

Abstract
The translocation t(8;21)(q22;q22) is one of the most frequent chromosome translocations in acute myeloid leukemia (AML). AML1/RUNX1 at 21q22 is involved in t(8;21), t(3;21), and t(16;21) in de novo and therapy-related AML and myelodysplastic syndrome as well as in t(12;21) in childhood B cell acute lymphoblastic leukemia. Although DNA breakpoints in AML1 and ETO (at 8q22) cluster in a few introns, the mechanisms of DNA recombination resulting in t(8;21) are unknown. The correlation of specific chromatin structural elements, i.e., topoisomerase II (topo II) DNA cleavage sites, DNase I hypersensitive sites, and scaffold-associated regions, which have been implicated in chromosome recombination with genomic DNA breakpoints in AML1 and ETO in t(8;21) is unknown. The breakpoints in AML1 and ETO were clustered in the Kasumi 1 cell line and in 31 leukemia patients with t(8;21); all except one had de novo AML. Sequencing of the breakpoint junctions revealed no common DNA motif; however, deletions, duplications, microhomologies, and nontemplate DNA were found. Ten in vivo topo II DNA cleavage sites were mapped in AML1, including three in intron 5 and seven in intron 7a, and two were in intron 1b of ETO. All strong topo II sites colocalized with DNase I hypersensitive sites and thus represent open chromatin regions. These sites correlated with genomic DNA breakpoints in both AML1 and ETO, thus implicating them in the de novo 8;21 translocation.
The translocation t(8;21)(q22;q22) is one of the most frequent chromosome translocations in acute myeloid leukemia (AML), primarily in the M2 subtype (1). AML1 (also called RUNX1, CBFA2,) at 21q22 is involved in t(8;21) and other recurrent chromosome translocations, such as t(3;21) and t(16;21) in de novo and therapy-related AML (t-AML) and myelodysplastic syndrome as well as in t(12;21), the most common aberration in childhood B cell acute lymphoblastic leukemia (2–8). These translocations result in novel chimeric genes that play an important role in leukemogenesis. In t(8;21), the 5′ part of the AML1 gene is fused with nearly the entire ETO (MTG8) gene at 8q22, resulting in an in-frame AML1–ETO fusion located on 8q22 (3, 4).
AML1 is a transcription factor and a critical regulator of hematopoietic cell development; it contains the Runt DNA binding domain at the N terminus and a transactivation domain at the C terminus (9, 10). The complete 260-kb genomic sequence of AML1 contains nine exons (11, 12). The genomic breakpoints in AML1 are located in intron 5 in t(8;21) and t(16;21) and in introns 6–7b in t(3;21). In t(12;21), in contrast, DNA breaks in AML1 occur within introns 1 or 2 (Fig. 1) (2–7, 11–13). ETO contains a region homologous to the Drosophila gene Nervy and several DNA-binding protein domains (3, 4). Unlike AML1, which has multiple partner genes, ETO is involved only with AML1 in t(8;21). ETO is about 150 kb in size and contains 13 exons. The breakpoints in ETO are located in introns 1b–2 (Fig. 2) (14).
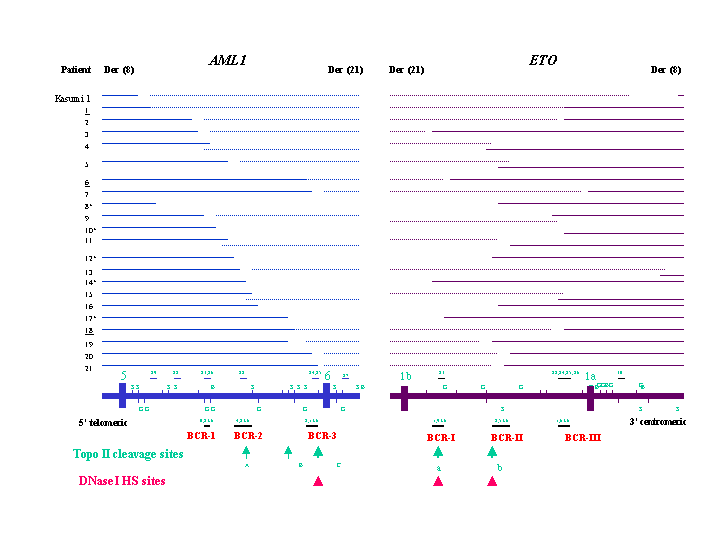
Figure 1.

In vivo topo II cleavage, DNase I HS sites, and genomic breakpoints in AML1 in t(8;21). The vertical boxes and lines represent exons, genomic DNA probes, and BamHI (B), BglII (G), and SacI (S) restriction sites. Large and small arrows at the bottom represent strong and weak topo II sites, and arrowheads indicate DNase I HS sites. The numbers represent the distance (bp) of the breakpoints to exon 6. Three AML1 lambda working clones (WC) were integrated in the map. The genomic breakpoints in intron 7a of AML1 in t(5;21) and t(12;21) (8) as well as in three t-AML with t(3;21) are indicated by arrows. K: Kasumi 1 cell line.
Figure 2.

In vivo topo II cleavage, DNase I HS sites and genomic breakpoints in ETO in t(8;21). The vertical boxes and lines represent exons, genomic DNA probes, and BamHI (B), BglII (G) and SacI (S) restriction sites. Arrows and arrowheads at the bottom represent topo II cleavage and DNase I HS sites. The numbers represent the distance (bp) of the breakpoints to exon 1a. K: Kasumi 1 cell line.
Some specific chromatin structural elements, i.e., topoisomerase II (topo II) DNA cleavage sites, DNase I hypersensitive (HS) sites, and scaffold-associated regions (SARs), at or near the breakpoints of genes have been implicated in chromosome recombination (15–21). Topo II binds preferentially to the scaffold and is essential for chromosome condensation, transcription, and replication (21–24). Many DNase I HS sites are associated with transcriptional regulatory DNA elements at gene boundaries or within genes; some of these sites colocalize with SARs and/or topo II sites (18–30). AT-rich DNA SARs define the attachment sites of chromatin loops in the DNA scaffold-loop model of chromosomes and are presumed to facilitate the entry of transcription, replication, or chromosome condensation protein factors to target sequences (17–22, 26–35).
