

Abstract
Presenilin heterodimers apparently contain the active site of γ-secretase, a polytopic aspartyl protease involved in the transmembrane processing of both the Notch receptor and the amyloid-β precursor protein. Although critical to embryonic development and the pathogenesis of Alzheimer's disease, this protease is difficult to characterize, primarily because it is a multicomponent complex of integral membrane proteins. Here the functional γ-secretase complex was isolated by using an immobilized active site-directed inhibitor of the protease. Presenilin heterodimers and nicastrin bound specifically to this inhibitor under conditions tightly correlating with protease activity, whereas several other presenilin-interacting proteins (β-catenin, calsenilin, and presenilin-associated protein) did not bind. Moreover, anti-nicastrin antibodies immunoprecipitated γ-secretase activity from detergent-solubilized microsomes. Unexpectedly, C83, the major endogenous amyloid-β precursor protein substrate of γ-secretase, was also quantitatively associated with the complex. These results provide direct biochemical evidence that nicastrin is a member of the active γ-secretase complex, indicate that β-catenin, calsenilin, and presenilin-associated protein are not required for γ activity, and suggest an unprecedented mechanism of substrate–protease interaction.
Sequential proteolysis of the amyloid-β precursor protein (APP) by β- and γ-secretases releases the amyloid-β peptide (Aβ), the major protein component of the cerebral plaques of Alzheimer's disease (1). Mutations in APP and the presenilins (PSs) PS1 and PS2 cause familial, early-onset Alzheimer's disease, and these mutations increase the production of Aβ in general or the highly fibrillogenic Aβ42 in particular (2). For these reasons, β- and γ-secretases are considered important therapeutic targets for Alzheimer's disease (3). β-Secretase is a membrane-tethered aspartyl protease in the pepsin family (4), whereas γ-secretase remains an enigmatic enzyme and has an unusual mode of action. This protease processes the transmembrane domain of APP, an otherwise water-excluded region of the substrate, and seems to be a complex of multiple integral membrane proteins (5–7).
The polytopic PSs are required for γ-secretase processing of APP (8, 9) and themselves are processed into two pieces, an N-terminal fragment (NTF) and a C-terminal fragment (CTF), that remain associated (6, 10). The NTF–CTF complexes are metabolically stable (11, 12), and their formation is tightly regulated by unidentified stoichiometric activators (13). Each subunit possesses one conserved transmembrane aspartate critical to γ-secretase activity (14–16), and aspartyl protease transition-state analogue inhibitors of γ-secretase bind directly and specifically to PS heterodimers (17, 18). Thus, heterodimeric PSs apparently comprise an unusual intramembranous active site and are among the founding members of a new class of polytopic membrane proteases (19).
PSs are required also for the transmembrane processing of the Notch receptor (20), part of a signaling pathway critical for embryonic development (21, 22). Knockout of PS1 in mice is lethal in utero or soon after birth, with a phenotype similar to that seen after deletion of Notch1 (23, 24). Cultured cells from PS1/PS2 double knockout mice have no γ-secretase activity with respect to either APP or Notch (8, 9), and mutation of either conserved PS aspartate (15, 25, 26) or treatment with γ-secretase inhibitors also prevents Notch proteolysis (20, 26). Moreover, APP and Notch can compete with one another for γ-secretase processing (27), and swapping the Notch transmembrane domain into APP allows efficient formation of Aβ-like peptides (J. Zhang and D.J.S., unpublished data).
Although PSs seem to be the catalytic component of γ-secretase, these proteins alone do not show γ-secretase activity, which is consistent with other evidence that γ-secretase is a high molecular weight, multiprotein complex (5–7). Identification of these other components, however, has been elusive. Immunoaffinity purification with PS antibodies led to the identification of nicastrin, a single-pass membrane protein involved in Notch signaling in Caenorhabditis elegans and Aβ production in human cell lines (28). The role of this protein in γ-secretase activity is not known. Here we report an activity-dependent purification method for γ-secretase using an immobilized transition-state analogue inhibitor and show that PS heterodimers, nicastrin, and an APP γ-secretase substrate (C83) bind to and elute from this matrix under conditions tightly correlating with γ-secretase activity. These results provide compelling biochemical evidence that nicastrin is a member of the active γ-secretase complex and suggest that substrates for this protease associate with an initial binding site before entry into the active site.
Methods
Preparation of Affinity Resins.
The compounds WPE-III–31C and WPE-III–112 were synthesized in solution phase by using standard methods (29). Details are described in Figs. 7 and 8, which are published as supporting information on the PNAS web site, www.pnas.org. The methyl esters were hydrolyzed by using LiOH in aqueous dioxane. The carboxylic acids then were coupled in DMSO by using 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (3 equiv) to the primary amine of a 6-atom hydrophilic linker present on the agarose resin affi-gel 102 (BioRad). Affi-gel 102 was empirically selected because it showed no background binding to PS1 NTF, PS1 CTF, or nicastrin at pH 7.0. The reaction mixtures were incubated with continuous gentle inversion for >12 h. The resins were washed extensively with DMSO and exchanged into aqueous buffer.
In Vitro γ-Secretase Assays.
