

Abstract
Both in yeast and humans, DNA polymerase (Pol) η functions in the error-free replication of UV-damaged DNA, and Polη has the unique ability to efficiently replicate through a cis-syn thymine–thymine (T–T) dimer by inserting two As opposite the two Ts of the dimer. Although human DINB1-encoded Polκ belongs to the same protein family as Polη, Polκ shows no ability to bypass this DNA lesion and its biological function has remained unclear. Here, we examine Polκ for its ability to extend from primer-terminal mispairs opposite nondamaged and damaged DNA templates. We find that Polκ is a promiscuous extender of primer-terminal mispairs opposite nondamaged DNA templates, and interestingly, it is also very efficient at extending from a G opposite the 3′T of a T–T dimer. These observations provide biochemical evidence for a role of Polκ in the extension of mismatched base pairs during normal DNA replication, and in addition, they implicate Polκ in the mutagenic bypass of T–T dimers. In its proficient mismatch extension ability, Polκ is more similar to the unrelated DNA polymerase ζ than it is to the phylogenetically related Polη or Polι. Thus, in humans, Polκ would compete with Polζ for the extension of mismatched base pairs on damaged and undamaged DNAs.
DNA lesions often block replication, and bypass of such lesions requires the additional participation of translesion synthesis DNA polymerases. In both prokaryotes and eukaryotes, DNA polymerases belonging to the UmuC/DinB/Rad30 protein family promote replication through lesion sites (1, 2). In eukaryotes, the RAD30-encoded DNA polymerase η functions in the error-free replication of UV-damaged DNA, and mutations in DNA polymerase (Pol) η in humans cause the cancer-prone syndrome, the variant form of xeroderma pigmentosum (3, 4). Polη is a low fidelity enzyme, and steady-state kinetic analyses have shown that on undamaged DNA, yeast and human Polη misinsert nucleotides with a frequency of 10−2 to 10−3 (5, 6). Importantly, both yeast and human Polη incorporate As opposite the two Ts of the cis-syn thymine–thymine (T–T) dimer, and they do so with the same efficiency and fidelity as for the incorporation of As opposite undamaged Ts (6, 7). Genetic studies in yeast have also implicated Polη in the error-free bypass of cyclobutane dimers formed at 5′-TC-3′ and 5′-CC-3′ sites (8), and additionally, Polη can promote the bypass of UV-induced (6-4) photoproducts by inserting a nucleotide opposite the 3′ nucleotide of the photoproduct. Another DNA polymerase, Polζ, then extends from the nucleotide inserted by Polη, thus completing the bypass process (9).
In addition to Polη, humans contain another related polymerase, Polι, which is a very low fidelity polymerase, and which misincorporates a G opposite the T template residue ≈10-fold better than it incorporates the correct nucleotide A (10, 11). On its own, Polι shows no tendency to bypass DNA lesions such as a cis-syn T–T dimer or a (6-4) T–T photoproduct; however, it is able to insert nucleotides opposite the 3′T of the (6-4) T–T photoproduct, from which Polζ can then extend (10).
By contrast to Polη and Polι, which are present only in eukaryotes, homologs of the DinB protein exist in both prokaryotes and eukaryotes, and UmuC is found only in prokaryotes (1, 2). The umuC-encoded Pol V is the main lesion bypass polymerase in Escherichia coli (12, 13) and it is responsible for the majority of mutations that result from exposure of E. coli cells to DNA-damaging agents, including UV light. In E. coli, dinB-encoded Pol IV shows no propensity for bypassing a cis-syn T–T dimer, a (6-4) photoproduct, or an abasic site (14). Thus, the role of Pol IV in lesion bypass has remained unclear. Overproduction of the DinB protein in E. coli, however, leads to an increase in the frequency of spontaneous frameshift mutations (15).
