

Abstract
The iron-sulfur flavoenzyme adenylylsulfate (adenosine 5′-phosphosulfate, APS) reductase catalyzes reversibly the reduction of APS to sulfite and AMP. The structures of APS reductase from the hyperthermophilic Archaeoglobus fulgidus in the two-electron reduced state and with sulfite bound to FAD are reported at 1.6- and 2.5-Å resolution, respectively. The FAD-sulfite adduct was detected after soaking the crystals with APS. This finding and the architecture of the active site strongly suggest that catalysis involves a nucleophilic attack of the N5 atom of reduced FAD on the sulfur atom of APS. In view of the high degree of similarity between APS reductase and fumarate reductase especially with regard to the FAD-binding α-subunit, it is proposed that both subunits originate from a common ancestor resembling archaeal APS reductase. The two electrons required for APS reduction are transferred via two [4Fe-4S] clusters from the surface of the protein to FAD. The exceptionally large difference in reduction potential of these clusters (−60 and −500 mV) can be explained by interactions of the clusters with the protein matrix.
Sulfur compounds are widely used for energy conservation by several bacterial lineages and one hyperthermophilic order of Archaea, the Archaeoglobales. In the dissimilatory pathway the terminal electron acceptor sulfate becomes reduced to hydrogen sulfide, a process that is used for energy conservation (1). This pathway has to be distinguished from the assimilatory sulfate reduction in plants and bacteria, where the hydrogen sulfide is used for the biosynthesis of cysteine and S-containing cofactors. In the sulfur-oxidizing pathway sulfide, elemental sulfur, and thiosulfate serve as electron donors, and sulfate is the final product.
The pathways of dissimilatory sulfate reduction and sulfur oxidation involve three key enzymes localized in the cytoplasm or at the cytoplasmic aspect of the inner membrane. Sulfate has to be activated to adenylylsulfate (adenosine 5′-phosphosulfate, APS) by ATP-sulfurylase (EC 2.7.7.4) at the expense of ATP, APS reductase (EC 1.8.99.2) converts APS to sulfite and AMP, and sulfite is reduced to sulfide by sulfite reductase (EC 1.8.7.1): AdeO3POSO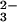 (APS) + 2e− → AdeOPO
(APS) + 2e− → AdeOPO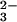 (AMP) + SO
(AMP) + SO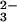 . In the sulfur-oxidizing pathway these three enzymes operate in the reverse direction, liberating electrons and ATP.
. In the sulfur-oxidizing pathway these three enzymes operate in the reverse direction, liberating electrons and ATP.
The molecular parameters including mass, subunit composition, and cofactor stoichiometry of APS reductase, which had been characterized from diverse sulfate-reducing and sulfur-oxidizing organisms, have been a matter of debate. Most recently, highly active APS reductase from sulfate-reducing and sulfur-oxidizing bacteria and archaea was isolated as a 1:1 αβ-heterodimer with a molecular mass of ≈95 kDa (2). The α-subunit (≈75 kDa) contains a noncovalently bound flavin-adenine dinucleotide (FAD), and the β-subunit (≈20 kDa) contains two [4Fe-4S] clusters of the ferredoxin type (2, 3). The spectroscopic properties of these APS reductases, especially UV/visible and EPR spectra, are practically identical. Thus, the redox cofactors appear to be embedded into a strictly conserved protein matrix. Sequence comparisons of enzymes from mesophilic eubacteria and hyperthermophilic archaea support the hypothesis that both APS reductase and sulfite reductase were present in the last common ancestors of archaea, bacteria, and Eukarya (4). Here we describe the three-dimensional structure of the iron-sulfur flavoenzyme APS reductase from the hyperthermophilic archaeon Archaeoglobus fulgidus (5). The protein was purified and crystallized under strict exclusion of dioxygen. Under these conditions the FAD cofactor is in the two-electron reduced state (APSR-red), whereas both Fe-S centers are in the oxidized state. Soaking of these crystals with APS resulted in the reductive cleavage of APS and formation of a FAD-sulfite adduct (APSR-sulfite).
Protein Purification and Crystallization.
