

Abstract
Recognition by transcription factors of the regulatory DNA elements upstream of genes is the fundamental step in controlling gene expression. How does the necessity to provide stability with respect to mutation constrain the organization of transcription control networks? We examine the mutation load of a transcription factor interacting with a set of n regulatory response elements as a function of the factor/DNA binding specificity and conclude on theoretical grounds that the optimal specificity decreases with n. The predicted correlation between variability of binding sites (for a given transcription factor) and their number is supported by the genomic data for Escherichia coli. The analysis of E. coli genomic data was carried out using an algorithm suggested by the biophysical model of transcription factor/DNA binding. Complete results of the search for candidate transcription factor binding sites are available at http://www.physics.rockefeller.edu/∼boris/public/search_ecoli.
The accumulation of knowledge on control of transcription in simple and complex organisms (1–4) poses many questions regarding its system-level function and organization. What aspects of control network architectures insure their stability with respect to mutation along with their ability to adapt and acquire new function (see ref. 1)? In its turn, could better understanding of the general organization of these networks help to dissect specific systems? Motivated by these questions, we formulate a model of transcription control that captures many of the essential features of the process and can be tested against data. This model is applied to the study of evolutionary stability of bacterial regulons (5), each involving a transcription factor that controls multiple genes by binding to multiple regulatory sequence elements. Evolutionary stability, or stability with respect to mutation, provides a sensible quantitative definition of robustness (1, 6, 7). Factors with highly sequence-specific binding impose severe constraint on regulatory sequences, increasing the probability of failure due to mutation, whereas low specificity of binding increases the probability of spurious interactions. Robustness is maximized by the compromise between these two effects. From this follows a quantitative prediction relating the number of elements in a regulon and the degree of binding site sequence variability. Our model also suggests a method of identifying candidate transcription factor binding sites, which we use in the analysis of the genomic data for Escherichia coli (8). Genomic data provides support for the prediction of the theory.
Model of Transcription Factor/DNA Interaction
In modeling the transcription control network, let us concentrate on the flow of information from factors to genes. Active transcription factors bind to the regulatory response element (RE) subsequences associated with genes (2). A given gene may be controlled by multiple repressing or activating factors acting through multiple REs (see Fig. 1). It will suffice for now to assume that all of the controlling factors and REs are nonredundant so that the loss of factor/RE recognition results in a significant detriment of fitness, due to the loss of regulatory linkage.§
Figure 1.

Schematic model of transcription control. Fs are active transcription factor proteins, xs are response element subsequences upstream of the coding regions of the genes, G. Arrows indicate regulatory interactions.
Binding of a factor to an RE depends on the factor concentration and the binding energy of the pair which together determine the binding probability. The interaction of a transcription factor with a DNA sequence x (of length L) may be approximated (9, 10) by the binding energy E(x) = x⋅ɛ ≡ ∑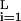 ∑
∑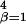 ɛ
ɛ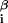 x
x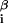 where ɛ
where ɛ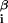 is the interaction energy with base β at position i = 1, … , L of the DNA string and x
is the interaction energy with base β at position i = 1, … , L of the DNA string and x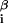 = 1 if the sequence x contains base β at position i and is 0 otherwise. Thus DNA binding properties of a factor are parametrized by ɛ
= 1 if the sequence x contains base β at position i and is 0 otherwise. Thus DNA binding properties of a factor are parametrized by ɛ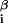 .
.
It is useful to consider the distribution of binding energies E(x) among all possible sequences, which is described by a histogram (or density of states) ρ(E) as shown in Fig. 2. The vast majority of random sequences fall into the approximately Gaussian center of this distribution, whereas the strongest binding, or consensus, sequence defines the leftmost edge (see Appendix A). (Note: It is convenient to set the scale of energy by the standard deviation of the binding energy in a random sequence ensemble and to set the average energy—i.e. the energy of nonspecific interaction—as E = 0.)
Figure 2.

Typical energy histogram, ρ(E), for a transcription factor interacting with a random DNA subsequences. In equilibrium, strings corresponding to energies below the chemical potential, μ (set by factor concentration), bind the factor with high probability given explicitly by 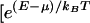 + 1]−1.
+ 1]−1.