We showed that human DINB1-encoded Polκ (originally called Polθ by us) is unable to bypass a cis-syn T–T dimer or a (6-4) T–T photoproduct and it does not even insert a nucleotide opposite either of these lesions (16). So far, there has been no biochemical evidence linking Polκ to a role in promoting the replication of undamaged DNA or the replication of UV-damaged DNA, and the biological function of Polκ has remained unclear. Here, we examine Polκ for its ability to extend from base mispairs. The impetus for this study came from the observations we had made earlier with Polζ, essential for the mutagenic bypass of DNA lesions in Saccharomyces cerevisiae. Although Polζ is a member of the Polα family of DNA polymerases and is unrelated to the human DinB1 protein or to the other members of this polymerase family, Polζ resembles Polκ in that both polymerases are very poor at inserting nucleotides opposite the 3′T of the T–T dimer or the (6-4) T–T photoproduct, and both polymerases are less prone to misincorporate nucleotides on undamaged DNA templates than is Polη (10, 16). Polζ differs from the other eukaryotic DNA polymerases in its proficient ability to extend from base mispairs on undamaged DNA as well as damaged DNAs (10). Whereas most DNA polymerases, including Polη, extend from base mispairs with nearly the same frequency at which they insert the respective mispair (17–19), Polζ is a very efficient extender of mispairs. On undamaged DNAs, Polζ extends from mismatched bases with a frequency of 10−1 to 10−2, and on DNAs containing a cis-syn T–T dimer or a (6-4) T–T photoproduct, Polζ extends from a G inserted opposite the 3′T of either of these lesions even more efficiently than from an A (10). Polζ acts similarly in the bypass of an abasic site, where it efficiently extends from the nucleotide inserted opposite the abasic site (20). For all these lesions, Polζ efficiently extends from the nucleotides incorporated opposite the lesion site by another DNA polymerase. Consequently, the mutagenic bypass of these various DNA lesions requires the action of two DNA polymerases, wherein one inserts the nucleotide opposite the lesion and Polζ extends from the nucleotide inserted.
Here, we show that Polκ efficiently extends from base mispairs on undamaged DNA, and it is also very efficient at extending from a G placed opposite the 3′T of a cis-syn T–T dimer. These observations provide biochemical evidence for a role of Polκ in the mutagenic replication of undamaged DNA, and in addition, they implicate Polκ in the mutagenic bypass of T–T dimers. The similar ability of Polζ and Polκ to proficiently extend from mispairs on undamaged DNA and on T–T dimer containing DNA supports the premise that these polymerases compete for the extension of mismatched termini on undamaged DNA and on T–T dimer-containing DNA.
Materials and Methods
DNA Substrates.
For measuring mispair extension, 4 52-nt oligomers were used as templates, and have the sequence:5′-TTCGT ATNAT GCCTA CACTG GAGTA CCGGA GCATC GTCGT GACTG GGAAA AC, where N is G, A, T, or C. Four 45-nt oligomers were used as primers, and they have the sequence 5′-GTTTT CCCAG TCACG ACGAT GCTCC GGTAC TCCAG TGTAG GCATN, where N is G, A, T, or C. For measuring incorporation and mismatch extension opposite the cis-syn T–T dimer or the (6-4) T–T photoproduct, 2 75-nt templates were used. The nondamaged template sequence was 5′-AGCAA GACAC CAATG TCTAA GAGTT CGTAT TATGC CTACA CTGGA GTACC GGAGC ATCGT CGTGA CTGGG AAAAC, and the damaged template sequences were identical except for a T–T dimer or a 6-4 (T–T) lesion at the underlined T residues. A 44-nt primer was used to measure incorporation opposite the 3′ T of a T–T dimer or the 6-4 (T–T) lesion and it had the sequence 5′-GTTTT CCCAG TCACG ACGAT GCTCC GGTAC TCCAG TGTAG GCAT. The 4 45-nt primers listed above were also used to measure extension of a mismatch opposite the 3′T of the T–T dimer or the 6-4 (T–T) lesion. Primers were 5′ end-labeled by using ATP[γ-32P] (Amersham Biosciences) and polynucleotide kinase (Roche Diagnostics). Labeled primers (1 μM) were annealed to templates (1.5 μM) in 50 mM Tris⋅HCl, pH 7.5/100 mM NaCl by heating to 90°C for 2 min and slowly cooling to room temperature over several hours.
Proteins.
Polκ was expressed in and purified from yeast strain BJ5464 as described (16).
Steady-State Kinetics Assays.
Polκ (1 nM) was incubated with DNA substrates (10 nM) containing either a correct base pair or a mispair at their primer terminus in 25 mM Tris⋅HCl (pH 7.5), 5 mM MgCl2, 5 mM DTT, 100 μg/ml of BSA, 10% glycerol, and various concentrations of dATP. For paired primer termini, 0–0.2 μM dATP was used, and for mispaired primer termini, 0–2 μM dATP was used. Reactions were carried out at 25°C and quenched after 10 min with 10 volumes of formamide loading buffer (80% deionized formamide/10 mM EDTA, pH 8.0). Samples were boiled for 2 min, placed on ice, and run on a 10% polyacrylamide sequencing gel containing 6 M urea. Gel band intensities were determined by using the PhosphorImager (Molecular Dynamics) and used to determine the rate of nucleotide incorporation at each dATP concentration. For each DNA substrate, the rate of incorporation was plotted as a function of dATP concentration, and the kcat and Km steady-state parameters were obtained by using nonlinear regression (sigma plot 4.0). These parameters were used to calculate the intrinsic efficiency of mispair extension, f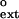 , for each mispair by using the following equation (17, 18, 21):
, for each mispair by using the following equation (17, 18, 21):
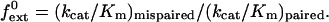 |
1 |
In this analysis, we assume that the binding affinities for the paired and mispaired substrates are similar, a situation which has been demonstrated to hold for other DNA polymerases (21–23).