Solubilized γ-secretase was prepared essentially as described by Li et al. (7) except that membranes were washed in 0.1 M sodium carbonate, pH 11.3, to remove nonintegral membrane proteins before solubilization in 1% 3-[(3-cholamidopropyl)dimethylammonio]-2-hydroxy-1propanesulfonate (CHAPSO). To monitor γ-secretase activity, the solubilized preparation (at ≈0.2 mg of protein/ml) was incubated with either a Flag-tagged APP-based substrate (C100Flag) or Notch-based substrate (N100Flag) at 37°C for 0 or 4 h. The substrates and C-terminal cleavage products were detected by Western blot using anti-Flag antibody M2 (Sigma) or anti-Aβ antibody 6E10 (Signet Labs, Dedham, MA). The recombinant substrate C100Flag was prepared as described by Li et al. (7). Plasmid encoding N100Flag in vector pET21a(+) (Novagen) was prepared by cloning the Notch sequence from Val-1711 to Glu-1809 from mouse Notch1 ΔE (21) using PCR (simultaneously incorporating an N-terminal methionine and a C-terminal Flag epitope). The BL21(DE3) expression host was transformed with the purified constructs. After a 2-h induction (1 mM isopropyl β-D-thiogalactoside at 37°C), bacteria were lysed in 10 mM Tris, pH 7.0/200 mM NaCl/1% Nonidet P-40 by using a French press. The N100Flag was purified by MonoQ anion exchange chromatography. The protein elutes between 195 and 240 mM NaCl. Alternatively, the proteins were purified by using an M2 anti-Flag affinity column using 100 mM glycine, pH 2.7, with 1% Nonidet P-40 for elution.
Affinity Chromatography.
For batch mode experiments, the solubilized γ-secretase preparation (adjusted to 1% CHAPSO) was incubated with compound resin for 2 h at room temperature with gentle rocking, and the resin was washed twice with several resin volumes of buffer (50 mM Pipes or Hepes, pH 7.0/150 mM NaCl/1% CHAPSO). The bound proteins then were released as described in the text for each given experiment. For preparative scale experiments, 2-ml columns of each resin were prepared and run on a Pharmacia AKTA-FPLC chromatography system. After sample injection, the columns were washed with 10 column volumes of binding buffer, and the bound material was eluted in either 1% Brij-35 or 100 mM glycine, pH 2.7, containing 1% CHAPSO. To measure γ-secretase activity in Brij-35 eluates, the PS complex was immunoprecipitated as described below.
Western Blotting and Immunoprecipitation.
For western analysis of PS1 NTF, PS1 CTF, and nicastrin, the samples were run on 8–16% Tris-glycine PAGE gels, transferred to polyvinylidene difluoride, and probed with Ab14 (for PS1 NTF, 1:2,000), 13A11 (for PS1 CTF, 5 μg/ml), and nicastrin antibodies (1:2,500). In addition, polyclonal antisera directed against PS2 NTF (2972), PS-associated protein (PSAP), and calsenilin and monoclonal antibodies to β-catenin (BD Transduction Laboratories, Lexington, KY) and APP (8E5 and 13G8) were used to probe selected polyvinylidene difluoride membranes. Samples from the γ-secretase activity assays were run on 4–20% Tris-glycine gels and transferred to polyvinylidene difluoride. The Flag epitope (in both the substrate and CTF generated by γ-secretase proteolysis) was detected by using anti-Flag M2 or anti-Aβ 6E10 antibodies. PS1 or nicastrin coimmunoprecipitations were performed by using anti-sera X81 (raised to the first 81 residues of the PS1 NTF) or R302 (raised to the last 12 residues of nicastrin), respectively, in 1% CHAPSO or 1% Brij-35 and were analyzed for PS1 NTF and PS1 CTF as described above. Activity measured after PS1 or nicastrin precipitation was performed in 0.25% CHAPSO buffer containing 0.05% phosphatidylethanolamine.
Results and Discussion
A (Hydroxyethyl)urea Transition-State Analogue Inhibits γ-Secretase Activity and Retains PS1 Heterodimers.
Cleavage of the scissile bond by an aspartyl protease proceeds through a transient gem-diol intermediate, or “transition state,” in the substrate. Compound WPE-III–31C (Fig. 1a) is a (hydroxyethyl)urea peptidomimetic, a transition-state analogue that mimics this gem-diol intermediate. This compound is similar structurally to a hydroxyethylene transition-state analogue recently reported to inhibit γ-secretase activity in vitro and to bind directly to heterodimeric PSs (7, 17). Compound 31C inhibited Aβ production at the γ-secretase level in whole cells with an IC50 of 300 nM (data not shown). The ability of this compound to inhibit cleavage of a Notch-based and an APP-based substrate also was examined in cell-free assays. The APP-based substrate, C100Flag, is the endogenous γ-secretase substrate C99 plus an N-terminal methionine start site and a C-terminal Flag epitope (7). The Notch-based substrate, N100Flag, contains 99 residues of the Notch1 sequence beginning from the ligand-dependent S2 cleavage site and likewise includes an N-terminal methionine and C-terminal Flag sequence. Bicarbonate-washed HeLa cell membranes were solubilized with the detergent CHAPSO and incubated with C100Flag (7) or N100Flag in the presence or absence of various concentrations of inhibitor. Compound 31C inhibited cleavage of both N100Flag and C100Flag in the low nM range (Fig. 1b), as determined by anti-Flag Western blot (N100Flag and C100Flag) or anti-Aβ Western blot (C100Flag). We also examined the inhibitory activity of a control compound, WPE-III–112 (Fig. 1a), which is of similar structure to 31C and has the same empirical formula but lacks the transition-state mimic. In contrast to 31C, the control peptide 112 did not inhibit cleavage of N100Flag or C100Flag (Fig. 1b) at all concentrations tested (up to 1 μM).
