

Abstract
MYC is frequently overexpressed in human cancers, but the downstream events contributing to tumorigenesis remain incompletely understood. MYC encodes an oncogenic transcription factor, whose target genes presumably contribute to cellular transformation. Although Myc regulates about 15% of genes and combinations of target genes are likely required for tumorigenesis, we studied in depth the expression of the Myc target gene, JPO1/CDCA7, in human cancers and its ability to provoke tumorigenesis in transgenic mice. JPO1/CDCA7 is frequently overexpressed in human cancers, and in particular, its expression is highly elevated in chronic myelogenous leukemia blast crisis as compared with the chronic phase. In murine lymphoid tissues, ectopic human JPO1/CDCA7 expression resulted in a 2-fold increased risk of lymphoid malignancies at 1 year. The transgene, which was driven by the H2K promoter, exhibited leaky expression in non-lymphoid tissues such as kidney. We observed a significant increased incidence of transgenic animal solid tumors, which were not seen in littermate controls. These observations suggest that JPO1/CDCA7 may contribute to Myc-mediated tumorigenesis.
Keywords: chronic myelogenous leukemia, Myc, oncogene, tumorigenesis, gene expression, transgenic mice
Introduction
In addition to the chromosomal translocations, which are hallmarks of Burkitt’s lymphoma (1–4), involving MYC, its expression is frequently increased in many different tumor types including breast, colon, lung and prostate (for a compilation of 93 studies see www.myc-cancer-gene.org) (5–7). In contrast to normal MYC, which displays tightly regulated expression, deregulated MYC in cancer cells may contribute to the process of transformation by altering the expression of a set of target genes regulated by the c-Myc transcription factor (8, 9). These targets affect a variety of functions including control of cell growth and apoptosis, as well as cellular metabolism, adhesion and differentiation (6, 10–12). Extensive studies have identified thousands of Myc target genes that may cooperate to promote tumorigenesis (www.myc-cancer-gene.org) (7). The current challenge is to identify those critical target genes that play significant roles in human cancers, as well as in Myc-mediated tumorigenesis in experimental models.
Although the Myc target gene network involves about 3000 human genes, which participate in cell cycle regulation, glucose metabolism, ribosome biogenesis as well as other cellular processes, how de-regulated MYC expression contributes to tumorigenesis through targets genes remains poorly understood. In particular, the contributions of large numbers of target genes with unknown functions to Myc-mediated tumorigenesis remain undelineated. Here, we sought to determine the expression pattern of JPO1/CDCA7, which encodes a nuclear protein with unknown function, in human cancers and its transforming activity in transgenic mice.
We identified JPO1 as a novel transcript that is differentially regulated in Myc transformed fibroblasts in a representational difference analysis screen (13). The gene has since been officially termed CDCA7 by the Human Genome Nomenclature Committee based on its periodic expression in the cell cycle, peaking at the G1 to S phase transition (14). JPO1/CDCA7 is encoded by a nine exon gene located at 2q31 that produces a 2.5 kb transcript expressed in the adult thymus and small intestine and an alternatively spliced transcript. Our previous work confirmed JPO1/CDCA7 as a direct Myc target through the use of an inducible Myc system, and showed that its expression is strongly correlated with Myc in experimental systems that have been examined, including human Burkitt’s lymphoma cell lines (15). Moreover, chromatin immunoprecipitation assays verified that Myc directly binds the JPO1/CDCA7 genomic locus (16).
The 47 kDa JPO1 protein encoded by JPO1/CDCA7 consists of 371 amino acids and is localized to the nucleus (15). JPO1 contains two putative functional domains: a leucine zipper motif in the N terminal region of the protein, and a cysteine-rich region that is homologous to a class of DNA-binding proteins in plants (Figure 1 A). JPO1/CDCA7 is conserved across species and has orthologs in mouse, rat, and xenopus, among others. Early experiments by Prescott et. al. showed that over-expression of JPO1/CDCA7 in rat fibroblasts conferred some transforming properties, although the ability of JPO1/CDCA7 to induce colony formation in soft agar assays was diminished and variable compared to Myc (15). Although the same JPO1 overexpressing cells were unable to form tumors when injected into nude mice, JPO1/CDCA7 complemented the hypomorphic W135E mutant MYC allele in transforming Rat1A cells (15).
Figure 1.
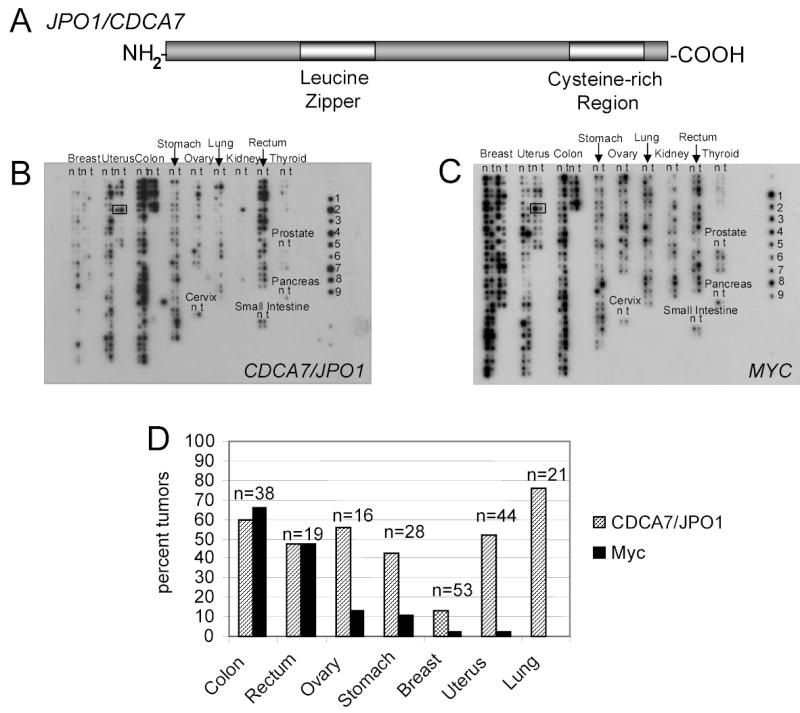
A. Schematic representation of the JPO1/CDCA7 protein. Analysis of amino acid sequence predicts a leucine zipper in the amino terminal region and a cysteine rich region toward the carboxy terminus.