In previous studies by us and others, these structural elements have been localized in MLL and AF9 (17–20, 36–39). Like AML1, MLL at 11q23 is also often involved in de novo acute leukemia and t-AML/myelodysplastic syndrome. Little is known about the location of these structural elements in AML1 and ETO, or the precise genomic breakpoints in either gene (13, 14, 40); thus the correlation of genomic breakpoints in AML1 and ETO with these structural elements is largely unknown. Our mapping studies of a large series of leukemia patients with de novo t(8;21) reveal a strong correlation of genomic breakpoints in AML1 and ETO with in vivo topo II DNA cleavage and DNase I HS sites.
Materials and Methods
Cell Lines.
We used the BV173 cell line with t(9;22)(q34;q11), the Mono Mac 6 monocytic cell line with t(9;11)(p22;q23), the ML-2 myeloid cell line with t(6;11)(q27;q23), the UoC-M1 myeloid cell line with a complex karyotype, the Kasumi 1 cell line with t(8;21)(q22;q22), and the CD34+ KG-1A cell line (18, 41).
Patients.
Thirty-one leukemia patients, including one t-AML patient, were studied with informed consent as described in an Institutional Review Board-approved protocol. Except for three patients aged 3 and 12 years, all patients were between 20 and 79 years old (Table 1). Cytogenetic analysis identified t(8;21)(q22;q22) as the sole chromosome aberration in seven patients, with loss of an X or Y chromosome in 12 patients, and with other or complex chromosome aberrations in 12 patients.
Table 1.
Leukemia patients with diagnosis, cytogenetic, and sequence results
| No. | Age/sex | Diagnosis | Materials | Karyotype
and percentage of clones with t(8;21) by cytogenetic
analysis
|
Duplications (+)/deletions (−)
|
||
|---|---|---|---|---|---|---|---|
| Karyotype | % | AML1 (bp) | ETO (bp) | ||||
| 1* | 20/M | AML-M2 | BM, DX | 46,XY,t(4;5)(p14;p15),t(8;21)(q22;q22)[18]/46,idem,−9,+mar[2] | 100 | No change | No change |
| 2 | 36/M | AML | BM, RL | 46,XY,der(5)t(1;5)(5;?)(q25;p11q31;?), t(8;21)(q22;q22)[19]/44,idem,−4,−11,del(1)(p32p36)[1] | 100 | +104 | −6 |
| 3 | 56/M | AML-M2 | BM,DX | 46,XY,t(8;21)(q22;q22)[13]/45,idem,−Y[6]/46,XY[4] | 83 | −44 | −60 |
| 4* | 25/F | AML-M2/M4 | BM, DX | 45,X,−X,t(8;21)(q22;q22)[8]/46,idem,+15[11]/46,XX[1] | 95 | +87 | +157 |
| 5* | 27/F | AML-M2 | BM, RL | 45,X,−X,t(8;21)(q22;q22)[20]/46,XX[2] | 91 | −35 | +122 |
| 6 | 60/M | AML-M2 | BM, RL | 45,X,−Y,t(8;21)(q22;q22),del(9)(q11or12q31)[28]/46,XY[1] | 97 | No change | −45 |
| 7 | 3/F | AML-M2 | BM, DX | 46,XX,t(8;21)(q22;q22)[27]/46,XX[4] | 87 | −46 | −225 |
| 8 | 47/M | AML-M2 | BM, DX | 46,XY,t(6;21;8)(p23;q22;q22),inv(7)(q31q32orq22q34)x2, inv(9)(p11q13)c[20] | 100 | Unknown | Unknown |
| 9* | 30/F | AML-M2 | BM, DX | 46,XX,t(8;21)(q22;q22),del(9)(q22q32)[15]/45,idem,−X[7] | 100 | −305 | +143 |
| 10 | 61/F | AML-M2 | BM, RL | 46,XX,t(8;21)(q22;q22)[12]/46,XX[10] | 54 | +2 | +302 |
| 11* | 21/M | AML-M2 | BM, DX | 45,X,−Y,t(8;21)(q22;q22)[21]/46,XY[3] | 88 | +133 | −16 |
| 12* | 35/F | AML-M2 | BM,DX | 46,XX,t(8;21)(q22;q22)[3]/47,idem,+4[17] | 100 | Unknown | Unknown |
| 13* | 20/M | AML-M2 | BM, DX | 46,XY,t(8;21)(q22;q22)[21]/46,idem,del(9)(q13q31)[2] | 100 | −13 | +161 |
| 14 | 43/F | AML-M2 | BM, DX | 46,XX,t(8;21)(q22;q22)[19]/46,XX[1] | 95 | Unknown | Unknown |
| 15 | 75/M | AML-M2 | PB, DX | 45,X,−Y,t(8;21)(q22;q22)[24]/46,XY[1] | 96 | +63 | +51 |
| 16* | 32/M | AML-M2 | PB, DX | 46,XY,t(8;21)(q22;q22)[21] (in BM) | 100 | Unknown | Unknown |
| 17* | 49/M | AML-M2 | PB, DX | 45,X,−Y,t(8;21)(q22;q22)[31] | 100 | −1316 | +190 |
| 18 | 27/F | AML-M2 | BM, DX | 46,XX,t(8;21)(q22;q22)[21] | 100 | +1 | −12 |
| 19 | 12/F | AML-M2 | BM, DX | 46,XX,t(8;21)(q22;q22)[19]/46,idem,del(9)(q22q31)[3] | 100 | −556 | −191 |
| 20* | 12/M | AML-M2 | BM, DX | 46,XY,t(8;21)(q22;q22),del(9)(q13q22)[29]/46,XY[1] | 97 | −98 | −201 |
| 21 | 56/F | AML-M2 | PB, RL | 46,X,−X,der(2)t(2;8)(q31;q11),t(8;21)(q22;q22)[5]/45, idem,t(8;15)(q13;q15)[4]45,idem,t(12;15)(q22;q13)[3]/ 45,idem,t(1;1)(p32;q36),t(12;15)[9]/90,XX,−X,−X,t(1;1)x2, +der(2)t(2;8)x2,t(8;21)x2,t(12;15)x2[2] (in BM) | 100 | −5 | −15 |
| 22 | 43/F | HD, t-AML | BM, DX | 46,XX,t(8;21)(q22;q22)[20]/46,idem,add(11)(q21or22)[1]/45,idem,−X,add(11)[2]/46,XX[3] | 89 | Unknown | Unknown |
| 23* | 25/F | AML-M2 | BM, RL | 46,XX,t(8;21)(q22;q22)[8]/46,XX[13] | 38 | Intron 5 | Intron 1b |
| 24 | 21/M | AML-M2 | BM, RL | 45,X,−Y,t(8;21)(q22;q22)[1]/45,idem,add(19)(p13.3)[1]/46,XX[18] | 10 | Intron 5 | Intron 1b |