APS reductase from A. fulgidus was purified and crystallized as described (6). Briefly, crystals were grown at 4°C with the hanging-drop method by using 4–6% polyethylene glycol 4000/0.1 M NaAc, pH 4.8/0.2 M NaCl as reservoir solution. The crystals had the space group P212121 with cell parameters of a = 72.4 Å, b = 113.2 Å, and c = 194.0 Å and one α2β2-heterotetramer per asymmetric unit.
Redox Potentials.
EPR-monitored potentiometric titrations of iron-sulfur centers were performed according to Dutton (7). The redox potential of FAD was determined according to Massey (8).
Structure Determination.
Diffraction data were collected at BW6 DESY, Hamburg and processed with the HKL- (9) and CCP4 (10) program suites. The data sets APSR-red and APSR-sulfite were based on crystals of APS reductase after purification and on crystals after soaking with 5 mM APS (Table 1), respectively. Phases were determined for APSR-red by a combination of the multiple isomorphous replacement and multiple anomalous dispersion methods. After soaking a crystal with 1 mM thimerosal for 24 h, the data were collected at the mercury and iron absorption edge (Table 1). The [4Fe-4S] cluster positions were extracted manually or with the help of SHELXS (11) from the anomalous difference Patterson map. The mercury sites were detected by using phases that were calculated from the [4Fe-4S] clusters positions by using MLPHARE (10) and SHARP (12). The quality of the generated solvent flattened electron density map, and the position of the heavy atoms allowed to mask each αβ-heterodimer and to determine the noncrystallographic symmetry operator. After 2-fold molecular averaging using DM (10) a nearly complete polypeptide chain including most of the side chains could be incorporated into the electron density map. The refinement of the APSR-red and APSR sulfite structures was performed by using CNS and converged to an Rcryst of 17.6%/16.0% and Rfree of 19.8%/17.6%. The figures were produced by using BOBSCRPIT (13), DINO (www.bioz.unibas.ch/∼xray/dino; ref. 14), and RASTER3D (15).
Table 1.
Data collection and structure determination
| Data set | APSR-red | APSR-sulfite | APSR-phas* | Thim1† | Thim2† | Thim3† | Thim4† |
|---|---|---|---|---|---|---|---|
| Wavelength | 1.06 | 1.06 | 1.07 | 1.005 | 1.009 | 1.733 | 1.740 |
| Resolution, Å | 1.6 | 2.5 | 2.5 | 2.6 | 2.5 | 2.7 | 2.6 |
| Completeness, % | 92.6 | 79.2 | 96.5 | 91.1 | 90.7 | 95.3 | 88.9 |
| Multiplicity | 3.3 | 2.6 | 3.1 | 5.9 | 6.1 | 6.2 | 6.0 |
| Rsym, %‡ | 6.3 | 8.0 | 3.2 | 4.6 | 4.8 | 6.2 | 5.2 |
| Phasing power§ | −/− | 0.3/0.3 | 2.1/3.2 | 1.5/2.4 | |||
| (centrics/acentrics) [ano] | −/2.2 | −/2.2 | −/2.7 | −/2.4 | |||
| Rcullis, %¶ | −/− | 0.6/0.6 | 0.4/0.3 | 0.5/0.4 | |||
| (centrics/acentrics) [ano] | −/0.7 | −/0.7 | −/0.6 | −/0.7 | |||
| Rcryst, %‖ | 17.6 | 16.0 | |||||
| Rfree, %** | 19.8 | 19.9 | |||||
| rms deviation bond length, Å | 0.010 | 0.008 | |||||
| rms deviation bond angles, Å | 1.4 | 1.5 |
APSR-phas, native data set used for phase determination.
Thim, thimerosal, 0.5 mM, 24 h. The cryobuffer was used for heavy-atom soaking experiments.
Rsym = ∑hkl∑i|Ii − 〈I〉|/∑〈I〉. Ii, intensity of the ith measurement per reflection hkl; 〈I〉, average intensity for a reflection.
Phasing power = rms. F(H)/E, F(H) is the heavy-atom structure factor amplitudes, and E is the lack of closure error.
Rcullis = ∑hkl(|FPH(obs)| − |FPH(calc)|)/∑hkl(|FPH(obs)| − |FP(obs)|).
Rcryst = ∑hkl(|F(obs)| − |F(calc)|)/∑hkl|F(obs)|.