Results
Efficient Mispair Extension on Undamaged DNA by Human Polκ.
To examine the ability of human Polκ to extend from mismatched primer-template termini on undamaged DNAs, we used steady-state kinetics assays and measured the rate of extension from a matched or mismatched primer terminus over a broad range of nucleotide concentrations. We incubated Polκ with various concentrations of dATP and nondamaged DNA substrates with either a matched C⋅G primer terminus or a mismatched G⋅G, A⋅G, or T⋅G primer terminus as described in Materials and Methods (Fig. 1A). For the matched primer terminus, we used 0–0.2 μM dATP, and for the mismatched primer termini, we used 0–2 μM dATP. The rate of nucleotide incorporation was plotted as a function of nucleotide concentration (Fig. 1B). The apparent kcat and Km values for extension of each primer terminus were determined from the best fit to the Michaelis–Menten equation by using nonlinear regression, and these values are listed in Table 1. The frequency of mispair extension, f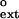 , which is a constant indicating the relative efficiency of extending a mismatched primer terminus in competition with an equal concentration of a matched primer terminus, was determined (17, 18, 21) by using the following equation:
, which is a constant indicating the relative efficiency of extending a mismatched primer terminus in competition with an equal concentration of a matched primer terminus, was determined (17, 18, 21) by using the following equation:
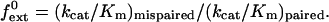 |
2 |
As shown in Table 1, the f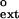 values for extending the G⋅G, A⋅G, and T⋅G mispairs are 3.4 × 10−2, 6.5 × 10−2, and 9.2 × 10−2, respectively. We determined the f
values for extending the G⋅G, A⋅G, and T⋅G mispairs are 3.4 × 10−2, 6.5 × 10−2, and 9.2 × 10−2, respectively. We determined the f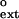 values for all primer terminal mismatched base pairs, and the f
values for all primer terminal mismatched base pairs, and the f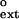 values range from 2.8 × 10−2 for the T⋅T and C⋅T mismatches to 1.3 × 10−1 for the C⋅C mismatch (Table 1).
values range from 2.8 × 10−2 for the T⋅T and C⋅T mismatches to 1.3 × 10−1 for the C⋅C mismatch (Table 1).
Figure 1.

Kinetics of mispair extension by human Polκ. (A) Mispair extension by hPolκ on nondamaged DNA. Polκ (1 nM) was incubated with various concentrations of dATP and DNA substrates (10 nM) containing a template T residue following a C⋅G base pair and a G⋅G, A⋅G, or T⋅G mispair. Reactions were carried out as described in Materials and Methods. (B) Quantitation of mispair extension. The rate of extension is graphed as a function of dATP concentration, and the data are fit to the Michaelis–Menten equation. The kcat and Km parameters obtained from the fit are listed in Table 1.
Table 1.
Steady-state kinetic parameters for mispair extension by Polκ
| Primer terminus* | kcat, min−1 | Km, μM | kcat/Km, μM−1min−1 |
f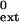
|
|---|---|---|---|---|
| G⋅G | 0.27 ± 0.03 | 0.82 ± 0.23 | 0.33 | 3.4 × 10−2 |
| A⋅G | 0.26 ± 0.02 | 0.42 ± 0.10 | 0.62 | 6.5 × 10−2 |
| T⋅G | 0.22 ± 0.03 | 0.25 ± 0.11 | 0.88 | 9.2 × 10−2 |
| C⋅G | 0.47 ± 0.03 | 0.049 ± 0.008 | 9.6 | 1.0 |
| G⋅A | 0.18 ± 0.01 | 0.33 ± 0.08 | 0.55 | 7.9 × 10−2 |
| A⋅A | 0.12 ± 0.01 | 0.53 ± 0.15 | 0.23 | 3.3 × 10−2 |
| T⋅A | 0.42 ± 0.03 | 0.060 ± 0.012 | 7.0 | 1.0 |
| C⋅A | 0.36 ± 0.03 | 0.73 ± 0.14 | 0.49 | 7.0 × 10−2 |
| G⋅T | 0.36 ± 0.02 | 1.0 ± 0.1 | 0.36 | 3.0 × 10−2 |
| A⋅T | 0.63 ± 0.05 | 0.054 ± 0.011 | 12 | 1.0 |
| T⋅T | 0.19 ± 0.03 | 0.57 ± 0.22 | 0.33 | 2.8 × 10−2 |
| C⋅T | 0.090 ± 0.004 | 0.27 ± 0.03 | 0.33 | 2.8 × 10−2 |
| G⋅C | 0.57 ± 0.04 | 0.052 ± 0.011 | 11 | 1.0 |
| A⋅C | 0.40 ± 0.04 | 0.58 ± 0.13 | 0.69 | 6.3 × 10−2 |
| T⋅C | 0.20 ± 0.02 | 0.47 ± 0.14 | 0.43 | 3.9 × 10−2 |
| C⋅C | 0.20 ± 0.01 | 0.14 ± 0.03 | 1.4 | 1.3 × 10−1 |
Primer extension was measured in the presence of dATP, the next correct nucleotide.