| 25 | 28/M | AML-M2 | PB, DX | 45,X,−Y,t(8;21)(q22;q22)[16]/46,XY[9] (in BM) | 64 | Intron 5 | Intron 1b |
| 26* | 79/M | AML-M2 | BM, DX | 46,XY,del(13)(q12q14)[9]/46,idem,t(8;21)(q22;q22)[2]/ 46,XY,der(8)t(8;21)(q22;q22),del(13),der(21)t(8;21) (q22;q22)t(15;21)(q15;q24)[18]/46,XY[2] | 65 | Intron 5 | Intron 1b |
| 27* | 22/M | CML accelerated phase | BM,DX | 46,XY,t(9;22)(q34;q11.2)[1]/46,idem,t(8;21)(q22;q22)[18]/47, idem,del(2)(q11.2;q13.3),+8,t(8;21)[5]/48,idem, t(8;21),+der(8)t(8;8)(p11;q21)x2[4]/46,XY[1] | 94 | Intron 6 | Not detected |
| 28 | 66/M | AML-M2 | BM, DX | 47,XY,+8,t(8;21)(q22;q22)[14]/48,idem,+12[12]/46,XY[3] | 90 | Intron 5 | Not detected |
| 29* | 57/F | AML-M2 | BM, DX | 45,X,−X,t(8;21)(q22;q22)[30] | 100 | Intron 5 | Not detected |
| 30 | 58/M | AML-M2 | BM | 46,XY,t(8;21)(q22;q22)[15]/46,XY[1] (in a sample 1 month earlier) | 94 | Not detected | Intron 1a |
| 31* | 44/M | AML-M2 | BM, RL | Complex karyotypes with t(8;21)(q22;q22)† | 57 | Not detected | Not detected |
BM: bone marrow. PB: peripheral blood. DX: diagnosis. RL: relapse. HD: Hodgkin's disease. M: male. F: female. In patients 16, 21, and 25, a t(8;21) was detected by cytogenetic analysis in bone marrow, but only peripheral blood obtained at the same time was available for the study. In patient 30, cytogenetic analysis detected a t(8;21) in a bone marrow sample 1 month earlier than the sample used in this study, which included no cytogenetic information. In patients 8, 12, 16, and 22, AML1-ETO fusion junctions only were cloned, whereas in patient 14 ETO-AML1 fusion only was sequenced. In patient 23–26, genomic fusions between AML1 and ETO have not yet been sequenced.
Patients included in our previous study (46).
The karyotype of patient 31 is 92,XXYY,+Y,del(1)(p3?3p3?5),+2,t(2;2)(q35;q37),der(6)t(6;15)(p23;q21.2 or p21;q15),t(7;16)(p1?2;p13),t(8;21)(q22;q22)x2,−11, der(12)t(12;15)(p13;q1?3),−15,−15,add(16)(p13),−17,del(17)(q2?1q2?3),+18,der(19)t(11;19)(q13;q13),t(21;22)(q11;q13)x2,+mar1[9]/94,idem,+mar2,+mar3[4].
DNA Primers and Probes.
Genomic sequences of AF000125 and AF181450 obtained from the GenBank database were aligned with published cDNA sequences for determination of exon and intron boundaries of AML1 and ETO, respectively. Eighty-two and 98 DNA primers of 19–33 bp specific for introns 4–7a of AML1 and for introns 1b and 1a of ETO, respectively, were purchased from Integrated DNA Technologies, Coralville, IA. Twenty-three and 14 genomic DNA probes of AML1 and ETO were produced with these primers.
In Vivo Topo II Cleavage of DNA.
An in vivo topo II DNA cleavage assay was carried out according to Strissel et al. (18, 20). Cells were treated for 6 h with 5 μM–50 μM etoposide (VP16) (Sigma) or 0.2 μM–1.0 μM doxorubicin (Dox, Sigma). Cell viability measured by trypan blue staining was more than 85%. For cell lines that showed topo II DNA cleavage resistance, the drug concentrations tested were 25 μM–200 μM for VP16 and 1 μM–5 μM Dox for 16 h. For these cell lines, cell viability did not differ from that of controls.
In Vivo Topo II Reversibility Analysis.
Topo II-induced DNA cleavage was reversed by two different methods (17, 42, 43). Briefly, the BV173 cells were incubated with 25 μM and 50 μM VP16 for 6 h and 16 h, followed by heat treatment (42) or drug removal (43).
DNase I Treatment of Nuclei.
For each hematopoetic cell line, nuclei were isolated and stored according to Mirkovitch et al. (28) and our previous studies (18, 20, 44). Nuclei were treated with 2–10 units of DNase I (Sigma) according to Kas and Laemmli (45) and Strissel et al. (19).
Southern Blot Analysis.
Genomic DNA isolation and restriction digestion was performed as described (17–20). Genomic DNA probes were randomly primed with 32P-dCTP. DNA rearrangements in AML1 and ETO were identified with at least two different DNA restriction digests and different genomic probes.
Long-Distance PCR, Direct Sequencing.
Long-distance PCR was performed by using the Taqplus system (Stratagene). PCR products were directly sequenced on an Applied Biosystems Prism 377 sequencer. The blast search, macvector (Oxford Molecular Group, Campbell, CA), and sequencher (Gene Codes, Ann Arbor, MI) software were used for identifying genomic junctions between AML1 and ETO and for searching for specific recombination-related DNA sequence motifs, including V(D)J recombination sequences, topo II consensus sites, translin binding sequences, x-like sequences, and purine/pyrimidine repeat regions.
Results
Cloning Genomic DNA Breakpoints in AML1 and ETO in Leukemia Patients with t(8;21).