Rfree = ∑hkl(|F(obs)| − |F(calc)|)/∑hkl|F(obs)|, where 5% of the observed structure factor amplitudes are not used for refinement.
Overall Molecular Structure.
The structures of two-electron reduced APS reductase (APSR-red) and of enzyme reacted with APS (APSR-sulfite) were solved at 1.6 and 2.5 Å resolution, respectively. The APSR-sulfite structure contained an FAD-sulfite adduct, showing that APS was cleaved by APSR in the crystalline state. The formed AMP could not be assigned unambiguously in the electron density. There are two αβ-heterodimers in the asymmetric unit that form a tight α2β2-heterotetramer (Fig. 1). Investigations of the enzyme using dynamic light scattering and gel filtration indicated that APS reductase from several organisms formed an αβ-heterodimer in solution (2). The difference between solution and crystal state is most likely caused by the protein concentration and buffer conditions. Thus, the functionally essential unit is the αβ-heterodimer. The heterodimer has a compact shape with dimensions of 56 × 65 × 70 Å. The globular part of the β-subunit is embedded into a shallow hollow of the α-subunit, whereas its long tail wraps around the α-subunit (Fig. 1).
Figure 1.

The domain structure of APS reductase. Stereo view of the αβ-heterodimer. The α-subunit is colored according to the domain structure. The FAD-binding domain that binds a noncovalently attached FAD is shown in blue, the capping domain in cyan, and the helical domain in light blue. The β-subunit that harbors two [4Fe-4S] clusters is shown in red.
Structure of the Two Subunits.
The structure of the α-subunit can be grouped into the so-called FAD-binding (A2–261, A394–487), helical (A488–643), and capping domains (A262–393; Fig. 1). The global fold of the α-subunit is reminiscent to that of the fumarate reductase structure family. The rms deviations between the α-subunit of APS reductase and the corresponding parts in aspartate oxidase of Escherichia coli (16) and fumarate reductase of E. coli (17), fumarate reductase of Wolinella succinogenes (18), and fumarate reductase of Shewanella frigidimarina (19) are 3.9, 2.6, 2.9, and 3.1 Å, respectively, calculated for 70, 81, 82, and 58% of the Cα atoms of APS reductase (20). The sequence identities between the aligned residues are just 21, 20, 19, and 15%, respectively. Because the anaerobic sulfur metabolism evolved very early during evolution, the three-dimensional structure of the α-subunit of APS reductase most likely resembles the ancestor of this group of flavoenzymes.
The structure of the β-subunit can be subdivided into three segments (Fig. 1). The fold of the first segment (B1–68) is highly similar to that found in bacterial ferredoxins (21) and envelopes two [4Fe-4S] clusters. The rms deviations between this segment and ferredoxins from Clostridium acidiurici (ref. 22; Fig. 2A), Desulfovibrio gigas (23), and Sulfolobus acidocaldarius (24) are 1.1, 1.7, and 1.4 Å for 85, 85, and 94% of the Cα atoms, respectively. In contrast, the second and especially the third segment are quite unusual and have not been observed yet in combination with a ferredoxin-like protein. The second segment (B69–104) of the β-subunit consists of a three-stranded antiparallel β-sheet, which constitutes an interface to the α-subunit. The third segment is composed of 44 amino acids forming a tail with a length of 50 Å that wraps around the α-subunit. This segment increases the contact surface between the α- and β-subunit to 4,300 Å (2) compared with an interface of 2,000 Å (2) formed by the first two segments. Although this C-terminal tail exhibits no secondary structure, the temperature factors of its residues are remarkably low, revealing that this segment is anchored firmly to the α-subunit. Obviously the tight interaction of the third segment of the β-subunit to the α-subunit establishes a stable heterodimer formation (Fig. 1). Such a tightening of the subunit interaction by a rather unordered segment is observed also in the structure of the human electron transfer flavoprotein (25). By comparison with most ferredoxins the two [4Fe-4S] centers of APS reductase differ significantly in their reduction potentials, −60 mV for cluster I and ≈−500 mV for cluster II (ref. 26; G.F., T.B., and P.M.H.K., unpublished data). Both [4Fe-4S] clusters have the shape of a distorted cube and the iron-iron, iron-sulfur, and sulfur-sulfur distances are similar compared with each other and with values of other ferredoxins (22, 24). However, the environment of the two clusters is significantly different (Fig. 2B). Recent studies indicate that local dipoles in close proximity to the cluster largely modulate the reduction potential. After electron uptake the extra negative charge will be localized predominantly on both the acid-labile sulfur and cysteinyl sulfur atoms (27). The reduced state is stabilized by NH–S hydrogen bonds (28) and backbone amide dipoles (29) shifting the reduction potential to more positive values. The sulfur atoms of cluster I exhibit 17 interactions with backbone amides at a distance of less than 3.5 Å vs. 7 amides in the proximity of the sulfur atoms of cluster II. In addition, one acid-labile sulfur of cluster II comes very close (3.0 Å) to the carboxylate oxygen of Asp-B11, which provides a negatively charged surrounding, appropriate to stabilize the oxidized state (Fig. 2B). This feature has not been observed thus far in the known structures of ferredoxins. Thus, the substantially increased number of polar interactions between cluster I compared with cluster II and the protein matrix can explain the high reduction potential of cluster I of −60 mV and the low potential of cluster II of ≈−500 mV.