The first base is in the primer strand and the second base is in the template strand.
For most DNA polymerases, including the replicative polymerases, as well as Polη, the relative efficiency of extending a given mispair (f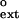 ) is approximately the same as the relative efficiency of incorporating the incorrect nucleotide to make the same mispair (finc) (17–19). Fig. 2 compares the f
) is approximately the same as the relative efficiency of incorporating the incorrect nucleotide to make the same mispair (finc) (17–19). Fig. 2 compares the f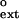 and finc values for each mispair for Polκ, wherein the finc values are taken from ref. 16. Points lying above the line represent mispairs with a higher efficiency of extension than formation, whereas points lying below the line indicate those mispairs where insertion is more efficient than extension. Because all of the data points lie above the line, Polκ is much more efficient at mispair extension than mispair formation. By contrast, for human Polη, most of the points lie below the line (see Fig. 2 Inset).
and finc values for each mispair for Polκ, wherein the finc values are taken from ref. 16. Points lying above the line represent mispairs with a higher efficiency of extension than formation, whereas points lying below the line indicate those mispairs where insertion is more efficient than extension. Because all of the data points lie above the line, Polκ is much more efficient at mispair extension than mispair formation. By contrast, for human Polη, most of the points lie below the line (see Fig. 2 Inset).
Figure 2.

Graphical representation of finc and f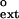 values for nondamaged DNA for human Polκ. The dashed line corresponds to finc = f
values for nondamaged DNA for human Polκ. The dashed line corresponds to finc = f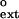 . The finc values are from ref. 16. (Inset) Graphical representation of finc and f
. The finc values are from ref. 16. (Inset) Graphical representation of finc and f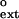 values for undamaged DNA for human Polη taken from ref. 19. [Reproduced with permission from ref. 19 (Copyright 2001, Am. Soc. Biochem. Mol. Biol.).]
values for undamaged DNA for human Polη taken from ref. 19. [Reproduced with permission from ref. 19 (Copyright 2001, Am. Soc. Biochem. Mol. Biol.).]
Efficient Mispair Extension on T–T Dimer Containing DNA by Human Polκ.
Previous studies with human Polκ have shown that it is unable to bypass a cis-syn T–T dimer or a (6-4) T–T photoproduct, two prominent DNA lesions formed by UV light (16). We have now examined the ability of Polκ to incorporate each of the four nucleotides opposite the 3′T of these lesions over a broad range of nucleotide concentrations, but we have detected no nucleotide incorporation opposite these lesions up to 200 μM nucleotide concentration (data not shown). Thus, assuming a kcat value less than 0.05 min−1 and a Km greater than 200 μM, we have determined an upper limit for the efficiency (kcat/Km) of nucleotide incorporation opposite these lesions by Polκ to be 2.5 × 10−4 μM−1min−1. The average efficiency for correct nucleotide incorporation opposite nondamaged template residues under these conditions, 10 μM−1min−1, was at least 40,000 times more efficient than nucleotide incorporation opposite these lesions. The average efficiency for incorrect nucleotide incorporation opposite nondamaged template residues, 2 × 10−3 μM−1min−1 (16), was at least 10 times more efficient than nucleotide incorporation opposite these lesions.