In equilibrium, the probability of any string x to bind a factor is given explicitly by f(E(x)) = [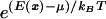 + 1]−1, where μ is the chemical potential set by factor concentration. Provided that the characteristic scale of binding energy is large compared to kBT [e.g., for lac-repressor (11) Em/kBT ∼ 10] one may as a first approximation replace f(E) by a step function; i.e. an “all or nothing” binding condition. The binding condition identifies the subset of all possible sequences (of length L) that at a given physiological concentration of a factor bind to it strongly: sequence, x, belongs to such a subset for factor Fi if it satisfies Ei(x) < μi. On Fig. 3, sequence subsets binding to different factors are pictured as nonoverlapping discs. Points within a given disk denote response elements controlled by Fi: their number, ni, is the degree of pleiotropy of the factor. A given factor, present in certain concentration, may be characterized by its “binding specificity,” σ ≡ ln v−1, defined in terms of the fraction, v, of random sequences (of length L) that bind strongly. These sequences lie in the “tail” of energy distribution ρ(E) below μ (see Fig. 2). In Fig. 3 binding specificity is represented by disk area.
+ 1]−1, where μ is the chemical potential set by factor concentration. Provided that the characteristic scale of binding energy is large compared to kBT [e.g., for lac-repressor (11) Em/kBT ∼ 10] one may as a first approximation replace f(E) by a step function; i.e. an “all or nothing” binding condition. The binding condition identifies the subset of all possible sequences (of length L) that at a given physiological concentration of a factor bind to it strongly: sequence, x, belongs to such a subset for factor Fi if it satisfies Ei(x) < μi. On Fig. 3, sequence subsets binding to different factors are pictured as nonoverlapping discs. Points within a given disk denote response elements controlled by Fi: their number, ni, is the degree of pleiotropy of the factor. A given factor, present in certain concentration, may be characterized by its “binding specificity,” σ ≡ ln v−1, defined in terms of the fraction, v, of random sequences (of length L) that bind strongly. These sequences lie in the “tail” of energy distribution ρ(E) below μ (see Fig. 2). In Fig. 3 binding specificity is represented by disk area.
Figure 3.

Response elements (dots) and factor binding subsets (disks) in sequence space. RE located within a disk binds the corresponding factor. Black dots lying outside the discs represent potential cis-regulatory sites, which must not bind transcription factors in order to avoid interference with transcription control. Arrows represent random changes in the sequence of REs (and of potential cis-regulatory sites) due to mutation.
In addition to regulatory response elements, DNA regions upstream of genes contain stretches of sequence without direct function. Some of these nonfunctional stretches are truly passive; others, however, may become “active” if through mutation they acquire a binding site for some transcription factor that can then interfere, positively or negatively, with transcription. In Fig. 3, such “potential cis-regulatory sites” are represented as points outside the disks.
Mutation Load in Transcription Control
Let us now introduce mutations. An accumulation of point mutations in a response element sequence corresponds to a random walk of the RE in sequence space (Fig. 3). A mutation in the DNA binding domain of the transcription factor changes its interaction energy with the sequence, ɛ. This change would appear in Fig. 3 as a random shift of the disc representing the binding subset in the sequence space. Either process could lead to an RE moving out of the binding subset of sequences, disrupting the interaction with the factor. In our model of a nonredundant network involving only essential genes, such failure is assumed to be “lethal.” Another form of regulatory failure involves a mutation of the potential regulatory site, causing it to enter the domain of binding with a wrong factor (see Fig. 3). For simplicity, this process is assumed to be lethal as well. To discuss the robustness of any given network, we must be able to calculate the probability (per mutation) of both modes of failure.
For a single factor/RE link, the binding probability depends only on the interaction energy E(x) = ɛ⋅x which changes randomly with mutations in x and ɛ. Although this is a discrete process, most qualitative features could be understood in the limit where the energy changes continuously. This approximation is accurate when the binding energy has contribution from many sites, i.e. L is large, and mutation at an individual site results in only a small change in the total energy. Consider a mutating ‘population’ of single factor/RE links with n(E, t) denoting the number of links with binding energy in the E, E + dE interval. It is possible to derive (Appendix B) an equation which governs the time evolution of n(E, t). It has the form of biased diffusion in the energy variable:
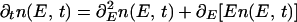 |
1 |
where unit of time is set by the point mutation rate. In addition we impose the boundary condition n(E, t)|E=μ = 0 which implements the assumption that “escape” of the RE from the domain of binding represents a lethal failure.¶ The first term on the right hand side represents “diffusion” in E arising from small random changes of the binding energy, whereas the second term describes the drift toward energies corresponding to the larger number of sequences; i.e., toward higher density of states ρ(E) (see Fig. 2). In the absence of selection (i.e., without the boundary condition) Eq. 1 is solved by n(E) ∼ exp(−E2/2)—the Gaussian distribution that approximates the distribution of E in random sequence ensemble (Fig. 2 and Appendix A).