B. Multiple tumor array probed for JPO1/CDCA7. cDNAs are spotted in pairs, with normal samples spotted on the left in each column (N) and matched tumor sample (T) from the same patient spotted on the right in each column. An example from one patient pair is boxed in the uterus tumor section. Tissue type is indicated above each group of samples. Controls derived from cell lines are on the right and indicate: 1, HeLa; 2, Daudi (Burkitt’s lymphoma); 3, K562 (CML); 4, HL-60 (PML); 5, Melanoma; 6, Lung Carcinoma; 7, Lymphoblastic leukemia; 8, SW480 (colon); 9, Raji (Burkitt’s lymphoma).
C. Multiple tumor arrays probed for MYC. Blot is identical to that shown in B.
D. Quantitated JPO1/CDCA7 and MYC expression in solid tumors. Values represent the percentage of tumors that have a greater than 2-fold tumor to normal ratio for either JPO1/CDCA7 or MYC. All values were normalized to ubiquitin.
Given its remarkable increased expression in Myc-transformed cells and JPO1’s in vitro complementation with a weak mutant MYC allele in transformation, we sought to determine the potential in vivo role of JPO1/CDCA7 in human cancers by assessing its expression levels in primary samples and its transforming potential in the T and B cell compartments in a transgenic mouse model. We measured JPO1/CDCA7 expession in human tumors and found that its expression is elevated in a significant fraction of human colon, rectum, ovary, lung, uterus and stomach cancers. Its expression is also highly elevated in acute myelogenous leukemia and in particular, in chronic myelogenous leukemia (CML) blast crisis as compared with chronic phase CML. The tumorigenic role of JPO1/CDCA7 is further supported by a 2-fold increase risk of developing lymphoid malignancies, and surprisingly a dramatic increase in solid tumors, in transgenic mice ectopically expressing JPO1/CDCA7 in T and B cells.
Materials and Methods
Multiple tumor array hybridization and quantitation
Multiple tumor and blood disease arrays were obtained from Clontech/BDBioScience (Mountain View, CA) and hybridized using cDNA probes to JPO1/CDCA7, c-Myc and ubiquitin. Probes were random primed labeled (Stratagene, LaJolla, CA) with α-32P dATP, and 3×106 cpm per mL of hybridization solution (ExpressHyb, Clontech, Mountain View, CA) was added to the arrays and hybridized overnight. Arrays were washed according to the manufacturers’ recommendations and exposed overnight to a phosphorimager screen, followed by quantitation using ImageQuant software.
Real time analysis on patient tumor samples
Samples of white blood cells were obtained from patients with CML or AML and RNA was extracted using the RNEasy method (Qiagen, Valencia, CA). Real time PCR primers and a Fam-labeled probe (Applied Biosystems, Foster City, CA) were designed to detect the human sequence of JPO1/CDCA7 at the junction of exons 7 and 8. Probes for MYC were commercially available (Applied Biosystems, Foster City, CA). Transgenic RNA was amplified in a one step RT-PCR reaction (Applied Biosystems, Foster City, CA). Expression was normalized to human β-actin (Applied BioSystems, Foster City, CA). Studies on de-identified patient tumor samples obtained from UCLA, Hopkins and University of Maryland Medical Center were approved by the Johns Hopkins Medicine Institutional Review Board.
Creation of transgenic mice and breeding.
A transgene contruct was created by cloning the coding sequence of JPO1 (Hs.470654) into the SalI sites of the vector pHSE3’ (a gift of S. Desiderio, Department of Molecular Biology and Genetics, Johns Hopkins University School of Medicine, Baltimore, MD) (17). The resulting plasmid was then linearized using XhoI to create a fragment of DNA containing the JPO1 coding sequence, along with the H2-K promoter, exons 2 and 3 of the β-globin gene, and the Ig μ heavy chain enhancer. This fragment was then injected into fertilized eggs from C57Bl6 or B6SJL female mice (transgenic core) and maintained by crossing hemizygous mice to wild-type C57Bl6 mice. Presence of the transgene was assessed by PCR using one primer in the H2-K sequence and another in the coding sequence of JPO1. Initial litters were confirmed using Southern blot analysis.
Analysis of transgene expression by real time PCR.
Following autopsy, sections of spleen and thymus were frozen in liquid nitrogen, crushed and homogenized. RNA was extracted using the Trizol method (Invitrogen). Real time PCR was carried out as described above. RNA expression was normalized to mouse18S (Applied Biosystems).
Immunohistochemistry.
During necropsy, all organs were removed, preserved in formalin and then embedded in paraffin blocks. Five-micron unstained sections were cut onto poly-lysine slides which were then deparaffinized in xylene for 30 minutes, followed by ethanol hydration and washes in PBS. The slides were boiled in antigen unmasking solution (Vector Laboratories, Burlingame, CA) for 10 minutes, and left in the solution at room temperature for an additional 20 minutes. Following two more PBS washes, slides were quenched in 0.6% hydrogen peroxide/methanol, washed again in PBS and then blocked overnight in a humidifying chamber at 4° C using normal blocking serum. (Vector Laboratories, VectaStain Elite ABC kit) After washing in PBS three times, slides were incubated with either primary antibodies to JPO1 (15) or rabbit IgG (Vector Labs) for at least one hour. Following another three PBS washes, slides were incubated with 2° antibody for 30 min – 1 hr and then detected with the VectaStain Elite ABC reagents and counterstained with haemotoxylin (Vector Laboratories).
Analysis of phenotype.
Transgenic mice and normal littermates were sacrificed by CO2 asphyxiation and necropsy was performed. Mouse tissue was preserved in formalin following necropsy, paraffin embedded and H&E stained. Slides were prepared and then evaluated for neoplastic phenotype without knowledge of transgene status.