Next, we examined the ability of Polκ to extend from paired or mispaired primer termini opposite the 3′T of the T–T dimer and the nondamaged T–T sequence by monitoring the incorporation of dATP opposite the 5′T. The kcat, Km, and f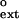 values with these DNA substrates are listed in Table 2. Although the efficiency (kcat/Km) of extension from the T⋅T and C⋅T mispairs decreased ≈100- to 200-fold on the T–T dimer-containing DNA relative to the extension of these mispairs on undamaged DNAs, the efficiency of extension of the correct A⋅T base pair decreased about 3-fold on the T–T dimer-containing DNA relative to the nondamaged DNA substrate. Interestingly, the efficiency of extension from a G placed opposite the 3′T of the T–T dimer was over 3-fold higher than the extension from the G⋅T mispair on the nondamaged DNA substrate. Furthermore, on the T–T dimer-containing DNA, the G⋅T mispair was extended about 2-fold more efficiently than the A⋅T base pair. Thus, Polκ is highly proficient in extending from a G nucleotide placed opposite the 3′T of the T–T dimer.
values with these DNA substrates are listed in Table 2. Although the efficiency (kcat/Km) of extension from the T⋅T and C⋅T mispairs decreased ≈100- to 200-fold on the T–T dimer-containing DNA relative to the extension of these mispairs on undamaged DNAs, the efficiency of extension of the correct A⋅T base pair decreased about 3-fold on the T–T dimer-containing DNA relative to the nondamaged DNA substrate. Interestingly, the efficiency of extension from a G placed opposite the 3′T of the T–T dimer was over 3-fold higher than the extension from the G⋅T mispair on the nondamaged DNA substrate. Furthermore, on the T–T dimer-containing DNA, the G⋅T mispair was extended about 2-fold more efficiently than the A⋅T base pair. Thus, Polκ is highly proficient in extending from a G nucleotide placed opposite the 3′T of the T–T dimer.
Table 2.
Mispair extension parameters for Polκ on nondamaged and T–T dimer-containing DNA
| Primer⋅terminus* | kcat, min−1 | Km, μM | kcat/Km, μM−1min−1 |
f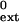
|
|---|---|---|---|---|
| G⋅T (nondamaged) | 0.42 ± 0.03 | 0.28 ± 0.05 | 1.5 | 1.5 × 10−1 |
| A⋅T (nondamaged) | 0.44 ± 0.03 | 0.045 ± 0.009 | 9.8 | 1.0 |
| T⋅T (nondamaged) | 0.25 ± 0.03 | 0.39 ± 0.11 | 0.64 | 6.5 × 10−2 |
| C⋅T (nondamaged) | 0.19 ± 0.007 | 0.19 ± 0.03 | 1.0 | 1.0 × 10−1 |
| G⋅T (T–T dimer) | 0.18 ± 0.008 | 0.036 ± 0.012 | 5.0 | 1.7 |
| A⋅T (T–T dimer) | 0.21 ± 0.01 | 0.072 ± 0.014 | 2.9 | 1.0 |
| T⋅T (T–T dimer) | 0.12 ± 0.009 | 17 ± 4 | 7.1 × 10−3 | 2.4 × 10−3 |
| C⋅T (T–T dimer) | 0.10 ± 0.006 | 18 ± 4 | 5.6 × 10−3 | 1.9 × 10−3 |
Primer extension was measured in the presence of dATP, the next correct nucleotide.
The first base is in the primer strand and the second base is the 3′T of a template T–T dimer or an identical nondamaged sequence.
We also examined the ability of Polκ to extend from a G, an A, a T, or a C nucleotide placed opposite the 3′T of the (6-4) T–T photoproduct by measuring the incorporation of dATP opposite the 5′T of the lesion. No nucleotide incorporation was observed with any of the (6-4) T–T photoproduct substrates. Consequently, the upper limit for the efficiency of extension from nucleotides opposite the 3′T of the (6-4) T–T product is 2.5 × 10−4 μM−1min−1. Thus, the efficiency of extension from any of the nucleotides placed opposite the 3′T of the (6-4) T–T photoproduct is at least 4,000-fold less than extension from the same nondamaged base pair.
Discussion
Here, we show that Polκ is highly adept at extending from base mispairs on nondamaged DNA, and it also extends proficiently from a G opposite the 3′T of a cis-syn T–T dimer. These observations provide biochemical evidence for a role of Polκ in the mutagenic replication of nondamaged DNA, and they also implicate the involvement of Polκ in the mutagenic bypass of T–T dimers.