After a long time the distribution of binding energies behaves as n(E, t) ≈ 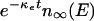 with κl being the smallest eigenvalue of Eq. 1. Thinking of the population of factor/RE links (or more generally, transcription control networks) as a population of “organisms” one can draw on the ideas from population genetics (12). The asymptotic “death rate,” κl, determines the minimal rate of replication that would be necessary to maintain a stable population and is therefore identified as mutation load (12).
with κl being the smallest eigenvalue of Eq. 1. Thinking of the population of factor/RE links (or more generally, transcription control networks) as a population of “organisms” one can draw on the ideas from population genetics (12). The asymptotic “death rate,” κl, determines the minimal rate of replication that would be necessary to maintain a stable population and is therefore identified as mutation load (12).
The lowest eigenvalue of Eq. 1 can be computed (Appendix B) as a function of binding specificity σ, yielding κl(σ) ≈ σ/2, accurate for σ ≫ 1. This computation makes quantitative the intuitive expectation that more specific interaction is more sensitive to mutation. The same calculation (Appendix B) determines n∞(E), which gives the distribution of factor/RE binding energies in a population after many mutations. This distribution exhibits a sharp maximum near the boundary of the domain of binding, E = μ, meaning that most of the response elements are about as far from the consensus sequence (the sequence which binds most strongly) as the binding condition permits. The reason is that there are many more possible sequences (i.e., higher density of states) near the boundary of the domain than at its center. The entropic, or sequence-space volume effects dominate the evolution of response elements!
To calculate the rate with which a factor can acquire a spurious regulatory site, we compute the rate with which a sequence outside the domain of binding, E > μ would “diffuse” in. This requires solving Eq. 1 with the same boundary condition as before, but in the range E > μ. The probability (per mutation), κsp(σ), with which a spurious site enters the domain of binding is given by (Appendix C) κsp(σ) ≈ (π/2)σe−σ (computed as before in the high specificity limit). This rate is exponentially small, because now most of the sequence space is outside the domain of binding. However, because the number of potential cis-regulatory sites, NcR, is large, the total probability for the factor to acquire a spurious regulatory target, NcRκsp(σ), may not be negligible. The two modes of failure, κsp and κl have the opposite dependence on binding specificity and their balance will determine the optimal choice for the latter.
Robustness Optimization for a Regulon
Let us now estimate the total rate of mutation induced failure, γ, in a control network consisting of Nf factors with factor i controlling ni genes (or operons) by the same number of response elements. It is given by the sum of failure rates‖ for all of the links plus the total “spurious site” acquisition rate
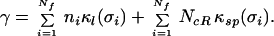 |
2 |
As we already noted, the total rate of failure, γ, or “death rate” per mutation in a population of network “organisms” evolving under a selection constraint, is also known as mutation load (12). It sets the minimal rate of replication that would sustain a steady population. An organism (or network architecture) more stable, or robust, with respect to mutation has lower mutation load and has an evolutionary advantage. In fact we can quantitatively define evolutionary robustness as the inverse mutation load, γ−1. (Note that the connection with the lowest eigenvalue of the evolution operator means that this definition is readily generalizable.) Evolutionary interpretation makes it meaningful to minimize the total mutation load γ in Eq. 2 with respect to binding specificities, σi. This yields
 |
3 |
which relates the optimal binding specificity of a factor with the number of its regulatory targets—its degree of pleiotropy—ni. Because σi = ln v , Eq. 3 implies that up to logarithmic corrections, ni is proportional to the volume fraction vi. Note that a linear scaling would also hold for the number of binding sites within a segment of random DNA of length N, n
, Eq. 3 implies that up to logarithmic corrections, ni is proportional to the volume fraction vi. Note that a linear scaling would also hold for the number of binding sites within a segment of random DNA of length N, n = Nvi. The similarity is accidental: in deriving Eq. 3 we considered nonrandom response elements, which evolve under selection, and in contrast with the random case, the proportionality constant ni/(viNcR) is not equal to 1. The decrease of the optimal specificity with the increasing degree of pleiotropy is forced by the need to reduce the mutation induced failure rate per RE, which is achieved by allowing corresponding REs to occupy a larger fraction of sequence space.