DNA rearrangements in AML1 were detected in the Kasumi l cell line and in 29 of the 31 patients. These leukemia patients included 24 adults and three children with de novo AML, one adult with t-AML, and one adult with chronic myelogenous leukemia (CML) in an accelerated phase. Genomic breakpoints in AML1 were identified in intron 5 in 28 patients and intron 6 in the CML patient (Fig. 1). Two DNA rearrangements were detected with probes A4 and A6 on BamHI blots in the Kasumi 1 cell line and patient 5, indicating that the breakpoints in these patients must fall within the region corresponding to the probes. In six patients, including patient 22 with t-AML, genomic breakpoints were located in a 0.8-kb breakpoint cluster region (BCR) (named BCR-1) at the 5′ end of the 18.7-kb BamHI fragment. In eight other patients, breakpoints occurred in a 4.2-kb BCR (BCR-2) in the middle of intron 5. In seven patients, the breakpoints were clustered in a 2.1-kb BCR (BCR-3) 0.6 kb 5′ of exon 6.
In ETO, DNA rearrangements were detected in the Kasumi 1 cell line and 27 leukemia patients (Fig. 2). Two DNA rearrangements resulting from splitting of the probes were detected on BamHI, HindIII, or SacI blots with probe E2 in patients 6 and 17, probe E4 in patients 5 and 19, probe E5 in patient 4, probe E8 in patients 9 and 15, and probe E9 in patient 7. The genomic breakpoints in five patients, including patient 22 with t-AML were clustered in a 1.9-kb BCR (BCR-I) ≈3 kb 3′ of exon 1b. Genomic breakpoints in nine other patients were clustered in a 2.5-kb BCR (BCR-II) in the middle of intron 1b. The third BCR (BCR-III) involving 10 patients was located within a 1.6-kb region 1.6 kb 5′ of exon 1a. Thus, genomic DNA breakpoints were detected in both AML1 and ETO in the Kasumi 1 cell line and 26 leukemia patients. In patients 27 (CML), 28, 29, and 30, DNA rearrangements were detected in AML1 or ETO only. In patient 31, no DNA rearrangement was detected in AML1 and ETO with multiple probes on different DNA digests.
Using long-distance PCR and direct sequencing we cloned genomic junctions between AML1 and ETO in 23 patients. In the Kasumi 1 cell line and 17 patients, both AML1-ETO and the reciprocal fusion were sequenced, whereas AML1-ETO or ETO-AML1 only was cloned in five patients (Table 2, which is published as supporting information on the PNAS web site, www.pnas.org). Among these 18 patients, deletions ranging from 5 to 18,098 bp were identified in AML1 in 10 patients and in ETO in 10 patients, whereas duplications of 1 to 302 bp were detected in AML1 in six patients and in ETO in seven patients, respectively (Table 2 and Fig. 5, which is published as supporting information on the PNAS web site). Of 41 DNA fusion junctions involving AML1 and ETO in 23 patients, nine had microhomologies of 1–5 bp and 23 contained nontemplate DNA sequences of 1–18 bp (Table 2). In patient 1, the genomic breakpoint in AML1 was located within a thymidine repeat region. Genomic breakpoints in ETO in patient 14 and in AML1 in patient 16 occurred in an Alu sequence. In these three patients, the breakpoints in the partner gene were not located in another repeat region. Using the macvector and sequencher programs with a minimal match of 70%, we did not identify any specific recombination sequence motifs near the breakpoint junctions. A 14/18 match to in vitro topo II consensus sequences was identified at 666 bp 5′ of exon 6, which is located within the BCR-3.
Localizing in Vivo Topo II DNA Cleavage Sites and DNase I HS Sites in AML1 and ETO.
Because DNA breakpoints in AML1 in chromosome translocations are clustered in introns 5–7b and in ETO in introns 1b–2, we first mapped topo II and DNase I HS sites in these regions, which comprise about 88 kb of AML1 and 66 kb of ETO, using 23 and 14 genomic DNA probes for AML1 and ETO, respectively (Figs. 1 and 2).
Using probe A7, we detected a 4.8-kb HindIII topo II cleavage fragment in intron 5 of AML1 (Fig. 3A). This site (site A) at 10 kb 5′ of exon 6 is located within BCR-2 and was confirmed with probe A6 as a 7.2-kb BamHI DNA fragment (Fig. 1). Using probe A10 or A11, an extra 4.8-kb BamHI DNA fragment was detected. We assume that this topo II site (site B) is located between BCR-2 and BCR-3 at 5 kb 5′ of exon 6 near the breakpoints in patients 17 and 18 (Fig. 1). Using probe A12, A13, or A14, another topo II cleavage site (site C) was detected about 0.7 kb 5′ of exon 6, which colocalizes with BCR-3 (Figs. 1 and 3B). Topo II site C was confirmed with probe A12 or A13 as a 2.1-kb HindIII DNA fragment. Topo II site C was stronger than sites A and B in Southern blot analysis; topo II sites A and C detected in the BV173 cell line were confirmed in the Mono Mac 6 and UoC-M1 cell lines.
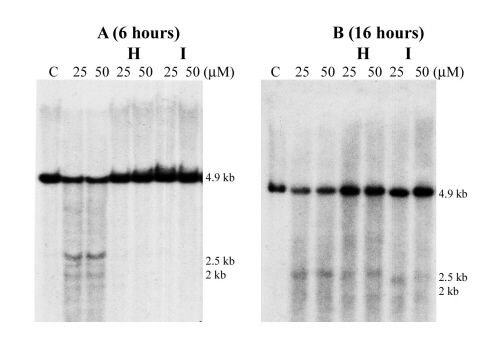
Figure 3.

Topo II DNA cleavage sites in AML1 (A–E) and ETO (F and G). (A) HindIII digestion of the BV173 DNA and hybridization with probe A7. A 4.8-kb DNA fragment was detected (topo II site A). (B) BamHI digestion and hybridization with probe A14. A 2.2-kb DNA fragment was detected (topo II site C). (C) BglII digestion and hybridization with probe A20. Four DNA fragments of 1.2 kb, 1.4 kb, 1.5 kb, and 1.8 kb were detected (topo II sites D, E, F, and G clustering in a 0.6-kb region). (D) BamHI digestion of the ML-2 DNA and hybridization with A21. A 3.2-kb DNA fragment was detected (topo II site H). (E) BamHI digestion and hybridization with probe A23. Two 2.5-kb and 2-kb DNA fragments were detected (topo II sites I and J). (F) HindIII digestion and hybridization with probe E1. A 0.9-kb DNA fragment was detected (topo II site a). (G) BglII digestion and hybridization with probe E4. Two 0.6-kb and 2.1-kb DNA fragments were found, indicating topo II site b and splitting of probe E4. Note topo II sites A, G, H, and J are weaker than other sites. C: control of the BV173 or ML-2 cell line without drug treatment.
