligar S G, Chien E Y T, Dlott D D, Fayer M D. J Phys Chem. 1997;101:1468–1475. [Google Scholar]
- 5.Rector K D, Engholm J R, Hill J R, Myers D J, Hu R, Boxer S G, Dlott D D, Fayer M D. J Phys Chem B. 1998;102:331–333. [Google Scholar]
- 6.Silvestri S D, Weiner A M, Fujimoto J G, Ippen E P. Chem Phys Lett. 1984;112:195–199. [Google Scholar]
- 7.Joo T, Albrecht A C. Chem Phys. 1993;176:233–247. [Google Scholar]
- 8.Joo T, Jia Y, Fleming G R. J Chem Phys. 1995;102:4063–5068. [Google Scholar]
- 9.Fleming G R, Cho M. Annu Rev Phys Chem. 1996;47:109–134. [Google Scholar]
- 10.Joo T, Jia Y, Yu J-Y, Jonas D M, Fleming G R. J Phys Chem. 1996;100:2399–2409. [Google Scholar]
- 11.Groot M-L, Yuj Y, Agarwal R, Norris J R, Fleming G R. J Phys Chem. 1998;102:5923–5931. [Google Scholar]
- 12.Leeson D T, Wiersma D A, Fritsch K, Friedrich J. J Chem Phys. 1997;101:6331–6340. [Google Scholar]
- 13.Hamm P, Lim M, Hochstrasser R M. In: Ultrafast XI. Elsaesser T, Wiersma D, Zinth W, Fujimoto J G, editors. Berlin: Springer; 1998. , in press. [Google Scholar]
- 14.Hamm, P., Lim, M. & Hochstrasser, R. M. (1998) Phys. Rev. Lett., in press.
- 15.Moore J N, Hansen P A, Hochstrasser R M. Proc Natl Acad Sci USA. 1988;85:5062–5066. doi: 10.1073/pnas.85.14.5062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Anfinrud P A, Han C, Hochstrasser R M. Proc Natl Acad Sci USA. 1989;86:8387–8391. doi: 10.1073/pnas.86.21.8387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Locke B, Lian T, Hochstrasser R M. Chem Phys. 1991;158:409–419. [Google Scholar]
- 18.Hill J R, Tokmakoff A, Peterson K A, Sauter B, Zimdars D, Dlott D, Fayer M D. J Chem Phys. 1994;98:11213–11219. [Google Scholar]
- 19.Owrutsky J C, Li M, Locke B, Hochstrasser R M. J Phys Chem. 1995;99:4842–4846. [Google Scholar]
- 20.Lim M, Jackson T A, Anfinrud P A. Science. 1995;269:962–966. doi: 10.1126/science.7638619. [DOI] [PubMed] [Google Scholar]
- 21.Lim M, Jackson T A, Anfinrud P A. J Chem Phys. 1995;102:4355–4366. [Google Scholar]
- 22.Maiti S, Walker G C, Cowen B R, Pippenger R, Moser C C, Dutton P L, Hochstrasser R M. Proc Natl Acad Sci USA. 1994;91:10360–10364. doi: 10.1073/pnas.91.22.10360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hamm P, Zurek M, Mäntele W, Meyer M, Scheer H, Zinth W. Proc Natl Acad Sci USA. 1995;92:1826–1830. doi: 10.1073/pnas.92.6.1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Diller R, Maiti S, Walker G, Cowen B R, Pippenger R, Bogomolni R A, Hochstrasser R M. Chem Phys Lett. 1995;241:109–115. [Google Scholar]
- 25.Szabo A. J Chem Phys. 1984;81:150–167. [Google Scholar]
- 26.Mukamel S. Principles of Nonlinear Optical Spectroscopy. New York: Oxford Univ. Press; 1995. [Google Scholar]
- 27.Fourkas J T, Kawashima H, Nelson K A. J Chem Phys. 1995;103:4393–4407. [Google Scholar]