= Nvi. The similarity is accidental: in deriving Eq. 3 we considered nonrandom response elements, which evolve under selection, and in contrast with the random case, the proportionality constant ni/(viNcR) is not equal to 1. The decrease of the optimal specificity with the increasing degree of pleiotropy is forced by the need to reduce the mutation induced failure rate per RE, which is achieved by allowing corresponding REs to occupy a larger fraction of sequence space.
The predicted, approximately linear, scaling of vi with ni, which follows from Eq. 3 arises largely from the exponential dependence of κsp on σ, which in its turn is due to the fact that potential regulatory sites have an exponentially large fraction of sequence space to “diffuse” in without interfering with any of the transcription factors. We expect this conclusion to persist if the sharp “viability” boundary defined by the “lethal failure” threshold in our model is replaced by a smoother fitness landscape.
Specificity and Pleiotropy for E. coli Transcription Factors
The optimal specificity argument, presented above, may be applied in the context of prokaryotic regulons (2, 5). In E. coli a single operon is positively or negatively controlled by a small number of transcription factors. A single factor regulates a variable number of operons ranging from one (e.g., LacI) to perhaps hundreds, as in the case of cAMP-receptor protein (Crp) (5). Thus, factors have different degrees of pleiotropy. From the known functional binding sites one deduces that factors bind with different specificity: e.g., the dimeric target sites of LacI are very specific, whereas Crp sites are not (an effect due in large degree to a difference in intracellular concentration of the respective factors). A collection of known chromosomal binding sites for a set of 55 transcription factors has been assembled (8). This data set gives us the opportunity to look for an empirical correlation between specificity and degree pleiotropy indicated by Eq. 3.
We use an algorithm (described in Appendix D) to estimate the characteristic parameters (ɛj, μj) of each factor (dimer or monomer, as appropriate) from the binding sites in E. coli transcription factor database (8). We then search the intergenic regions of E. coli genome for sequences, s, satisfying ɛj⋅s < μj condition. Assuming that the number of these candidate sites, nj, is not vastly different from the number of the true binding sites, we use it as an estimate of the degree of pleiotropy. The binding specificity σj is computed from the fraction of random background sites satisfying the same binding condition (see supporting information, http://www.physics.rockefeller.edu/∼boris/public/search_ecoli). Fig. 4 presents the number of (candidate) target sites versus the binding specificity (or the sequence volume fraction v = exp(−σ). For comparison, the red line on Fig. 4 gives, as a function of specificity, the number of binding sites, Nrand, expected in a stretch of random DNA of length (N ≈ 5.4 × 105) equal to the length of noncoding part of the E. coli genome examined in the search. The extent to which n exceeds Nrand is a measure of statistical significance of the results of the sequence search: we have set the cutoff at 3 standard deviations (i.e., we have excluded six cases where n is within 3 standard deviations of Nrand) so that the counts above the red line plausibly correspond to functional sites.** Green line is a possible asymptotic fit to Eq. 3 (with NcR ≈ N/12). Note, that although NcR—the number of potential regulatory sites in the promoter regions—is a property of the biological system, it is not known and is determined here only as a fitting parameter. For high specificity/low pleiotropy factors, where very few example binding sites are known, our algorithm overestimates specificity because of overfitting. However, despite the very considerable scatter (note the logarithmic scale of the plot) there is a clear correlation between the two quantities that plausibly follow Eq. 3 in the high pleiotropy regime where our considerations apply. This provides empirical support to the prediction of the robustness optimization argument. Future experimental determination of functional response elements in the E. coli will allow more direct and precise determination of both the binding specificity and the degree of pleiotropy of transcription factors.
Figure 4.

The number of (candidate) response element sites, n, obtained from the E. coli genomic data versus factor binding specificity σ (circles). Note that exp(−σ) is the fraction of random sequences which bind the factor. Red line: expected number of binding sites for the random sequence background (reproducing the base frequency and nearest neighbor correlations of the noncoding segments). Green line: asymptotic fit to the predicted specificity/pleiotropy relation, Eq. 3.
Coevolution of Factors and Response Elements
So far we have focused on the mutation of response elements. As long as mutations in the DNA-binding domain of the factor produce only small random changes in ɛ, their effect on factor/RE recognition is still described by Eq. 1 with appropriately adjusted rate of “diffusion” in energy E. For a factor interacting with n REs comprising a regulon, the contribution of factor mutations is small compared to that due to RE mutations and has been neglected in Eq. 2. Factor mutation however is the limiting step in the evolutionary drift of the consensus sequence of the REs of the regulon. It is possible to estimate how the rate of coevolution of the factor together with its n regulatory targets depends on their number. A calculation of the effective rate of diffusion in sequence space of the consensus RE of a regulon (Appendix E) predicts that it should decrease as 1/n. It is well recognized that more pleiotropic factors are more conserved. Our result however makes a falsifiable quantitative prediction (distinct from the result of ref. 13), which can be checked by comparing orthologous factors between different species of prokaryotes (N. Rajewsi, N. Socci, M. Zapotocki, and E. D. Siggia, unpublished work; ref. 15).