Results
Expression of JPO1/CDCA7 and MYC in solid tumors
Overexpression of Myc and its target genes are frequent events in human cancers. As a first approximation, we sought to determine whether JPO1/CDCA7 participates in human tumorigenesis by assessing the level of its expression in human malignancies. To determine the expression of JPO1/CDCA7 in a large number of human tumors, we used filter arrays that have been quantitatively spotted with cDNAs to reflect the original mRNA levels in a number of different types of solid tumors (Figures 1 B and C). Arrays were probed with random-primed, radio-labeled JPO1/CDCA7 or MYC cDNAs and quantitated by a phosphorimager. All values were normalized to ubiquitin according to manufacturer’s instructions and represent the average of two independent hybridizations on separate arrays. To obtain values for the relative expression of JPO1/CDCA7 and MYC in tumor tissue compared to normal tissue, the value for each tumor spot on the array was compared to the matched normal tissue provided from the same individual. Tumors that showed a greater than 2-fold tumor to normal ratio were considered to overexpress JPO1/CDCA7. Figure 1D shows a graphical representation of the percentage of solid tumors overexpressing JPO1/CDCA7 and MYC.
Expression of JPO1/CDCA7 and MYC in hematologic malignancies
To determine the expression of JPO1/CDCA7 in human hematologic malignancies, we probed an array (Clontech) similar to the multiple human cancer array, which allows for the analysis of multiple blood fractions from patients with no disease (designated as normal), acute myeloid leukemia (AML), Hodgkin’s disease (HD), von Willebrand’s disease (vWD), chronic myeloid leukemia (CML) or non-Hodgkin’s lymphoma (NHL). Seven normal lymph node samples were also included. Blots were probed for both JPO1/CDCA7 and MYC (Figures 2A and 2B). For the hematologic malignancies, normal samples from patients are unavailable, and hence the values for each sample were compared to the corresponding averaged values from the normal donors spotted in the upper left of the array.
Figure 2.
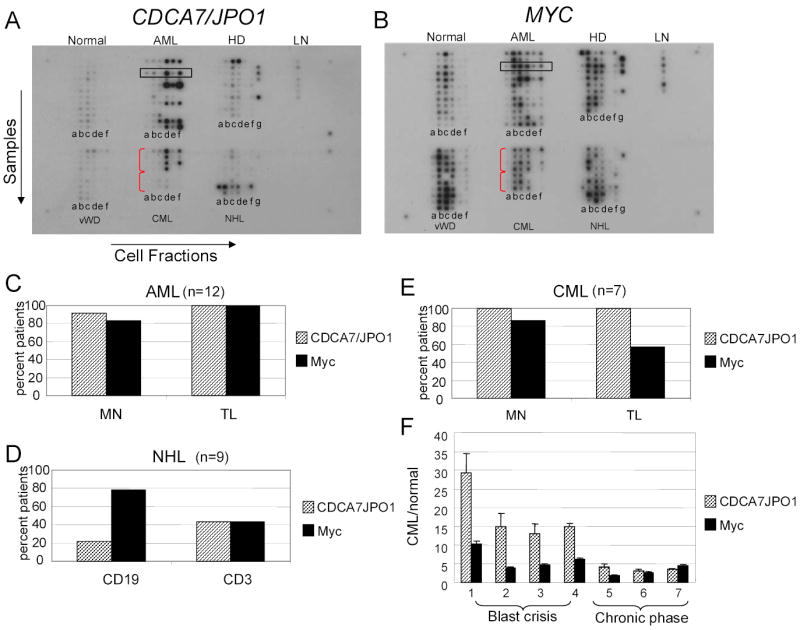
A. Blood disease profiling array probed for JPO1/CDCA7. Each horizontal row represents cDNA from 6 cell fractions derived from a single patient, as indicated by the box in the AML section. Each vertical column represents samples for a given fraction as indicated by a letter below each column. The disease is indicated above each section. Abbreviations are: AML, acute myeloid leukemia; HD, Hodgkin’s disease; LN, normal lymph nodes; vWD, von Willebrand disease; CML, chronic myeloid leukemia; NHL, non-Hodgkin’s lymphoma. The blood fractions are as follows: a, CD14 (monocyte/macrophage); b, CD19 (B-cell); c, CD3 (T-cell); d, mononuclear cells; e, polymorphonuclear; f, total leukocyte; g, lymph node.
B. Blood disease profiling array probed for MYC. The array is identical to the one used in A.
C-E. Quantitation of CDCA7/JPO1 and MYC expression in hematologic malignancies. For each disease, the sorted fractions most likely to contain the neoplastic cells were analyzed. Note that the percent of patients with a greater than 2-fold disease to normal ratio based on array quantification is plotted for each fraction. The array included twelve patients with AML (A), seven with CML (B), and nine with NHL (C).
F. Expression of JPO1/CDCA7 and MYC in the individual CML patient samples. Plotted on the Y-axis is the normalized signal for JPO1/CDCA7 and MYC in each patient’s mononuclear sample, divided by the averaged signal detected in normal monocytes.
For each type of hematologic malignancy, except NHL and HD, we looked specifically at the expression of JPO1/CDCA7 in the fractions that are likely to contain the malignant cells for each disease. Data are shown for AML, CML and non-Hodgkins B-cell lymphoma in Figures 2C, 2D and 2E. Since JPO1/CDCA7 is normally expressed in T lymphocytes, it is surprising that JPO1/CDCA7 and MYC are most consistently upregulated in acute myeloid leukemia patients, and not those with lymphomas. It should be noted however, that the blood fractions are less likely to reveal neoplastic cells in the case of lymphomas. The one lymph node available (second case down from the top of the NHL series) did not have elevated JPO1/CDCA7 expression as compared to normal lymph nodes (LN). By contrast, all four available lymph nodes from Hodgkin’s disease have high JPO1/CDCA7 expression.