Genetic studies in both prokaryotes and eukaryotes have suggested a role for the DinB polymerase in the mutagenic replication of undamaged DNA. Deletion of the dinB gene in E. coli reduces the rates of frameshift and base substitution mutations, and this decrease in the dinB mutant is most evident in the presence of a dnaE mutation that affects the α-subunit of the replicative polymerase, Pol III, and which confers a mutator phenotype (24). Further, the effect of deleting dinB becomes more striking in a dnaE mutS double mutant, where the mismatch repair system has been inactivated in a dnaE mutant background (24). These data have indicated a role for the E. coli dinB-encoded polymerase in generating spontaneous base substitution and frameshift mutations in the presence of normal replicative polymerase, and this role of dinB in generating spontaneous mutations becomes even more pronounced in the presence of a partially disabled replicative polymerase and a mismatch repair deficiency. Genetic studies in eukaryotes have also suggested a role for the DINB1-encoded DNA polymerase in generating spontaneous mutations. Transient expression of mouse DINB1 cDNA in cultured mouse cells confers an ≈10-fold increase in the frequency of spontaneous mutations, and among the mutants recovered, ≈70% of the mutations were base substitution mutations, which included both transition and transversion mutations, and the remainder, about 30%, of the mutations were single base additions or deletions (25). The proficient ability of Polκ to extend from mismatched primer termini now provides biochemical evidence for a role of this enzyme in chain extension from mismatched termini during normal DNA replication.
In its ability to extend from base mispairs, Polκ resembles Polζ. Polκ is as proficient, and in some cases even more proficient, at extending from mismatched primer termini, as is Polζ. Because S. cerevisiae contains Polζ but not Polκ, in this yeast species, only Polζ would contribute to chain extension from mismatched primer termini during normal DNA replication, whereas in humans, both Polζ and Polκ would contribute to this process. In this role, we expect the two polymerases to act independently and to compete for the mismatched primer termini (Fig. 3).
Figure 3.

Model of spontaneous and UV-induced mutagenesis by Polκ and Polζ. Both Polκ and Polζ efficiently extend from mispairs in nondamaged DNA and from a G⋅T mispair at the 3′T of the T–T dimer. Polζ also efficiently extends from a G⋅T mispair at the 3′T of the (6-4) T–T lesion, but Polκ does not. Although not shown here, we assume that subsequent to the insertion of the nucleotide following the mismatched base pair, or soon thereafter, Polζ and Polκ dissociate from DNA and Polδ takes over.
Genetic studies in S. cerevisiae have indicated the requirement of Polζ in the mutagenic bypass of DNA lesions (26, 27). For three lesions, a cis-syn T–T dimer, a (6-4) T–T photoproduct, and an abasic site, for which extensive steady-state kinetic data are available for the ability of Polζ to insert nucleotides opposite these DNA lesions and for extending from the nucleotides inserted opposite these lesion sites, it has emerged that Polζ functions in translesion DNA synthesis primarily at the extension step, wherein it efficiently extends from the nucleotides inserted opposite these lesions by another DNA polymerase. For instance, Polζ extends from a G opposite the 3′T of the T–T dimer or the (6-4) T–T photoproduct as efficiently as it extends from a G⋅T mispair in undamaged DNA. In fact, Polζ is more efficient at extending from a G opposite the 3′T of either of these lesions than from an A (10). Here, we show a similar propensity for Polκ in its ability to extend from a G nucleotide opposite the 3′T of the T–T dimer. On dimer-containing DNA, Polκ extends from the G⋅T mispair over 3-fold better than the extension of this mispair in nondamaged DNA, and it is about 2-fold more efficient at extending when a G is opposite the 3′T of the T–T dimer than when an A is present (Table 2). From these results, we propose a role for Polκ in the mutagenic bypass of cis-syn T–T dimers, and because both Polζ and Polκ proficiently extend from the G⋅T mispair on dimer-containing DNA, we expect these polymerases to compete for extending from this mispair (Fig. 3).
In human cells, replication through a cis-syn T–T dimer would primarily involve Polη, which bypasses this lesion fairly accurately by inserting two As opposite the two Ts of the dimer. Although Polη inserts the wrong nucleotides opposite the T–T dimer, it does so with a frequency of ≈10−2 to 10−3 (6, 7). Also, because human Polη inserts a G opposite the 3′T of the T–T dimer with a frequency of 4.2 × 10−3 (6) and it extends from this mispair with a frequency of 4.4 × 10−2 (28), we expect Polη to contribute little to the mutagenic bypass of a T–T dimer resulting from the insertion of a G. The almost absolute dependence of UV mutagenesis in S. cerevisiae on Polζ also attests to the indispensability of this enzyme for the mutagenic bypass of UV-induced DNA lesions. Thus, in S. cerevisiae, Polζ would be the enzyme solely responsible for extending from nucleotides placed opposite the 3′T of the T–T dimer or the (6-4) T–T photoproduct by another DNA polymerase. On the other hand, in human cells, the mutagenic bypass of a T–T dimer would involve Polκ, in addition to Polζ (Fig. 3). However, because of the inability of Polκ to extend from the nucleotides placed opposite the 3′T of the (6-4) T–T photoproduct, only Polζ would function in the bypass of this photoproduct (Fig. 3).