Analysis of 21.8 kb of intron 7a of AML1 with probe A20 revealed four topo II sites (sites D-G) that clustered in a 0.6-kb region 8.6 kb 5′ of exon 7b (Fig. 3C). However, topo II site G was weaker than the others. Using probe A21, a topo II site (site H) was identified about 5.4 kb 5′ of exon 7b (Fig. 3D). Using probe A23, topo II sites I and J were detected as 2.5-kb and 2-kb BamHI DNA fragments (Figs. 1 and 3E). However, site I is stronger than sites H and J. Using probe A22, topo II site I was confirmed as a 2.4-kb BamHI DNA fragment. No topo II cleavage sites were detected in intron 4 (21 kb), intron 6 (12.7 kb), or intron 7b (6.7 kb). Treatment of the Kasumi 1 and KG-1A cell lines with VP16 and Dox failed to detect any topo II cleavage in AML1. Similarly, we did not detect any topo II sites in MLL and AF9 in the KG-1A cell line (18, 20), indicating they are resistant to VP16- or Dox-induced topo II DNA cleavage in these genes.
In ETO, extra 0.8-kb BglII and 0.9-kb HindIII DNA fragments were detected with probe E1 (Figs. 2 and 3F), indicating a topo II site (site a) about 3 kb 3′ of exon 1b. Topo II site a colocalizing with BCR-I was confirmed by probe E2 as 1.9-kb BglII and 3.3-kb HindIII DNA fragments. Another topo II site (site b) located within BCR-II was revealed with probes E5 and E6 as a 1.9-kb HindIII and a 4.3-kb BamHI DNA fragment, respectively. Moreover, topo II site b is covered by probe E4, because hybridization with probe E4 detected two 0.6-kb and 2.1-kb BglII fragments and two 0.7-kb and 2.2-kb HindIII DNA fragments (Fig. 3G). As in AML1, these topo II sites were also detected in the Mono Mac 6 and UoC-M1 cell lines. No topo II cleavage sites were detected in the 3′ portion of intron 1b, intron 1a (45.2 kb), or intron 2 (2.4 kb) of ETO.
In in vivo topo II DNA cleavage reversibility experiments, topo II sites A-C and H-J in AML1 and topo II sites a and b in ETO were examined. After initial incubation with 25 μM and 50 μM VP16 for 6 h, followed by either heat treatment or drug removal, no DNA breakage was detected (Fig. 6A, which is published as supporting information on the PNAS web site); thus, all of these strong and weak topo II sites in both genes were completely reversible. However, these topo II sites were not reversible after treatment with VP16 for 16 h (Fig. 6B). These results indicate that, during the drug treatment of the first 6 h, topo II is responsible for DNA cleavage and that DNA religation can occur by means of topo II after heat treatment or drug removal. However, after 16 h treatment, DNA cleavage may then be occurring because of apoptotic DNases.
We also examined the entire 88-kb region in AML1 and the 66-kb region of ETO for DNase I HS sites. Using probes A12 and A15, a DNase I HS site was detected at 0.7 kb 5′ of exon 6 of AML1, which colocalizes with topo II site C (Fig. 4A). Hybridization with probe 20 revealed three DNase I HS sites colocalizing with topo II sites D, E, and F (Fig. 4B). A DNase I HS site located at the same region of topo II site I was detected with probe 22 as a 2.7-kb BamHI fragment (Fig. 4C). In ETO, two DNase I HS sites that colocalize with topo II sites a and b, respectively, were detected with probe E1 as 1.9-kb and 5.7-kb BamHI DNA fragments (Fig. 4D). These two DNase I HS sites were confirmed by probes E2 and E6 as a 3.5-kb EcoRI and a 4.2-kb BamHI fragment, respectively (Fig. 4 E and F).
Figure 4.

DNase I HS sites in AML1 (A–C) and ETO (D–F). (A) EcoRI digestion and hybridization with A12. A 3.9-kb DNA fragment was detected, indicating a DNase I HS site colocalizing with topo II site C in intron 5. (B) EcoRI digestion and hybridization with probe A20. Three DNA fragments of 1.5, 1.6, and 1.8 kb were detected, indicating three DNase I HS sites colocalizing with topo II sites D, E, and F. (C) BamHI digestion and hybridization with A22. A 2.7-kb DNA fragment was identified, indicating one DNase I HS site colocalizing with topo II site I. (D) BamHI digestion and hybridization with probe E1. Two DNA fragments of 1.9 kb and 5.7 kb were detected, thus two DNase I HS sites colocalize with topo II sites a and b, respectively. (E) EcoRI digestion and hybridization with probe E2. A 3.5-kb DNA fragment was identified, confirming the DNase I HS site that colocalizes with topo II site a. (F) BamHI digestion and hybridization with E6. A 4.2-kb DNA fragment was detected, confirming the DNase I HS site colocalizing with topo II site b.
Discussion
Clustering of Genomic DNA Breakpoints in AML1 and ETO in the Kasumi 1 Cell Line and Leukemia Patients.
Using Southern blot, long-distance PCR and DNA sequencing, we showed that genomic DNA breakpoints in AML1 in t(8;21) leukemia clustered in three BCRs in intron 5. In our previous study DNA rearrangements in AML1 were detected in 12 of 15 patients with t(8;21) (46). With multiple digestions and many genomic probes, we defined these breakpoints more accurately in all 12 patients. Moreover, we detected genomic breakpoints in AML1 in patients 17 and 27, in whom no rearrangement was found in our previous study. In a study of 21 patients with t(8;21), Shimizu et al. (13) detected genomic breakpoints in AML1 in eight and 10 patients in the middle and the 3′ part of intron 5, respectively, which overlap with BCR-1 and BCR-3. Recently, in 16 childhood leukemia patients and two leukemia cell lines with t(8;21), Xiao et al. (47) mapped genomic breakpoints in AML1 in nine patients in the 3′ part of intron 5 that covers BCR-3.