Among the CML patient samples, it is remarkable that patients in the acute or blast crisis stages of disease (samples 1–4) expressed very high levels of JPO1/CDCA7 in the mononuclear fraction (row d), compared to patients in the chronic phase of disease (samples 5–7). The level of expression of JPO1/CDCA7 and MYC for each individual CML patient is shown in Figure 2F. In order to extend these initial observations, samples were obtained from 9 patients with AML and 20 patients with CML (9 patients in blast crisis and 11 patients in the chronic phase from three cancer centers). Levels of JPO1/CDCA7 mRNA were analyzed by real-time PCR and a tumor to normal ratio was obtained by comparison to a normal WBC control (Figure 3). As expected, AML samples show varying degrees of JPO1/CDCA7 overexpression. In the CML samples, although the level of JPO1/CDCA7 expression varies widely and overlaps between blast crisis and chronic phase patients, there is a statistically significant higher JPO1/CDCA7 expression in the blast crisis samples as compared to chronic phase. Comparison of blast vs. chronic CML patients using a T-test (two tailed, type 3) gave a p value of 0.038, for chronic vs. AML (two tailed, type 2 T-test) p=0.012, and for AML vs. blast (two tailed, type 2 T-test) p=0.318. A p value of <0.05 was considered significant.
Figure 3.
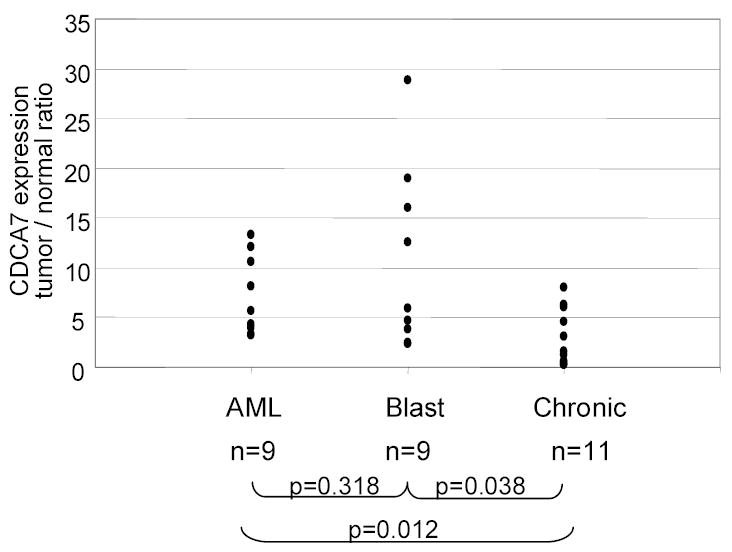
JPO1/CDCA7 expression in additional leukemia samples as detected by real-time RT-PCR. Nine additional AML samples and 20 CML samples (11 in blast crisis and 9 in the chronic phase) were obtained and analyzed for JPO1/CDCA7 expression. RNA was extracted from peripheral blood samples. Each point represents one patient and the level of JPO1/CDCA7 expression relative to a non-diseased control. P values represent the results of a two tailed T test pairwise comparison of the different groups.
JPO1/CDCA7 Transgenic Mice
Given that we previously found JPO1/CDCA7 transforming activity in vitro and that its expression is elevated in a number of human cancers in this study, we sought to determine the potential transforming activity of JPO1/CDCA7 in vivo in transgenic mice. We began these studies knowing that JPO1/CDCA7 is expressed in the thymus and small intestine, and that it is overexpressed in a number of human Burkitt’s lymphoma cell lines. As such, we chose to target the murine lymphoid compartment by expressing human JPO1/CDCA7 in transgenic mice under the control of the murine promoter H2-K and Eμ enhancer (Figure 4A). This expression vector has been successfully used to express N-myc and BAP130/TFII-I in transgenic T and B lymphocytes (18). An alternatively spliced form JPO1/CDCA7 was not known at the time we constructed the mice and hence the shorter spliced form of the human JPO1/CDCA7 coding sequence was used.
Figure 4. Creation of JPO1/CDCA7 transgenic animals.
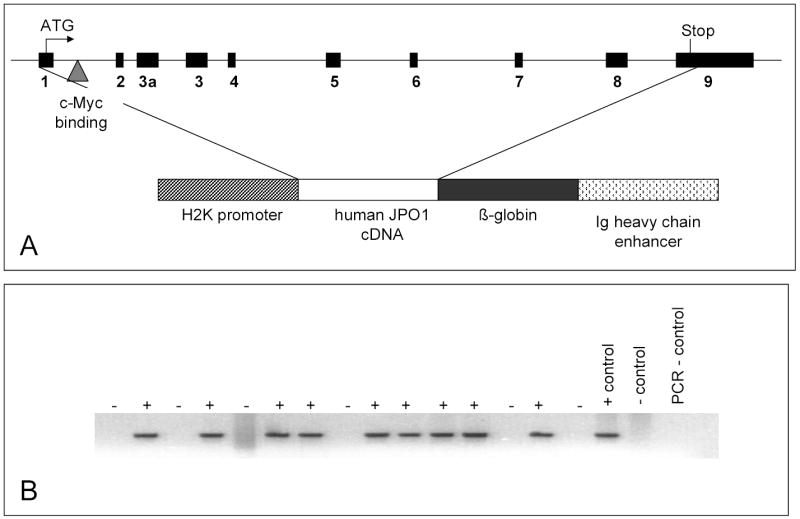
A. Schematic showing the structure of the JPO1/CDCA7 transgene construct. The upper diagram represents the genomic structure of human JPO1/CDCA7 with black boxes depicting exons. The human JPO1 cDNA is depicted in the transgene construct which is driven by the H2K promoter and regulated by the Ig heavy chain enhancer.
B. Ethidium bromide stained gel showing PCR amplified DNA to detect presence of transgene in a new litter of mice.
Four transgenic lines were created, and the presence of the transgene was confirmed by PCR analysis of DNA extracted from tail clippings (Figure 4B). From the original four founders, two lines of transgenic mice were successfully established; one on the C57Bl6 background and the other on the B6SJL hybrid background. B6SJL founders were bred against C57Bl6 for at least 5 generations. In order to determine the expression pattern of the transgene, RNA was extracted from various organs and analyzed by real-time PCR for several mice derived from each of the two founders from which we were able to propagate lines. Figure 5A shows representative expression data for two of the mice. As expected, expression of the transgene is high in both the T and B cell compartments. A common incidental finding in these animals was cystic kidneys, leading us to examine the expression of the transgene in the kidney (Figure 5A). Since littermates with and without the transgene developed cystic kidneys, which appear to be secondary to hydronephrosis, at equal frequency, it is not likely that leaky expression of the transgene could cause this phenotype. Expression of the transgene in lymphoid tissues at the protein level was confirmed by immunohistochemical staining using polyclonal antibodies raised against JPO1. Figure 5B shows a representative transgenic spleen section that stained positively with anti-human Jpo1 as compared to one that did not stain with an isotype control IgG. Sections from non-transgenic littermates were also stained with anti-JPO1 antibody and IgG, and these were both negative (data not shown).