Although the polymerases belonging to the UmuC/DinB/Rad30 protein family contain the same five highly conserved motifs I-V, and Rad30 and DinB proteins have the same basic structure (29–31), the functional differences among them are quite remarkable. Of the eukaryotic polymerases, Polη, Polι, and Polκ of this family, although Polη can efficiently replicate through a cis-syn T–T dimer (6, 7), Polι is unable to insert a nucleotide opposite the 3′T of this lesion or to extend from nucleotides placed opposite this lesion site (10). By contrast to both these enzymes, Polκ is unable to insert nucleotides opposite the 3′T of the T–T dimer, but it can efficiently extend from a G opposite this site. The three polymerases differ also in their fidelity of nucleotide incorporation and in their ability to extend from mismatched primer termini on undamaged DNA. Although Polη and Polκ misincorporate nucleotides with a frequency of 10−2 to 10−3 and 10−3 to 10−4, respectively (5, 6, 16), Polι misincorporates a G opposite the template T about 10-fold better than it incorporates an A (10). By contrast to the proficient ability of Polκ to extend from base mispairs, Polη extends mispairs with nearly the same frequency at which it inserts the respective mispair (see Fig. 2 Inset and ref. 19). Thus, although Polκ is phylogenetically related to Polη and Polι, it is most similar to unrelated Polζ in its biochemical properties. The inability of Polζ and Polκ to insert nucleotides opposite lesions sites, but their proficient ability to extend from mismatched base pairs on nondamaged and damaged DNAs would suggest that both these enzymes are particularly sensitive to geometric distortions at the site of the templating base opposite the incoming nucleotide, but they are quite insensitive to the geometric distortions conferred on DNA by the presence of mismatched base pairs at the primer terminus. However, by contrast to Polζ, which can efficiently extend from mismatched bases opposite a T–T dimer, or a (6-4) T–T photoproduct, or from nucleotides placed opposite an abasic site, Polκ is able to do so only on dimer-containing DNA. The more restricted ability of Polκ to extend from mispaired bases opposite DNA lesions than of Polζ would suggest that compared with Polζ, Polκ is more limited in the degree of geometric distortions it can tolerate at the primer terminus.
Acknowledgments
This work was supported by National Institutes of Health Grants GM19261 and CA80882.
Abbreviations
- Pol
DNA polymerase
- T–T
thymine–thymine
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Johnson R E, Washington M T, Prakash S, Prakash L. Proc Natl Acad Sci USA. 1999;96:12224–12226. doi: 10.1073/pnas.96.22.12224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Goodman M F, Tippin B. Curr Opin Gen Dev. 2000;10:162–168. doi: 10.1016/s0959-437x(00)00057-5. [DOI] [PubMed] [Google Scholar]
- 3.Johnson R E, Kondratick C M, Prakash S, Prakash L. Science. 1999;285:263–265. doi: 10.1126/science.285.5425.263. [DOI] [PubMed] [Google Scholar]
- 4.Masutani C, Kusumoto R, Yamada A, Dohmae N, Yokoi M, Yuasa M, Araki M, Iwai S, Takio K, Hanaoka F. Nature (London) 1999;399:700–704. doi: 10.1038/21447. [DOI] [PubMed] [Google Scholar]
- 5.Washington M T, Johnson R E, Prakash S, Prakash L. J Biol Chem. 1999;274:36835–36838. doi: 10.1074/jbc.274.52.36835. [DOI] [PubMed] [Google Scholar]
- 6.Johnson R E, Washington M T, Prakash S, Prakash L. J Biol Chem. 2000;275:7447–7450. doi: 10.1074/jbc.275.11.7447. [DOI] [PubMed] [Google Scholar]