Discussion
In this paper we analyzed the mutation load of the regulon control architecture as a function of specificity of transcription factor binding and found that minimization of the mutation load predicts a correlation between specificity and pleiotropy of the factor. Provided that the sequence dependent contribution to factor/DNA interaction energy is much larger that kBT, binding specificity can be optimized simply by changing factor concentration.
Our analysis was based on the “all or nothing” fitness model, which associated each violation of the E < μ binding condition with lethal failure. This model is an abstraction from a more realistic situation where fitness is determined by the ability of a given gene to be switched “on” or “off” by a change in the concentration of the controlling transcription factor. This means that the binding energy of the RE in question must lie in between the μon, μoff values—the chemical potentials corresponding to the on and off concentrations. Thus, one expects the fitness to be a smooth function of E with a peak at some finite value. Eq. 1 can be extended to this case by effectively replacing each “disk” in Fig. 3 with an annulus. Mutation load on a single RE then still decreases as σ with decreasing specificity, because, as in the analysis above, the mutation load is dominated by the outward drift due to larger number of sequences with lower binding energy. The analysis may be readily extended to include mutation-induced variation in transcription factor concentration levels, by including in the evolution Eq. 1 diffusion in μ. This extension would introduce into the model consideration of robustness with respect to concentration fluctuations. However, the genetic mechanisms controlling transcription factor levels were not presently included in the model.
Another realistic complication arises from the fact that different genes even in the same regulon may be required to turn on at different levels of the transcription factor. For example, operons for metabolism of preferred alternatives of glucose have stronger binding sites for CRP (16) and therefore get turned on at lower concentrations of activated CRP. Hence, REs of different genes comprising the same regulon may have individual constraints on their binding energy. The mutation load on such a factor is still a sum of contributions of regulated genes, but is no longer given simply by niκl(σi) term as it was in Eq. 2. The measure of specificity of such a factor would be defined by some weighted average over different thresholds. Yet the scaling of mutation load with (inverse) specificity should still hold because of the phase space considerations—i.e., mutation load on any of the REs being determined by the number of distinct sequences with binding energy E near the corresponding switching threshold.
The key role of the multiplicity of genetic states corresponding to the same phenotype has been emphasized in the “neutral” evolution theory (17) and specifically in the context of RNA folding (6, 18). In the context of transcription control, the neutrality of mutations that preserve the binding energy is plausible to the extent that the regulatory phenotype is determined entirely by the probability of factor/RE binding determined by the binding energy. The multiplicity of response element sequences corresponding to the same binding energy allows the “population” of factor/RE links to spread out over the “equi-fit” (i.e., same binding energy) manifold. This spreading generates an entropic contribution to fitness. Of two network architectures with the same phenotype (and replication rate), the one with larger neutral volume will have the lower mutation load and thus higher rate of growth. Estimating the mutation rate per nucleotide at 10−9 per generation in bacteria, we expect the mutation load effects to act on the time scale of 10,000 years. Much work has been done on the effect of finite size of populations on evolution (19). In small populations, random events can dominate over a small selection pressure. The condition for this to happen is that the difference of mutation load is smaller than the inverse of the population size. We assume that over sufficiently long time separate bacterial colonies exchange genetic information, so that the effective size of bacterial population is large enough (>109 to 1010) for the finite size effects, or “genetic drift” (19), to be ignored. It will be very interesting to investigate the diversity of regulatory binding sites between different strains of E. coli and between related bacterial species.
Our quantitative definition of the “effective fitness” or robustness of a transcription factor network by mutation load (and the lowest eigenvalue of the operator describing the evolution of a population of networks) can be extended to complex networks mapping factor activation patterns into patterns of gene expression. Mutations tend to spread the distribution of network parameters (e.g., RE sequences) over the whole domain consistent with the selected phenotype (i.e., factor/gene activity patterns). This domain will in general have very complex structure dependent on the network: different network architectures will have different mutation load (which can be determined numerically) and hence most robust network architecture for a given regulatory task can be identified on a firmly quantitative basis. The term “robustness” has been used with different meanings and is often interpreted as complete insensitivity to parameters, leaving an ambiguity as to which parameters should be considered (or excluded from the consideration) and what the quantitative measure should be. Identifying robustness with (inverse) mutation load solves both difficulties. All and only the parameters subject to mutation enter robustness consideration. Quantitative measure of robustness is provided by comparison of mutation loads for different architectures.