Figure 2.

The [4Fe-4S] electron transfer sites of APS reductase. (A) Structural alignment of the FeS-binding domain of the β-subunit of APS reductase with the ferredoxin from Clostridium acidiurici. The β-subunit is shown in red, and the ferredoxin is shown in green. The β-subunit of APS reductase contains an elongated loop (shown in magenta) that presumably represents the docking site for the physiological electron donor. Electron transfer over a distance of ≈30 Å proceeds from the protein surface to FAD via the two [4Fe-4S] clusters and conserved Trp-B48 to the C8 methyl group of FAD. (B) Schematic representation of the two [4Fe-4S] cluster binding sites. Clusters I and II are linked covalently to the sulfhydryl group of four cysteines. The substantially increased number of polar interactions between cluster I compared with cluster II and the protein matrix can explain the differences in redox potential.
The APSR-Red and APSR-Sulfite States.
A pronounced feature in the APSR-red structure is the substantial bend of the isoalloxazine ring along the N5–N10 axis caused by a shift of the dimethyl and pyrimidine rings toward the si-face of FAD by an angle of 25° (Fig. 3A). This butterfly arrangement is better compatible with FAD in the reduced state (30). According to molecular orbital calculations, the optimal bending angle is 15–30° for the reduced and 0–10° for the oxidized isoalloxazine ring. Distortions from this optimal bending angle are not expensive energetically, in particular not for the reduced form of FAD (31, 32), but they can affect the reduction potential (33). Thus, the adjacent protein matrix presumably can exert a considerable influence on the bending angle whereby a flat conformation of FAD favors the oxidized state, and a bent conformation favors the reduced state. The butterfly conformation of the isoalloxazine ring in APS reductase is enforced by several polar and hydrophobic van der Waals contacts to the polypeptide chain. On the re-face, Asn-A74 and Trp-A234 push the pyrimidine ring and dimethylbenzene ring of isoalloxazine, whereas Leu-A70 on the si-face of FAD points toward the pyrazine ring. The induced stabilization of the reduced form of FAD agrees well with the reduction potential of −45 mV of FAD in APS reductase (G.F., T.B. and P.M.H.K., unpublished data) vs. ≈−220 mV of free FAD. A comparison between the APSR-sulfite and the APSR-red structures revealed roughly identical overall structures and geometries of the [4Fe-4S] clusters. Again, the isoalloxazine ring in the APSR-sulfite state shows the butterfly conformation (27°). The APSR-sulfite structure shows a sulfite molecule covalently linked to the N5 atom of the isoalloxazine ring. Because the N5 atom is in an sp3 configuration, the sulfite is positioned out of the ring plane toward the re-face of FAD (Fig. 3B). The sulfite moiety presumably is unprotonated, which is compatible with its sulfate character and the type of the contacting atoms (Fig. 3B). Sulfite oxygen atom O1 is hydrogen-bonded to the side chain amide nitrogen of Asn-A74, the oxygen atom O2 forms a strong salt bridge to His-A398, and the oxygen atom O3 is connected via a water molecule to Arg-A265 and Trp-A234. The covalent binding of sulfite hardly induces any conformational change in the surrounding polypeptide chain, except the guanidyl group of Arg-A265 moves ≈0.3 Å toward the sulfite moiety, and the imidazole ring of His-A398 moves ≈0.3 Å away from it.