- 28.Hamm P, Lim M, Hochstrasser R M. J Chem Phys. 1997;107:10523–10531. [Google Scholar]
- 29.Levenson M D, Kano S S. Introduction to Nonlinear Laser Spectroscopy. Boston: Academic; 1988. [Google Scholar]
- 30.Jimenez R, Fleming G R, Kumar P V, Maroncelli M. Nature (London) 1994;369:471–473. [Google Scholar]
- 31.Nair S K, Christianson D W. Eur J Biochem. 1993;213:507–515. doi: 10.1111/j.1432-1033.1993.tb17788.x. [DOI] [PubMed] [Google Scholar]
- 32.Jönsson B M, Hakansson K, Liljas A. FEBS Lett. 1993;322:186–190. doi: 10.1016/0014-5793(93)81565-h. [DOI] [PubMed] [Google Scholar]
- 33.Gora T, Kemmey P J. J Chem Phys. 1972;57:3579–3581. [Google Scholar]
- 34.Merz K M, Jr, Banci L. J Phys Chem. 1996;100:17414–17420. [Google Scholar]
- 35.Polak M, Gruebele M, Saykally R J. J Am Chem Soc. 1987;109:2884–2887. [Google Scholar]
- 36.Derewenda Z, Dodson G, Emsley P, Harris D, Nagai K, Perutz M, Reynaud J-P. J Mol Biol. 1990;211:515–519. doi: 10.1016/0022-2836(90)90262-k. [DOI] [PubMed] [Google Scholar]
- 37.Lin S-H, Yu N T, Tame J, Shih D, Renaud J-P, Pagnier J, Nagai K. Biochemistry. 1990;29:5562–5566. doi: 10.1021/bi00475a021. [DOI] [PubMed] [Google Scholar]
- 38.Braunstein D P, Chu K, Egeberg K D, Frauenfelder H, Mourant J R, Nienhaus G U, Ormos P, Sligar S G, Springer B A, Young R D. Biophys J. 1993;65:2447–2454. doi: 10.1016/S0006-3495(93)81310-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li T, Quillin T L, Phillips G N, Olson J S. Biochemistry. 1994;33:1433–1446. doi: 10.1021/bi00172a021. [DOI] [PubMed] [Google Scholar]
- 40.Ray G B, Li X-Y, Ibers J A, Sessler J L, Spiro T G. J Am Chem Soc. 1994;116:162–176. [Google Scholar]
- 41.Maurus R, Bogumil R, Nguyen N T, Mauk A G, Brayer G. Biochem J. 1998;332:67–74. doi: 10.1042/bj3320067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Austin R H, Beeson K W, Eisenstein L, Frauenfelder H, Gunsalus I C. Biochemistry. 1975;14:5355–5373. doi: 10.1021/bi00695a021. [DOI] [PubMed] [Google Scholar]
- 43.Jones C M, Henry E R, Hu Y, Chan C-K, Luck S D, Bhuyan A, Roder H, Hofricher J, Eaton W A. Proc Natl Acad Sci USA. 1993;90:11860–11864. doi: 10.1073/pnas.90.24.11860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lim M H, Jackson T A, Anfinrud P A. J Biol Inorg Chem. 1997;2:531–536. [Google Scholar]
- 45.Seilmeier A, Kaiser W. In: Ultrashort Laser Pulses. Kaiser W, editor. New York: Springer; 1993. pp. 279–317. [Google Scholar]
- 46.Owrutsky J C, Raftery D, Hochstrasser R M. Annu Rev Phys Chem. 1994;45:519–555. doi: 10.1146/annurev.pc.45.100194.002511. [DOI] [PubMed] [Google Scholar]
- 47.Raftery D, Gooding E, Romanovsky A, Hochstrasser R M. J Chem Phys. 1994;101:8572–8579. [Google Scholar]
- 48.Li M, Owrutsky J, Sarisky M, Culver J P, Yodh A, Hochstrasser R M. J Chem Phys. 1993;98:5499–5507. [Google Scholar]