Our present analysis illustrates how modeling in conjunction with genomic data can be used to extract general features of control network organization. Many more of the fundamental principles underlying the organization of genetic networks are yet to be discovered.
Abbreviation
- RE
response element
Calculating ρ(E) Distribution
Consider the probability of finding a random oligomer that binds to the factor with free energy between E and E + dE:
 |
4 |
where 〈⋯〉s denotes an unrestricted average over the sequences x. To calculate ρ(E) let us introduce a Laplace transform
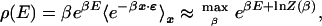 |
5 |
where the latter expression is the leading term of the saddle-point approximation to the β integral. We have introduced a “partition function”
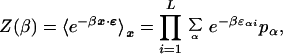 |
6 |
where pα is the frequency of base α. The analogy with the canonical and microcanonical ensembles is evident with the thermodynamic limit implicit in this analogy corresponding to L → ∞. To evaluate ρ(E) from Eq. 5 one must determine corresponding β from the saddle-point condition
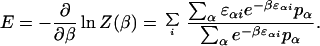 |
7 |
We also have the x ensemble analogue of “entropy,” ln ρ(E), consistent with the definition β = ∂ ln ρ/∂E.
Quite generally, for large L, ρ(E) near its peak at E = 0 is well approximated by a Gaussian ρ(E) ∼ exp(−E2/2χ2) with χ2 = ∂2/∂β2 ln Z(β)|β=0 = ∑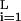 ∑α pαɛ
∑α pαɛ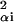 , which provides a measure of sequence specificity of the factor in question. (Note that addition of sequence sites with small ɛ
, which provides a measure of sequence specificity of the factor in question. (Note that addition of sequence sites with small ɛ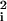 does not contribute much to χ!) Away from the center deviations from Gaussianity appear. In fact, the support of ρ(E) is finite with the bottom of the “band” Em = ∑i minα ɛiα (and the top at EM = ∑i maxα ɛiα). Note that to simplify notation in the main text we have chosen the energy units so that χ2 = 1. However, when comparing weakly specific factors (i.e., χ/kBT ≈ 1) the relative magnitude of their χ's becomes important.
does not contribute much to χ!) Away from the center deviations from Gaussianity appear. In fact, the support of ρ(E) is finite with the bottom of the “band” Em = ∑i minα ɛiα (and the top at EM = ∑i maxα ɛiα). Note that to simplify notation in the main text we have chosen the energy units so that χ2 = 1. However, when comparing weakly specific factors (i.e., χ/kBT ≈ 1) the relative magnitude of their χ's becomes important.
For the purpose of establishing the significance of genomic search results it is useful to redo the calculation of ρ(E) in the random ensemble, which reproduces not only the single base statistics of the genome, but includes the correlations between neighboring bases and hence provides an improved representation of genomic “background.” Fig. 4 uses specificity σ estimated on the basis of the latter ensemble. The details of this calculation can be found at http://www.physics.rockefeller.edu/∼boris/public/search_ecoli.
Derivation of the Evolution Equation
Let n(E, t) denote the number of sites to have binding energy in the E, E + dE interval, which is expressed in terms of the number of occurrences of site x: η(x, t).
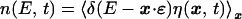 |
8 |
As a result of mutations occurring with probability α per unit time the sequence changes by Δx = x′ − x and we have
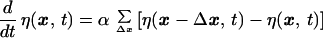 |
9 |
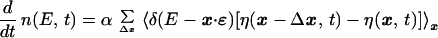 |
10 |
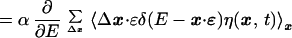 |
11 |
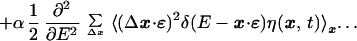 |
12 |
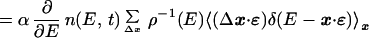 |
13 |
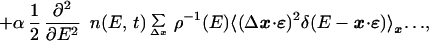 |
14 |
where the last line was derived by assuming that η(x, t) is the same for all x's with E(x) within a narrow shell, E, E + dE. Observing that
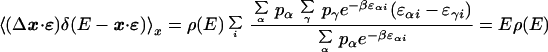 |
15 |
and defining the effective “diffusivity” in energy
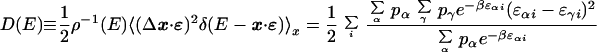 |
16 |
we arrive at the evolution equation
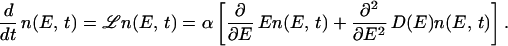 |
17 |
In the region where E ∼ χ ∼ 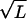 , β ≈ E/χ2 ∼ 1/
, β ≈ E/χ2 ∼ 1/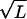 , we have D(E) ≈ D(0) = χ2 (the corrections being order of β, which is small when L is large). To simplify notation in the main text we have rescaled energy to eliminate χ and time to absorb α. Thus we have derived Eq. 1.