Figure 1.

A (hydroxyethyl)urea peptidomimetic inhibits γ-secretase and binds the PS–γ-secretase complex. (a) Structures of WPE-III–31C and WPE-III–112. The transition-state mimicking hydroxyethyl moiety is shown in red. (b) In vitro γ-secretase assays were performed by using CHAPSO-solubilized HeLa cell membranes and the substrates C100Flag and N100Flag in the presence or absence of indicated concentrations of 31C or 112. Shown are M2 anti-Flag Western blots that detect the Flag-tagged substrate (upper bands) and the C-terminal cleavage product that retains the Flag epitope (arrow). Note that C100Flag is highly prone to aggregation, forming SDS-stable oligomers. Also shown are anti-Aβ Western blots using antibody 6E10. (c) Immobilized 31C and 112 were incubated with CHAPSO-solubilized microsomes. Bound or unbound PS1 NTF, PS1 CTF, and nicastrin were detected by Western blotting. (d) Coimmunoprecipitation (Co-IP) using either anti-PS1 NTF antibody X81 (left lanes) or anti-nicastrin antibody R302 (right lanes) from CHAPSO-solubilized HeLa cell microsomes brings down γ-secretase activity using C100Flag as substrate. Protease activity is blocked by inhibitor 31C in both cases.
Inhibitor 31C was covalently linked to a solid support suitable for use in affinity chromatography to enrich for the active γ-secretase complex. The compound was covalently linked to the free primary amine of a 6-atom hydrophilic spacer on an agarose affinity support (Affi-gel 102, BioRad). To control for nonspecific hydrophobic interactions between the immobilized compound and the proteins of interest, we also prepared a resin with the inactive 112. We then evaluated their ability to deplete PS1 NTF, PS1 CTF, and nicastrin from a CHAPSO-solubilized HeLa microsome preparation that retains γ-secretase activity. The γ-secretase preparation was incubated with the affinity resins in batch format, and the PS1 NTF, PS1 CTF, and nicastrin remaining in the supernatant after incubation (“unbound”) were detected via Western blotting. The resins were washed extensively, and bound proteins were released with Laemmli sample buffer. PS1 NTF, PS1 CTF, and nicastrin released from each resin (“bound”) were detected likewise. At a loading concentration of 5 mg of 31C/ml of resin, PS1 NTF, PS1 CTF, and nicastrin were retained quantitatively on the resin (Fig. 1c). In clear contrast, PS1 NTF, PS1 CTF, and nicastrin remained completely unbound in the presence of 112 resin (Fig. 1c). This striking difference strongly supports a specific inhibitor-active site interaction on the 31C matrix that is required for binding of PS1 NTF, PS1 CTF, and nicastrin.
The finding that nicastrin specifically associated with the immobilized transition-state analogue prompted us to ask whether immunoprecipitation with anti-nicastrin antibodies could bring down γ-secretase activity. As reported previously by Li et al. (7), γ-secretase activity was precipitated with anti-PS1 NTF antibodies from CHAPSO-solubilized HeLa cell microsomes (Fig. 1d, left lanes). CHAPSO solubilization not only retains γ-secretase activity but also keeps PS NTF–CTF complexes together (7). Immunoprecipitation with the anti-nicastrin antibody R302 (to the C terminus of nicastrin) under these conditions likewise brought down γ-secretase activity, as assessed by using either C100Flag (Fig. 1d) or N100Flag (data not shown) as substrate. This precipitated activity was blocked fully by the inhibitor 31C (Fig. 1d), confirming specific proteolysis by γ-secretase. Anti-PS1 NTF antibodies precipitated PS1 CTF and nicastrin, and antinicastrin antibodies brought down PS1 NTF and CTF under these same conditions (data not shown). Together, these findings implicate nicastrin as a member of the active γ-secretase complex along with PS NTF and PS1 CTF (see also below).
Binding of PS1 Heterodimers and Nicastrin to the Inhibitor Matrix Requires Active γ-Secretase.
To probe the specificity of the interaction of the affinity resin with PS1 NTF, PS1 CTF, and nicastrin further, we reasoned that pretreatments that disrupt enzymatic activity should interfere also with binding to the resin. We first explored treatment with the detergent Triton X-100, which is known to eliminate γ-secretase activity (7) and dissociate PS1 heterodimers (6). We therefore treated a solubilized γ-secretase preparation with 2% Triton for 20 min. Because this detergent also could alter hydrophobic interactions, we removed the Triton by exchanging it with CHAPSO through extensive dialysis. As a control, a similar sample was treated identically except that no Triton detergent was added. To document the effects of Triton, we first tested the two preparations for γ-secretase activity and the ability to coimmunoprecipitate PS1 heterodimers. Treatment with Triton irreversibly disrupted γ-secretase activity, whereas the control preparation retained the ability to cleave N100Flag (Fig. 2a Left). To confirm that the detergent dissociated the PS complex, we coimmunoprecipitated PS1 by using an N terminus-specific antibody (X81). Although X81 immunoprecipitated PS1 NTF under both conditions, PS1 CTF was coimmunoprecipitated only in the CHAPSO control (Fig. 2a Center). Therefore, these two preparations are identical except that the PS1–γ-secretase complex is disrupted in the Triton-treated sample. Incubating the two samples with 31C resin demonstrated that only active, complexed γ-secretase can bind (Fig. 2a Right); PS1 NTF, PS1 CTF, and nicastrin bound only in the control sample.