Figure 5. Expression of the JPO1/CDCA7 construct in transgenic animals.
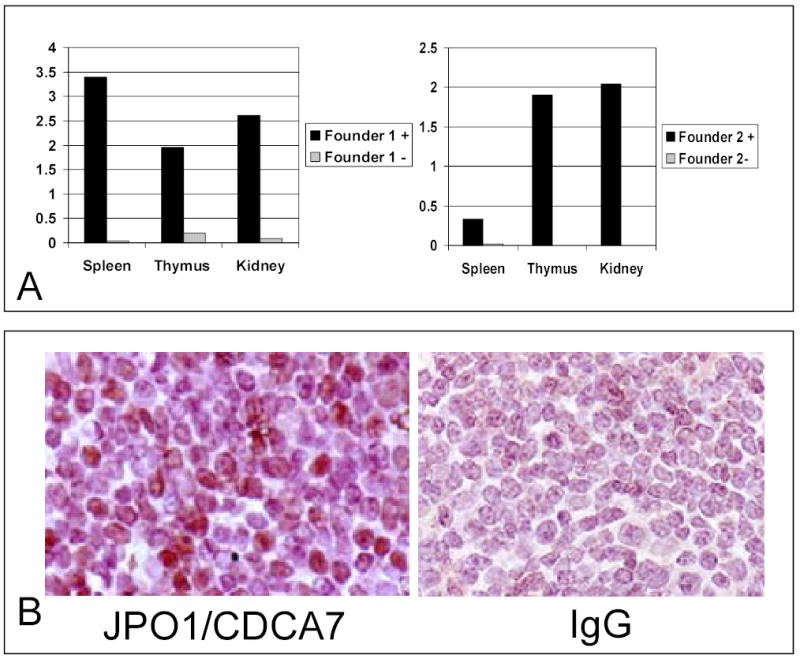
A. Real time PCR analysis measured expression of human JPO1/CDCA7 transcript in RNA extracted from mouse tissues as indicated. Expression levels are shown relative to levels of murine 18S rRNA.
B. Immunohistochemistry staining of spleen from a JPO1/CDCA7 transgenic animal. Sections were stained with anti-JPO1 antibody or matched isotype IgG used as a negative control, incubated with 2° antibody, and then detected with the VectaStain Elite ABC reagents and counterstained with haemotoxylin.
Although young JPO1 transgenic mice appeared normal and healthy and early sacrifices of JPO1 transgenic animals showed no abnormalities, one founder at 8 months developed a gait disorder that rapidly progressed to hindlimb paralysis. This animal could not be propagated and was found to have a lymphoma that infiltrated the thoracic spine. The animals were, therefore, allowed to attain 1 year of age before sacrifice, at which time they were necropsied and analyzed for evidence of neoplasia. Table 1 lists the phenotypes that were observed in the 1 year old animals. The types of tumors observed and the rates at which they occurred are shown in Figure 6A. A total of 45 animals that contained the JPO1 transgene (a combination of the four founder lines with two that were able to be propagated) were analyzed and compared to 28 non-transgenic age-matched littermates. In addition to an increased incidence of lymphomas (see representative tumor histology, Figure 6B), other types of solid tumors also intriguingly significantly increased in frequency. In fact, no solid tumors were observed in littermate controls. Since this effect was specific to the presence of the transgene, and we have shown that the transgene is expressed in the kidneys of transgenic animals, it is possible that there is some level of JPO1/CDCA7 expression due to the H2-K promoter in the tissues from which the tumors arose. In order to confirm expression of the JPO1/CDCA7 transgene in the tumors, sections were stained with anti-JPO1 antibodies. Specific staining was observed in both a pancreatic lymphoma and a splenic histiocytic sarcoma (Figure 7). Statistical analysis using binomial sampling revealed a trend for increased tumorigenesis in JPO1/CDCA7 transgenic mice; however, significance was not reached with a 2-sided p-value of 0.296, Z score=−1.045.
Table 1.
Phenotypes observed in transgenic mice at one year of age
| Spleen with lymphoid hyperplasia |
| Follicular fusion |
| Follicular expansion |
| Marginal zone expansion |
| Lymph node hyperplasia |
| Intestine: mesenteric lymph node and Gut-Associated Lymphoid Tissue (GALT) |
| Lung: bronchial and mediastinal lymph node |
| Kidney: renal lymph node |
| Other Lesions |
| Uterus: cystic endometrial hyperplasia |
| Kidney: renal cysts secondary to hydronephrosis |
| Malignancies: |
| Solid tumors |
Figure 6.