, we have D(E) ≈ D(0) = χ2 (the corrections being order of β, which is small when L is large). To simplify notation in the main text we have rescaled energy to eliminate χ and time to absorb α. Thus we have derived Eq. 1.
Calculating Mutation Load
To calculate the probability of failure we must impose the binding condition E < μ as the selection constraint. This effect is included by imposing an “absorbing” boundary condition n(μ, t) = 0. In the long time limit, solution of Eq. 1 has the form n(E, t) ∼ e−κ(μ)tn∞(E), where κ is the smallest magnitude eigenvalue of the evolution operator ℒ (17). κ is the rate at which sequences move outside the binding region. n∞(E) is given in terms of the parabolic cylinder function U(x, a) (20)
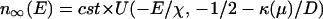 |
18 |
κ(μ) as a function of μ is given by
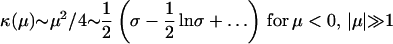 |
19 |
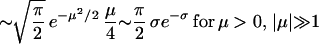 |
20 |
derived from asymptotics of parabolic cylinder functions. Specificity in terms of μ is given by σ = ln v−1 = μ2/22 + ½ ln(2πμ2).
Thus, rate of losing a response element from inside the binding region is γloss(μ) = κ(μ). We can similarly calculate the rate of a string outside the binding region to diffuse in. This process controls the rate of spurious activation (see text). The rate is γsp(μ) ≈ κ(−μ) when μ is not too far from the modal value of energy. One interesting thing about the asymptotic distribution of energy n∞(E), for μ < 0, is that most of the weight is concentrated near the boundary, E = μ. Hence most response elements found in the organism would be the “marginally” bound elements, rather different from the sequence that binds most strongly. This is one more example of how the entropy/phase-space effects dominate the evolution of response elements.
Empirical Determination of ɛ, μ Parameters
Given a set of known binding sites (8) for the jth factor, we find parameters ɛ(j) and μj such that all the known binding site sequences for this factor (the “example sequences”) s(k) satisfy s(k)⋅ɛ(j) ≤ μj < 0 with the maximal possible μ (and ɛ(j)⋅ɛ(j) = 1 constraint). Because specificity increases for large negative thresholds, μ, maximizing μ2 insures that our model of the factor [i.e., ɛ(j) and μj] has the highest specificity consistent with the data. This procedure minimizes the probability for a random sequence to satisfy the binding condition and therefore minimizes the number of “false-positive” sites in a genomic search for possible binding sites.
(and ɛ(j)⋅ɛ(j) = 1 constraint). Because specificity increases for large negative thresholds, μ, maximizing μ2 insures that our model of the factor [i.e., ɛ(j) and μj] has the highest specificity consistent with the data. This procedure minimizes the probability for a random sequence to satisfy the binding condition and therefore minimizes the number of “false-positive” sites in a genomic search for possible binding sites.
Restated in terms of a scaled variable ɛ̃(j) ≡ ɛ(j)/|μ|, the problem becomes that of minimization of a quadratic form ɛ̃(j)⋅ɛ̃(j) = 1/μ2 subject to a set of linear constraints ɛ̃(j)⋅s(k) < −1. This problem is solved by “quadratic programming” (14). Our string search algorithm is different from the widely used weight matrix (9, 10) procedure. Its key advantage is the parsimony in the number of the candidate binding sites it generates for the low specificity factors where the weight-matrix approach (8) produces too many genomic “hits.” The detailed results of the search are available electronically at http://www.physics.rockefeller.edu/∼boris/public/search_ecoli.