Figure 2.

Disrupting γ-secretase activity and PS heterodimers prevents binding of the PS complex to the affinity resin. (a) CHAPSO-solubilized γ-secretase was treated with Triton X-100 (final concentration 2%) and dialyzed into 0.25% CHAPSO buffer to remove Triton. Controls are dialyzed, but lysates are untreated. These preparations were assayed for γ-secretase activity by using the N100Flag substrate (Left). Li et al. previously showed that Triton disrupts γ-secretase cleavage of C100Flag (7). PS1 NTF-specific X81 antibody was used to coimmunoprecipitate PS1 CTF from the samples (Center). These samples also were incubated with 31C resin, and the amounts of PS1 NTF, PS1 CTF, and nicastrin bound to the resin were assessed (Right). (b) Solubilized γ-secretase was heated to 65°C for 0, 10, 30, and 60 min. Samples were assayed for protease activity by using N100Flag (anti-Flag western) or C100Flag (anti-Aβ western) (Left) and for nicastrin binding to the affinity resin (Right).
We also considered heating as a method to test specificity of binding to the resin. When the solubilized γ-secretase preparation was heated at 65°C for 10, 30, and 60 min, a time-dependent loss in N100Flag cleavage (γ-secretase activity) was observed (Fig. 2b Left). A parallel loss in Aβ production from C100Flag also was seen (Fig. 2b Left). We then took the same preparations and tested for binding to 31C resin. Because extended heating aggregates PSs (W. Xia, unpublished observations), we monitored only nicastrin binding after this treatment. In agreement with its effect on γ-secretase activity, heating reduced the ability of nicastrin to bind the affinity matrix in a time-dependent manner (Fig. 2b Right). The greater the loss in enzymatic activity, the greater was the reduction in nicastrin binding to the resin. Thus, γ-secretase activity correlates with binding to 31C resin, confirming the specificity of this matrix for active γ-secretase complexes.
Alterations That Disrupt γ-Secretase Activity Can Elute the Bound PS Complex from the Affinity Resin.
Because we found a tight correlation between γ-secretase activity and binding to the immobilized inhibitor, we attempted to identify methods for specific elution off the resin. Li et al. (7) have demonstrated that γ-secretase cleavage of C100Flag has a broad pH range with an optimum of ≈pH 7.0. N100Flag proteolysis displayed a similar pH optimum and no detectable proteolysis at pHs 2.7 and 12.0 (Fig. 3a Upper). To assess the ability of these different pHs to elute proteins off the affinity resin, CHAPSO-solubilized HeLa microsomes were incubated with the immobilized inhibitor at pH 7.0 and then washed extensively. The resin subsequently was exchanged into phosphate buffers adjusted to the various pHs. Only the pHs that eliminated γ-secretase activity completely, 2.7 and 12.0, were able to elute PS1 NTF, PS1 CTF, and nicastrin (Fig. 3a Lower).
Figure 3.

Conditions that abolish γ-secretase activity elute the PS complex from the affinity resin. (a) CHAPSO-solubilized cell membranes were diluted with 50 mM phosphate buffer to alter the pH as indicated and examined for γ-secretase activity (Upper). Solubilized γ-secretase at pH 7.0 was incubated with the affinity resin. PS1 NTF, PS1 CTF, and nicastrin eluted at the indicated pHs (Lower). The pH dependence of C100Flag cleavage has been reported (7). Some proteins nonspecifically associate to the resin at pH 2.7 (data not shown); apparently, nicastrin likewise associates nonspecifically at this pH, accounting for its diminished release. (b) In vitro γ-secretase assays monitored cleavage of N100Flag (or C100Flag, data not shown) in the presence of various detergents (at 1%; Upper). Elution of the PS complex from the affinity resin was examined also (Lower). Solubilized γ-secretase was incubated with affinity resin, and the ability of detergents (at 1%) to elute PS1 NTF, PS1 CTF, and nicastrin was determined.
We also examined the effect of several detergents on γ-secretase activity and their ability to elute the PS complex from the affinity matrix. A CHAPSO-solubilized γ-secretase preparation was spiked with various detergents, and the in vitro cleavage of N100Flag (Fig. 3b Top) and C100Flag (data not shown) were analyzed. The presence of 1% Triton X-100, Nonidet P-40, or Brij-35 abolished γ-secretase cleavage of both substrates, whereas treatment with CHAPS or BIGCHAP [N,N-bis(3-d-gluconamidopropyl)cholamide] had little or no effect relative to the CHAPSO-treated control. When we examined the ability of these five detergents to elute PS1 NTF, PS1 CTF, and nicastrin from the affinity resin, only those detergents that disrupted γ-secretase activity (Triton, Nonidet P-40, and Brij-35) eluted the PS complex (Fig. 3b Bottom). In every case, for both the pH alterations (Fig. 3a) and the detergent treatments (Fig. 3b), release from the affinity matrix was similar for all three proteins, consistent with an interaction as a complex, and correlated with the presence of γ-secretase activity.