The clustering of genomic breakpoints in ETO in t(8;21) is even stronger. The breakpoints in ETO in 24 patients clustered in three BCRs in intron 1b. Tighe and Calabi (14) identified two BCRs in intron 1b and one BCR in intron 1a in 18 patients. These two BCRs in intron 1b may overlap BCR-I and BCR-II, respectively. In the study by Xiao et al. (47), a large BCR was defined spanning the middle and the 3′ portion of intron 1b, thus covering our BCR-II and BCR-III.
Cloning of genomic fusion junctions between AML1 and ETO in the Kasumi 1 cell line and 22 leukemia patients revealed frequent deletions and duplications in AML1 and ETO as well as microhomology and nontemplate DNA at the breakpoint junctions. There were no site-specific or homologous recombination motifs near the junctions. Similar findings were noted in childhood AML with t(8;21) (47); microhomologies were detected in 14 patients and nontemplate DNA in seven patients. In addition, genomic breakpoints in MLL, AF9, and AF4 have been cloned in many patients, and similar changes also were observed at breakpoint junctions (1, 38, 48–50). Thus, our results suggest that nonhomologous DNA recombination and repair occurred between AML1 and ETO after DNA double-strand breakage.
Correlation of Topo II Cleavage and DNase I HS Sites with Genomic DNA Breakpoints in AML1 and ETO.
In this study, we mapped 10 and two in vivo topo II DNA cleavage sites in introns 5 and 7a of AML1 and in intron 1b of ETO, respectively. Based on the genomic sequences of AML1 and the putative in vitro topo II consensus sequence, 22 in vitro topo II sites were predicted in the entire AML1 (12). The in vivo topo II site B in intron 5 and topo II site H or I in intron 7a identified in our study may colocalize with some of these predicted in vitro topo II sites. These results are similar to our finding in MLL; seven topo II cleavage sites were predicted, but only one was confirmed by in vivo analysis (17–19). Therefore, topo II appears to recognize DNA structure rather than a DNA-specific sequence (51). Topo II site C is most likely the same site identified earlier by Stanulla et al. (40) in a T-acute lymphoblastic leukemia patient and in several lymphoid and myeloid cell lines. Notably, these topo II DNA cleavage sites in AML1 are correlated with genomic BCRs in leukemia patients. Topo II sites A and C are located within BCR-2 and BCR-3, respectively, whereas topo II site B is within a 0.2-kb region surrounded by the breakpoints of patients 17 and 18 (Fig. 1). In a patient with CML in blast crisis with t(9;22) and t(3;21), the genomic breakpoint in AML1 was mapped to intron 5, 5 kb 5′ of exon 6, at the same region of topo II site B (52).
Seven topo II DNA cleavage sites are clustered in the 3′ portion of intron 7a of AML1. In our ongoing study of t(3;21) patients, genomic breakpoints in AML1 in three t-AML patients mapped in the regions of topo II sites D-G and I and J (Fig. 1). In our previous fluorescence in situ hybridization study, genomic breakpoints in AML1 in t(5;21) and t(12;21) were located in the same regions (Fig. 1) (8). Thus, genomic breakpoints in AML1 in t(3;21) and these less common translocations may also be clustered in and thus correlated with topo II cleavage sites.
A stronger correlation of topo II DNA cleavage sites with genomic breakpoints was observed in intron 1b of ETO (Fig. 2). Topo II site a is located at the same region as the 1.9-kb BCR-I, whereas topo II site b is within the 2.5-kb BCR-II.
We have also identified five and two DNase I HS sites in AML1 and ETO, respectively, all colocalizing with all strong in vivo topo II sites in both genes. Similarly, in MLL, AF9, and BCR involved in t(9;22) in CML, all DNase I HS sites colocalized only with strong in vivo topo II sites (refs. 18–20 and 36–38; unpublished data). These results indicate that topo II preferentially cleaves at specific open chromatin sites that correlate with stronger topo II DNA cleavage sites.
Based on our results and those of others, and by analogy to MLL and AF9, we propose that our nonhomologous chromosome translocation model, namely, a common mechanism for both de novo and t-AML leukemia, is also applicable to the t(8;21) (17,20). No common DNA motifs have been found thus far at DNA breakpoint junctions in MLL, AF9, and now in AML1 and ETO; moreover, many DNA breakpoints do not colocalize exactly with, but map close to or at a variable distance from the in vivo topo II cleavage sites. Stanulla et al. (37) identified that the in vivo topo II DNA cleavage sites in MLL and AML1 are also targets for apoptotic nucleases. These findings are important for our understanding of a two-step pathway involving both topo II and the apoptotic nucleases (15, 17). We showed that as with MLL and AF9 (17, 20), topo II sites A-C and H-J of AML1 and a and b of ETO were reversible at 6 h but not at 16 h of VP16 treatment (Fig. 6). Thus, for four independent gene loci tested in reversibility experiments, topo II–DNA cleavage complexes formed by drug/topo II/DNA can be repaired and religated. Therefore, DNase I, presumably cellular apoptotic DNases and topo II recognize and cleave at the same DNA sites; however, the timing of topo II- and apoptotic DNase-induced DNA cleavages by VP16 and Dox may be different and only partly overlap. Thus, the first target of VP16 and Dox and other topo II-inhibiting drugs is topo II bound to specific DNA sites, which is reversible; after some time, apoptotic DNases cleave DNA, which is no longer reversible.
We propose that topo II normally functions at these in vivo sites to monitor the helicity of DNA during replication, transcription, repair, and condensation. DNA damage and repair at these sites, which could lead to chromosome translocations, is a rare occurrence, but perhaps could be caused by the natural topo II inhibitors such as some bioflavonoids, which we recently proposed as one mechanism for de novo infant leukemia involving MLL (17). Indeed, we found that the bioflavonoid quercetin induces the same level of in vivo topo II DNA cleavage in both AML1 and ETO as the chemotherapy drugs (data not shown). In addition, other DNA-damaging agents such as pesticides and organic chemicals could act at these topo II sites by either inhibiting topo II or cleaving open chromatin (53). Therefore, DNA cleavage is induced at the topo II cleavage sites, followed by complex DNA repair mechanisms including 3′ to 5′ exonucleolytic processing, which may lead to illegitimate recombination between AML1 and ETO in t(8;21). Recently, Eguchi-Ishimae et al. (54) reported induction of topo II cleavage sites in AML1 and TEL in immature B cell lines after treatment with VP16 and subsequent detection of TEL-AML1 fusion. Analysis of the higher-order chromatin structures at BCRs in these genes is necessary to clarify their role in chromosome translocations.