Estimate of Coevolution Rate
Consider a transcription factor controlling n binding sites. How fast can its consensus sequence and the whole domain of binding drift through sequence space? Suppose a mutation of the factor causes a change in its DNA interaction so that ɛ → ɛ + δɛ with probability P(δɛ). Such a mutation may cause each of the n RE links to fail with probability 1 − e−q(δɛ), so that the survival probability per mutation is given by p = ∫d(δɛ)P(δɛ)e−nq(δɛ). The effective diffusivity in ɛ is given by the variance of the “nonlethal” δɛ shift per mutation: Dɛ = p−1 ∫ d(δɛ)P(δɛ)e−nq(δɛ)(δɛ)2. Assuming that mutation induces only small shifts in ɛ, we can approximate q(δɛ) ≈ cnst × (δɛ)2 and for n ≫ 1 we arrive at Dɛ ∼ n−1 scaling. This provides a falsifiable quantitative prediction for the rate of evolutionary drift as a function of the degree of pleiotropy.
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
The case of more complex and redundant regulatory interactions can be considered within the same framework.
This evolution equation may be readily generalized to include binding-energy-dependent “fitness” term V(E)n(E,t).
Assuming for simplicity that different transcription factors do not bind the same REs.
The number of candidate binding sites found for DnaA, GlpR, Hns, MetJ, RpoD, RpoS, and SoxS is within 3 standard deviations of that expected in the random ensemble. Their significance must be established by further work. We note however that excess over random “background” is not required for the functionality of sites, but is rather an internal check on the bioinformatic approach to motif discovery.
References
- 1.Gerhart J, Kirschner M. Cells, Embryos and Evolution. Oxford: Blackwell Scientific; 1997. [Google Scholar]
- 2.Lewin B. Genes VI. Oxford: Oxford Univ. Press; 1997. p. 812. [Google Scholar]
- 3.Ptashne M. A Genetic Switch. 2nd Ed. Oxford: Blackwell Scientific; 1992. [Google Scholar]
- 4.Yuh CH, Bolouri H, Davidson E H. Science. 1998;279:1896–1902. doi: 10.1126/science.279.5358.1896. [DOI] [PubMed] [Google Scholar]
- 5.Neidhardt F C, editor. E. coli and Salmonella: Cellular and Molecular Biology. Washington, DC: Am. Soc. Microbiol.; 1996. [Google Scholar]
- 6.Barkai N, Liebler S. Nature (London) 1997;387:913–917. doi: 10.1038/43199. [DOI] [PubMed] [Google Scholar]
- 7.van Nimwegen E, Crutchfield J P, Huynen M. Proc Natl Acad Sci USA. 1997;96:9716–9720. doi: 10.1073/pnas.96.17.9716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Robison K, McGuire A M, Church G M. J Mol Biol. 1998;284:241–254. doi: 10.1006/jmbi.1998.2160. [DOI] [PubMed] [Google Scholar]
- 9.von Hippel P H. In: Biological Regulation and Development. Goldberger R F, editor. Vol. 1. New York: Plenum; 1979. pp. 279–347. [Google Scholar]
- 10.Stormo G D, Fields D S. Trends Biochem Sci. 1998;23:109–113. doi: 10.1016/s0968-0004(98)01187-6. [DOI] [PubMed] [Google Scholar]
- 11.Muller-Hill B. The lac Operon. Berlin: de Gruyter; 1996. [Google Scholar]
- 12.Smith J M. Evolutionary Genetics. Oxford: Oxford Univ. Press; 1998. [Google Scholar]
- 13.Waxman D, Peck J R. Science. 1998;279:1210–1213. [PubMed] [Google Scholar]
- 14.Fletcher R. Practical Methods of Optimization. New York: Wiley; 1987. [Google Scholar]
- 15.McCue L, et al. Nucleic Acids Res. 2001;29:774–782. doi: 10.1093/nar/29.3.774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Berg O G, von Hippel P H. J Mol Biol. 1988;200:709–723. doi: 10.1016/0022-2836(88)90482-2. [DOI] [PubMed] [Google Scholar]
- 17.Kimura M. Molecular Evolution and the Neutral Theory. Chicago: Univ. of Chicago Press; 1994. [Google Scholar]
- 18.Fontana W, Schuster P. Science. 1998;280:1451–1455. doi: 10.1126/science.280.5368.1451. [DOI] [PubMed] [Google Scholar]
- 19.Ohta T. Annu Rev Ecol Syst. 1992;23:263–286. [Google Scholar]
- 20.Abramowitz M, Stegun I A, editors. National Bureau of Standards Applied Mathematical Series. Washington, DC: U.S. Government Printing Office; 1964. Handbook of Mathematical Functions with Formulas, Graphs, and Mathematical Tables. [Google Scholar]