We found that of the five conditions capable of eluting PS1 and nicastrin from the inhibitor matrix, only Brij-35 substantially retained the association of PS1 NTFs and PS1 CTFs as determined by coimmunoprecipitation with X81 (Fig. 4a). Because 1% Brij-35 is incompatible with our N100Flag or C100Flag assays (Fig. 3b), we coimmunoprecipitated γ-secretase complexes from the Brij-35 eluate off a preparative 31C column and washed the beads with CHAPSO to exchange it into optimal detergent. After the addition of either N100Flag or C100Flag, the coimmunoprecipitated material showed clear γ-secretase activity (Fig. 4b), demonstrating that the active γ-secretase complex could be isolated from the inhibitor matrix. Interestingly, Brij-35 was reported to support γ-secretase activity in microsomes isolated from APP-transfected cells that had been pretreated with a γ-secretase inhibitor (30). Thus, Aβ produced in this assay was generated from cell-derived substrate. Brij-35 apparently interferes with the initial interaction of a recombinant substrate with the enzyme but does not affect a preassembled enzyme-substrate complex. Nevertheless, in our hands the active γ-secretase complex clearly is maintained in Brij-35, because column elution and immunoprecipitation out of this detergent isolated the protease activity (Fig. 4b).
Figure 4.
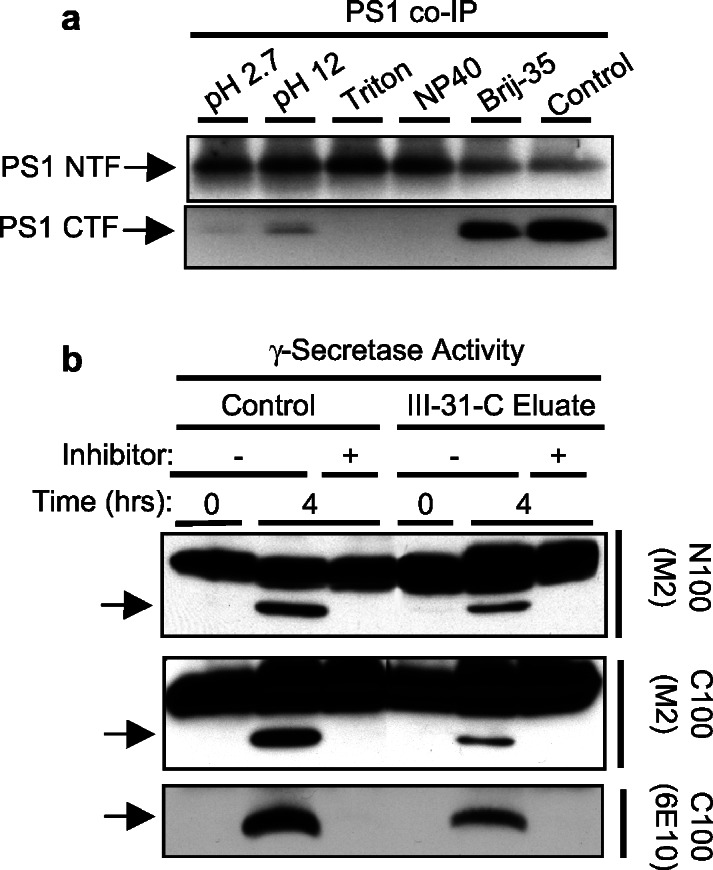
The PS complex and γ-secretase activity can be eluted from the affinity resin. (a) Coimmunoprecipitates (Co-IP) were used to determine whether elution conditions disrupt PS heterodimers. CHAPSO-solubilized cell membranes were treated as indicated and then immunoprecipitated with X81 (to PS1 NTF). Precipitated PS1 NTF and PS1 CTF were detected by Western blotting. NP40, Nonidet P-40. (b) Solubilized γ-secretase was passed over a 31C affinity column, washed in binding buffer, and eluted in 1% Brij-35. The PS complex was coimmunoprecipitated with X81 and exchanged into 0.25% CHAPSO. This material was compared with the starting material in γ-secretase assays using N100Flag and C100Flag in the presence (+) or absence (−) of γ-secretase inhibitor 31C.
Partial Characterization of the Enriched γ-Secretase Complex.
Several proteins have been proposed to be PS-interacting proteins. To test whether these proteins are involved in the PS–γ-secretase complex, we performed a preparative-scale affinity separation by using 31C or 112 resins and probed for the presence or absence of these other PS binding partners. Using a column format, the majority of all protein (as monitored by UV280 absorbance) from a CHAPSO-solubilized γ-secretase preparation passed through the 31C column without binding. After thorough washing, the bound material was eluted by using acidic glycine (Fig. 5a) or 1% Brij 35 (data not shown). In both cases, a single UV elution peak was observed. To determine the elution profiles of PS1 NTF, PS1 CTF, and nicastrin, the starting material and all the fractions were Western-blotted (Fig. 5a). Very little PS1 NTF, PS1 CTF, and nicastrin were detected in the flow-through peak (fractions 1–5), indicating that these proteins were almost quantitatively bound to the column. These three proteins eluted from the column in parallel and were found primarily in fractions 10–11, corresponding to the elution peak detected by UV. Consistent with the results in Fig. 1c, when the 112 column was run under identical conditions, all the PS1 NTF, PS1 CTF, and nicastrin were found in the flow-through fractions (Fig. 5b).
Figure 5.

Preparative-scale affinity isolation and characterization of the components of the γ-secretase complex. (a) Solubilized γ-secretase was passed over a 31C affinity column and eluted with glycine, pH 2.7 (or Brij-35, data not shown). In the UV chromatogram, the first arrow is the injection point, the first (larger) peak is the flow-through, the second arrow is the start of the elution, and the second (smaller) peak corresponds to the proteins eluted from the column. Western blotting demonstrates that essentially all the PS1 NTF, PS1 CTF, and nicastrin were bound to the column and eluted in the fractions corresponding to the UV elution peak. The area under the UV elution peak corresponds to 5% of the total area. (b) The flow-through and elution fractions from the 31C column or a similarly run 112 column were collected and probed for the presence of the indicated proteins using Western blotting.