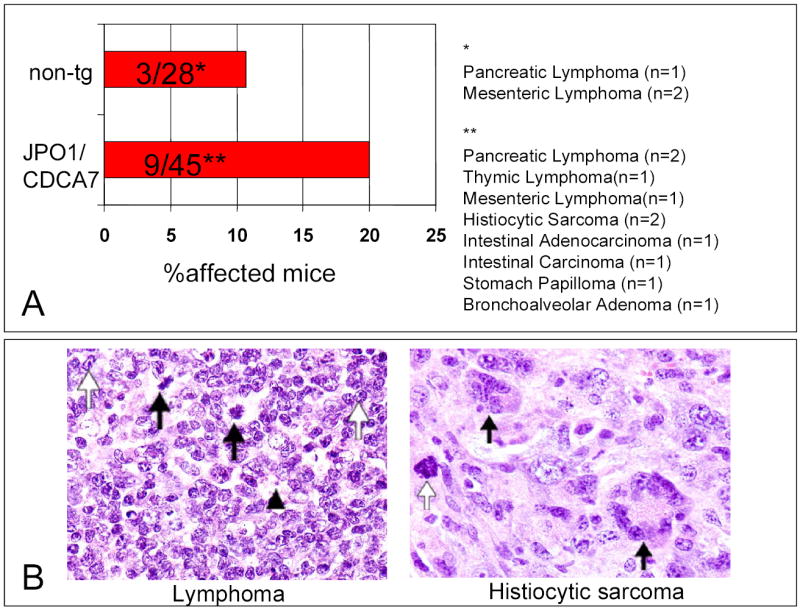
A. The rate of tumor formation in one year old JPO1/CDCA7 transgenic mice as compared to non-transgenic, age-matched littermates. Data is compiled from all 4 founders. Tumors observed were, for non-transgenic animals: pancreatic lymphoma (n=1), mesenteric lymphoma (n=2); for transgenic animals: pancreatic lymphoma (n=2), thymic lymphoma (n=1), mesenteric lymphoma(n=1), histiocytic sarcoma (n=2), intestinal adenocarcinoma (n=2), stomach papilloma (n=1), and bronchoalveolar adenoma (n=1). Note that one transgenic mouse had both a pancreatic and a mesenteric lymphoma, which were combined as one for determination of incidence.
B. Representative histology from necropsy of JPO1/CDCA7 transgenic mice. The left panel shows a lymphoma in pancreatic lymph node. Mitoses are indicated by black arrows, prominent nucleoli by white arrows, and a vesicular nucleus by the arrowhead. The panel on the right shows histiocytic sarcoma present in spleen. Black arrows indicate giant cells on a background of histiocytes, and the white arrow points out mitosis.
Figure 7.
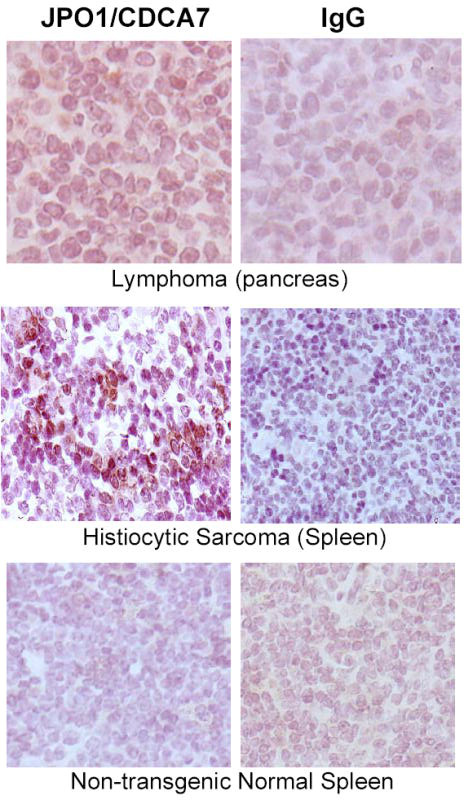
Immunohistochemistry for JPO1/CDCA7 in transgenic mouse tumors. Pancreatic lymphoma stained with JPO1 antibodies (left) and IgG isotype control (right). 200x Histiocytic Sarcoma from spleen stained with antibodies against JPO1 (left) and IgG isotype control (right). 200x Normal spleen from non-transgenic mouse stained with JPO1 antibodies as a negative control. 200x
Discussion
Although the molecular function of the JPO1 nuclear protein remains largely unknown, the fact that it is a direct Myc target that is highly upregulated when cells grow independently of anchorage suggests that it may play an important role in tumorigenesis (13, 15). When the amino acid sequence of JPO1 is compared via NCBI BLAST search to other proteins, several functional domains are recognized, including a leucine zipper motif and a cysteine rich region that is present in plant DNA-binding proteins. The presence of these functional domains suggests that JPO1 either homo-oligomerizes or may have an unknown binding partner that would allow it to bind DNA and perhaps modulate transcription.
Until recently, there were no human proteins in the database that showed significant homology to JPO1. A recent sequence entry (EAL24271; Locuslink 55536), termed transcription factor RAM2 or JPO2 (Huang et al., companion manuscript) which shows high homology to JPO1 in both the leucine zipper and cysteine rich region, has been identified as a transcription factor that modulates genomic integration of adeno-associated virus (as annotated in Genbank entry AY161169; T. Cathomen and M. Weitzman, personal commun.). While we observed (unpublished) that JPO1 does not associate with JPO2 by coimmunoprecipitation, both proteins can interact with the Myc N-terminal domain (Huang et al., personal commun.). In addition to the finding that JPO2 is transforming in medulloblastoma cells (Huang et al., personal commun.), we also found that JPO2 can induce colony formation in soft-agar when overexpressed in Rat1a fibroblasts, whereas JPO1 display a weaker transforming phenotype (Othus et al., unpublished observation). Moreover, JPO2 is able to transform human medulloblastoma cells and collaborates with a mutant Myc (W135E), as does JPO1 (15), to transform Rat1a cells (Huang et al., personal commun.). The observation that JPO1 could bind the Myc transactivation domain (Huang et al., personal commun.) suggest a possible positive feed-forward loop, such that Myc transactivates JPO1, which in turn collaborate with Myc in transformation. These observations on JPO2 taken together with our observations that JPO1 has limited transforming activity suggest that these structurally related proteins could contribute to tumorigenesis.
To begin to understand the potential role of JPO1/CDCA7 in human cancers, we measured the expression of MYC and JPO1/CDCA7 in human malignancies simply to determine whether they are concordantly overexpressed as compared with normal tissues. Although both MYC and JPO1/CDCA7 are frequently overexpressed in human malignancies, their magnitudes of induction are not always concordant. For example, while the expression of MYC and JPO1/CDCA7 are concordant in colorectal carcinomas, only JPO1/CDCA7 is overexpressed in lung cancers. These observations suggest that while high MYC may be sufficient to induce JPO1/CDCA7 expression, MYC is not necessary and other oncogenic events may induce JPO1/CDCA7 independently of MYC. A similar phenomenon has been observed with the Myc target gene HMG I/Y in breast cancer cell lines. A comparison of HMG I/Y and Myc levels in multiple breast cancer lines revealed that those cell lines with the highest levels of the target gene (HMG I/Y) are not necessarily correlated with the highest levels of Myc (19).