Figure 3.

The FAD of APS reductase. (A) The isoalloxazine of FAD is present in a bent conformation (25°) that is enforced mainly by the protein environment, in particular Asn-A74, Leu-A70, and Trp-A234. The view is along the N5–N10 axis of FAD; the FAD and residues are shown in stick type and as transparent van der Waals spheres. (B) Interactions between the sulfite moiety of the FAD-sulfite adduct and protein. All three sulfite oxygens are strongly hydrogen-bonded to the polypeptide surrounding; His-A398 and Arg-A265 are the key residues involved in substrate binding and catalysis.
The Catalytic Reaction.
The reduction of APS requires two electrons that have to be transferred to the buried FAD over 30 Å via cluster II at the surface of the protein and cluster I (Fig. 4). Electron transfer between the unknown physiological electron donor and cluster II requires the docking of the donor to the protein surface adjacent to cluster II. Sequence comparisons indicate that the potential interface region that includes a flexible loop between Cys-B13 and Arg-B18 is conserved in APS reductases of the sulfate-reducing organisms but not in the enzyme of the sulfur-oxidizing Allochromatium vinosum in which the loop is absent. This observation supports the view that different redox partners interact with APS reductase depending on whether APS reduction or oxidation of sulfite and AMP is catalyzed. The distances between the redox centers in APS reductase are appropriate for effective electron transfer (34). The [4Fe-4S] clusters I and II have an edge-to-edge distance of 9.7 Å; the distance between the S3 of cluster I and the methyl group C8M of FAD is 12.4 Å (Fig. 4). The strictly conserved Trp-B48 is located between the two cofactors in van der Waals contact to both centers (Fig. 2A). The indole ring of Trp-B48 is locked in its position by a hydrogen bond to the carbonyl oxygen of Thr-A233 and by aromatic interactions to Arg-A232. Tryptophan residues between two redox centers are suited especially for electron transfer, as documented in the photosynthetic reaction center (35) and cytochrome peroxidase-cytochrome c complex (36). The active site of APS reductase is buried deeply in the protein interior and is accessible only from the outside through a 17-Å-long channel with a diameter of ≈10 Å (Fig. 4). The channel is formed at the interface between the FAD-binding and capping domains. The opening of the channel is lined up by an excess of positively charged residues (Arg-A83, Lys-A281, Lys-A283, Arg-A317) that might provide a selection for anions such as sulfite, AMP, or APS. The bottom of the channel accommodates the binding pocket of the isoalloxazine ring (Fig. 4). The substrate-binding site is formed in front of the re-face of FAD by His-A398, Trp-A234, Arg-A265, Val-A273, Gly-A274, Glu-A141, Gln-A145, Asn-A74, and Phe-A448. The analysis of the active sites of APSR-red and APSR-sulfite structures supports the proposed mechanism of the APS reduction (ref. 26; G.F., T.B., and P.M.H.K., unpublished data) outlined in Fig. 5. The reaction cycle is initiated by reduction of FAD to FADH2 and subsequent binding of APS. Atom N5 FADH2 attacks the sulfur of the APS to form a FAD-APS adduct. The proposed intermediate decomposes spontaneously to AMP and the APSR-sulfite, and sulfite becomes liberated. Presumably, the key step in the reaction cycle is the formation of the FAD-APS intermediate that would be facilitated if the atom N5 of FAD becomes more nucleophilic and the sulfate sulfur becomes more electrophilic. Furthermore, this first step could be driven through electrostatic stabilization of the negatively charged FAD-APS intermediate by the surrounding polypeptide matrix.
Figure 4.

The active site channel of APS reductase. Shown is a cut through the molecular surface of APS reductase to show the active site channel (blue) and the position of the cofactors. The active site channel is lined up by a number of conserved positively charged residues. Almost only the N5 atom of FAD is accessible to the solvent.
Figure 5.