To identify other candidates in the PS–γ-secretase complex, fractions corresponding to the flow-through and elution peaks were probed for the presence or absence of proteins proposed to be PS-interacting partners (Fig. 5b). Although there are many candidates that interact with PS1, we focused on several lead candidates including APP holoprotein, C83, β-catenin, PSAP, and calsenilin. β-Catenin, PSAP, and calsenilin were not retained by the immobilized inhibitor. Indistinguishable results were obtained by using glycine or Brij-35 for elution. These results suggest that these candidates are not required for activity, although they may have other indirect or modulatory roles. PS2 NTF, however, was quantitatively bound, which is consistent with the fact that PS2 is a functional homolog of PS1 and is associated with proteolytic activity (16, 31, 32). Only a very small amount of full-length APP associated with the complex, which is in agreement with previous results that it coimmunoprecipitates inefficiently with PS1 in endoplasmic reticulum-enriched vesicles (33). However, essentially all the C83 APP CTF (the sole APP γ-secretase substrate detectable in HeLa cells) was retained on the affinity resin and coeluted with PS1 and nicastrin. This result agrees with prior work that showed that C83 and C99 coimmunoprecipitated with PS1, in particular in subcellular fractions that exhibit in vitro γ-secretase activity (33). In this earlier study, our coimmunoprecipitations were not quantitative; however, they were not carried out in the presence of a γ-secretase inhibitor and were performed before the discovery of conditions ideal for isolation of the active γ-secretase complex. Thus, differences in isolation conditions likely account for the current observation that C83 is recovered quantitatively from the immobilized 31C column.
Why would a substrate and enzyme copurify after inhibitor affinity chromatography? Because the affinity resin binds to the active site of γ-secretase (17, 18), we interpret the invariant presence of C83 as evidence for a two-step mechanism for substrate–protease interaction. Specifically, we propose the existence of an initial binding site for the substrate on the surface of the γ-secretase complex before its entry into the active site. This hypothesis makes particular sense in the two-dimensional context of a lipid bilayer, in which the water-containing active site of the protease should be sequestered from the hydrophobic environment. The substrate first must contact the outer surface of the complex before its entry into and cleavage at the active site (Fig. 6). The presence of the immobilized inhibitor in the active site therefore would stabilize the C83 substrate at the initial binding site. This idea is corroborated by evidence that C83 and C99 coimmunoprecipitated with wild-type PS1 using lysates from cells pretreated with transition-state analogue inhibitors (33). In these coimmunoprecipitation experiments, however, C83 and C99 levels were increased pharmacologically by treating the cells with inhibitors. We show here that endogenous levels of C83 copurify with PS heterodimers and nicastrin, suggesting that this is a biologically relevant interaction and not simply a result of elevated protein levels. In contrast, full-length APP apparently does not interact with the γ-secretase complex efficiently under these conditions, perhaps because the large APP ectodomain sterically interferes with binding. Identifying compounds that block the initial substrate binding site on the γ-secretase complex might be an alternative means of inhibiting this important therapeutic target.
Figure 6.
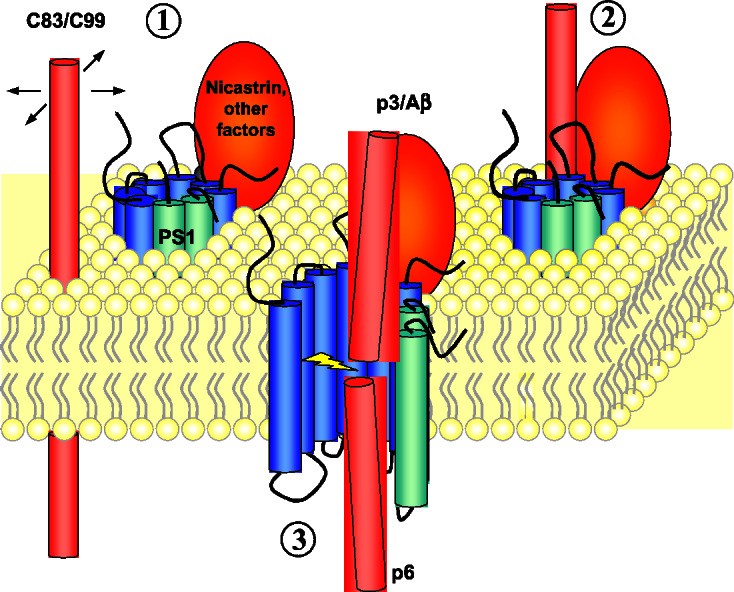
A model for the interaction of γ-secretase with its substrate. The γ-secretase complex apparently includes PS NTF (blue), PS1 CTF (green), and nicastrin (orange). The water-containing active site in PS is sequestered from the hydrophobic membrane lipids, whereas substrate C83 or C99 (red) can move only in two dimensions within the confines of the bilayer (step 1). The substrate first interacts on the outer surface of the protease (step 2) before accessing the active site (step 3).