Not only is JPO1/CDCA7 overexpressed in solid tumors, it is also strikingly overexpressed in myeloid leukemias. Intriguingly, the expression pattern of JPO1/CDCA7 on the human hematologic malignancies array shows an elevation of JPO1/CDCA7 expression in both acute and chronic myeloid leukemia. Increased expression of MYC has been previously observed in CML patients(20). Trisomy 8 and amplification of the MYC locus on chromosome 8 are also observed in CML (21). In addition, the Bcr-Abl oncogenic fusion protein, a hallmark of CML, has been shown to increase MYC expression and increase Myc protein stability through interaction with Jak2 (22). Expression of Myc was also demonstrated to be essential for transformation by Bcr-Abl (23). These data in aggregate imply that downstream targets of Myc may play a role in mediating the growth phenotype observed in CML. The fact that JPO1/CDCA7 is highly expressed in the blast or terminal phases of CML, but not in the chronic phase suggests that JPO1/CDCA7 could be a marker for disease progression. Microarray analysis experiments that compare gene expression profiles in blast versus chronic phase samples have failed to identify a common set of genes that are specifically expressed in all samples in a given phase (24). Instead those studies suggest that while the expression pattern changes with disease progression, CML is a heterogeneous disease and there are no specific changes that can be used to monitor the transition from chronic to blast phase (24). The overlap in JPO1/CDCA7 expression levels between chronic and terminal phase leukemia could reflect this phenomenon.
Both our previous in vitro study of cell lines (15) and current study of JPO1/CDCA7 expression in human cancers support the notion that JPO1/CDCA7 contributes to tumorigenesis. To further establish the role of JPO1/CDCA7 in tumorigenesis, we constructed transgenic mice in which JPO1/CDCA7 is overexpressed in T and B lymphocytes. Studies of the transgenic mice suggest that JPO1/CDCA7 has a mild tumorigenic effect in vivo. The lag time required for tumor formation indicates that there are multiple hits that must be acquired in order for tumorigenesis to take place. This is not unexpected, and is also seen in the Eμ-myc transgenic mouse model, where mice expressing c-Myc under the control of an immunoglobulin enhancer element develop lymphomas and lymphoblastic leukemias with a lag time of 3 to 6 months. The tumors that develop in that mouse model are clonal, suggesting that additional genetic lesions are necessary to initiate tumor formation even in the presence of high levels of Myc (25, 26). The observation that other solid tumors can also be induced in the JPO1/CDCA7 transgenic mice suggests that the H2-K promoter construct with an Eμ-enhancer is leaky, a phenomenon that is also observed with HMGY(I) transgenic mice driven by the same promoter-enhancer expression vector (L. Resar, personal commun.). These observations are compatible with the finding that JPO1/CDCA7 may be dysregulated in various types of human malignancies. Indeed, JPO1/CDCA7 is upregulated in a large percentage of solid tumors derived from colon, rectum, ovary, stomach, uterus and lung.
In summary, the Myc oncogenic transcription factor contributes to tumorigenesis through regulation of thousands of genes with many novel ones whose functions are unknown. Among these, we identified JPO1/CDCA7 as one that encodes a nuclear protein JPO1 without a known function, although its high homology with the transcription factor JPO2 or RAM2 suggests that JPO1 is also a transcription factor. To begin to understand the JPO1/CDCA7 gene we took a multi-pronged approach. In aggregate, our transformation studies with cell lines in vitro, expression analysis of JPO1/CDCA7 in human cancers and in vivo tumorigenic studies in transgenic mice all support a role for JPO1/CDCA7 in tumorigenesis. As compared with MYC, JPO1/CDCA7 is weakly transforming in vitro and in vivo; however, the widespread overexpression JPO1/CDCA7 in human cancers suggests a more profound role.
Acknowledgments
We would like to thank Drs. Charles Sawyers, Neil Shah and John Nicoll from the David Geffen School of Medicine at UCLA for the additional CML and AML samples. We also thank Dr. Adele Mitchell for help with the statistical analysis. This work was partially supported by NIH grants CA51497, CA57341 and RR00171. Clinical specimens were provided by the Sidney Kimmel Cancer Center at Johns Hopkins Tumor and Cell Procurement Bank, supported by the Regional Oncology Research Center Grant # 5 P30 CA06973. C. Dang is The Johns Hopkins Family Professor in Oncology Research.