Scheme of the catalytic mechanism of APS reductase. The mechanism is based on a nucleophilic attack of the atom N5 of FAD on the sulfate sulfur of APS, thus forming a FAD-APS intermediate that decays to AMP and a FAD-sulfite intermediate; the latter was structurally characterized. It is unclear whether the proton of N5 remains in proximity of the active site. It might be transferred to the residues Glu-A141 or Asp-A361.
The nucleophilicity of the N5 atom of FAD might be increased as a consequence of the deprotonation of the atom N1 in the APSR-red state. The resulting negative charge becomes delocalized primarily over the N1—C⩵O2 group (37) but also over the entire isoalloxazine ring including atom N5. The counterbalancing positive charge necessary to maintain the unprotonated state is provided by two hydrogen bonds donated from the polypeptide to the O2 atom of FAD and by the large dipole of a 30-Å-long helix that is pointing with its N terminus directly toward the N1 atom.
Active site residues around the sulfate moiety stabilize the FAD-APS and FAD-sulfite states. The structural analysis attributes a crucial role to the charged side chains of Arg-A265 and His-A398. The guanidyl group of Arg-A265 protrudes from the capping domain into the active site channel and is ideally suited to bind both the sulfate and phosphate groups of APS. The movement of the side chain toward the sulfite moiety in the APSR-sulfite compared with the APSR-red state indicates that the position of this conformationally flexible arm might be tuned to compensate optimally for the negative charges. The positively charged side chain of His-A398 is flanked by the two aromatic rings of Trp-A234 and Phe-A448 thereby imposing a specific conformation of the imidazole ring (Fig. 3B). The imidazole ring points directly to the sulfite moiety of the APSR-sulfite and forms a nearly ideal hydrogen bond to one sulfite oxygen. Furthermore, His-A398 might act as a proton donor to sulfite during cleavage (Fig. 5). The proposed formation of the covalent FAD-APS intermediate (Fig. 5B) is facilitated by the release of the proton at the atom N5 of FAD. However, there is no attractive proton acceptor in the vicinity. It is possible that a firmly bound water molecule (water 7001) located 3.8 Å away from the N5 atom acts as the proton acceptor. The basicity of this water molecule is significantly increased because of the hydrogen bond network comprising the side chain amide of Asn-A74 and the carboxylate side chain of Glu-A141 and Asp-A361 (Fig. 3B).
Comparison of the α-Subunit of APS Reductase with the Flavoprotein Subunit of Fumarate Reductase.
Structural alignments of the αsubunit of APS reductase with several members of the fumarate reductase family revealed a highly similar overall fold between the two classes of enzymes. Major changes were observed in the loops carrying the active site residues of both enzymes, which are responsible for substrate binding, catalysis, and conformation of FAD (Fig. 6). In particular, the isoalloxazine in fumarate reductase is planar compared with the bent isoalloxazine in APS reductase. In view of the similarity of the α-subunits of APS reductase and fumarate reductase, it is tempting to suggest that both subunits have a common ancestor resembling archaeal APS reductase. The insertion of a seven-residue-long loop that is strictly conserved in fumarate reductases into the active site of an ancestral APS reductase might have a pronounced impact on the conformation of the isoalloxazine ring. In APS reductase from A. fulgidus, such an inserted loop would cause His-A398 to move toward a position occupied by His-A369 in the case of fumarate reductase (numbering according to W. succinogenes). Consequently, movement of His-A398 would displace Trp-A234, one of the residues in APS reductase, which keeps the isoalloxazine moiety in its bent conformation (Fig. 6). Thus, the introduction of a seven-amino acid loop into the active site of an ancestral APS reductase could result in a planar isoalloxazine ring, concomitant with a negative shift in reduction potential as required for fumarate reduction (38). The comparison of APS and fumarate reductases provides an instructive example, by which means different biochemical reactions are accomplished by highly similar protein scaffolds mainly through the redesign of loop structures.
Figure 6.

Comparison of the α-subunit of APS reductase with fumarate reductase (stereo view). The structural alignment of the α-subunit of APS reductase and the flavoprotein subunit of fumarate reductase from W. succinogenes is shown. For APS reductase, the residues of the FAD-binding domain are shown in blue, the residues of the capping domain are shown in cyan and the FAD in blue-green. For fumarate reductase, the residues of the FAD-binding domain are shown in red and the insert in orange, and the residue of the capping domain is shown in light red and the FAD in magenta.