The present study also confirms the importance of nicastrin, providing direct biochemical evidence that nicastrin is associated with the active γ-secretase complex in wild-type (nontransfected) human cells. The presence of protease activity correlated tightly with the binding of PS1 and nicastrin to the immobilized inhibitor. Conversely, conditions that abolished activity released bound PS1 heterodimers and nicastrin from the resin. Moreover, anti-nicastrin antibodies precipitated γ-secretase activity. We conclude that the γ-secretase complex apparently contains PS1 NTF and PS1 CTF, nicastrin, and one or more additional proteins. It is likely that there is at least one other critical member of the complex, because overexpression of nicastrin and PS1 neither generates increased PS1 NTF and PS1 CTF nor augments γ-secretase activity (28). The unknown stoichiometric activator(s) of PS endoproteolysis are expected to remain a part of the PS complex, because these factors otherwise would continue to allow PS heterodimer formation (i.e., overexpressed PS would lead to an increased level of NTF–CTF complexes, which is not observed). The identification of these unknown partners must await complete purification of the γ-secretase complex. Further work toward the purification of this enzyme will yield additional insights into its function and should reveal new targets for the treatment and prevention of Alzheimer's disease.
Supplementary Material
Acknowledgments
We thank J. Vandrovec for preparation of the N100Flag expression host, R. Kopan for mouse Notch1 ΔE, S. Gandy for Ab14, P. St. George-Hyslop and D. Miller for antibodies to nicastrin, C. Haass for 2972, X. Xu for PSAP antibody, W. Wasco for calsenilin antibodies, and P. Seubert and D. Schenk for 8E5 and 13G8. We also thank D. Walsh, M. LaVoie, and W. Xia for helpful discussions. This work was supported by National Institutes of Health Grants AG 17574, NS 41355 (to M.S.W.), and AG 12749 (to D.J.S.) and a Pioneer Award (to D.J.S.) and a New Investigator Award (to W.P.E.) from the Alzheimer's Association.
Abbreviations
- APP
amyloid-β precursor protein
- Aβ
amyloid-β peptide
- PS
presenilin
- NTF
N-terminal fragment
- CTF
C-terminal fragment
- PSAP
PS-associated protein
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Selkoe D J. Nature (London) 1999;399:23–31. doi: 10.1038/399a023. [DOI] [PubMed] [Google Scholar]
- 2.Hardy J. Proc Natl Acad Sci USA. 1997;94:2095–2097. doi: 10.1073/pnas.94.6.2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wolfe M S. J Med Chem. 2001;44:2039–2060. doi: 10.1021/jm0004897. [DOI] [PubMed] [Google Scholar]
- 4.Howlett D R, Simmons D L, Dingwall C, Christie G. Trends Neurosci. 2000;23:565–570. doi: 10.1016/s0166-2236(00)01647-7. [DOI] [PubMed] [Google Scholar]
- 5.Yu G, Chen F, Levesque G, Nishimura M, Zhang D M, Levesque L, Rogaeva E, Xu D, Liang Y, Duthie M, et al. J Biol Chem. 1998;273:16470–16475. doi: 10.1074/jbc.273.26.16470. [DOI] [PubMed] [Google Scholar]
- 6.Capell A, Grunberg J, Pesold B, Diehlmann A, Citron M, Nixon R, Beyreuther K, Selkoe D J, Haass C. J Biol Chem. 1998;273:3205–3211. doi: 10.1074/jbc.273.6.3205. [DOI] [PubMed] [Google Scholar]
- 7.Li Y M, Lai M T, Xu M, Huang Q, DiMuzio-Mower J, Sardana M K, Shi X P, Yin K C, Shafer J A, Gardell S J. Proc Natl Acad Sci USA. 2000;97:6138–6143. doi: 10.1073/pnas.110126897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Herreman A, Serneels L, Annaert W, Collen D, Schoonjans L, De Strooper B. Nat Cell Biol. 2000;2:461–462. doi: 10.1038/35017105. [DOI] [PubMed] [Google Scholar]
- 9.Zhang Z, Nadeau P, Song W, Donoviel D, Yuan M, Bernstein A, Yankner B A. Nat Cell Biol. 2000;2:463–465. doi: 10.1038/35017108. [DOI] [PubMed] [Google Scholar]
- 10.Thinakaran G, Borchelt D R, Lee M K, Slunt H H, Spitzer L, Kim G, Ratovitsky T, Davenport F, Nordstedt C, Seeger M, et al. Neuron. 1996;17:181–190. doi: 10.1016/s0896-6273(00)80291-3. [DOI] [PubMed] [Google Scholar]
- 11.Ratovitski T, Slunt H H, Thinakaran G, Price D L, Sisodia S S, Borchelt D R. J Biol Chem. 1997;272:24536–24541. doi: 10.1074/jbc.272.39.24536. [DOI] [PubMed] [Google Scholar]