Supplementary Material
Acknowledgments
We thank Nancy Zeleznik-Le and Dong-Er Zhang for their helpful comments. This study is supported in part by National Institutes of Health/National Cancer Institute Grants CA40046 and CA42557 (to J.D.R.), CA84405 (to J.D.R., M.M.L., and R.A.L.), and CA67189 and CA72675 (to G.N.), a grant from the G. Harold and Leila Y. Mathers Charitable Foundation (to J.D.R.), and a grant from the Spastic Paralysis Research Foundation of the Illinois-Eastern Iowa Chapter of Kiwanis International (J.D.R.). G.N. is a Scholar of The Leukemia and Lymphoma Society.
Abbreviations
- AML
acute myeloid leukemia
- t-AML
therapy-related AML
- topo II
topoisomerase II
- HS
hypersensitive
- DOX
doxorubicin
- CML
chronic myelogenous leukemia
- BCR
breakpoint cluster region
References
- 1.Rowley J D. Semin Hematol. 1999;36:59–72. [PubMed] [Google Scholar]
- 2.Nucifora G, Rowley J D. Blood. 1995;86:1–14. [PubMed] [Google Scholar]
- 3.Miyoshi H, Shimizu K, Kozu T, Maseki N, Kaneko Y, Ohki M. Proc Natl Acad Sci USA. 1991;88:10431–10434. doi: 10.1073/pnas.88.23.10431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Erickson P, Gao J, Chang K S, Look T, Whisenant E, Raimondi S, Lasher R, Trujillo J, Rowley J D, Drabkin H. Blood. 1992;80:1825–1831. [PubMed] [Google Scholar]
- 5.Nucifora G, Birn D J, Espinosa R, III, Erickson P, Le Beau M M, Roulston D, McKeithan T W, Drabkin H, Rowley J D. Blood. 1993;81:2728–2734. [PubMed] [Google Scholar]
- 6.Gamou T, Kitamura E, Hosoda F, Shimizu K, Shinohara K, Hayashi Y, Nagase T, Yokoyama Y, Ohki M. Blood. 1998;91:4028–4037. [PubMed] [Google Scholar]
- 7.Golub T R, Barker G F, Bohlander S K, Hiebert S W, Ward D C, Bray-Ward P, Morgan E, Raimondi S C, Rowley J D, Gilliland D G. Proc Natl Acad Sci USA. 1995;92:4917–4921. doi: 10.1073/pnas.92.11.4917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roulston D, Espinosa R, III, Nucifora G, Larson R A, Le Beau M M, Rowley J D. Blood. 1998;92:2879–2885. [PubMed] [Google Scholar]
- 9.Okuda T, van Deursen J, Hiebert S W, Grosveld G, Downing J R. Cell. 1996;84:321–330. doi: 10.1016/s0092-8674(00)80986-1. [DOI] [PubMed] [Google Scholar]
- 10.Yergeau D A, Hetherington C J, Wang Q, Zhang P, Sharpe A H, Binder M, Marin-Padilla M, Tenen D G, Speck N A, Zhang D E. Nat Genet. 1997;15:303–306. doi: 10.1038/ng0397-303. [DOI] [PubMed] [Google Scholar]
- 11.Miyoshi H, Ohira M, Shimizu K, Mitani K, Hirai H, Imai T, Yokoyama K, Soeda E, Ohki M. Nucleic Acid Res. 1995;23:2762–2769. doi: 10.1093/nar/23.14.2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Levanon D, Glusman G, Bangsow T, Ben-Asher E, Male D A, Avidan N, Bangsow C, Hattori M, Taylor T D, Taudien S. Gene. 2001;262:23–33. doi: 10.1016/s0378-1119(00)00532-1. [DOI] [PubMed] [Google Scholar]
- 13.Shimizu K, Miyoshi H, Kozu T, Ohki M. Cancer Res. 1992;52:6945–6948. [PubMed] [Google Scholar]
- 14.Tighe J E, Calabi F. Clin Sci. 1995;89:215–218. doi: 10.1042/cs0890215. [DOI] [PubMed] [Google Scholar]
- 15.Liu L F. Annu Rev Biochem. 1989;58:351–375. doi: 10.1146/annurev.bi.58.070189.002031. [DOI] [PubMed] [Google Scholar]
- 16.Sperry A O, Blasquez V C, Garrard W T. Proc Natl Acad Sci USA. 1989;86:5497–5501. doi: 10.1073/pnas.86.14.5497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Strick R, Strissel P L, Borgers S, Smith S L, Rowley J D. Proc Natl Acad Sci USA. 2000;97:4790–4795. doi: 10.1073/pnas.070061297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Strissel P L, Gill-Super H, Thirman M J, Pomykala H, Yonebayashi Y, Tanabe S, Zeleznik-Le N, Rowley J D. Blood. 1996;87:1912–1922. [PubMed] [Google Scholar]
- 19.Strissel P L, Strick R, Rowley J D, Zeleznik-Le N. Blood. 1998;92:3793–3803. [PubMed] [Google Scholar]
- 20.Strissel P L, Strick R, Tomek R J, Roe B A, Rowley J D, Zeleznik-Le N. Hum Mol Genet. 2000;9:1671–1679. doi: 10.1093/hmg/9.11.1671. [DOI] [PubMed] [Google Scholar]
- 21.Laemmli U K, Kas E, Poljak L, Adachi Y. Curr Opin Genet Dev. 1992;2:275–285. doi: 10.1016/s0959-437x(05)80285-0. [DOI] [PubMed] [Google Scholar]
- 22.Strick R, Laemmli U K. Cell. 1995;83:1137–1148. doi: 10.1016/0092-8674(95)90140-x. [DOI] [PubMed] [Google Scholar]
- 23.Adachi Y, Luke M, Laemmli U K. Cell. 1991;64:137–148. doi: 10.1016/0092-8674(91)90215-k. [DOI] [PubMed] [Google Scholar]