In conclusion, the three-dimensional structure of APS reductase presented here adds important information to our understanding of how the reduction potential and reactivity of FAD and FeS centers are finely tuned by the protein structure.
Acknowledgments
We thank H. Michel for continuous support and C. Vonrhein and Sandro Ghisla for valuable discussions. This work was supported in part by Deutsche Forschungsgemeinschaft (P.M.H.K.), Volkswagenstiftung (P.M.H.K.), and Fonds der Chemischen Industrie (P.M.H.K.).
Abbreviations
- APS
adenylylsulfate
- APSR-red
two-electron reduced state
- APSR-sulfite
FAD-sulfite adduct
Footnotes
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.rcsb.org (PDB ID codes 1JNR and 1JNZ).
References
- 1.Hansen T A. Antonie Leeuwenhoek. 1994;66:165–185. doi: 10.1007/BF00871638. [DOI] [PubMed] [Google Scholar]
- 2.Fritz G, Büchert T, Huber H, Stetter K-O, Kroneck P M H. FEBS Lett. 2000;473:63–66. doi: 10.1016/s0014-5793(00)01500-3. [DOI] [PubMed] [Google Scholar]
- 3.Lampreia J, Pereira A S, Moura J J G. Methods Enzymol. 1994;243:241–260. [Google Scholar]
- 4.Wagner M, Roger A J, Flax J L, Brusseau G A, Stahl D A. J Bacteriol. 1998;180:2975–2982. doi: 10.1128/jb.180.11.2975-2982.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stetter K O, Lauerer G, Thomm M, Neuner A. Science. 1987;236:822–824. doi: 10.1126/science.236.4803.822. [DOI] [PubMed] [Google Scholar]
- 6.Roth A, Fritz G, Büchert T, Huber H, Stetter K O, Ermler U, Kroneck P M H. Acta Crystallogr D. 2000;56:1673–1675. doi: 10.1107/s0907444900013366. [DOI] [PubMed] [Google Scholar]
- 7.Dutton P L. Methods Enzymol. 1978;54:411–435. doi: 10.1016/s0076-6879(78)54026-3. [DOI] [PubMed] [Google Scholar]
- 8.Massey V. Flavins and Flavoproteins. New York: Walter de Gruyter; 1991. pp. 59–66. [Google Scholar]
- 9.Otwinowski Z. In: Proceedings of the CCP4 Study Weekend. Wolf W, Evans P R, Leslie A G W, editors. Warrington, U.K.: Daresbury Laboratory; 1991. pp. 80–86. [Google Scholar]
- 10.Collaborative Computational Project No. 4. Acta Crystallogr D. 1994;50:760–763. [Google Scholar]
- 11.Sheldrick G H. In: Proceedings of the CCP4 Study Weekend. Wolf W, Evans P R, Leslie A G W, editors. Warrington, U.K.: Daresbury Laboratory; 1991. pp. 23–38. [Google Scholar]
- 12.DeLaFortelle E, Bricogne G. Methods Enzymol. 1997;276:472–494. doi: 10.1016/S0076-6879(97)76073-7. [DOI] [PubMed] [Google Scholar]
- 13.Esnouf R M. J Mol Graph Model. 1997;15:132–134. doi: 10.1016/S1093-3263(97)00021-1. [DOI] [PubMed] [Google Scholar]
- 14.Philippsen A. DINO. University of Basel, Switzerland: Biozentrum; 2001. [Google Scholar]
- 15.Merrit E A, Bacon D J. Methods Enzymol. 1997;277:505–524. doi: 10.1016/s0076-6879(97)77028-9. [DOI] [PubMed] [Google Scholar]
- 16.Iverson T M, Luna-Chavez C, Cecchini G, Rees D C. Science. 1999;284:1961–1966. doi: 10.1126/science.284.5422.1961. [DOI] [PubMed] [Google Scholar]
- 17.Mattevi A, Tedeschi G, Bacchella L, Coda A, Negri A, Ronchi S. Structure (London) 1999;7:745–756. doi: 10.1016/s0969-2126(99)80099-9. [DOI] [PubMed] [Google Scholar]