- 7.Washington M T, Johnson R E, Prakash S, Prakash L. Proc Natl Acad Sci USA. 2000;97:3094–3099. doi: 10.1073/pnas.050491997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yu S-L, Johnson R E, Prakash S, Prakash L. Mol Cell Biol. 2001;21:185–188. doi: 10.1128/MCB.21.1.185-188.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Johnson R E, Haracska L, Prakash S, Prakash L. Mol Cell Biol. 2001;21:3558–3563. doi: 10.1128/MCB.21.10.3558-3563.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Johnson R E, Washington M T, Haracska L, Prakash S, Prakash L. Nature (London) 2000;406:1015–1019. doi: 10.1038/35023030. [DOI] [PubMed] [Google Scholar]
- 11.Tissier A, McDonald J P, Frank E G, Woodgate R. Genes Dev. 2000;14:1642–1650. [PMC free article] [PubMed] [Google Scholar]
- 12.Reuven N B, Arad G, Maor-Shoshani A, Livneh Z. J Biol Chem. 1999;274:31763–31766. doi: 10.1074/jbc.274.45.31763. [DOI] [PubMed] [Google Scholar]
- 13.Tang M, Shen X, Frank E G, O'Donnell M, Woodgate R, Goodman M F. Proc Natl Acad Sci USA. 1999;96:8919–8924. doi: 10.1073/pnas.96.16.8919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tang M, Pham P, Shen X, Taylor J-S, O'Donnell M, Woodgate R, Goodman M F. Nature (London) 2000;404:1014–1018. doi: 10.1038/35010020. [DOI] [PubMed] [Google Scholar]
- 15.Kim S-R, Maenhaut-Michel G, Yamada M, Yamamoto Y, Matsui K, Sofuni T, Nohmi T, Ohmori H. Proc Natl Acad Sci USA. 1997;94:13792–13797. doi: 10.1073/pnas.94.25.13792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Johnson R E, Prakash S, Prakash L. Proc Natl Acad Sci USA. 2000;97:3838–3843. doi: 10.1073/pnas.97.8.3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goodman M F, Creighton S, Bloom L B, Petruska J. Crit Rev Biochem Mol Biol. 1993;28:83–126. doi: 10.3109/10409239309086792. [DOI] [PubMed] [Google Scholar]
- 18.Mendelman L V, Petruska J, Goodman M F. J Biol Chem. 1990;265:2338–2346. [PubMed] [Google Scholar]
- 19.Washington M T, Johnson R E, Prakash S, Prakash L. J Biol Chem. 2001;276:2263–2266. doi: 10.1074/jbc.M009049200. [DOI] [PubMed] [Google Scholar]
- 20.Haracska L, Unk I, Johnson R E, Johansson E, Burgers P M J, Prakash S, Prakash L. Genes Dev. 2001;15:945–954. doi: 10.1101/gad.882301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Creighton S, Bloom L B, Goodman M F. Methods Enzymol. 1995;262:232–256. doi: 10.1016/0076-6879(95)62021-4. [DOI] [PubMed] [Google Scholar]
- 22.Wong I, Patel S S, Johnson K A. Biochemistry. 1991;30:526–537. doi: 10.1021/bi00216a030. [DOI] [PubMed] [Google Scholar]
- 23.Huang M-M, Arnheim N, Goodman M F. Nucleic Acids Res. 1992;20:4567–4573. doi: 10.1093/nar/20.17.4567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Strauss B S, Roberts R, Francis L, Pouryazdanparast P. J Bacteriol. 2000;182:6742–6750. doi: 10.1128/jb.182.23.6742-6750.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ogi T, Kato T, Jr, Kato T, Ohmori H. Genes Cells. 1999;4:607–618. doi: 10.1046/j.1365-2443.1999.00289.x. [DOI] [PubMed] [Google Scholar]
- 26.Lawrence C W, Hinkle D C. Cancer Surveys. 1996;28:21–31. [PubMed] [Google Scholar]
- 27.Johnson R E, Torres-Ramos C A, Izumi T, Mitra S, Prakash S, Prakash L. Genes Dev. 1998;12:3137–3143. doi: 10.1101/gad.12.19.3137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Washington M T, Johnson R E, Prakash L, Prakash S. Proc Natl Acad Sci USA. 2001;98:8355–8360. doi: 10.1073/pnas.121007298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Trincao J, Johnson R E, Escalante C R, Prakash S, Prakash L, Aggarwal A K. Mol Cell. 2001;8:417–426. doi: 10.1016/s1097-2765(01)00306-9. [DOI] [PubMed] [Google Scholar]
- 30.Zhou B-L, Pata J D, Steitz T A. Mol Cell. 2001;8:427–437. doi: 10.1016/s1097-2765(01)00310-0. [DOI] [PubMed] [Google Scholar]
- 31.Ling H, Boudsocq F, Woodgate R, Yang W. Cell. 2001;107:91–102. doi: 10.1016/s0092-8674(01)00515-3. [DOI] [PubMed] [Google Scholar]