- 12.Podlisny M B, Citron M, Amarante P, Sherrington R, Xia W, Zhang J, Diehl T, Levesque G, Fraser P, Haass C, et al. Neurobiol Dis. 1997;3:325–337. doi: 10.1006/nbdi.1997.0129. [DOI] [PubMed] [Google Scholar]
- 13.Thinakaran G, Harris C L, Ratovitski T, Davenport F, Slunt H H, Price D L, Borchelt D R, Sisodia S S. J Biol Chem. 1997;272:28415–28422. doi: 10.1074/jbc.272.45.28415. [DOI] [PubMed] [Google Scholar]
- 14.Wolfe M S, Xia W, Ostaszewski B L, Diehl T S, Kimberly W T, Selkoe D J. Nature (London) 1999;398:513–517. doi: 10.1038/19077. [DOI] [PubMed] [Google Scholar]
- 15.Steiner H, Duff K, Capell A, Romig H, Grim M G, Lincoln S, Hardy J, Yu X, Picciano M, Fechteler K, et al. J Biol Chem. 1999;274:28669–28673. doi: 10.1074/jbc.274.40.28669. [DOI] [PubMed] [Google Scholar]
- 16.Kimberly W T, Xia W, Rahmati T, Wolfe M S, Selkoe D J. J Biol Chem. 2000;275:3173–3178. doi: 10.1074/jbc.275.5.3173. [DOI] [PubMed] [Google Scholar]
- 17.Li Y M, Xu M, Lai M T, Huang Q, Castro J L, DiMuzio-Mower J, Harrison T, Lellis C, Nadin A, Neduvelil J G, et al. Nature (London) 2000;405:689–694. doi: 10.1038/35015085. [DOI] [PubMed] [Google Scholar]
- 18.Esler W P, Kimberly W T, Ostaszewski B L, Diehl T S, Moore C L, Tsai J-Y, Rahmati T, Xia W, Selkoe D J, Wolfe M S. Nat Cell Biol. 2000;2:428–434. doi: 10.1038/35017062. [DOI] [PubMed] [Google Scholar]
- 19.Brown M S, Ye J, Rawson R B, Goldstein J L. Cell. 2000;100:391–398. doi: 10.1016/s0092-8674(00)80675-3. [DOI] [PubMed] [Google Scholar]
- 20.De Strooper B, Annaert W, Cupers P, Saftig P, Craessaerts K, Mumm J S, Schroeter E H, Schrijvers V, Wolfe M S, Ray W J, et al. Nature (London) 1999;398:518–522. doi: 10.1038/19083. [DOI] [PubMed] [Google Scholar]
- 21.Schroeter E H, Kisslinger J A, Kopan R. Nature (London) 1998;393:382–386. doi: 10.1038/30756. [DOI] [PubMed] [Google Scholar]
- 22.Huppert S S, Le A, Schroeter E H, Mumm J S, Saxena M T, Milner L A, Kopan R. Nature (London) 2000;405:966–970. doi: 10.1038/35016111. [DOI] [PubMed] [Google Scholar]
- 23.Wong P C, Zheng H, Chen H, Becher M W, Sirinathsinghji D J, Trumbauer M E, Chen H Y, Price D L, Van der Ploeg L H, Sisodia S S. Nature (London) 1997;387:288–292. doi: 10.1038/387288a0. [DOI] [PubMed] [Google Scholar]
- 24.Shen J, Bronson R T, Chen D F, Xia W, Selkoe D J, Tonegawa S. Cell. 1997;89:629–639. doi: 10.1016/s0092-8674(00)80244-5. [DOI] [PubMed] [Google Scholar]
- 25.Ray W J, Yao M, Mumm J, Schroeter E H, Saftig P, Wolfe M, Selkoe D J, Kopan R, Goate A M. J Biol Chem. 1999;274:36801–36807. doi: 10.1074/jbc.274.51.36801. [DOI] [PubMed] [Google Scholar]
- 26.Berezovska O, Jack C, McLean P, Aster J C, Hicks C, Xia W, Wolfe M S, Kimberly W T, Weinmaster G, Selkoe D J, et al. J Neurochem. 2000;75:583–593. doi: 10.1046/j.1471-4159.2000.0750583.x. [DOI] [PubMed] [Google Scholar]
- 27.Berezovska O, Jack C, Deng A, Gastineau N, Rebeck G W, Hyman B T. J Biol Chem. 2001;276:30018–30023. doi: 10.1074/jbc.M008268200. [DOI] [PubMed] [Google Scholar]
- 28.Yu G, Nishimura M, Arawaka S, Levitan D, Zhang L, Tandon A, Song Y Q, Rogaeva E, Chen F, Kawarai T, et al. Nature (London) 2000;407:48–54. doi: 10.1038/35024009. [DOI] [PubMed] [Google Scholar]
- 29.Getman D P, DeCrescenzo G A, Heintz R M, Reed K L, Talley J J, Bryant M L, Clare M, Houseman K A, Marr J J, Mueller R A, et al. J Med Chem. 1993;36:288–291. doi: 10.1021/jm00054a014. [DOI] [PubMed] [Google Scholar]
- 30.McLendon C, Xin T, Ziani-Cherif C, Murphy M P, Findlay K A, Lewis P A, Pinnix I, Sambamurti K, Wang R, Fauq A, et al. FASEB J. 2000;14:2383–2386. doi: 10.1096/fj.00-0286fje. [DOI] [PubMed] [Google Scholar]
- 31.Rogaev E I, Sherrington R, Rogaeva E A, Levesque G, Ikeda M, Liang Y, Chi H, Lin C, Holman K, Tsuda T, et al. Nature (London) 1995;376:775–778. doi: 10.1038/376775a0. [DOI] [PubMed] [Google Scholar]
- 32.Levy-Lahad E, Wasco W, Poorkaj P, Romano D M, Oshima J, Pettingell W H, Yu C E, Jondro P D, Schmidt S D, Wang K, et al. Science. 1995;269:973–977. doi: 10.1126/science.7638622. [DOI] [PubMed] [Google Scholar]
- 33.Xia W, Ray W J, Ostaszewski B L, Rahmati T, Kimberly W T, Wolfe M S, Zhang J, Goate A M, Selkoe D J. Proc Natl Acad Sci USA. 2000;97:9299–9304. doi: 10.1073/pnas.97.16.9299. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}