- 24.Watt P M, Hickson I D. Biochem J. 1994;303:681–695. doi: 10.1042/bj3030681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bouilikas T. Int Rev Cytol. 1995;162:279–388. doi: 10.1016/s0074-7696(08)61234-6. [DOI] [PubMed] [Google Scholar]
- 26.Phi-Van L, Stratling W H. EMBO J. 1988;3:655–664. doi: 10.1002/j.1460-2075.1988.tb02860.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Razin S V, Shen K, Ioudinkova E, Scherrer K. J Cell Biochem. 1999;74:38–49. [PubMed] [Google Scholar]
- 28.Mirkovitch J, Mirault M, Laemmli U K. Cell. 1984;39:223–232. doi: 10.1016/0092-8674(84)90208-3. [DOI] [PubMed] [Google Scholar]
- 29.Bode J, Maass K. Biochemistry. 1988;27:4706–4711. doi: 10.1021/bi00413a019. [DOI] [PubMed] [Google Scholar]
- 30.Cockerill P N, Garrard W T. Cell. 1986;44:273–282. doi: 10.1016/0092-8674(86)90761-0. [DOI] [PubMed] [Google Scholar]
- 31.Strissel P L, Dann H A, Pomykala H M, Diaz M O, Rowley J D, Olopade O I. Genomics. 1998;47:217–229. doi: 10.1006/geno.1997.5103. [DOI] [PubMed] [Google Scholar]
- 32.de Lange T. EMBO J. 1992;11:717–724. doi: 10.1002/j.1460-2075.1992.tb05104.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Betz A G, Milstein C, Gonzalez-Fernandez A, Pannell R, Larson T, Neuberger M S. Cell. 1994;77:239–248. doi: 10.1016/0092-8674(94)90316-6. [DOI] [PubMed] [Google Scholar]
- 34.Forrester W C, van Genderen C, Jenuwein T, Grosschedl R. Science. 1994;265:1221–1225. doi: 10.1126/science.8066460. [DOI] [PubMed] [Google Scholar]
- 35.Jenuwein T, Forrester W C, Fernandez-Herrero L A, Laible G, Dull M, Grosschedl R. Nature (London) 1997;385:269–272. doi: 10.1038/385269a0. [DOI] [PubMed] [Google Scholar]
- 36.Aplan P D, Chervinsky D S, Stanulla M, Burhans W C. Blood. 1996;87:2649–2658. [PubMed] [Google Scholar]
- 37.Stanulla M, Wang J, Chervinsky D S, Thandla S, Aplan P D. Mol Cell Biol. 1997;17:4070–4079. doi: 10.1128/mcb.17.7.4070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Domer P H, Head D R, Renganathan N, Raimondi S C, Yang E, Atlas M. Leukemia. 1995;9:1305–1312. [PubMed] [Google Scholar]
- 39.Zhou R H, Wang P, Zou Y, Jackson-Cook C K, Povirk L F. Cancer Res. 1997;57:4699–4702. [PubMed] [Google Scholar]
- 40.Stanulla M, Wang J, Chervinsky D S, Aplan P D. Leukemia. 1997;11:490–498. doi: 10.1038/sj.leu.2400632. [DOI] [PubMed] [Google Scholar]
- 41.Allen R J, Smith S D, Moldwin R L, Lu M M, Giordano L, Vignon C, Suto Y, Harden A, Tomek R, Veldman T, et al. Leukemia. 1998;12:1119–1127. doi: 10.1038/sj.leu.2401002. [DOI] [PubMed] [Google Scholar]
- 42.Hsiang Y H, Liu L F. J Biol Chem. 1989;264:9713–9715. [PubMed] [Google Scholar]
- 43.Borgnetto M E, Zunino F, Tinelli S, Kas E, Capranico G. Cancer Res. 1996;56:1855–1862. [PubMed] [Google Scholar]
- 44.Strissel P L, Espinosa R, III, Rowley J D, Swift H. Chromosoma. 1996;105:122–133. doi: 10.1007/BF02509522. [DOI] [PubMed] [Google Scholar]
- 45.Kas E, Laemmli U. EMBO J. 1992;11:705–716. doi: 10.1002/j.1460-2075.1992.tb05103.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nucifora G, Birn D J, Erickson P, Gao J, Le Beau M M, Drabkin H A, Rowley J D. Blood. 1993;81:883–888. [PubMed] [Google Scholar]
- 47.Xiao Z, Greaves M F, Buffler P, Smith M T, Segal M R, Dicks B M, Wiencke J K, Wiemels J L. Leukemia. 2001;15:1906–1913. doi: 10.1038/sj.leu.2402318. [DOI] [PubMed] [Google Scholar]
- 48.Atlas M, Head D, Behm F, Schmidt E, Zeleznik-Le N, Roe B A, Burian D, Domer P H. Leukemia. 1998;12:1895–1902. doi: 10.1038/sj.leu.2401223. [DOI] [PubMed] [Google Scholar]
- 49.Reichel M, Gillert E, Nilson I, Siegler G, Greil J, Fey G H, Marschalek R. Oncogene. 1998;17:3035–3044. doi: 10.1038/sj.onc.1202229. [DOI] [PubMed] [Google Scholar]
- 50.Super H G, Strissel P L, Sobulo O M, Burian D, Reshmi S C, Roe B, Zeleznik-Le N, Diaz M O, Rowley J D. Genes Chromosomes Cancer. 1997;20:185–195. [PubMed] [Google Scholar]
- 51.Udvardy A, Schedl P. Mol Cell Biol. 1991;11:4973–4984. doi: 10.1128/mcb.11.10.4973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hirai H, Ogawa S, Kurokawa M, Yazaki Y, Mitani K. Genes Chromosomes Cancer. 1999;26:92–96. [PubMed] [Google Scholar]
- 53.Alexander F E, Patheal S L, Biondi A, Brandalise S, Cabrera M E, Chan L C, Chen Z, Cimino G, Cordoba J C, Gu L J, et al. Cancer Res. 2001;61:2542–2546. [PubMed] [Google Scholar]
- 54.Eguchi-Ishimae M, Eguchi M, Ishii E, Miyazaki S, Ueda K, Kamada N, Mizutani S. Blood. 2001;97:737–743. doi: 10.1182/blood.v97.3.737. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}