- 18.Lancaster C R D, Kröger A, Auer M, Michel H. Nature (London) 1999;402:377–385. doi: 10.1038/46483. [DOI] [PubMed] [Google Scholar]
- 19.Bamford V, Dobbin P S, Richardson D J, Hemmings A M. Nat Struct Biol. 1999;6:1104–1107. doi: 10.1038/70039. [DOI] [PubMed] [Google Scholar]
- 20.Holm L, Sander C. J Mol Biol. 1993;233:123–138. doi: 10.1006/jmbi.1993.1489. [DOI] [PubMed] [Google Scholar]
- 21.Sticht H, Rösch P. Prog Biophys Mol Biol. 1998;70:95–136. doi: 10.1016/s0079-6107(98)00027-3. [DOI] [PubMed] [Google Scholar]
- 22.Dauter Z, Wilson K S, Sieker L C, Meyer J, Moulis J-M. Biochemistry. 1997;36:16065–16073. doi: 10.1021/bi972155y. [DOI] [PubMed] [Google Scholar]
- 23.Kissinger C R, Sieker L C, Adman E T, Jensen L H. J Mol Biol. 1991;219:693–715. doi: 10.1016/0022-2836(91)90665-s. [DOI] [PubMed] [Google Scholar]
- 24.Fujii T, Hata Y, Wakaki T, Tanaka N, Oshima T. Nat Struct Biol. 1996;3:834–837. doi: 10.1038/nsb1096-834. [DOI] [PubMed] [Google Scholar]
- 25.Roberts D L, Freeman F E, Kim J-J P. Proc Natl Acad Sci USA. 1996;93:14355–14360. doi: 10.1073/pnas.93.25.14355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fritz G, Büchert T, Kroneck P M H. In: Flavins and Flavoproteins. Ghisla S, Kroneck P M H, Macheroux P, Sund H, editors. Berlin: R. Weber Agency for Scientific Publications; 1999. pp. 767–774. [Google Scholar]
- 27.Li J, Nelson M R, Peng C Y, Bashford D, Noodleman L. J Phys Chem A. 1998;102:6311–6324. [Google Scholar]
- 28.Denke E, Merbitz-Zahradnik T, Hatzfeld O M, Snyder C H, Link T A, Trumpower B L. J Biol Chem. 1998;273:9085–9093. doi: 10.1074/jbc.273.15.9085. [DOI] [PubMed] [Google Scholar]
- 29.Chen K, Tilley G J, Sridhar V, Prasad G S, Stout C D, Armstrong F A, Burgess B K. J Biol Chem. 1999;274:36479–36487. doi: 10.1074/jbc.274.51.36479. [DOI] [PubMed] [Google Scholar]
- 30.Lennon B W, Williams C H, Ludwig M L. Prot Sci. 1999;8:2366–2379. doi: 10.1110/ps.8.11.2366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dixon D A, Lindner D L, Branchaud B, Lipscomb W N. Biochemistry. 1979;18:5770–5775. doi: 10.1021/bi00593a004. [DOI] [PubMed] [Google Scholar]
- 32.Hall L H, Bowers M L, Durfor C N. Biochemistry. 1987;26:7401–7409. doi: 10.1021/bi00397a031. [DOI] [PubMed] [Google Scholar]
- 33.Trickey P, Wagner M A, Schuman J, Matthews F S. Structure (London) 1999;7:331–345. doi: 10.1016/s0969-2126(99)80043-4. [DOI] [PubMed] [Google Scholar]
- 34.Moser C C, Keske J M, Warnke K, Farid R S, Dutton P L. Nature (London) 1992;355:796–802. doi: 10.1038/355796a0. [DOI] [PubMed] [Google Scholar]
- 35.Deisenhofer J, Michel H. EMBO J. 1989;8:2149–2170. doi: 10.1002/j.1460-2075.1989.tb08338.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pelletier H, Kraut J. Science. 1992;258:1748–1755. doi: 10.1126/science.1334573. [DOI] [PubMed] [Google Scholar]
- 37.Ghisla S, Massey V. Biochem J. 1986;239:1–12. doi: 10.1042/bj2390001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Turner K L, Doherty M K, Heering H A, Armstrong F A, Reid G A, Chapman S K. Biochemistry. 1999;38:3302–3309. doi: 10.1021/bi9826308. [DOI] [PubMed] [Google Scholar]