Footnotes
Running title: Myc target JPO1/CDCA7 in human cancers
References
- 1.Boxer LM, Dang CV. Translocations involving c-myc and c-myc function. Oncogene. 2001;20:5595–5610. doi: 10.1038/sj.onc.1204595. [DOI] [PubMed] [Google Scholar]
- 2.Shen-Ong GL, Keath EJ, Piccoli SP, Cole MD. Novel myc oncogene RNA from abortive immunoglobulin-gene recombination in mouse plasmacytomas. Cell. 1982;31:443–452. doi: 10.1016/0092-8674(82)90137-4. [DOI] [PubMed] [Google Scholar]
- 3.Dalla-Favera R, Bregni M, Erikson J, Patterson D, Gallo RC, Croce CM. Human c-myc onc gene is located on the region of chromosome 8 that is translocated in Burkitt lymphoma cells. Proc Natl Acad Sci U S A. 1982;79:7824–7827. doi: 10.1073/pnas.79.24.7824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dalla-Favera R, Martinotti S, Gallo RC, Erikson J, Croce CM. Translocation and rearrangements of the c-myc oncogene locus in human undifferentiated B-cell lymphomas. Science. 1983;219:963–967. doi: 10.1126/science.6401867. [DOI] [PubMed] [Google Scholar]
- 5.Nesbit CE, Tersak JM, Prochownik EV. MYC oncogenes and human neoplastic disease. Oncogene. 1999;18:3004–3016. doi: 10.1038/sj.onc.1202746. [DOI] [PubMed] [Google Scholar]
- 6.Dang CV. c-Myc target genes involved in cell growth, apoptosis, and metabolism. Mol Cell Biol. 1999;19:1–11. doi: 10.1128/mcb.19.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zeller KI, Jegga AG, Aronow BJ, O'Donnell KA, Dang CV. An integrated database of genes responsive to the Myc oncogenic transcription factor: identification of direct genomic targets. Genome Biol. 2003;4:R69. doi: 10.1186/gb-2003-4-10-r69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Levens DL. Reconstructing MYC. Genes Dev. 2003;17:1071–1077. doi: 10.1101/gad.1095203. [DOI] [PubMed] [Google Scholar]
- 9.Levens D. Disentangling the MYC web. Proc Natl Acad Sci U S A. 2002;99:5757–5759. doi: 10.1073/pnas.102173199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grandori C, Cowley SM, James LP, Eisenman RN. The Myc/Max/Mad network and the transcriptional control of cell behavior. Annu Rev Cell Dev Biol. 2000;16:653–699. doi: 10.1146/annurev.cellbio.16.1.653. [DOI] [PubMed] [Google Scholar]
- 11.Cole MD, McMahon SB. The Myc oncoprotein: a critical evaluation of transactivation and target gene regulation. Oncogene. 1999;18:2916–2924. doi: 10.1038/sj.onc.1202748. [DOI] [PubMed] [Google Scholar]
- 12.Oster SK, Ho CS, Soucie EL, Penn LZ. The myc oncogene: MarvelouslY Complex. Adv Cancer Res. 2002;84:81–154. doi: 10.1016/s0065-230x(02)84004-0. [DOI] [PubMed] [Google Scholar]
- 13.Lewis BC, Shim H, Li Q, Wu CS, Lee LA, Maity A, Dang CV. Identification of putative c-Myc-responsive genes: characterization of rcl, a novel growth-related gene. Mol Cell Biol. 1997;17:4967–4978. doi: 10.1128/mcb.17.9.4967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Whitfield ML, Sherlock G, Saldanha AJ, Murray JI, Ball CA, Alexander KE, Matese JC, Perou CM, Hurt MM, Brown PO, Botstein D. Identification of genes periodically expressed in the human cell cycle and their expression in tumors. Mol Biol Cell. 2002;13:1977–2000. doi: 10.1091/mbc.02-02-0030.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Prescott JE, Osthus RC, Lee LA, Lewis BC, Shim H, Barrett JF, Guo Q, Hawkins AL, Griffin CA, Dang CV. A novel c-Myc-responsive gene, JPO1, participates in neoplastic transformation. J Biol Chem. 2001;276:48276–48284. doi: 10.1074/jbc.M107357200. [DOI] [PubMed] [Google Scholar]
- 16.Haggerty TJ, Zeller KI, Osthus RC, Wonsey DR, Dang CV. A strategy for identifying transcription factor binding sites reveals two classes of genomic c-Myc target sites. Proc Natl Acad Sci U S A. 2003;100:5313–5318. doi: 10.1073/pnas.0931346100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dildrop R, Ma A, Zimmerman K, Hsu E, Tesfaye A, DePinho R, Alt FW. IgH enhancer-mediated deregulation of N-myc gene expression in transgenic mice: generation of lymphoid neoplasias that lack c-myc expression. Embo J. 1989;8:1121–1128. doi: 10.1002/j.1460-2075.1989.tb03482.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Malek SN, Dordai DI, Reim J, Dintzis H, Desiderio S. Malignant transformation of early lymphoid progenitors in mice expressing an activated Blk tyrosine kinase. Proc Natl Acad Sci U S A. 1998;95:7351–7356. doi: 10.1073/pnas.95.13.7351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dolde CE, Mukherjee M, Cho C, Resar LM. HMG-I/Y in human breast cancer cell lines. Breast Cancer Res Treat. 2002;71:181–191. doi: 10.1023/a:1014444114804. [DOI] [PubMed] [Google Scholar]
- 20.Handa H, Hegde UP, Kotelnikov VM, Mundle SD, Dong LM, Burke P, Rose S, Hsu WT, Gaskin F, Raza A, Preisler HD. The effects of 13-cis retinoic acid and interferon-alpha in chronic myelogenous leukemia cells in vivo in patients. Leuk Res. 1997;21:1087–1096. doi: 10.1016/s0145-2126(97)00090-8. [DOI] [PubMed] [Google Scholar]
- 21.Jennings BA, Mills KI. c-myc locus amplification and the acquisition of trisomy 8 in the evolution of chronic myeloid leukaemia. Leuk Res. 1998;22:899–903. doi: 10.1016/s0145-2126(98)00097-6. [DOI] [PubMed] [Google Scholar]
- 22.Xie S, Lin H, Sun T, Arlinghaus RB. Jak2 is involved in c-Myc induction by Bcr-Abl. Oncogene. 2002;21:7137–7146. doi: 10.1038/sj.onc.1205942. [DOI] [PubMed] [Google Scholar]
- 23.Sawyers CL, Callahan W, Witte ON. Dominant negative MYC blocks transformation by ABL oncogenes. Cell. 1992;70:901–910. doi: 10.1016/0092-8674(92)90241-4. [DOI] [PubMed] [Google Scholar]
- 24.Nowicki MO, Pawlowski P, Fischer T, Hess G, Pawlowski T, Skorski T. Chronic myelogenous leukemia molecular signature. Oncogene. 2003;22:3952–3963. doi: 10.1038/sj.onc.1206620. [DOI] [PubMed] [Google Scholar]
- 25.Adams JM, Cory S. Transgenic models of tumor development. Science. 1991;254:1161–1167. doi: 10.1126/science.1957168. [DOI] [PubMed] [Google Scholar]
- 26.Adams JM, Harris AW, Pinkert CA, Corcoran LM, Alexander WS, Cory S, Palmiter RD, Brinster RL. The c-myc oncogene driven by immunoglobulin enhancers induces lymphoid malignancy in transgenic mice. Nature. 1985;318:533–538. doi: 10.1038/318533a0. [DOI] [PubMed] [Google Scholar]